

Abstract
Cells require tight regulation of the intracellular redox balance and consequently of reactive oxygen species for proper redox signaling and maintenance of metal (e.g., of iron and copper) homeostasis. In several diseases, including cancer, this balance is disturbed. Therefore, anticancer drugs targeting the redox systems, for example, glutathione and thioredoxin, have entered focus of interest. Anticancer metal complexes (platinum, gold, arsenic, ruthenium, rhodium, copper, vanadium, cobalt, manganese, gadolinium, and molybdenum) have been shown to strongly interact with or even disturb cellular redox homeostasis. In this context, especially the hypothesis of “activation by reduction” as well as the “hard and soft acids and bases” theory with respect to coordination of metal ions to cellular ligands represent important concepts to understand the molecular modes of action of anticancer metal drugs. The aim of this review is to highlight specific interactions of metal-based anticancer drugs with the cellular redox homeostasis and to explain this behavior by considering chemical properties of the respective anticancer metal complexes currently either in (pre)clinical development or in daily clinical routine in oncology.
I. Introduction
Since ancient times, metal compounds have been successfully used for the treatment of a variety of diseases. Already the ancient Egyptians knew about the therapeutic potential of gold salts (272). In traditional Chinese medicine, arsenic drugs, like arsenic trioxide (ATO), were used as antiseptic agents or in the treatment of rheumatoid diseases, syphilis, and psoriasis (93, 370). Indeed, ATO was one of the first compounds that was suggested for anticancer therapy, and during the 18th and 19th century ATO represented the main treatment for leukemia. The modern era of metal-based anticancer drugs began with the discovery of the platinum(II) complex cisplatin by Barnett Rosenberg in the 1960s (323). Nowadays, cisplatin and its successors carboplatin and oxaliplatin are among the most important chemotherapeutics used against a wide variety of different cancers (189, 323). Stimulated by the success of cisplatin, also other coordination compounds based on ruthenium, gold, titanium, copper, rhodium, vanadium, and cobalt were tested for their anticancer activity and several promising candidates are currently in (pre)clinical evaluation (79, 100, 106, 149, 188, 202, 203, 285, 343).
One of the characteristics of metals is their potential to undergo redox processes, as determined by their redox potentials. Especially, transition metal ions are usually able to switch between several oxidation states. However, not all oxidation states are observed under physiological conditions in the living organism. Due to the redox activity of metals and, therefore, a possible disturbance of the sensitive cellular redox homeostasis, a tight regulation of the metal and redox balance is crucial for health and survival (15, 17, 19, 127, 134, 158).
Cancer cells are known to differ distinctly in their redox metabolism from healthy tissues (134, 381). Thus, enhanced levels of intracellular reactive oxygen species (ROS) are often observed in tumor cells and the specific milieu of the solid tumor is characterized by high metabolic activity, hypoxia, and, in general, reductive conditions. Consequently, interference with the cellular redox homeostasis of cancer cells seems an attractive and promising approach for cancer therapy (a general overview on the role of ROS in the activity of metal anticancer drugs is summarized in Fig. 1). Indeed, many of the currently used chemotherapeutic drugs have been shown to exert some interaction with the cellular redox balance and there are several attempts to specifically target the altered redox conditions in cancer cells (9, 74, 77, 134, 138, 149). Due to their redox properties, especially metal compounds often directly interact with and disturb the cellular redox homeostasis. This review aims to evaluate and summarize the current knowledge on the role of redox processes in the modes of action of metal compounds used in anticancer therapy or being in (pre)clinical development.
FIG. 1.
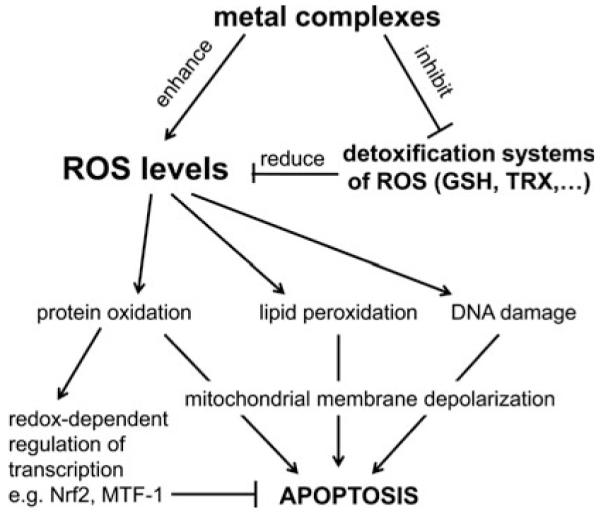
General overview on the role of ROS in the activity of anticancer metal drugs.
II. Redox Processes in Living Organisms
A. Mammalian redox metabolism
To understand the intracellular behavior of redox-(inter) active anticancer metal compounds, it is useful to consider the mechanisms responsible for the physiological cellular redox balance. Generation of ROS in general is a normal physiological process with several important functions for the living organism in metabolism, signal transduction, regulation of cellular functions, as well as in host defense (388). The most important ROS with physiological relevance are superoxide (O2• −), hydrogen peroxide (H2O2), as well as the hydroxyl radical (OH•) (detailed characteristics are given in Table 1). These species have been shown to be directly involved in the regulation of diverse signal transduction pathways important for cell proliferation, differentiation, and cell death (127, 388).
Table 1.
Overview of Physicochemical and Biological Properties of the Most Important Reactive Oxygen Speciesa
| Reactivity | Reactions in cells | Eo’ [V]b | Antioxidative defense | |
|---|---|---|---|---|
| OH• | Most reactive oxygen radical, which reacts immediately at its origin |
Reacts immediately with almost every molecule found in living cells, including sugars, amino acids, phospholipids, and DNA bases |
+ 2.31 [OH• + e− +H+↔H2O] |
Glutathione |
| O2• − | Low reactivity in aqueous solution at pH 7.4, damage is based on reactions with other radicals or metal ions; membrane impermeable but can cross cell membranes via anion channels (379) |
Reaction with [Fe-S] clusters and radicals such as NO• generating peroxynitrit (ONOO−) |
+ 0.94 [O2• − + e− +2H+↔H2O2] or −0.16 [O2 + e−↔O2• −](336) |
Superoxide dismutase; glutathione; nonenzymatic dismutation |
| H2O2 | Weak oxidizing and reducing agent; generally poorly reactive; very diffusible between cells |
Oxidation of cysteine and methionine; can be reduced to OH• by transition metals like FeII (Fenton reaction) |
+ 0.32 [H2O2 + e− +H+↔H2O+OH•] |
Catalase; peroxidases; peroxiredoxins (319) |
Unless otherwise stated the data are from ref. (140).
Redox potentials versus NHE at pH 7, with 1 M concentrations of oxidized and reduced form.
The redox environment within a cell strongly differs in diverse intracellular compartments (127). The most redox-active parts of the cell are the mitochondria, which consequently are also the major intracellular generators of ROS (221). In contrast, the cytoplasm is characterized by low levels of ROS and a less redox-active milieu. Thus, it might be hypothesized that the cytoplasm on the one hand functions as redox buffer zone between the cellular organelles and on the other hand allows specific ROS signaling (127). The high reactivity of ROS makes their tight regulation necessary for cell survival. This is also indicated by the wide range of redox-associated diseases, which include, besides diverse neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases, also several types of cancer (134). Consequently, the living organism constantly maintains a complex oxidant–antioxidant homeostasis system with diverse ROS generating and degrading systems in different compartments of the cell. There are several regulatory levels for maintenance of redox balance in the cell involving enzymatic (such as superoxide dismutases, catalase, thioredoxin reductases [TrxR], glutathione reductases [GR], and glutathione peroxidases [GPx]) as well as nonenzymatic antioxidants (such as glutathione [GSH], thioredoxin [Trx], and several vitamins) (Fig. 2).
FIG. 2.

Main interaction sites of anticancer metal complexes with cellular redox and oxidative stress pathways. Several metal compounds produce directly reactive oxygen species (ROS) and activate several ROS-dependent signaling and protection pathways (e.g., mediated by stress responsive transcription factors Nrf2, NF-κB, and AP-1). Sustained stress can induce apoptosis, for example, via the intrinsic mitochondrial pathway resulting in caspase-mediated cell death. Beside ROS-induced DNA damage, lipid peroxidation and protein oxidation also direct interactions with redox-regulatory mechanisms can disturb cellular redox homeostasis. Examples are the interaction of metal complexes with the thioredoxin (Trx) and glutathione (GSH) systems in the cytosol as well as in other cellular compartments such as mitochondria and endoplasmic reticulum (ER). Further, direct DNA damage by metal complexes and induction of ER stress due to accumulation of misfolded proteins can again lead to apoptosis (e.g., mediated by the transcription factors p53 and CHOP, respectively, as well as Ca2+ release after ER stress) and/or p53-mediated cell cycle arrests. In general, the different pathways are highly cross-linked and metal compounds target different sites. Metal complexes are indicated in bold face; cellular compartments in italic face; TrxR, thioredoxin reductase; TPx, thioredoxin peroxidases; GPx, glutathione peroxidases; GR, glutathione reductase; SOD, superoxide dismutase.
Superoxide dismutases (SOD) catalyze the dismutation of O2• − to O2 and to the less reactive but very diffusible H2O2. In humans, there are three kinds of SOD: the cytosolic Cu/Zn-SOD, the mitochondrial Mn-SOD, and the extracellular SOD (again containing a Cu/Zn core) (248). Although these forms of SOD exert similar functions, they distinctly differ—besides their metal centers—also in chromosomal localization, genomic sequence, and protein structure. Basically, the Mn-SOD does not share any substantial homology with the Cu/Zn-SODs. Nevertheless, regulatory elements for several redox-responsive transcription factors, including Nrf2, NF-κB, AP-1, AP-2, and Sp1, have been described in the promoter regions of most if not all SOD genes (248).
The peroxisome-located catalase very effectively promotes the conversion of H2O2 to H2O and O2. Notably, this enzyme has one of the highest turn over rates known, as one protein is able to convert ~6 million molecules H2O2 per minute.
GPx is the general name for a family of multiple isozymes. So far, five GPx have been identified in humans (all containing selenium) that catalyze the reduction of H2O2 or organic hydroperoxides to water (or corresponding alcohols) using reduced GSH as an electron donor (48).
With regard to nonenzymatic antioxidants ascorbate (the monodeprotonated form of ascorbic acid), GSH, and Trx seem to be the most important molecules inside cells (Fig. 3). Especially in case of ascorbate and GSH, intracellular levels in the millimolar range have been reported (22, 81). However, in contrast to GSH which is produced by the human body, ascorbate is an essential nutrient, which has to be ingested via food. Ascorbate is a very good reducing agent (50). Consequently, oxidizing free radicals, including OH•, RO•, ROO•, or GS•, have higher reduction potentials and can be scavenged by ascorbate. Such, potentially very damaging radicals are replaced by the less reactive ascorbate radical (50), which is also the reason why ascorbate is termed as “antioxidant.” However, ascorbate also reduces several redox-active metals such as iron and especially copper (50, 222, 234), thereby inducing redox cycling and ROS generation of these metals via Fenton chemistry (compare Section II.C.). Nevertheless, as most transition metals exist in inactive, protein-bound form in vivo (Compare Section III.), the relevance of reaction with ascorbate under normal physiological conditions has been questioned. Moreover, it is widely unexplored whether the intracellular ascorbate levels impact the anticancer therapy with metal compounds in the in vivo situation.
FIG. 3.
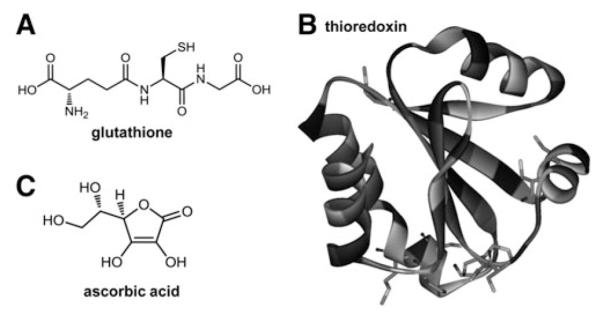
Major cellular nonenzymatic antioxidants. Structures of (A) the tripeptide glutathione (built from L-glutamic acid, L-cysteine, and glycine), (B) thioredoxin (1AIU) (16), and (C) ascorbic acid.
Besides its direct radical scavenging properties, ascorbic acid serves as crucial cofactor in several enzymatic reactions, including various hydroxylation reactions (234). Consequently, ascorbate was found to be essential for the biosynthesis of collagen as well as L-carnitine, and the conversion of dopamine to norepinephrine (217, 316).
The second important low-molecular-weight antioxidant inside the cell is the tripeptide GSH (113, 388). GSH is synthesized in the cytosol in a two-step process catalyzed by the glutamate cysteine synthetase followed by GSH ligase. Its degradation occurs exclusively in the extracellular space (22). Similar to ascorbate, GSH is highly abundant in most intra-cellular compartments with concentrations in the mM range, whereas in blood plasma only μM concentrations were detected (22). Notably, GSH is not only used in several processes directly involved in the cellular redox balance but has also diverse additional functions. Thus, GSH was found to play an important role in cell death regulation and depletion of GSH seems to be crucial for the execution of apoptosis (115). Moreover, GSH contains several potential coordination sites for diverse metal ions, including arsenic, copper, zinc, as well as cadmium. Elevated cellular GSH levels have been frequently associated with resistance of cells to metal compounds treatment (155). Additionally, GSH is an essential component of the phase II detoxification system, where it conjugates or is conjugated by glutathione-S-transferases (GSTs) to diverse endo- and xenobiotics to enhance their hydrophilicity and to facilitate their elimination. In general, GSH-conjugates are excellent substrates for diverse ATP-driven efflux pumps (especially of the multi-drug resistance [MRP, ABCC] protein family) (22), which are responsible for the final extrusion of GSH-metabolites out of the cell. For most metal-containing compounds interaction with GSH has been described, but with different results. For example enhanced GSH pools are associated with detoxification of and resistance to PtII or AsIII drugs (155). In contrast, there are several metal compounds such as PtIV,CoIII, and RuIII where GSH-mediated reduction is believed to be crucial for activation of their anticancer potential.
With respect to its role in redox balance, GSH has several functions (388): (i) scavenging of hydroxyl and superoxide radicals, (ii) cofactor for several detoxifying enzyme reactions (concerning, e.g., GPx, peroxiredoxins, and glutaredoxins), and (iii) involvement in the regeneration of other important antioxidants such as vitamins C and E. In course of these reactions, two GSH molecules are oxidized to GSSG, which then accumulates inside the cell (388). As GSSG is able to react with protein thiol groups forming protein adducts, cells physiologically contain high levels of GR, which maintains most of the GSH in its reduced form.
In addition to GSH and ascorbate, the Trx system represents the third major antioxidant defense system in human cells (37). Trx are small polypeptides with a size of 12 kDa harboring in close vicinity two cysteine residues in the active sites. In the transfer of electrons to respective substrates (e.g., proteins containing a so-called Trx fold), Trx undergo reversible oxidation of the two cysteine residues by formation of disulfide bonds leading to the oxidized Trx-S2. The reduction back to the dithiol form [Trx-(SH2)] is catalyzed by the selenium-containing TrxR and for this reaction NADPH serves as electron donor (15):
| 1 |
| 2 |
In humans, three different TrxR isoenzymes have been identified. Besides the cytoplasmic Trx1 and TrxR1 couple, mitochondria harbor a separate Trx mechanism executed by Trx2 and TrxR2. A third system was predominantly found in the testis (TrxR3). This reductase is capable of reducing GSH in addition to Trx and was consequently termed thioredoxin glutathione reductase (TGR).
Interestingly, knock-out mice for all Trx/TrxR genes are lethal during embryogenesis (240, 275), indicating the widespread and essential regulatory functions of the Trx/TrxR system in mammalian cells and tissues. Comparable to GSH, in addition to mere protection against oxidative stress, this cellular redox system regulates several other biological processes. Such Trx, together with the glutaredoxin system, is delivering electrons for the substrate turn-over cycle of the ribonucleotide reductase (compare Section III.A.2.). Additionally, the Trx system has been shown (in analogy to the GSH system) to protect cells from apoptosis induction (37). Several antioxidant defense systems are directly affected by and/or depending on reduction by Trx/TrxR: (i) Peroxiredoxins are a family of thiol-containing peroxidases that are oxidized by peroxides and reduced back to the reactive state by Trx. Peroxiredoxins are very abundant (up to 1% of soluble proteins) in the cytoplasm and diverse cell organelles and are key players in resistance against oxidative stress and regulation of H2O2-mediated signal cascades (82, 269, 270). (ii) Also, the antioxidant heme oxygenase-1 (HO-1), which catalyzes the conversion of the pro-oxidant molecule heme into the products biliverdin, iron ions, and CO, is regulated by the Trx/TrxR system. HO-1 is expressed ubiquitously in many cell types, and transcription is activated by numerous prooxidant molecules like heme, metal ions, proinflammatory cytokines, and ROS (287). Cell-type dependently both a positive and negative effect of TrxR activity on HO-1 expression was reported (102, 259, 383). (iii) Trx is also involved in the reduction of methionine sulfoxide formed during radical scavenging by oxidation of methionine residues of proteins (226). The reduction of methionine sulfoxide by Trx allows repeated scavenging of potentially damaging oxygen and nitrogen species (403). (iv) Additionally, to these important protein regulators of oxidative stress, diverse low-molecular-weight antioxidant systems, including ascorbate and flavonoids are regulated by the Trx/TrxR system (378).
Notably, both GSH as well as Trx1 are important in the redox-dependent regulation of several proteins, including important transcription factors as well as receptor and sensor proteins. There is, for example, increasing evidence for redox-sensing switches in protein structure based on two so-called critical cysteine residues (263). Oxidizing conditions induce the formation of a disulfide bond between these cysteine residues resulting in a conformational change of the protein structure. Subsequently, these alterations in the secondary protein structure lead to changed protein function. As an example, the DNA binding of redox-sensitive transcription factors AP-1, NF-κB, Nrf2, and p53 is only possible under reducing conditions when the critical cysteines are free (127). In general, cleavage of the disulfide bond is mainly performed by cellular reductants including Trx1/2 and GSH (263). Another mechanism of redox-dependent protein modifications is based on S-glutathionylation (88, 249). In the cell notable amounts of GSH are reversibly bound to −SH groups of diverse cysteinyl residues generating S-glutathionylated proteins. Interstingly, GSTs have been recently shown to catalyze the forward reaction of S-glutathionylation extending the protective role of this enzyme family toward drugs that are not substrates for phase II detoxification (380). This results in altered protein conformation and consequently—depending on the targeted protein—either in activation or inactivation. In mammalians a large panel of proteins targeted by S-glutathionylation has been identified by redox proteomics (88). This list includes diverse protein classes/families such as several mitochondrial and glycolytic enzymes, heat shock proteins, as well as many transcription factors (88).
When generally considering the interaction of metals with the cellular redox homeostasis, it has to be kept in mind that the cell harbors an extended and very complex arsenal of control mechanisms to ensure tight regulation of its redox balance. Consequently, it is not surprising that also the impact of anticancer metal compounds upon the cellular redox balance will be complex and not always easy to predict.
B. Cellular response to oxidative stress and resistance to metal compounds
Disturbance of the oxidant–antioxidant balance favoring oxidizing environment is called oxidative stress. Elevated levels of oxidative stress are known to induce cell damage and cell death by interference with multiple important cellular molecules. ROS can be produced by extracellular stress, such as irradiation, air pollutants, and exposure to toxic agents. Additionally, some intracellular metabolic and/or signaling pathways generate ROS as byproducts of oxygen-dependent enzymatic reactions. Examples for these processes are the mitochondrial respiratory chain, glucose oxidation, the cytochrome P450 family, and protein folding in the endoplasmic reticulum (ER). Most important ROS-induced damages include (i) DNA single-strand breaks, (ii) disruption of the mitochondrial inner membrane causing mitochondrial dys-function, (iii) lipid peroxidation leading to disturbed cell membranes, and (iv) oxidation of cysteine residues to sulfenic (SOH), sulfinic (SO2H), or sulfonic acid (SO3H) resulting in changes in the secondary protein structure (388) (Fig. 1). However, these oxidative stress-induced damages do not necessarily always result in cell death, but the induced DNA damage can also lead to genomic instability and hence tumor initiation and/or progression (134). Moreover, low levels of oxidative stress were shown to promote cell proliferation and induce diverse protection and survival pathways.
Surviving oxidative stress is only possible by activation of a coordinated effort to get rid of the stressors and to avoid destructive damages (Fig. 2). Consequently, transcription factors are central to oxidative stress response allowing simultaneous activation of an array of diverse genes involved in metabolism, detoxification, export of xenobiotics, as well as in the repair of the induced cellular damages. As anticancer metal drugs are redox-active substances interfering with the cellular redox status and supporting ROS generation by different mechanism, such protective response mechanisms are almost generally activated as a consequence of cell exposure. While in the nonmalignant tissues these responses are important for reducing unwanted adverse effects, they might counteract the cancer cell-damaging effect of drugs such causing therapy failure (155).
Within the respective transcription factors several are known for their redox-sensitive regulation often based on critical cysteins (compare Section II.A.) and the presence of antioxidant responsive elements in the promoter regions. This list includes AP-1, NF-κB, p53, and Nrf2. The AP-1 transcription factor is important in regulating genes involved in cell cycle progression, inflammation, and apoptosis. With regard to its protein structure, AP-1 exists either in the form of homo- or heterodimers consisting of Jun (c-Jun, Jun B, and Jun D) and Fos (c-Fos, FosB, Fra-1, and Fra-2) family members, which interact via their basic leucine-zipper domains (249, 262). Oxidative stress is known to activate the MAP kinase pathway, which in turn leads to increased transcription of c-fos and c-jun (127, 249). However, AP-1 is also negatively regulated by oxidative conditions. The critical cystein residues essential for the inhibition of AP-1-mediated transcription are found in the DNA-binding domain (Cys269) as well as close to the leucine-zipper domain (Cys320) (262). It is believed that upon changes in the GSH/GSSG ratio, S-glutathionylation of the Cys269 residue occurs, which sterically blocks binding of AP-1 to DNA (249, 262). Thus, redox regulation of AP-1 seems to be dependent on several opposing mechanisms.
Many forms of cellular stress induced by different stimuli, including ROS but also inflammatory cytokines (TNF-α, IL-6), bacterial toxins, and radiation are known to activate NF-κB (394). Thus, it is not surprising that regulation of this stress-responsive transcription factor is rather complex involving opposing mechanisms at multiple levels of the NF-κB signaling pathway. In a nutshell, there are five known members of the NF-κB family (p50, RelA (p65), c-Rel, p52, and RelB), which form homo- and heterodimers. In unstressed cells, these dimers are inactivated by binding to IκB proteins (249, 262, 394). Upon oxidative stimulation, these IκB proteins are rapidly phosphorylated (at Ser32 and Ser 36) by IκB kinase α (IKKα) and β (IKKβ) and degraded via the ubiquitin-proteasome pathway. The resulting free NF-κB dimers translocate to the nucleus and activate transcription of diverse genes involved in stress response, inflammation, and apoptosis (249, 262, 394).
The Nrf2-Keap1-ARE system plays a central role in the protection of cells and tissues against oxidative stress as recently reviewed by Singh et al. (355) and Hayes et al. (151). It consists of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor), which is tightly bound to the actin-binding protein Keap1 (kelch-like ECH-associated protein) in unstressed cells (170). This protein fixes, on the one hand, Nrf2 in the cytosol and, on the other hand, is an adaptor for an E3 ligase-mediating ubiquitination and in turn proteosomal degradation of Nrf2. Consequently, Nrf2 has a short half-life in unstressed cell. This situation is dramatically changed by the impact of ROS interacting with multiple reactive cysteines in the Keap1 molecule leading to loss of Nrf2 binding and/or Nrf2 degradation. Consequently, enhanced amounts of Nrf2 are imported into the nucleus where it binds to so-called ARE or EpRE (antioxidant or electrophilic response elements) present in the promoter or enhancer regions of multiple genes involved in oxidative and electrophilic stress response (151). The efficiency of target gene activation might thereby be modulated by dimerization of Nrf2 with other early response gene products like AP-1 family members and MAF proteins. Surprisingly, strong evidence suggests that constitutive activation of Nrf2 based on mutations in Keap1 or Nrf2 is frequent in several cancer types and contributes to chemoresistance (390). Interestingly, the list of genes with ARE promoter elements contains mainly those proteins that are also involved in the resistance of tumor cells against anticancer metal compounds (151, 355). First, several protection mechanisms regulating cellular redox balance are upregulated by Nrf2, including GSH, Trx, and peroxiredoxins (compare Section II.A.). In case of GSH, enzymes involved in synthesis (glutamate-cysteine ligase and glutathione synthetase), in redox recycling (GPx and GR), and in conjugation (several GSTs) are activated in response to Nrf2. In case of Trx, both the gene coding for Trx and the one for TrxR contain ARE sequences. As outlined in this review, multiple metal drugs cause oxidative stress by Fenton-like reactions and interaction with the cellular iron homeostasis. Interestingly, also several genes involved in iron metabolism are responsive to Nrf2 like ferritin H and HO-1 (compare Section III.A.).
While Nrf2 is a general alert and protection system for all forms of oxidative and electophilic stress, also more specific transcription factor responses to disturbance of metal homeostasis (compare Section III.) exist. Thus, the metal-responsive transcription factor (MTF-1), a zinc finger protein, and its cognate DNA binding site, the metal-response element (MRE), regulate cellular responses to heavy metals, ionizing radiation, and oxidative stress and control expression of components involved in metal homeostasis, such as zinc (ZnT-1) and copper (CTR1) transporters (351).
Additionally, both Nrf2 and MTF-1 bind to the promoter regions and activate several members of the important cellular metal-binding metallothioneines (MT). Mammalian MTs are small cysteine-rich proteins of 6–7 kDa, which are able to bind monovalent as well as divalent metal ions (70, 295). All cysteines in these molecules occur in reduced form and are coordinated to the metal ions to form metal-thiolate clusters with bridging sulfur groups. Although this allows binding of a range of metals (under cell-free conditions), mammalian MTs contain mostly zinc under physiological conditions (295). Moreover, MT genes have been shown to be highly inducible by metals such as Zn, Cu, or Cd and induction of MT and ZnT-1 expression via MTF-1 was shown to protect cells against zinc and cadmium toxicity (70, 288). Consequently, it is generally accepted that MTs are necessary, on the one hand, for detoxification of potentially toxic metal ions and, on the other hand, are involved in the regulation of metabolically essential trace elements (especially Zn) (70, 295). However, the involvement of this signaling pathway in regulation of the effects of metals (besides Zn, Cu, and Cd) is widely unknown. Recently, microarray studies revealed that gallium nitrate-resistant lymphoma cells displayed a marked increase in MTF-1, MT-2A, and ZnT-1 (415). Consequently, it has been suggested that under specific conditions MT might be involved in acquired resistance against metallodrugs.
In addition to the transcription factor-mediated protection from oxidative stress, also several other important signaling pathways exist to cope with ROS-induced cellular damages. Thus, ROS also induce ER stress and in turn the unfolded protein response (UPR) (Fig. 2). Under unstressed conditions, protein folding in the ER is catalyzed by the protein disulfide isomerase (PDI) and the ER oxidase 1 (ERO1). During this process ROS are produced, which are normally detoxified by, for example, the GSH system. Metal compounds can disturb this protein folding pathway, for example, by inhibition of chaperons like heat-shock proteins or by inhibition of ROS detoxification pathways, consequently rising the number of misfolded proteins, which leads to ER stress. Moreover, ROS-induced protein oxidation by metal complexes plays a major role in the accumulation of misfolded proteins and consequently ER stress and UPR. ER stress is recognized by three main sensors (PERK, IRE1a, and ATF6), which mediate signals to induce expression of specific UPR or ER-associated degradation (ERAD) proteins, such as chaperons and heat-shock proteins. In a nutshell, PERK signaling leads to a specific stop of mRNA translation, thereby attenuating the accumulation of newly synthesized proteins. IRE1a has an endonuclease site that activates X-box binding protein 1 (XBP1), a transcription factor for UPR and ERAD-related genes, by alternative splicing. Finally, ATF6 acts in its cleaved form as transcription factor similar to XBP1. In general, it is believed that these pathways are an adaptive response to cope with oxidative stress and to preserve cell function and survival. However, continuous stress and protein misfolding can lead to the activation of CHOP, a central transcription factor in ER stress, which induces proapoptotic proteins, such as Bim, and inhibits antiapoptotic ones such as bcl-2. Consequently, prolonged ER stress can induce not only survival pathways but also apoptosis [detailed reviews on protein folding and ER stress (129, 194, 232, 352)].
With regard to systemic cancer therapy, it has to be kept in mind that all the cellular responses to disturbance of the redox balance and oxidative stress described above significantly impact on the anticancer activity of, for example, metal compounds. Most of the concerted protection mechanisms activated by, for example, Nrf2 or UPR significantly reduce the sensitivity of malignant cells toward oxidative stress-inducing compounds, including anticancer metal drugs. This can result in (i) reduced drug uptake; (ii) enhanced efflux of drugs or conjugates via ABC transporters; (iii) enhanced drug metabolization; (iv) drug binding by MTs; (v) protection from oxidative stress by, for example, the above-mentioned anti-oxidative molecules (compare section II.A.); (vi) enhanced repair of metal drug-mediated damages, for example, of DNA or proteins; and (vii) activation of antiapoptotic programs involving, for example, bcl-2 and IAP family members. These chemotherapy resistance mechanisms against anticancer metal compounds have been reviewed recently by others and our group (155, 390) and are, thus, not in focus of this article.
Overall, it has to be kept in mind that, in general, cancer cells are characterized by an imbalance in redox homeostasis, leading to enhanced intracellular ROS generation (134, 381). The mechanisms underlying these redox alterations in tumor cells are diverse and very complex. For example, increased metabolic activity, mitochondrial malfunction and changes in virtually all antioxidant molecules are typically observed in cancer cells (134). Consequently, the interference with the cellular redox homeostasis of cancer cells seems an attractive and promising target for cancer therapy (9, 74, 77, 134, 138, 149). Indeed, many of the currently used chemotherapeutic drugs interact with the cellular redox balance und there are several attempts to specifically target the altered redox conditions in cancer cells. Thus, it is not surprising that—due to their redox properties—especially metal-containing compounds or drugs interfering with the cellular metal homeostasis by metal chelation (134) are in the focus of interest.
C. Fenton chemistry in biological context
In 1876, Henry John Horstman Fenton discovered the strong oxidative effects of FeII and H2O2 on some organic substrates (109), and later the occurrence of OH• in this reaction was suggested by Haber and Weiss (136). The “Fenton reaction” is defined as:
| 3 |
Thus, the reaction of FeII and H2O2 can produce the highly reactive OH• which is able to damage biological molecules like nucleic acids, lipid membranes, and proteins. The generated FeIII can then be reduced back to FeII by the superoxide radical O2• −
| 4 |
Together with the Fenton reaction this leads to an iron-catalyzed production of OH•, the so-called Haber-Weiss reaction, where iron cycles between its ferrous FeII and ferric FeIII form (Fig. 4) (396). In addition to the superoxide radical, also biological reductants like ascorbate and several thiols (e.g., GSH) are able to reduce FeIII to FeII (220). Consequently, not only OH•, but also reactive organic species such as peroxyl (ROO•), alkoxyl (RO•), and thiyl (RS•) radicals are formed via the Haber-Weiss reaction (289). Following the stepwise one-electron reduction cascade of molecular oxygen:
| 5 |
both the superoxide radical and H2O2 are constantly produced under physiological conditions in healthy cells (compare Section II.A.). The responsible mitochondrial and microsomal biomolecules include several oxidases, fumarate reductase, flavins, tetrahydropterins, and catecholamines (220). In some reactions, like that of glucose oxidase and urate oxidase, O2 is directly reduced to H2O2. However, in most cases O2 is first reduced to O2• − and subsequently dismutated by SOD to H2O2 and O2. The generated H2O2 is further processed by catalases, peroxidases, or peroxiredoxins (319). In general, the concept of ROS generation by reaction of a metal ion with H2O2 is not limited to FeII. Thus, the term “Fenton-like reactions” is also used in context with other metal ions like copper, cobalt, and vanadium that can substitute iron.
FIG. 4.

Iron-catalyzed production of hydroxyl radicals. The Haber-Weiss reaction is shown, whereby the left part depicts the Fenton reaction.
III. Homeostasis of Redox-Active Metals in Mammalians
A. Iron homeostasis
A crucial feature of the biological activity of iron is the possibility to readily switch in a one-electron oxidation– reduction reaction between the ferrous form, FeII, and the ferric form, FeIII. Under aerobic conditions, FeII is readily oxidized in solution to FeIII, which is virtually insoluble at physiological pH (289). Consequently, the bioavailability of iron is generally limited. To maintain iron in a soluble form and perform iron uptake, utilization, and storage diverse proteins binding Fe with high affinity (e.g., transferrin and ferritin) have evolved in biological systems.
1. Iron transport
In the blood stream iron is bound in its ferric state to the serum proteins transferrin and albumin. Human transferrin (Tf) is a large nonheme monomeric glycoprotein with a molecular mass of ~80 kDa and in blood plasma the concentration is 2–3.6 mg/ml (~35 μM) (8). At the slightly alkaline pH of 7.4, Tf can bind one or two ferric ions with an overall blood iron load of 30% (69). The cellular uptake of iron via the transferrin-dependent pathway is well investigated and has been extensively reviewed (214, 215, 304) (Fig. 5A). In a nutshell, two iron-loaded Tf molecules bind to one dimeric Tf-receptor (TfR1), whereas the binding constant of iron-free Tf to the receptor is distinctly lower. This Tf-TfR1 complex is then endocytosed into the cell. The acidic pH of the endosomal lumen induces a conformational change in Tf leading to release of the bound iron from its carrier. The Tf molecule itself remains tightly bound to the TfR1 under these conditions. The complex is then relocated to the cell surface, where the extracellular pH leads to dissociation of the apo-Tf molecules from the receptor. After reduction by a ferrireductase, FeII is transferred into the cytosol by the divalent metal transporter (DMT1) (277, 278).
FIG. 5.

Metal homeostasis in human cells. (A) Iron homeostasis: iron is accumulated in cells via transferrin-mediated endocytosis. Upon acidification iron is released from endosomal vesicles and becomes part of the labile iron pool (LIP) in the cytosol. Iron is utilized as cofactor, for example, in ribonucleotide reductases or proteins with Fe-S-clusters. Excess iron is stored in ferritin. (B) Copper homeostasis: a model of cellular copper transport and chaperoning is shown. Copper is taken up at the plasma membrane by diverse transporters (e.g., CTR1, CTR2, and DMT1). Once in the cell, copper is further distributed by intracellular chaperons. For example, copper is transported to the mitochondrial inner membrane via cox11. ATOX1 delivers excess copper to the trans-Golgi network where it is packed into vesicles by ATP7A/B and bound to ceruloplasmin for excretion. Finally, CCS chaperons copper for use in Cu/Zn-SODs.
Once in the cytosol, iron becomes part of the labile iron pool (LIP). This low-molecular-weight pool of weakly chelated iron rapidly passes through the cell. Under physiological conditions, the LIP represents only a minor fraction of the total cellular iron (3%–5%), but it is the crucial linkage between iron uptake and the permanent intracellular chelation by iron-dependent proteins (205). Thus, it has to be expected that all dietary iron should pass the LIP stage. The LIP harbors both FeII and FeIII associated with a variety of low-molecular-weight ligands with low affinity to iron ions, including citrate, phosphates, carbohydrates, carboxylates, and polypeptides. However, the actual nature of the LIP is still widely unexplored (183).
Cell damage associated with iron overload is attributed to increased levels of the LIP, which promotes the production of ROS via Fenton-like chemistry (compare Section II.C.) (126). Additionally, due to the only weak chelation of iron in the LIP, it is also the major coordination site for many therapeutic iron chelators (303). Chelation of the LIP-bound iron results—due to iron deprivation—also in prevention of iron redox-cycling and reduced ROS formation (46). With regard to metal compounds, it seems likely that interaction with the LIP also contributes to metal-induced intracellular ROS production.
2. Intracellular iron proteins
Iron is utilized as cofactor in several proteins, including aconitases, cytochromes, ribonucleotide reductase (RR), as well as heme complexes (214). With regard to anticancer therapy, the RR (199), as enzyme that provides dNTPs essential for proliferation and DNA repair, has been considered an ideal target for cancer therapy. This led to the (pre)clinical development of several RR inhibitors, including gemcitabine, hydroxyurea, the thiosemicarbazone Triapine, or the lanthanum compound KP772 (156, 341). Another important intracellular iron-binding protein is ferritin where excessive iron is stored (227, 372). Ferritin is a ubiquitous and highly conserved multimeric protein and consists in vertebrates of an apoprotein shell of 24 light and heavy subunits around a core of up to 4500 iron atoms (158, 416). As new iron is packed into the ferrihydrite mineral core, it is converted from FeII to FeIII by the inherent ferroxidase activity of the heavy ferritin subunits (416). Due to its iron-storage function ferritin prevents excess iron of the LIP from taking part in the Fenton reaction, which makes it crucial for the protection of the cell from ROS (227, 280).
B. Copper homeostasis
Copper is another redox-active metal, which is important in the biochemistry of every living organism. In biological systems copper exists mainly in two oxidation states: cuprous CuI and cupric CuII. Copper is used as cofactor in several redox reactions of enzymes with fundamental biological functions in growth and survival of cells such as the cytochrome c oxidase of the mitochondrial electron chain, the lysyl oxidase important for connective tissue formation, as well as the Cu/Zn-SODs (compare Section II.A.). However, due to its redox properties, copper (comparable to iron) has to be tightly regulated in the living organism to prevent formation of ROS. Thus, copper is constantly protein-bound and, for its distribution, always transferred directly from one protein to the other (Fig. 5B). The central structural requirement in Cu-binding proteins, which is necessary for these intimate protein–protein trans-chelation reactions, is the presence of unique cysteine, methionine, or histidine-rich domains, which bind CuI via metal–sulfur or metal–nitrogen bonds (166). Overall, there is virtually no free copper in the healthy organism. In the blood plasma, most copper is bound to ceruloplasmin (152), a cuprous oxidase, which is important in the body iron homeostasis by oxidizing FeII in the plasma, allowing iron binding to transferrin. However, the importance of ceruloplasmin in copper transport and homeostasis has been questioned (152). The remaining plasma copper (about 350 ng/ml) is bound to proteins of the exchangeable copper pool (258). This pool is composed primarily of albumin and α2-macroglobulin (transcuprein). In contrast to the extensively investigated and well-understood iron uptake using the transferrin receptor pathway, little is known how copper exactly enters mammalian cells. The main Cu uptake transporter in mammalian (liver) cells seems to be the copper transporter 1 (CTR1) (193). In addition, other metal transporters, including CTR2 and the divalent metal transporter 1 (DMT1), contribute to copper uptake of mammalian cells. In the cytoplasm, a highly specialized chaperone system assures the distribution of copper to the target proteins. There are three major functional groups of copper chaperones (17, 19): (i) ATOX1, which delivers copper to the P-type ATPases (ATP7A and B) of the secretory transgolgi network, (ii) CCS, which brings copper to the Cu/Zn-SOD in the cytoplasm, and (iii) cyclooxygenase 17 (Cox17), which transports copper to the inner mitochondrial membrane proteins Cox11 and Sco1 from which it is subsequently incorporated into cytochrome c oxidase.
Unlike iron, physiological storage of copper seems unnecessary as copper body levels are maintained primarily by balancing dietary absorption, distribution, and utilization (17). However, excess of copper (and other metals) stimulates the expression of metallothioneins, a protein family that is characterized by its outstanding metal binding capacity and is crucial in the protection of the body from toxic heavy metals (70, 295) (compare Section II.B.).
IV. From Electrochemistry to Cellular Redox Reactions and Anticancer Therapy
A. Oxidation and reduction: the principles of redox processes
In contrast to most organic cancer therapeutics being redox-inactive in the cellular environment, many metal-containing drugs can undergo redox processes. These changes significantly influence and alter the physicochemical properties of such complexes including geometry, charge, and reactivity. Consequently, the knowledge of the redox potential can be crucial for the understanding of the mode of action underlying the anticancer activity of metal compounds.
For each redox couple of metal ions Mn + /M(n − 1) + with adjacent oxidation states and for a variety of redox reactions standard electrode potentials (Eo) are available in literature (24, 162, 218). This potential is given for standard conditions of 298.15 K, 1 bar pressure, at pH 0, and at 1 M concentration of the reduced and oxidized forms. The Eo potentials are always referenced to the normal hydrogen electrode (NHE), which consists of hydrogen gas bubbled with 1 bar around a platinum electrode in an aqueous solution with pH 0. The potential of the NHE, according to the reaction
| 6 |
has been arbitrarily set to 0.00 V. Considering two different redox reactions, for example FeIII + e− ↔ FeII with a standard redox potential Eo = +0.77 V versus NHE and GSSG + 2H+ + 2e− 2GSH with Eo = + 0.18 V versus NHE (162, 336), it is directly possible to predict that under standard conditions FeIII will be reduced to FeII and GSH will be oxidized to GSSG. This is based on the thermodynamic principle that the redox couple with the more positive standard redox potential is always reduced and the one with the more negative potential is oxidized. However, apart from thermodynamics, which gives information, if a reaction is possible or not, also the kinetics have to be considered, which give information about the reaction rate. Thus, in principle a reaction that is possible from the thermodynamical point of view may not occur because of too slow kinetics.
However, when using redox potentials in a biological context, a range of additional factors have to be considered:
(i) the pH dependency
the majority of redox reactions, including all involving H+ ions, exhibit pH-dependent potentials. For example the potential of the redox reaction
| 7 |
is + 1.23 V versus NHE at pH 0, + 0.815 V at pH 7, and + 0.40 V at pH 14. Thus, for the physiologically relevant situation of pH 7 a separate denotation Eo′ has been defined. Depending on the number of electrons and protons involved in the redox reaction, the redox potential shifts when the cellular pH changes. For example, the potential of GSH (with a two electron/two proton couple) changes with a slope of −0.061 V/pH at 37oC (162, 336).
(ii) the proportion dependency of oxidized and reduced form
the standard redox potential Eo′ for the reaction
| 8 |
at −0.16 V versus NHE implies equal concentrations of O2 and O2• − (336). However, in the cellular environment a more realistic concentration of O2 is ~10− 5 M and of O2• − it is 10− 10 M. These differences in concentration result in a profound change of the redox potential of this reaction. The reason is a term in the Nernst equation (the underlying mathematical expression for estimation of redox potentials), which contains the proportion of oxidized to reduced species (e.g., O2 to O2• −). Thus, a change in the proportion strongly impacts on the redox potentials resulting in Eo′ ~ + 0.14 V versus NHE for O2/O2 in the cellular environment (336). This dependency on the concentrations is extremely important due to the lack of equilibrium conditions in biological systems.
(iii) the reference electrode
as the setup of the NHE is rather difficult to implement in routine measurements, in most cases other reference electrodes are used and the reported values are referred to them or converted to the NHE by addition of a constant value. In aqueous solution the most important references are the saturated silver/silver chloride electrode (+ 0.197 vs. NHE) and the saturated calomel electrode (+ 0.241 vs. NHE) (23). For nonaqueous solutions, ferrocenium/ferrocene is frequently used as internal reference with a conversion value that depends on the solvent (25, 293).
(iv) the biologically accessible redox potential window
in biological systems the accessible redox potential window ranges only from around −0.4 to +0.8 V versus NHE (197). The strongest reducing agent of the major redox active components in cells is the nicotinamide adenine dinucleotide phosphate couple (NADP+ + 2e− + H+ ↔ NADPH) with approximately −0.38 V versus NHE (336). On the other side, the strongest oxidizing agent is oxygen itself according to O2 + 4H+ + 4e− ↔ 2H2O at +0.815 V at pH 7.0. However, oxygen is kinetically inert and, thus, in vivo reactions involving molecular oxygen have to be catalyzed by enzymes (e. g. the above 4-electron reaction is catalyzed by cytochrome c oxidase). Usually, all redox reactions with higher or lower potentials than the biological window cannot occur in the cellular environment. However, it has to be mentioned that besides common biological reducing and oxidizing agents, also ROS like OH•, O2• −, and H2O2 (see also Table 1) as well as organic radicals such as RO•, ROO•, and RS• are present in cells. Especially, radicals are often characterized by very high Eo′ redox potentials (e.g., OH• [+ 2.31 V], RO• [+1.60 V], ROO• [~1.00 V], and RS• [e.g., cysteine + 0.92 V]) (140) and are able to oxidize far more compounds than the common cellular redox systems. Furthermore, oxidizing radicals like GS• can react with GS− to form strongly reducing GSSG• − radicals with redox potentials of −1.50 V (49). However, it has to be considered that in the cellular environment common redox agents like GSH are available in up to mM concentrations, whereas intracellular concentrations of radical species are generally very low and these highly reactive species often immediately react at their place of origin.
B. The impact of metal and ligand on redox potentials
Usually, the standard redox potentials of metal ion redox couples Mn + /M(n − 1) + are determined in aqueous solution without additional coordinating ligands. However, in biological systems as well as in synthetic metal complexes, coordinating ligands are frequently present, which often induce dramatic changes in the redox potential of a metal ion. One example is a series of investigational RuIII anticancer complexes (Table 2) (318). Starting with [RuIIICl6]3− at a redox potential of −1.36 V versus NHE the stepwise exchange of one chlorido ligand by indazole results in increasing redox potentials, ending up with trans-[RuIIICl2(Hind)4]+ at + 0.59 V versus NHE, nearly 2.0 V more positive then [RuIIICl6]3− Thus, the knowledge of the exact coordination sphere of a metal ion in the biological environment is necessary to draw conclusions about its redox properties. Moreover, the use of different ligands enables tuning of the redox potential of a selected metal ion, yielding in metal complexes with the desired redox properties.
Table 2.
Influence of Ligand Exchange on the Redox Potentiala
| Compound | E1/2 (RuIII/RuII) V vs. NHEb |
|---|---|
| [RuIIICl6]3 − | − 1.36c |
| [RuIIICl5(Hind)]2 − | − 0.87c |
| trans-[RuIIICl4(Hind)2]− | − 0.43 |
| mer-[RuIIICl3(Hind)3]0 | + 0.10 |
| trans-[RuIIICl2(Hind)4]+ | + 0.59 |
Values taken from ref. (318).
Redox potentials in V± 0.02, measured at a scan rate of 0.20 V/s in [n-Bu4N][BF4]/dimethylformamide.
Adequate detection was hampered by rearrangement of the complexes in dimethylformamide; therefore, the potentials were estimated using Lever’s parametrization approach (213): E1/2 = SM · ∑ELigand + IM (with SM= 1.14; ECl = −0.24 and EHind = 0.26; IM= −0.35).
Next to the ligands, the nature of the metal ion itself influences the redox properties of coordination compounds (see Fig. 6 for metal ions with an identical ligand set).
FIG. 6.

Impact of the central metal ion on the redox potential of metal complexes. As an example the cyclic voltammograms of complexes of the type M(Dp44mT)2 with different metal centers are shown (M = manganese, iron, cobalt, nickel, copper; Dp44mT = di-2-pyridylketone 4,4-dimethylthiosemicarbazone) (33). The figure illustrates the strong impact of the central metal ion on the redox potential of structurally similar complexes.
As example, the electrochemical response of the metal complexes [M(Dp44mT)2], with M = manganese, iron, cobalt, nickel, copper, and Dp44mT = di-2-pyridylketone 4,4-dimethylthiosemicarbazone is shown in Figure 6 (33). Although for each metal ion the MIII/II redox couple was investigated (for M = CuII/I), the complexes exhibit very different potentials. For example, [Ni(Dp44mT)2]+ with a redox potential of + 0.52 V versus NHE was found to be much easier to reduce than its cobalt analog [Co(Dp44mT)2]+ at −0.62 V.
C. Anticancer metal compounds and redox processes: overview
The interaction of transition metal complexes with the cellular redox balance is well investigated (140). For example, depletion of the GSH pools has been frequently described for many metal-containing anticancer drugs (253, 291, 414). However, the underlying modes of action strongly depend on the chemical/physical properties of the metal ion. Especially the hardness/softness of a metal ion seems to have a crucial impact on the intracellular reaction behavior of the complexes. Transition metals (“acids”) as well as the donor atoms of the potential ligands (“bases”) can be classified into soft (low charge/large ionic radius), intermediate, and hard (high charge/small ionic radius) according to the “hard and soft acids and bases” (HSAB) concept (294). Based on this concept, soft acids react faster and form stronger bonds with soft bases, whereas hard acids react faster and form stronger bonds with hard bases. Thus, the soft acids PtII, AsIII, or AuI easily react with soft bases like sulfur-containing GSH and other cysteine-rich molecules, such as TrxR and metallothioneins (compare Section II.A.). This leads to redox-independent formation of GSH conjugates and, consequently, cellular GSH pool depletion and sensibilization to ROS (34, 73, 239, 253, 358). In contrast, in case of intermediate to hard metal ions (such as VV, CoIII, CuII, or RuIII) with lower affinity for soft donor systems such as the thiol moiety in GSH (Compare Section II.A.), GSH pool depletion is caused by ROS generation via Fenton-like reactions, which leads in parallel to reduction of the metal and to oxidation of GSH to GSSG.
An important part of the mode of action of several metal-based drugs related to redox processes is widely known as the “activation by reduction” hypothesis (compare Section V.A.2., V.D., V.E., and V.H.). This concept is based on the idea to apply a less cytotoxic prodrug, which is then activated by intratumoral reduction. Especially, in case of PtIV,RuIII,CoIII, and CuII drugs activation by reduction is believed to be important in their modes of action (9, 74, 77, 134, 138, 149). Reduction results in increased reactivity of the metal center together with labilization/dissociation of the ligand. However, activation by reduction does not necessarily increase the intracellular activity of the metal drug per se, but may also contribute to selective transport and release of cytotoxic ligands within the tumor tissue as observed for several cobalt complexes.
V. Metal-Based Anticancer Drugs and Their Redox-Related Modes of Action
Anticancer metal complexes have been shown to strongly interact with or even disturb cellular redox homeostasis resulting in enhanced levels of oxidative stress (Fig. 2). In the following sections we summarize the current knowledge on Pt, Au, As, Ru, Rh, Cu, V, Co, Mn, Gd, and Mo complexes regarding the involvement of redox processes in their anti-cancer activity.
A. Platinum
Platinum (Pt) is used for many purposes in modern life. For example, it is applied as catalyst, used in electronics, and for jewelry. Further, it plays a decisive role in anticancer agents, such as cisplatin and oxaliplatin. The most common oxidation states of platinum are + 2 (d8) and + 4 (d6). According to the HSAB concept PtII is a “soft acid” and therefore readily reacts with “soft bases” like sulfur. In contrast, PtIV is a hard acid and prefers oxygen containing ligands. The oxidation states + 1 and + 3 are less common.
1. Platinum(II)
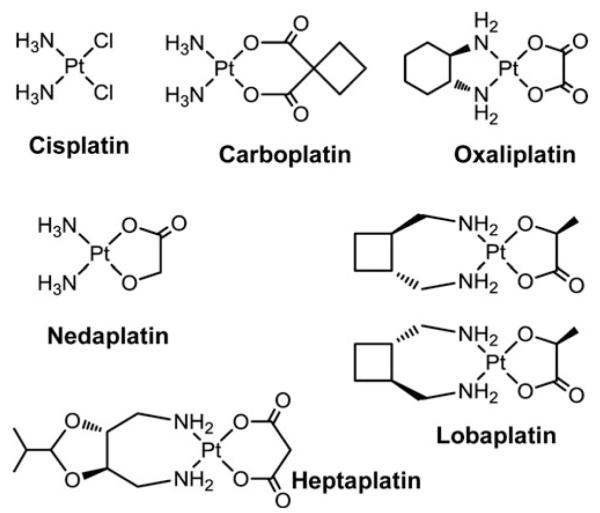
The era of metal-based anticancer drugs began with the discovery of the anticancer properties of the square-planar PtII cisplatin (cis-[PtCl2(NH3)2]) (Fig. 7) by Barnett Rosenberg in the 1960s (323). Nowadays, cisplatin is one of the most important chemotherapeutics used clinically against a wide variety of different solid tumors, including testicular, bladder, ovarian, as well as head and neck cancer (189). In general, it is accepted that the anticancer activity of cisplatin is based on the formation of platinum-DNA adducts. This coordination leads to a significant distortion of the helical DNA structure resulting in inhibition of DNA replication and transcription. Further, several signaling pathways are activated which—as a final consequence—lead to cell cycle arrest and/or apoptosis (189, 301).
FIG. 7.
Clinically approved PtII drugs.
Due to the Pt center of cisplatin, it is reasonable that the drug reacts not only with DNA but also with donor atom-containing proteins (compare Section II.A. and IV.C.), with particularly high affinity to sulfur and seleno amino acids. This is supported by the fact that less than 1% of intravenously administered cisplatin reaches DNA. Therefore, several other cellular targets have been suggested (130, 154, 315). Such DNA damage-independent mechanisms might involve, for example, alteration of cell membrane fluidity by inhibition of the Na+ / H+ membrane exchanger NHE1 and, consequently, activation of FAS-mediated apoptosis (314). Cisplatin detoxification is at least partially based on formation of cisplatin-GSH conjugates (100), which leads to intracellular GSH pool depletion (253), disturbance of the cellular redox homeostasis, and, consequently, increased levels of intracellular ROS (34, 73, 239, 358). Moreover, cisplatin treatment was found to deplete cellular NADPH pools (98, 238) resulting in altered mitochondrial redox status, which then causes hydroxyl radical generation. Further, recent studies suggest the ER as cytosolic target of cisplatin and induction of apoptosis also via ER stress (233). All these processes can lead to lipid peroxidation and oxidative protein damage, which contribute to the disruption of the mitochondrial membrane structures (146, 238) and consequently lead to apoptosis induction (compare Section II.B.).
Further, cisplatin directly reacts with TrxR, which has a redox-active disulfide/dithiol moiety in its active site and a reactive seleno-cysteine residue at the C-terminus (333). Cisplatin has been shown to irreversibly inhibit the activity of human TrxR in cell-free setting and in cell models in a dose- and time-dependent manner (16). Interestingly, in a cell-free system cisplatin inhibited the TrxR activity only in the presence of NADPH. It is therefore claimed that cisplatin interacts only with the reduced form of TrxR, which is generated by NADPH (16, 333) (compare Section II.A.). Notably, human GSH reductase, which has a strong homology to human TrxR and contains a similar redox-active disulfide/dithiol moiety but no seleno-cysteine residue, is not inhibited by cisplatin (16, 251, 404, 405). Therefore, the highly reactive seleno-cysteine residue at the C-terminal domain was suggested to be the TrxR target of cisplatin (405). These data were supported by a study investigating the ability of different modified forms of the cytosolic TrxR1 protein to induce apoptosis. As expected, the unmodified full-length TrxR1 with an intact selenocysteine residue did not promote cell death. In contrast, both a truncated selenocysteine-deficient TrxR1 form as well as a TrxR1, which was derivatized at the selenocysteine residue with cisplatin, were able to induce cell death in A549 lung cancer cells (11). Arnér et al. (16) showed that in addition to cisplatin, also different GSH-cisplatin conjugates inhibited the activity of TrxR. Interestingly, these GSH-adducts, in contrast to cisplatin alone, were able to reduce the activity of the GSH reductase system (16). Further, cisplatin resistance can be accompanied by overexpression of metallothioneins and GSTs (compare Section II.A.) (363). The latter enzymes catalyze the conjugation of GSH to the platinum complexes, which then can be excreted from the cells, for example, via the drug-conjugate efflux pump ABCC2 (67, 73). Several clinical studies showed that augmented expression and gene amplification of GSTs were unfavorable prognostic factors in ovarian cancer patients and could be associated with cisplatin resistance in head and neck squamous cell carcinoma (84, 366).
Thus, it can be summarized that the intracellular redox homeostasis is severely affected by cisplatin due to the disruption of the TrxR and GSH reductase systems. Therefore, it is not surprising that different studies have shown a correlation between Trx, TrxR, GSH, GSTs, and GR expression with cisplatin resistance (155, 363, 406). It has to be mentioned that cisplatin-induced oxidative stress participates not only in its cytotoxic effects against tumor cells, but is also responsible for unwanted effects such as nephrotoxicity (73) and hepatotoxicity (85, 146). Several studies demonstrated that the cisplatin-induced renal tubular injuries involve multiple signaling pathways, including ROS-mediated p53 signaling (179). Interestingly, it has been shown that γ-glutamyl-transpeptidase (γ-GT) expression plays a crucial role in cisplatin nephrotoxicity. While in the tumor tissue γ-GT expression was connected with resistance, kidney γ-GT expression rendered the cells sensitive to cisplatin toxicity, suggesting different mechanisms of apoptosis induction in tumor cells and proximal tubular cells. The authors further suggest that in the kidney excreted Pt-GSH conjugates are metabolized by γ-GT, reabsorbed, and further metabolized to reactive thiols, which primarily target mitochondria and thereby induce apoptosis and necrosis in the kidney tissue (144). Cisplatin-induced oxidative liver and renal damage and its possible protection by the hydroxyl radical scavenger dimethylthiourea (DMTU) were further studied in vivo in Wistar rats (98, 330). DMTU protected against decreased hepatic ATP levels, lipid peroxidation, cardiolipin oxidation, sulfhydryl protein oxidation, mitochondrial membrane rigidification, GSH oxidation, NADPH oxidation, and apoptosis (98).
In clinical use these severe side effects together with intrinsic and acquired resistance limit the application of cisplatin (155). To overcome these limitations, diverse novel metal-based anticancer drugs have been designed and around 30 compounds have so far been evaluated in clinical studies (65). From a plethora of newly synthesized square-planar four-coordinate cisplatin analogs (120) only two further PtII complexes have gained world-wide clinical approval, namely, the second- and third-generation derivatives carboplatin and oxaliplatin (Fig. 7). In addition, three other PtII-based drugs, namely, nedaplatin, lobaplatin, and heptaplatin (Fig. 7), have gained limited regional approval (172). These PtII drugs are believed to target DNA in analogy to cisplatin. Carboplatin is less toxic than cisplatin. This can be explained by the increased stability of carboplatin due to its dianionic biscarboxylato leaving group instead of the two chlorido ligands in the case of cisplatin, leading to a slower rate of aquation. After dissociation of the leaving group, carboplatin forms identical DNA adducts as cisplatin (198). Consequently, this drug is active in a comparable spectrum of tumors and cross-resistance to cisplatin is frequently observed (155). In contrast, oxaliplatin has been shown to be active against cisplatin-resistant tumor cell lines. However, in the clinical situation some cross-resistance between cisplatin and oxaliplatin has been observed (364). Differences in the activities of oxaliplatin and cisplatin can be explained by lower DNA adduct formation by oxaliplatin (408) and the more hydrophobic and bulkier (1R,2R)-cyclohexanediamine (Dach) ligand, which induces DNA bending different to cisplatin. Further, cisplatin and oxaliplatin adducts are recognized differently by mismatch repair proteins, DNA polymerases, and damage-recognition proteins (60).
For both oxaliplatin and carboplatin, only a few reports on the effects on cellular redox homeostasis are currently available. Laurent et al. investigated the impact of endogenous ROS production on tumor growth and the consequence of ROS modulation on oxaliplatin cytotoxicity (208). In this study, a dose-dependent increase of ROS production associated with a decrease in proliferation was detected after oxaliplatin treatment in a murine colon cancer model in vitro and in vivo (208). Moreover, addition of exogenous GSH or N-acetylcysteine (NAC) reduced oxaliplatin cytotoxicity, whereas depletion of GSH with buthionine sulfoximine (BSO) or cotreatment with SOD mimics (compare Section V.I.) increased the sensitivity toward oxaliplatin (7, 208). In accordance, in a cell-free system the levels of oxaliplatin-induced DNA damages were increased by the addition of SOD mimetics whereas NAC reduced them. The same effects were observed in combination studies in vivo (208).
Comparable to cisplatin, the Trx system is also influenced by oxaliplatin. This platinum drug inhibited the activity of TrxR in a cell-free system similar to cisplatin (405), whereas in a cellular environment TrxR was inhibited significantly stronger by cisplatin than oxaliplatin (157). In contrast, carboplatin had no effect on TrxR activity in cell-free systems (405), a rather unexpected result considering the similarities of carboplatin and cisplatin. However, the TrxR inhibitory activity of carboplatin has never been tested in live cells in vitro or in vivo. Therefore, two alternative hypotheses have been proposed. On the one hand, an intracellular activation might yield a more reactive carboplatin derivative that inhibits the TrxR similar to cisplatin. On the other hand, the lack of TrxR inhibition might be an explanation for the lower cytotoxicity of carboplatin compared to cisplatin (405).
Beside cisplatin, carboplatin, and oxaliplatin, only a few PtII drugs were investigated with respect to their impact on redox homeostasis of cancer cells. For example (2,2′:6,2″terpyridine)platinum(II) complexes (Fig. 8) exhibit their cytotoxic activity against different tumor cell lines (30, 224) not only by intercalating into DNA (176, 243) but also by inhibiting the human TrxR in a dose-dependent manner. TrxR activity was blocked with IC50 values in the low nM range, whereas the GSH reductase inhibitory concentrations were > 1000-fold higher (30). These results are again in accordance with the inhibition of TrxR and GSH reductase by cisplatin (16, 251, 404, 405). Two of the (2,2′:6,2″-terpyridine)platinum(II) complexes were further investigated in an orthotopic rat glioblastoma model. Both compounds had no effect on the blood redox parameters but reduced TrxR and GSH peroxidase activities significantly in the tumor tissue (3). For another set of terpyridine-platinum(II) complexes it has been shown by X-ray crystallography and MALDI mass spectroscopy that the complexes inhibit the TrxR activity by blocking the selenocysteines at the C-terminal active-site of the protein (223).
FIG. 8.

General structure for terpyridine-PtII complexes.
2. Platinum(IV)
The anticancer activity of PtIV complexes was discovered together with cisplatin in the 1960s (323), but these platinum drugs have been studied and developed less extensively than PtII compounds. The octahedrally coordinated PtIV compounds have a higher coordination number (six vs four) than the square-planar PtII complexes and therefore the possibility to introduce additional axial ligands. These ligands have a strong impact on diverse pharmacological properties of the compounds, such as lipophilicity, stability, and reduction potential (compare Section IV.B.). Furthermore, the ligands can be designed for targeting specific tumor sites or as additional bioactive components. PtIV complexes are kinetically more inert than their PtII counterparts and have a lower reactivity with biomolecules. These characteristics are the reason for reduced unwanted side effects, lower toxicities, as well as the possibility of oral administration (120, 139).
The first PtIV drugs in clinical trials were cis,trans, cis-[PtCl2(OH)2(isopropylamine)2] (JM9, iproplatin) and [PtCl4(d,l-cyclohexane-1,2-diamine)] (tetraplatin, ormaplatin; Fig. 9). The clinical development was abandoned due to the low activity in the case of iproplatin (382) and the severe neurotoxicity caused by tetraplatin (276, 337). Recently, another PtIV complex, namely, cis,trans-[PtCl2(OAc)2(NH3)(cyclohexylamine)] (JM-216, satraplatin) (Fig. 9), has been considered for approval by the FDA for the treatment of hormone-refractory prostate cancer in a combination regimen with prednisone, a synthetic corticosteroid. However, a phase III study did not achieve the anticipated endpoint of overall survival improvement (Agennix, http://agennix.com, ref. accessed 2010-09-15). Further clinical trials with satraplatin in a combination regime are ongoing (155).
FIG. 9.

PtIV drug candidates. Tetraplatin, iproplatin, and satraplatin, together with the major reduced PtII-metabolite of satraplatin (JM118) are shown.
Comparable to RuIII and CoIII drugs (compare Sections IV.C., V.D., and V.H.), PtIV complexes are considered as pro-drugs, which undergo reduction in the intracellular milieu. During this process the axial ligands are released and the corresponding anticancer active square-planar PtII analogs are formed. Therefore, the reduction potential of the PtIV complexes as well as the redox status of the tumor environment have strong impacts on the activity of PtIV anticancer drugs (124, 138). Several studies show that the reduction potential is influenced by the nature of the axial ligands and to a lesser extent by the equatorial ligands (compare Section IV.B.). For PtIV complexes with a given equatorial coordination pattern, reduction most easily occurs when chlorido ligands are in the axial position. Carboxylato ligands lead to an intermediate reduction potential, whereas hydroxido ligands possess strong electron donating properties resulting in low reduction potentials and therefore complexes that are difficult to be reduced (105, 120, 137, 141). In addition, Choi et al. showed that the reduction rates depend not only on the electron-withdrawing power of the axial ligands but also on the bulkiness of these ligands (74).
Several groups have investigated the correlation between the cytotoxicity and the reduction potential of PtIV compounds. It can be summarized that on the one side the cytotoxicity is mainly dependent on the activity of the resulting PtII complexes. On the other side, it depends on where and how readily the PtIV compounds are reduced. The clinical results of iproplatin and tetraplatin can be directly linked to their reduction properties. For iproplatin (axial hydroxido ligands, low redox potential) it was found that in vivo large amounts are not reduced, resulting in low toxicity but equally low activity (296). In contrast, tetraplatin (axial chlorido ligands, high redox potential) was very rapidly reduced and all detected biotransformation products were PtII analogs explaining the very high toxicity (62). The reduction of satraplatin (axial acetato ligands) is rapid but slowed down in vivo, resulting in at least six metabolites of which cis-amminedichlorido-(cyclohexylamine)platinum(II) (JM118) (Fig. 9) is the most abundant one. A comparatively mild toxicity was detected after satraplatin treatment (120, 312).
Even though there is a correlation between the activity of PtIV compounds and their reduction potential, it is difficult to predict their in vivo anticancer activity. One explanation of this disparity could be the early reduction of PtIV complexes in the blood stream, which can lead to lower lipophilicity and drug uptake (138).
One of the major questions regarding PtIV compounds concerns the in vivo kinetics and the mechanisms of reduction. Several cell-free and in vitro experiments investigated this problem, but still the reactions are not fully understood and in vivo analyses are incomplete. A large amount of molecules that are involved in the redox homeostasis of cells can reduce PtIV complexes, such as GSH, methionine, cysteine, ascorbate, and others. These reductants were mainly investigated with model compounds such as trans-[PtCl2(CN)4]2−, tetrachloridoam(m)ine platinum(IV) compounds and cis,-trans,cis-[PtCl2(OCOCH3)2(NH3)2] (68, 210, 345, 346).
As described previously (155), GSH possesses the ability to detoxify PtII drugs and enhanced GSH levels are associated with resistance (compare Section II.A.). With regard to PtIV complexes, GSH is believed to have an important role in activation (eq. 9). Eastman et al. showed that tetraplatin binds only very slowly to DNA whereas the addition of two stoichiometric equivalents of GSH markedly increased this reaction.
| 9 |
At higher GSH concentrations the DNA binding of tetraplatin decreased, indicating that the PtII analog of tetraplatin can be detoxified by reaction with GSH comparable to cisplatin (101). These data were confirmed by Kido et al. in a cell-free setting using salmon sperm DNA (191). Notably, levels of DNA platination after incubation of tetraplatin with GSH were similar to those of its reduction product [PtIICl2(Dach)] (61).
A sensitive leukemic L1210 cell model and two cisplatin- and oxaliplatin-resistant cell lines are sensitized toward tetraplatin by addition of GSH (191). A relationship between intracellular GSH levels, drug resistance, and cytotoxicity was shown for tetraplatin and iproplatin in several cell models (245, 297). However, in another study GSH cotreatment with tetraplatin of intraperitoneally inoculated cisplatin-sensitive and -resistant L1210 tumor cells in mice did not enhance the activity and reduced the platinum concentration in the plasma compared to tetraplatin alone (192).
One possible reduction mechanism (Fig. 10) of tetraplatin and other PtIV complexes with axial halogenido ligands by GSH is a halogenido-bridged electron transfer. Therefore, the thiol of GSH reacts with the highly polarized chlorido ligand of the platinum complex. From the resulting GS-Cl-PtIV transition state GSCl is eliminated, which can further react with GSH to GSSG and HCl. Expulsion of the trans ligand yields the square-planar platinum(II) complex (138).
FIG. 10.

Possible reduction mechanism of tetraplatin and other PtIV complexes. In the case of PtIV drugs like tetraplatin it is assumed that reduction with GSH occurs via a halide bridged electron transfer from GSH to PtIV resulting in GSCl and the corresponding PtII species. GSCl further reacts in aqueous solution with GSH yielding GSSG and HCl. Adapted from refs. (138, 210).
In addition to tetraplatin, also for iproplatin a relationship between intracellular GSH levels, drug resistance, and cytotoxicity was shown in several cell models (245, 297). Recently, Volckova et al. suggested a new mechanism for the reduction of iproplatin in which one GSH is coordinated to the metal center in equatorial position before the reduction of PtIV to PtII by additional equivalents of GSH. This reaction yields in chloridobis(isopropylamine)(glutathionato)platinum(II) and not the commonly believed cis-dichloridoplatinum(II) complex (392). Controversial data have been presented, whether GSH can reduce or detoxify satraplatin. In contrast to iproplatin, satraplatin was stable in vitro in GSH-containing solutions with and without NADH (55). On the one hand, GSH has been proposed as major deactivation pathway for satraplatin (112, 313). On the other hand, Mellish et al. found no correlation between GSH and satraplatin-induced cytotoxicity (246) and no increased GSH levels were found in JM118-resistant cells (327).
Beside the cysteine of GSH, also a range of other proteins/biomolecules possessing cysteine (containing a thiol moiety) or methionine (containing a thioether moiety) are able to interact with platinum complexes. The Cys thiol and the Met thioether are oxidized to disulfide-bridged cystine (compare Section II.A.) and methionine S-oxide, respectively. The cysteine/cystine system has a major structural function in biomolecules and the redox balance of cells. In general, thiols are stronger reductants and more pH-dependent than thioethers.
The model substance trans-[PtCl2(CN)4]2− is reduced by both cysteine and methionine at 2:1 and 1:1 molar ratios (amino acid: Pt complex), respectively (345, 346). There are only limited data available whether iproplatin or tetraplatin can be reduced by these amino acids. Pendyala et al. hypothesized that iproplatin can be reduced intracellularly by cysteine (296), but no mechanism of reduction has been suggested. In the case of tetraplatin in vivo biotransformation products are, next to PtII(Dach)Cl2, also Dach-Pt-methionine and Dach-Pt-cysteine species (374).
Next to GSH, also ascorbic acid (vitamin C) is considered to be a major low-molecular-weight antioxidant/reductant in the body (compare Section II.A.). A number of papers investigated the possible reduction of PtIV complexes by ascorbate. However, the investigations disagree in key aspects (42, 74, 211, 212, 402). Further, ascorbic acid has two pKa values with 3.95 and 11.24 (H2A ↔ HA− ↔ A2−). Thus, at physiological pH nearly all the ascorbic acid (H2A) is present as the singly deprotonated ascorbate anion (HA−) (138), which is therefore the major reductive species in the cellular environment.
Concerning the interaction of ascorbate with PtIV, Bose et al. suggested a complex mechanism for the reaction of iproplatin with ascorbate at pH 7 (42, 402). Therefore, the overall reactions are the expected two one-electron oxidations of ascorbate yielding dehydroascorbic acid and simultaneous reduction of PtIV to PtII. However, the direct reaction of iproplatin and ascorbate is very slow. Thus, a PtII catalyzed reduction of a PtIV-ascorbate complex by a second ascorbate molecule, with intermediate ascorbate radicals, is believed to take place. Choi et al. analyzed the reduction of PtIV complexes at pH 7 and confirmed the expected correlation between the reduction rate and the reduction potential (74). The investigations showed again that iproplatin is very slowly reduced by ascorbate and Choi supports the occurrence of an ascorbate radical. In contrast, Lemma et al. suggested for some model compounds like cis-[PtCl4(NH3)2] that not ascorbate or a PtII- catalyzed reaction is responsible for the PtIV reduction but the doubly deprotonated form of ascorbic acid A2− (211, 212), even though it represents less than 1% of ascorbic acid at pH 7. The authors assume that the electron transfer from ascorbate to the PtIV center involves a reductive attack by A2− /HA− on one of the halido ligands forming an activated halido-bridged complex with subsequent elimination of two trans ligands and formation of PtII (211). In a further study of this group, the reduction of satraplatin by ascorbate to JM118 was investigated (Fig. 9). It was found that only A2− and not HA− was able to reduce satraplatin at pH 7 with a suggested outer-sphere mechanism as described above (212). Recently, Gibson et al. analyzed the reduction of a doubly labeled cis,trans,cis-[PtIVCl2(OCO13CH3)2(15NH3)(n-butylamine)] complex by ascorbate at pH 7 with [1H,15N] and [1H,13C] HSQC NMR spectroscopy. Interestingly, the NMR pattern revealed that the elimination by ascorbate did not only lead to the expected product [PtIICl2(15NH3)(n-butylamine)] without the two axial acetato ligands, but also to complexes that have lost one axial acetato and one equatorial chlorido ligand, or two equatorial ligands, suggesting the existence of multiple reduction mechanisms (124, 268). These findings confirm that the reduction of PtIV by ascorbate might depend on several factors and that diverse reduction pathways can take place.
As discussed above, it is thought that sulfhydryl groups are major players in the reduction of PtIV compounds. However, in the case of satraplatin, there is so far no evidence for reduction by GSH, methionine, or cysteine. Recently, a new possible mechanism was suggested by Carr and colleagues (55). They investigated the reduction of satraplatin by heme-containing metalloproteins, such as cytochrome c and hemoglobin, and the role of their iron atoms. Satraplatin was stable in solutions containing hemoglobin or NADH alone. However, when hemoglobin and NADH were combined, satraplatin was reduced mainly to JM118. As this reaction could be inhibited with carbon monoxide, which inhibits heme-containing proteins by binding to the heme-iron, involvement of the heme ferrous iron was suggested. Similar results were obtained with cytochrome c. In contrast, reduction of satraplatin by cysteins in hemoglobin was, comparable to GSH, not observed, as shown by incubation with a sulfhydryl blocking agent (55). The role of redox-active proteins in the reduction of platinum(IV) complexes in the cellular environment is supported by differing reduction rates of cis,trans,cis-[PtIVCl2(OCO13CH3)2(NH3)2] in aqueous extracts measured for several cancer cell lines. Interestingly, kinetics found for the high-molecular-weight fraction (>3 kDa) of the extracts was very similar compared to the whole cell extracts, whereas the low-molecular-weight fraction (<3 kDa), including GSH, was nearly ineffective in reducing PtIV (267). However, biological data from several studies demonstrated an impact of GSH and other intracellular reductants on the activity of PtIV complexes. Nevertheless, it has to be considered that there are still major missing links to understand the intracellular mechanisms of these reactions. Also, the impact of intracellular enzymatic reduction by, for example, one-electron reductases is relatively unexplored (89, 90).
Consequently, “what do we really know about it?” asked Gibson critically in a recent review about the mechanism of action of platinum agents (124). He addressed the problem that most of the information on the mechanism of action of platinum compounds comes from cell-free analyses of biologically relevant molecules—for example, nucleosides—in aqueous solutions coincubated with platinum drugs using chemical methods, which lack the sensitivity and specificity necessary to characterize the platinum species in biological solutions. On the other hand, biochemical analyses of biological fluids, cells, or animals treated with the drugs have insufficient resolution to characterize platinum adducts at the molecular level (124).
B. Gold
The medical use of gold (Au) has a long history. Already the ancient Egyptians used gold compounds as therapeutic agents and alchemists made elixirs of “drinkable gold,” as it was believed that gold has immortalizing properties. The rational use of gold compounds in medicine started with the application of gold cyanide against tuberculosis in the 1920s. However, due to severe toxicities the treatment was changed to less toxic gold(I)thiolate complexes, namely, aurothiomalate and aurothioglucose (Fig. 11). These complexes were also applied against rheumatoid arthritis, an autoimmune inflammatory disease, which was thought to be a disease related to tuberculosis (250). In 1985, auranofin, [tetra-O-acetyl-β-D-(glucopyranosyl)thio](triethylphosphine) gold(I) (Fig. 11), was approved as orally available drug against rheumatoid arthritis, which was less toxic but also less efficient. However, auranofin still causes enormous side effects and only a subgroup of patients responds to the treatment. Due to this, only severe cases of rheumatoid arthritis are currently treated with AuI drugs (250, 272). The success of cisplatin in cancer therapy and a prospective long-term study which showed that rheumatoid patients treated with AuI compounds had a lower rate of malignancies than those treated with other drugs (117), led to a comprehensive search for AuI and AuIII complexes against cancer. However, beside auranofin no further gold compound was so far approved for the treatment of any disease. Currently, aurothiomalate is investigated in a phase I study against advanced nonsmall cell lung cancer (clinicaltrials.gov identifier: NCT00575393).
FIG. 11.
AuI drugs relevant for rheumatoid arthritis therapy additionally harboring anticancer activity.
1. Gold(I)
Gold in its elemental form is stable in a wide range of conditions. The oxidation states of gold range from −1 to + 5, out of which 0 (d10s1), + 1 (d10), and + 3 (d8) are the most important ones. The coordination geometry of the gold(I) complexes is usually linear (two ligands), even though there can also be a trigonal three-coordinate or a tetragonal four-coordinate sphere surrounding the gold center (343). Like PtII, AuI is regarded as a soft acid in the HSAB concept and prefers soft ligands (bases), as, for example, thiolates, cyanides, phosphines, and soft halides. Main representatives of AuI complexes with anticancer activity are aurothiomalate, aurothioglucose, auro(bis)thiosulfate, and auranofin (Figs. 11 and 12). With exception of the latter, these complexes form polymers with AuI connected via thiolate sulfur bridges (343). In general, AuI complexes are thought to be pro-drugs because they rapidly exchange their ligands and several gold-containing metabolites are formed. In the blood, for example, one of the major anchoring sites is the deprotonated cysteine-34 of serum albumin. In the case of auranofin, the binding leads to a release of the triethylphosphine ligand and consequently to oxidation to AuIII (76). For the cellular AuI uptake it has been postulated that the albumin-bound AuI and other metabolites can be transported into and out of cells via a thiol-shuttle (357).
FIG. 12.

Experimental AuI drugs.
Important metabolites of AuI complexes are dicyanoaurate(I) ([AuI(CN)2]−), metallothionein—and glutathione—AuI complexes (104, 207, 284). In general, it is believed that, due to their thermodynamical stability, AuI drugs do not change their oxidation state in vivo. However, there is evidence for the generation of AuIII species by powerful oxidants such as hypochlorite, an immunological oxidant at inflammation sites. Hypochlorite is involved in the generation of the metabolite [AuI(CN)2]−, which can be found in the blood and urine of gold-treated patients. During an oxidative burst in granulocytes and macrophages cyanide is generated from thiocyanate and hypochlorite, which can further react in vivo with AuI drugs to form [AuI(CN)2]− . Further, it has been shown that [AuI(CN)2]− can be oxidized by hypochlorite to AuIII species, such as tetracyanidoaurate ([AuIII(CN)4]−) (53). As shown by electrospray ionization-mass spectrometry, GSH can then reduce the generated AuIII species through the two intermediates [Au(CN)3(GS) −H]2− and [Au(CN)2(GS)2]3− back to [AuI(CN)2]− (417). In general, formation of [AuI(CN)2]− species leads to an enhanced gold uptake into red blood cells and has been connected to enhanced side effects. Therefore, a better understanding of the AuI/III redox cycling is of great interest for the clinical use of gold compounds (53, 343).
In several studies, a number of AuI complexes showed in vitro and in vivo anticancer activity. Most of the initially developed AuI compounds, including auranofin, are active in animal models against leukemia but not against solid tumors. The greatest activity was achieved when AuI was coordinated to phosphine- and thiosugar-ligands. Based on this knowledge, a series of AuI-phosphine complexes was synthesized. On the one hand, neutral two-coordinate complexes, such as auranofin and [chlorido(triethylphosphine)gold(I)] (Fig. 12) exist on the other hand, a group of cationic, tetrahedral AuI complexes with two chelating bis(diphenylphosphine)ethane (DPPE) or bis(di-2-pyridylphosphino)propane (D2PYPP) ligands (Fig. 12) have been developed.
Initially, it has been thought that AuI compounds target DNA similar to cisplatin. However, later it has been shown that DNA is not the primary target (202). In addition to the above described redox cycling of AuI compounds, an interaction with cellular redox processes by targeting mitochondria has been demonstrated (160, 161, 307). One of the earliest observed effects after [Au(DPPE)2]+ treatment in cisplatin-sensitive or -resistant murine P388 leukemia cells as well as in rat hepatocytes was the decrease of ATP concentration and stimulation of mitochondrial respiration. It has been suggested that [Au(DPPE)2]+ caused an uncoupling of oxidative phosphorylation, and thus inhibition of oxidative ADP phosphorylation (159, 356). One of the major impacts of AuI substances on redox homeostasis of cancer cells is the inhibition of the cytosolic and mitochondrial Trx system (compare Section II.A.) (122, 378, 389). Due to the high affinity of AuI to soft ligands (122), it is not surprising that AuI complexes might bind to the selenium atom of TrxR and thereby inhibit the activity of both cytosolic and mitochondrial TrxRs. In combination, TrxR inhibition and disturbance of mitochondrial respiration lead to increased ROS, mitochondrial swelling, a decrease in mitochondrial membrane potential, and subsequently to apoptosis. Additionally, it has been shown that auranofin inhibits the TrxR1 in a p53-independent manner (153). Similar to cisplatin (compare Section V.A.1.), AuI complexes effect the GR and Gpx system only at higher concentrations. Explanations for this are either the structure of the active site or the lability of the gold–ligand bonds (122, 209). Further, it has been shown that the generation of H2O2 by AuI complexes did not cause significant lipid peroxidation. Therefore, it was concluded that there is no generalized oxidative stress responsible for AuI-induced cell death. Additionally, no enhanced nitric oxide production and no alterations in GSH levels or its redox status were observed (320). Next to alterations of the GSH- and Trx-system, auranofin potently induces HO-1 expression by activating Keap1/Nrf2 signaling via Rac1/iNOS induction and MAPK activation (196) (Compare Section II.B.). Further, it has been shown that auranofin can inhibit the activation of STAT3, NF-κB, and the homodimerization of toll-like receptor 4 (177, 195, 420).
2. Gold(III)
AuIII is isoelectronic and isostructural with PtII and forms therefore also square-planar four-coordinate complexes. However, due to the high reactivity of AuIII complexes and reduction to AuI or Au0 under physiological conditions, it has been questioned whether they might be useful drugs. Nonetheless, there is a growing interest in AuIII complexes, as novel substances with improved stability are available (Fig. 13). AuIII complexes can be divided in four subgroups, namely, (i) classical square-planar mononuclear gold(III) complexes, most often with nitrogen or halide ligands, (ii) gold(III) porphyrins, (iii) organometallic gold(III) compounds with carbon-gold bonds, and (iv) oxo-bridged dinuclear gold(III) complexes (272). In contrast to AuI complexes, there is a greater affinity of AuIII for DNA and the binding can be both electrostatic and covalent. However, several studies suggest that the formed AuIII-DNA adducts are less stable than that formed by cisplatin (236, 322) presumably because of lower hydrolytic stability (57, 322).
FIG. 13.

Experimental AuIII drugs.
Similar to AuI complexes (and platinum(II) drugs), AuIII compounds are known to strongly target sulfur-containing amino acids (preferably cysteins), imidazole (His), and selenol groups (selenocysteine) of proteins. Therefore, it is not surprising that for a great number of AuIII complexes inhibition of the TrxR and disruption of the mitochondrial functions have been proposed as major modes of action (272). The GSH reductase system is only inhibited at higher concentrations of AuIII drugs, comparable to AuI compounds (321).
Two proteomic studies support the general idea that AuIII disturbs the cellular redox balance (229, 395). Next to the AuI complex auranofin, [Au2(6,6′-dimethyl-2,2′-bipyridine)(l-O)2] PF6 (Auoxo6) (Fig. 13), an oxo-bridged dinuclear AuIII complex, and AuIII porphyrin 1a (Fig. 13), alter proteins involved in the cellular redox homeostasis, including Trx and peroxiredoxin 1 and 3 (395). Based on these data, it has been proposed that Auoxo6 has a mode of action comparable to auranofin. The observations strongly suggest that Auoxo6 is reduced to a AuI species in the biological milieu (229). This hypothesis is supported by a previous study with a series of dinuclear AuIII complexes, including Auoxo6. In cell-free systems, ascorbic acid, and GSH, added at a slight excess, caused a relatively fast and complete reduction of the AuIII centers. Further, interactions with human serum albumin, horse heart cytochrome c, and bovine ubiquitin were analyzed spectrophotometrically. The spectral patterns suggested a progressive reduction of AuIII centers and a concomitant appearance of the respective free ligands. The authors concluded that all tested compounds retain significant oxidizing properties and, thus, may undergo important redox-driven transformations within a reducing biological environment (57).
Next to the inhibition of TrxR, a variety of mechanisms of action were proposed for AuIII complexes, such as the modulation of kinases and proteasome inhibition (272). Interestingly, inhibition of ROS production by NAC reversed the inhibition of the proteasome by a AuIII-dicarbamato complex (AUL12) (Fig. 13). Even though analyses with the AuI analog AUL15 resulted in a similar outcome, this substance did not induce the production of ROS. Therefore, the authors suggest that different redox-dependent and -independent mechanisms are responsible for the overall different effects of AuI and AuIII complexes (428).
C. Arsenic
Arsenic (As) has two biologically important oxidation states, AsIII and AsV. AsIII, as a soft metal ion (comparable to PtII and AuI), preferentially reacts with sulfur- and nitrogen-containing residues of proteins, such as thiols in cysteines and imidazole nitrogens in histidine residues (compare Section IV.C., V.A.1., and V.B.1.). The interaction with thiols can generate stable cyclic dithioarsinite complexes in which both sulfur atoms are bound to arsenic. These reactions can cause loss of function of the involved proteins and might be a key factor of arsenic cytotoxicity (93). AsIII compounds are known to interfere with and disturb the oxidation/reduction equilibrium through complex redox reactions involving the cellular oxidant/antioxidant systems, including GSH and TrxR (225) (compare Section II.A.). In contrast, AsV compounds, whose biological activity is mainly based on substitution for phosphate in molecules like ATP, are significantly less cytotoxic as compared to AsIII (271, 298).
Arsenic compounds have been used by humans in many respects since ancient times for example in various alloys, and as pesticides, herbicides, insecticides, and also for medical purposes [for review see (271)]. Thus, some of the oldest remedies known include arsenic. These compounds were empirically discovered as treatment for diverse diseases and in variable preparations, including external pastes, oral preparations, and injections. Also, in traditional Chinese medicine, arsenic acid and arsenic trioxide (ATO) (Fig. 14) were used as antiseptic agents or in the treatment of rheumatoid diseases, syphilis, and psoriasis. In the Western world the potassium bicarbonate-based Fowler’s solution of ATO for oral use developed in 1788 was frequently applied against aczema, asthma, and psoriasis but also against malignant diseases including leukemias like CML and Hodgkin’s disease. In fact, already Celsus in the first century AC had suggested activity of arsenic against solid tumors (271). During the 18th and 19th century ATO represented the main treatment for leukemia and its importance remained until the development of modern radio- and chemotherapy during the 20th century. Then ATO was replaced by novel chemotherapeutic regimens and in part was abandoned based on chronic toxicity in treated patients. Surprisingly, during the 1990ies a Chinese group reported an exceptionally high rate of complete, long-lasting remissions after ATO treatment in a small cohort of patient with acute promyelocytic leukemia (APL), a specific subtype of acute myeloid leukemia (AML) (66, 344, 431). These promising initial data were proofed in larger patient cohorts and international randomized studies (359, 360), leading to the approval of ATO for the treatment of APL in 2000. Concerns remained about arsenic poisoning and secondary malignancies known to result from long-term environmental exposure to inorganic arsenic mainly due to drinking water contamination (339). However, long-term observations (mean 70 months since treatment) in China did not indicate a higher risk for secondary malignancies in 85 alltrans retinoic acid/ATO-treated APL patients and urine arsenic levels had returned to levels far below the safety limit 24 months after the last treatment (167).
FIG. 14.
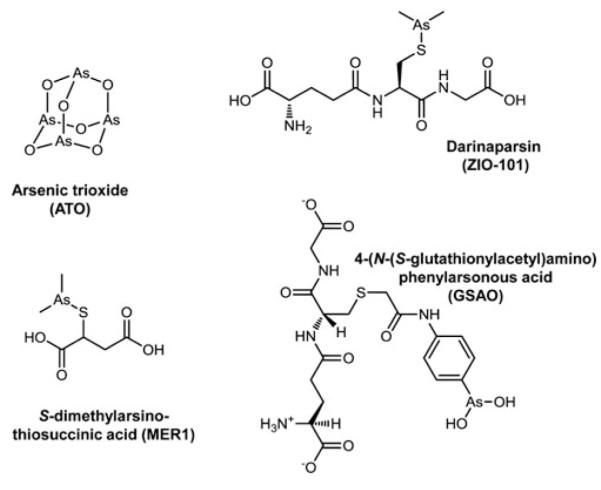
AsIII drugs. ATO is approved for treatment of acute promyelocytic leukemia, whereas the other compounds are in (pre)clinical development.
Based on the persistent environmental exposure, sophisticated metabolic pathways have developed during evolution allowing efficient detoxification of arsenic-containing compounds, which now also impact on ATO as clinically applied drug. The redox-driven metabolism has been studied extensively concerning environmental intake and toxicity, whereas specific studies on ATO as cancer therapeutic are comparably sparse. Immediately after dissolution of ATO in water, it forms arsenous acid (H3AsO3), the trivalent hydrolysis product of ATO (367), which is thought to be the pharmacologically active form of ATO. Arsenic is progressively methylated during its metabolism/detoxification involving a series of oxidation and reduction steps (Fig. 15). S-Adenosylmethionine represents the major methyl donor for these reactions. In general, only arsenic(III) species (e.g., inorganic arsenic(III) compounds or monomethyl arsenous acid) can be methylated by the arsenite methyltransferase to the respective arsenic(V) metabolites (monomethyl arsonic acid or dimethylarsinic acid). Thus, continual reduction steps are necessary to allow progressive methylation reactions. Several enzymes have been suggested to drive these reductions including most importantly glutathione-S-transferase omega (GSTO)—involving GSH as a reductant—(422, 423) and recently also a glyceraldehyde-3-phosphate dehydrogenase (132). As GSTO (−/−) mice are still able to reduce AsV (75), it was also suggested that arsenite methyltransferase might itself harbor the respective reductive activity, whereas Trx and NADPH are used as electron donors (339). Consequently, this enzyme would be sufficient for sustaining the whole methylation pathway which was experimentally confirmed at least in cell-free systems (216, 401).
FIG. 15.

Arsenic Metabolism. (A) The classical oxidative methylation pathway of arsenic is shown involving sequential reactions of reduction and oxidative methylation steps. (B) Alternative pathway scheme for methylation of arsenic involving generation of arsenic-glutathione (GSH) complexes. From ref. (373).
In addition to this well-described oxidative methylation pathway, recently a reductive methylation pathway was discovered (150), circumventing the need for subsequent oxidation/reduction steps and involving the formation of an arsenic triglutathione complex (Fig. 15). This complex is a direct substrate of arsenite methyltransferase catalyzing the formation of methylarsenic diglutathione and dimethylarsenic glutathione, which are hydrolyzed at low GSH concentrations followed by H2O2-mediated oxidation to monomethylarsonic and dimethylarsinic acids (13, 271). Little is known to what extent these methylation pathways are important during treatment of APL patients with ATO.
With regard to its anticancer activity, the mechanisms underlying the mode of action of ATO are complex and cell type-dependent (107). However, it has to be stated that in general DNA damage, which is frequently suggested for metal-containing anticancer agents, is not involved in the activity of ATO. Besides direct interaction with the APL-specific PML-RARα fusion protein (429), a multitude of studies in diverse cell types have indicated that ATO-induced cytotoxicity is at least in part based on the enhanced production of ROS including H2O2, superoxide anion, and hydroxyl radical in a Fenton-like reaction (compare Section IV.A.) and consequently in radical-mediated signals/damages (43, 180).
Paul et al. suggested that ROS production was mainly mediated via an electron transfer inhibition of complex I of the electron transport chain of the mitochondria, whereas no significant effects on complex II and III were detected (292). This is in good agreement with the fact that ATO-induced apoptosis is mainly characterized by progressive mitochondrial membrane depolarization, and enhanced radical stress. Accordingly, bcl-2 family members exerting their apoptosis-regulatory mechanisms mainly at the outer mitochondrial membrane have major impact on ATO-induced cytotoxicity. Additionally, mitochondrial protein translation by thiostrepton-sensitized melanoma cells against ATO (44). Moreover, ATO has been characterized as an “oxidative stress-sensitive drug,” meaning that cells under enhanced ROS-mediated stress are hypersensitive against ATO (419). Thus, the anticancer activity of ATO and the exerted side effects are strongly influenced by the cellular redox status and the functionality of radical-scavenging protection systems like the GSH and Trx systems (compare Section II.A.). Nevertheless, it has to be mentioned that several recent studies challenged the role of ROS in ATO-mediated apoptosis induction. Morales et al. demonstrated that the strong ATO-mediated antioxidant response, mainly mediated by the antioxidant-induced transcription factor Nrf2 (compare Section II.B.), is not required for ATO-induced apoptosis in four myeloma cell models (255). Neither Nrf2 down-modulation by siRNA nor ROS inhibition by butylated hydroxyanisole (BHA) protected cells from ATO. Surprisingly, ROS generation was even dispensable from Nrf2 activation. Interestingly, also the ATO-chelating cysteine-rich MTs (Compare Section II.B.), well-known to mediate protection against environmental metals, were inefficient to block ATO-mediated apoptosis.
In contrast to ROS, the role of GSH in the regulation of ATO cytotoxicity is beyond dispute. An inverse correlation between the cellular GSH content and the activity of ATO has been demonstrated in multiple cancer models (107). Stimulating the activity of GSH peroxidases by pretreatment with selenite-mediated ATO resistance in APL cells (180). Upregulation of GSH levels by, for example, N-acetylcysteine (NAC) or lipoic acid protected leukemic and solid tumor cells against ATO (87). Accordingly, GSH depletion by BSO (87, 107, 410, 414) or ascorbic acid (87, 125, 131) distinctly enhanced ATO cytotoxic activity against multiple cancer cell types. Moreover, treatment with ATO itself reduced the cellular GSH content (142). Consequently, ATO exerts synergistic activity with several other agents disturbing the cellular redox/ROS status, including, for example, substances of natural origin like isoflavones (329), the α-tocopherol (vitamin E) analog trolox (92), and cisplatin (427). Assuming a role of ROS in ATO-mediated cytotoxicity, GSH might exert its protective function mainly as a radical scavenging agent. Indeed, GSH binds arsenic to form a transient As(GS)3 complex (see above), thus preventing the inhibition of cellular redox-regulatory enzymes. Moreover, reduction of pentavalent to trivalent arsenic can occur nonenzymatically with GSH as electron donor, or via GSTO again involving GSH as a reductant. Additionally, as arsenic is believed to involve electrophilic attacks of cysteine residues in cellular proteins, GSH might function as a substrate sequestering arsenic from critical cysteine-containing cellular proteins (370, 386).
A second important cellular redox stabilization system influenced by ATO is Trx together with its reducing enzyme TrxR (compare Section II.A.). Also, overexpression of Trx-1 protected cancer cells against ATO-mediated mitochondrial apoptosis induction (375). When TrxR was inhibited by dinitrochlorobenzene (DNCB) or natural compounds (e.g., isoflavonoids), cells were sensitized toward ATO, again indicating that reduced Trx can counteract ATO-mediated cytotoxicity (178, 375). Consequently, it was shown that ATO itself is capable of inhibiting TrxR by interaction with the enzyme’s active site (225). Additionally, arsenites and the trivalent metabolite monomethylarsonic acid were identified as potent inhibitors of TrxR (219). Moreover, besides GPx, TrxR belongs to the most important cellular selenocysteine residue-containing proteins (compare Section II.A.) and arsenic is well known to interfere with the selenium metabolism in a redox-dependent manner. Accordingly, as an additional interaction between ATO and the cellular redox system, significant impacts of ATO and/or its metabolites on the expression of GSH and Trx have been reported (123, 369). Further, ATO was suggested to induce ER stress-mediated apoptosis in human neutrophils (38), again suggesting proteins as direct targets of ATO-mediated cytotoxicity.
In general, research on arsenic-containing anticancer drugs focused so far mainly on ATO. However, some other compounds were investigated with regard to their anticancer activities. Hence, darinaparsin (ZIO-101; Fig. 14)—a dimethylated arsenite compound linked to GSH—seems to be active against a wide variety of hematologic and solid tumors and to exert less severe side effects (387). Comparable to ATO, darinaparsin induces apoptosis via the mitochondrial pathway. However, it does not impact on bcl-2 and the oncogenic APL fusion protein. Moreover, this compound exerts even stronger ROS production as compared to ATO. In contrast to ATO, the cytotoxicity of darinaparsin is not dependent on intracellular GSH levels and it exerts activity against ATO-resistant tumors (235, 241). In general, mode of action data for this compound are very limited so far. Nevertheless, this novel arsenic compound has been evaluated in several phase I/II studies, whereby one phase II study with intravenous application of darinaparsin in hepatoma failed to show clinical benefit and consequently was terminated after the first stage of efficacy analysis (409). In contrast, a phase I study at a different schedule demonstrated promising activity in several therapy-refractory solid tumor types (387). Another example for an organic arsenic compound, S-dimethylarsinothiosuccinic acid (MER1, Fig. 14), demonstrated PML-RAR-independent, ROS-mediated cytotoxic activity against cancer cells in vitro and limited toxicity in vivo (128). However, clinical evaluation of this compound has not been reported so far. 4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid (GSAO, Fig. 14) is a small, synthetic mitochondrial poison containing trivalent arsenic that targets angiogenic endothelial cells (92) and is currently being tested in a phase I clinical trial (NCT01147029) (97) and first antivascular activities were reported from that study at ASCO 2010 (J Clin Oncol 28:15s, 2010; suppl; abstr TPS167). GSAO is believed to exert its antiangiogenic activity by interacting with two cysteines of the adenine nucleotide translocator (ANT) at the inner mitochondrial membrane. Inactivation of ANT by GSAO causes increase in superoxide levels based on mitochondrial damage, proliferation arrest, ATP depletion, mitochondrial depolarization, and apoptosis in endothelial cells. GSAO is processed at the cell surface and in the cytosol especially by γ-GT before reacting with mitochondria (94). Whether redox mechanisms are involved in the anticancer/antiangiogenic activities is widely unknown.
D. Ruthenium
Ruthenium (Ru) is a relatively rare element, which has, to current knowledge, no biological functions. Ru compounds occupy a wide variety of oxidation states (−2, 0, + 2, + 3, + 4, + 6 and + 8), of which RuII and RuIII are most relevant in biological environment, and different coordination geometries are known, that are, tetrahedral, square-pyramidal, and octahedral (162, 164).
Ruthenium complexes are among the best studied non-platinum metal complexes with anticancer activity, and two candidates, KP1019 and NAMI-A (Fig. 16), have recently been tested in clinical phase I trials. KP1019 was developed for solid tumors, whereas NAMI-A was developed as a purely antimetastatic drug. Both compounds proved to be tolerable with only minor side effects, especially in case of KP1019, whereas formation of blisters was considered as dose-limiting toxicity in case of NAMI-A (95, 149, 308). Additionally, 5/6 and 1/24 patients with solid tumors obtained a stable disease after treatment with KP1019 and NAMI-A, respectively (149, 308). Next to RuIII compounds, there are currently several promising organometallic RuII complexes with arene ligands (Fig. 16) in preclinical evaluation (45, 413).
FIG. 16.

Ruthenium drugs. KP1019 and NAMI-A have been already evaluated in clinical trials, whereas all others are under preclinical investigation.
With regard to their modes of action, ruthenium complexes have been assumed to target DNA comparable to platinum drugs and the DNA-binding properties of ruthenium compounds have been studied extensively mainly under cell-free conditions. However, although Ru has been detected in nuclei and bound to extracted DNA of cells after drug treatment, there is increasing evidence that the anticancer activity of some ruthenium compounds, like KP1019 and NAMI-A (77, 79, 154), but also of some RuII(arene) complexes (57), is not based on direct DNA damage. RuIII compounds are characterized by a high affinity to (serum) proteins, which has been suggested to be crucial for drug accumulation into the tumor tissue and to be responsible for the minor adverse effects observed in clinical trials with KP1019 (365, 377).
Ruthenium complexes can be divided into two major classes, namely, octahedral RuIII complexes and piano-stool RuII compounds. The classical octahedral RuIII coordination compounds, like KP1019 or NAMI-A, feature a ruthenium center, which is usually able to be reduced and reoxidized in the cellular environment. The ability of the cellular redox systems to reduce/oxidize the ruthenium complex strongly depends on the exact coordination sphere. For example, in an extensive study on the role of the number and nature of the azole ligands on the antiproliferative activity and their redox potentials, a significant correlation for these parameters was found for a series of mono-, bis-, tris-, and tetrakis indazole/imidazole complexes (14, 173, 317). Thus, for the bis(indazole) complex KP1339 (redox potential ~0.03 V vs. NHE) an IC50 of ~120 μM was observed, whereas for the bis(imidazole) complex KP418 (redox potential −0.24 V vs. NHE) even 300 μM did not induce 50% growth inhibition in SW480 cells (186). In contrast to the RuIII compounds, RuII(arene) “piano-stool” complexes are normally unable to change their +2 oxidation state due to stabilization by the -bonded arene ligand.
Comparable to PtIV and some CoIII compounds (compare Section V.A.2. and V.H.), the principle of “activation by reduction” is a central hypothesis in the mode of action of many RuIII drugs. However, there are some major differences between PtIV and RuIII complexes: reduction of the PtIV center to PtII induces profound changes in their coordination geometry (from octahedral to square-planar) and leads to ligand release, whereas the coordination geometry of Ru compounds remains widely unchanged upon reduction (318). However, for both RuIII and PtIV complexes reduction causes labilization and subsequent ligand exchange reactions, such as Cl to aqua in case of KP1019 (318, 338). Consequently, reduction facilitates and often increases reactivity with biomolecules and, in some cases, even determines the structure of the formed adducts. For example, in case of ethylenediaminetetraacetato (EDTA) Ru complexes, binding to the N3 and N7 atoms of GMP was found to be dependent of the Ru oxidation state (63).
The reduction of RuIII compounds by GSH and other biological reductants such as ascorbic acid has been extensively investigated, however, mainly in cell-free settings. Notably, due to the tight binding of Ru drugs to serum proteins the extracellular reduction of the Ru center seems improbable (300, 376). Consequently, it is assumed that reduction of ruthenium compounds takes place inside the cell after release of the Ru moiety from its biological carrier, which makes the Ru complex accessible for reduction (291). As DNA has been in the focus as major intracellular target for a long time, many experiments have been performed using DNA as reaction target (77, 80, 116, 334). Such studies show, for example, that the selectivity of [Ru(NH3)5Cl]2+ (Fig. 16) for DNA bases is influenced by GSH. The reaction with adenine and cytosine and the cleavage of such adducts is less affected by GSH, whereas the binding to guanine is significantly altered (116). Moreover, agarose gel electrophoresis studies with plasmid DNA and [RuIII(NH3)5] complexes revealed in presence of a reducing agent and O2 moderate DNA cleavage ability, potentially via a hydroxyl radical mechanism, whereas coordination of a [RuIII(NH3)5] to DNA did not cause DNA cleavage (78).
Several studies indicate that reduction of ruthenium facilitates reaction with biomolecules only at low GSH concentrations (116, 338). At higher GSH concentrations often decreased reactivity (149, 338) probably due to coordination of GSH to the reduced species and reoxidation to RuIII was observed. Interestingly, also in case of some RuII(arene) complexes (99, 393) redox reactions with GSH were reported in cell-free settings, although the RuII center itself is usually unable to participate in redox reactions (148). Notably, the kind of interactions with GSH differ between the diverse RuII(arene) complexes. For example, GSH conjugation to the Ru center by substitution of the chlorido ligand was reported in case of 1,2-ethylenediamine (en) complexes (393). In the case of phenylazopyridine RuII complexes the ligand is reduced causing catalytic oxidation of GSH to GSSG, in contrast to the metal-free ligand alone which is redox inactive (99). Also, for a Ru(arene)(en) complex bearing thiolato ligands such as isopropyl- and phenylthiolates, oxidation of the ligand as well as of GSH was observed in the presence of oxygen, which is reduced to ROS (299). Together, this indicates that already in cell-free systems the reaction pathways of ruthenium complexes are very complex and difficult to predict. As only a few studies have been performed on living cells, the in vivo situation is even less understood. Some of these experiments support the hypothesis of activation by GSH-mediated reduction also in vivo. Thus, enhanced activity of several RuIII compounds (including KP1019 and analogs) has been reported against the cisplatin-resistant cell model O-342/DPP, which is characterized by enhanced GSH levels (118, 426). In case of RuII(arene) drugs the activity against GSH-overproducing cisplatin-resistant A2780cis cells differed throughout the tested compound panel (6). However, there are also reports on the protective effects of intracellular GSH levels against RuIII drugs. For example, depletion of the intracellular GSH pools by pretreatment with BSO led to increased sensitivity of cancer cells to [Ru(NH3)5Cl]2+ (116) or KP1019 (155), and pretreatment with the radical scavenger and GSH precursor NAC protected human colon carcinoma cells against KP1019-induced ROS (185). Consequently, a comprehensive, detailed analysis of the in vivo interaction of intracellular GSH pools with ruthenium compounds and its impact on their activity seems urgently needed for better understanding of the mode of action of this class of compounds.
Besides GSH, there are also some recent reports on TrxR inhibition (compare Section II.A.) by RuIII as well as RuII(arene), in particular, RAPTA compounds (Fig. 16) (58, 261). In contrast to sodium arsenite which targets TrxR1 and TrxR2, the tested ruthenium compounds mainly inhibited the cytosolic TrxR1 in cell-free experiments. As both RuIII and RuII compounds display this inhibitory potential, it seems unlikely that redox interactions of the Ru core are responsible for the TrxR1 inhibition.
Interestingly, several RuIII compounds (including NAMI-A and KP1339, the sodium salt of KP1019) have been identified as direct nitric oxide (NO) scavengers by Moribelli et al. (256). Comparable to O2• −, the highly reactive NO• is known as intracellular and intercellular messenger for diverse physiological processes especially in vascular homeostasis and neurotransmission as well as inflammatory/immune response and tumor progression (31). Under serum-free conditions, RuIII drugs react with NO•, which lead to reduction of Ru and formation of a Ru-NO moiety (256). This NO• scavenging was shown to inhibit endothelial cell migration and angiogenesis especially in case of NAMI-A. Consequently, it seems likely that the antiangiogenic activity of NAMI-A might be related to this NO-scavenging activity.
In summary, although the exact modes of action of ruthenium compounds are still not fully understood, there is ample evidence that redox reactions and interference with the cellular redox balance play an important regulatory role in the anticancer activity of many ruthenium compounds.
E. Copper
Copper (Cu) is one of the most important transition metals in human physiology and, consequently, its uptake and distribution are tightly regulated (compare Section III.B.). Moreover, there is growing evidence that elevated copper levels are associated with cancer (134). There are currently several approaches to target cancer cells by diverse copper chelating agents, which include besides D-penicillamine, clioquinol, and trientine also the molybdenum-containing tetrathiomolybdate (compare Section V.I.). However, despite some rather early reports regarding the activity of copper complexes in vivo (331), the development of copper-containing compounds as anticancer agents remained in most cases at a very early stage of preclinical development and clinical studies are so far missing.
With regard to redox properties, the current knowledge on Cu compounds is primarily based on investigations using Cu complexes of α-N-heterocyclic carboxaldehyde thiosemicarbazonates (Cu-TSC) (52), 2,2′-bipyridyl-6-carbothioamide (Cu-BPYTA) (273, 274), and of 1,10-phenanthroline (Cu-phen) (Fig. 17). As BPYTA shares several structural and functional characteristics with thiosemicarbazones, it is not surprising that their modes of action seem to be widely similar. Both are well-known tridentate chelators and have been used for the synthesis of a wide range of metal complexes, including besides Cu also Fe, Co, Zn, Ni, or Ga (204, 274, 328, 331, 421). With regard to their modes of action, metal-free BPYTA (274) as well as thiosemicarbazones like triapine (342, 421) are known for their ribonucleotide reductase (RR) inhibitory potential (compare Section III.A.2.). The RR inhibition is based on the disruption of the R2-localized tyrosyl radical and is believed to be executed by an intracellularily formed redox-active Fe complex of BPYTA or TSC, able to generate ROS by redox cycling between FeIII/II. Comparably, also the copper complexes Cu-BPYTA (273) and Cu-TSC (264) were shown to inhibit the R2 tyrosyl radical, although it is widely unclear whether the underlying mechanisms are similar to their Fe complexes. Interestingly, addition of Cu to triapine significantly increased its RR inhibitory potential (111). It is not known whether 1,10-phenanthroline complexes are also able to inhibit the RR. However, we have recently revealed RR inhibition by the lanthanum 1,10-phenanthroline complex KP772, which was accompanied by the intracellular formation of an Fe-phen complex (156). In the light of these results interference with the RR tyrosyl radical by Cu-phen does not seem unlikely.
FIG. 17.

Ligands of the best investigated anticancer CuII complexes.
Cu complexes are well known for their redox activity, which seems to be at least involved if not responsible for most of their described biological activities (51, 242, 384). The redox cycling of Cu complexes is based on the reduction of CuII to CuI by intracellular thiols such as GSH under oxygen-containing conditions (compare Section II.A.) (12, 52, 72, 264, 332, 353). Schematically, the underlying reaction pathway for Cu-TSC is given in equations 10–12 (additional ligands like OH− or H2O are omitted for clarity; TSC = thiosemicarbazonato). Briefly, most CuII complexes rapidly form adducts with GSH (26, 230, 332), leading to CuI complexes and GS•. In the presence of oxygen, this CuI complex is able to generate a superoxide anion, which can induce ROS via a Fenton-like reaction (51, 52, 222, 384) (compare Section II.C.).
| 10 |
| 11 |
| 12 |
For dianionic thiosemicarbazonato ligands (52) it was shown that the resulting CuI complexes are also able to form GSSG via the following reaction:
| 13 |
| 14 |
These reactions lead to (transient) depletion of intracellular GSH pools, which has been frequently observed in cells after treatment with diverse Cu compounds (12, 190, 231, 242, 264). Elevated intracellular GSH levels and enhanced drug export by GSH-dependent multidrug-resistance transporters, such as MRP1 (ABCC1), are frequent handicaps for successful chemotherapy (compare Section II.A.) (155). Thus, the transient GSH depletion by Cu compounds came recently into focus of interest for overcoming of GSH-dependent drug resistance. Thus, an N-(2-hydroxyacetophenone)glycinato copper(II) complex CuNG (Fig. 17) was developed with the aim to reduce resistance of the MRP1-overexpressing and highly drug-resistant EAC/Dox cells to doxorubicin (231). Indeed, temporary GSH depletion by CuNG enhanced tumor response of these cells to doxorubicin against cancer cell lines and in a xenograft mouse experiment (230, 231). In these studies, a combination regimen consisting of 10 mg/kg CuNG and 2 mg/kg doxorubicin increased the mean survival of male Swiss albino mice from 19 to 87 days (230). Notably, CuNG treatment alone had no antitumor effects, although increased ROS levels in tumor, liver, and kidney tissue of the treated mice were observed (254). Accordingly, oxidative stress generation by CuNG led to stimulation of SOD and catalase activity, especially in heart and kidney tissue. In contrast, basal ROS levels in lung and heart tissue of EAC/Dox-bearing animals were significantly reduced by CuNG treatment (254). It has been recently reported that CuNG treatment significantly modulates the cytokine production of tumor-associated macrophages leading to decreased interleukin 10 and TGF-β production and increased interleukin 12 levels. As these effects were reversed by addition of the ROS scavenger tocopherol (vitamin E), it seems likely that the interplay of CuNG with redox homeostasis is responsible for these observations (64).
In a recent study it has been shown that a Cu2+ chelate of the novel thiosemicarbazone NSC689534 induces ROS and depletes GSH as well as protein thiols. Further, microarray analysis revealed the activation of several ROS connected pathways, such as oxidative and ER stress/UPR, autophagy, and metal metabolism by these compounds. In vitro studies confirmed an ER stress-dependent but autophagy-independent induction of apoptosis. Moreover, anticancer activity in a mouse in vivo model was demonstrated for this thiosemicarbazone copper complex (143).
In case of Cu-phen, the intercalation of its ligand, 1,10-phenanthroline, into the DNA minor groove allows DNA targeting, which enables redox reactions of the Cu core with DNA and RNA (309, 353). Consequently, Cu-phen has been used as footprinting reagent for the evaluation of protein-DNA interactions as well as a probe for DNA and RNA secondary structure. Thus, the redox-mediated interaction of Cuphen and derivatives with DNA in cell-free systems has been extensively investigated (21, 72, 121, 302, 309, 354, 425). In the presence of H2O2, the DNA-bound CuI(phen) complex is oxidized to form presumably CuII(oxo/hydroxo) species (237, 354). Thus, the reaction of Cu-phen with nucleic acids (especially B-DNA) is not via a diffusible species, such as hydroxyl radicals or freely diffusible chelates, but through the non-covalent, nondiffusible Cu-oxo/hydroxo intermediate (425). The main target was shown to be the DNA C-1 site of deoxyribose located in the minor groove (21, 206, 425), which leads to the production of 3′- and 5′-phosphomonoesters, free purine and pyrimidine, and 5-methylenefuranone (the oxygen source of the carbonyl group in the latter is water). A minor alternative reaction pathway involves DNA scission via C-4′ and C-5′oxidation (21, 354). However, it has to be kept in mind that all of these investigations have been performed under cell-free conditions and it is so far unknown, whether these interactions of Cu-phen with DNA have any relevance for its biological activity in living cells
F. Vanadium
Vanadium (V) is a transition metal existing in eight oxidation states, of which VIV and VV are the most important but also VIII and VII might occur in biological systems (106). Vanadium is a trace element and essential for diverse animals, but its importance as a micronutrient in humans is not entirely clear. Vanadium compounds have been shown to interact with numerous cellular signaling mechanisms by influencing key enzyme families starting with inhibition of protein tyrosine phosphatases, in turn activation of protein kinases, and regulation of intracellular signal pathways, which results in altered expression of multiple genes (168). Consequently, vanadium compounds exert diverse biological and physiological effects, including insulin-enhancing activity, regulation of oxygen affinity to hemo- and myoglobin, reduction of hyperlipidemia, obesity, and hypertension as well as cardioprotective properties (260). In combination with relatively minor toxicity, these characteristics open multiple possibilities for the use of vanadium drugs as medical remedies. Indeed, vanadium was already used at the beginning of the 20th century for treatment of anemia, tuberculosis, and diabetes (257). In contrast to many other metal compounds developed as potential anticancer drugs, vanadium exerts rather chemopreventive than carcinogenic activity as demonstrated in several chemically induced tumor models (39). These chemopreventive effects are believed to be based on several properties, including (i) reduced generation of carcinogen-derived reactive intermediates, (ii) specific modulation of the antioxidant capacity, and (iii) induction of phase I as well as phase II detoxifying enzymes.
Besides those cancer-preventive effects, vanadium compounds have also been shown to exert anticancer effects against already established tumors, for example, by inhibition of proliferation, apoptosis induction, blockage of invasion, as well as metastasis (106). Nevertheless, it needs to be mentioned that several vanadium(V) and vanadium(IV) compounds were characterized as genotoxic, which is probably based on the induction of oxidative stress or the inhibition of protein tyrosine phosphatases, leading, in addition to activated cell proliferation, to improper spindle formation in mitosis or meiosis and, thus, aneuploidy (35).
The chemopreventive and anticancer activities are distinctly influenced by redox processes based on the chemical and biochemical characteristics of vanadium as a transition metal. In aqueous solution, vanadium exists either as tetravalent vanadyl (VO2+) or pentavalent (meta)vanadate (VO43−;VO3−), whereby different monomeric and polymeric species can exist depending on pH and drug concentration. Both the redox reactions and the polymerization state seem to have a profound impact on the cytotoxic activity of vanadium compounds (106). In the human plasma, VIV and VV exist, though vanadyl predominates due to the efficient reduction of vanadate by several reductive components of the blood, such as ascorbic acid. The vanadium ions are bound to plasma proteins like transferrin and albumin and are taken up in this state into cellular compartments. Vanadium (V) might be reduced not only by GSH but also by flavoenzymes, for example, GR, or in microsomes both involving NADPH (eq. 15) and connected to the generation of hydroxyl radicals (347, 348, 350). As already mentioned in Sections II.A. and B., cancer cells are characterized by an altered pH, imbalance in the cellular redox homeostasis, and enhanced oxidative stress levels supporting radical generation reactions by vanadium compounds. Consequently, VIV might interact with oxygen generating a superoxide anion and VV in a Fenton-like reaction (Eqs. 16 and 17).
| 15 |
| 16 |
| 17 |
Peroxovanadium complexes, which can be formed during the above described reactions, are strong and irreversible inhibitors of most tyrosine phosphatases. In contrast, vanadate is mimicking phosphate and forms reversible bonds with the thiol groups of these enzymes (257). Several important components of the anticancer mode of action of vanadium compounds are, besides the deregulation of protein tyrosine phosphorylation, directly or indirectly depending on the generated radical species. Multiple vanadium compounds have been demonstrated to cause DNA damage (39, 106, 203, 349), whereas the cell cycle arrest in G2/M phase is believed to be caused by inhibition of cyclin-B complex dephosphorylation (108). Although at least for vanadocenes, adduct formation with DNA was demonstrated (18, 147), in most cases ROS and particularly the hydroxyl radicals generated in the cells are believed to be responsible for the induction of DNA damage of exposed cells (39, 106, 257). Additionally, considering the importance of tyrosine phosphorylation in multiple cellular signaling pathways, it is not surprising that vanadium compounds cause deregulation of cellular survival pathways and induce apoptosis. The involved pathways include for example the p38, JNK/SARK, and ERK/MAPK signal cascades via apoptosis signal-regulating kinase (ASK-1) and, probably in turn, the NF-κB pathway (175). Moreover, cellular survival pathways, including the PI3K/AKT/PKB pathway, and the antiapoptotic bcl-2 family members are deregulated by vanadium complex exposure (36, 311). The inhibition of phosphatases and the generation of oxidative stress seem to cooperate in these activities and even enhance each other (106, 257).
Given the vast array of vanadium compounds synthesized during the last decades and the broad knowledge delivered by studies concerning diabetes, it is surprising that no vanadium compound has been approved or is even close to clinical application for the treatment of cancer so far. It has to be mentioned that many vanadium compounds, including soluble aqueous peroxovanadates formed by the oxidation of vanadate with H2O2, are highly unstable in aqueous solution. Moreover, multiple vanadium species might be present in solution due to a series of hydrolysis and polymerization reactions, depending on pH and concentration of the vanadates, as well as rapid redox reactions (83). Moreover, based on a labile inner coordination sphere, vanadates tend to interact with electron pair donors. This makes the identification of an active species and/or metabolite almost impossible (257). In general, the presence of ancillary ligands in the complexes confer greater stability in aqueous solution than the pure vanadates or peroxovanadates.
Consequently, with regard to specific vanadium complexes, anticancer approaches have mainly focused on organometallic vanadocenes as well as vanadium/peroxovanadate coordination compounds (Fig. 18). The molecular anticancer mechanisms of vanadium complexes involve induction of oxidative stress (compare above) and were investigated in vitro using human cancer cell models, including leukemia, lymphoma, and solid tumor-derived cell lines (203). In contrast, most in vivo studies concerned the (chemo)preventive effects of vanadium complexes [for reviews see (39, 106)], whereas reports on therapeutic activity studies are limited. For example, activity of vanadocene dichloride and a [(2-methylaminopyridine) vanadium(IV)] complex against murine mammary tumor models was shown (103). [Bis(4,7-dimethyl-1,10-phenanthroline)sulfatooxovanadium(IV)], also termed Metvan (Fig. 18), induced potently apoptosis in tumor cell lines and demonstrated significant antitumor activity against human glioblastoma and breast cancer xenograft models in SCID mice (86). Another phenanthroline complex, namely, [(4,7-dimethyl-1,10-phenanthroline)bis(peroxo)oxovanadium(IV)] (Fig. 18), was active against transplanted breast cancer in vitro and in vivo (340). The situation in hematological malignancies seems more complicated. Vanadocene dichloride (201) and a series of vanadium(peroxo)(heteroligand) complexes (96) were demonstrated to prolong survival of lymphoid leukemia L1210-carrying mice. In contrast, only low doses of these vanadium complexes delayed progression of a lymphoma model, whereas higher doses enhanced malignant growth most likely due to an impact on drug-metabolizing enzymes (59). This complexity of pro- and anticancer activities as well as in mode of action and metabolism might be explanations why no vanadium complex is currently approved for anti-cancer therapy.
FIG. 18.

Vanadium drugs with anticancer potential.
G. Rhodium
Rhodium (Rh) complexes in the oxidation states + 1, + 2, and + 3 have been tested for their tumor-inhibiting potential and often the cisplatin-like binding to DNA was proposed essential for their modes of action (188). However, only a few studies have investigated anticancer Rh complexes in the context of biological redox processes.
For several RhI complexes in vivo anticancer activity against leukemic, solid, and metastasizing tumors in mice has been shown. However, it has to be considered that RhI complexes are inactivated by oxidation (335, 424).
Further, a number of RhIII analogs with similar structures of RuIII drug candidates (i.e., the MCl4L2− motif) have demonstrated antineoplastic activity. Whereas RuIII complexes are thought to be activated by reduction (compare Section IV.C.), reduction of RhIII compounds to more active + 2 species was suggested improbable (91, 188). In accordance, it has been shown that in vivo RhIII compounds do not alter biochemical pathways related to the GSH system and other enzymes involved in redox balance (56).
The discovery of the antitumor activity of RhII compounds led to various investigations of these complexes (188). It has been shown that RhII compounds have a high affinity to sulfhydryls and in particular RhII carboxylates of the general formula [Rh2(carboxylato)4(H2O)2] (Fig. 19) were found to be broken down in the presence of cysteine to liberate the carboxylates (165). This might be related to a redox process causing initial formation of RhI–RhII mixed-valence complexes, which are further reduced to RhI polynuclear species, for example, observed during the reaction with ceruloplasmin, cysteine, GSH, and coenzyme A. Complexes containing 1,10-phenanthroline or 2,2′-bipyridine ligands are readily reduced by sulfhydryl groups, whereas [Rh2(acetato)4(H2O)2]is relatively resistant to reduction (171).
FIG. 19.

General structure of RhII carboxylato complexes.
Interestingly, enzymes with sulfhydryl groups close to or in their active centers were inhibited by preincubation with Rh compounds. As the rate of enzyme inactivation correlated with toxicity and anticancer activity, the authors suggested that the activity of these RhII complexes is based on the reaction with enzymes or proteins containing sulfhydryl groups such as pyrovate kinase, aldolase, and LDH (165, 188). In contrast to RhI complexes, oxidation of dinuclear RhII carboxylates led to slightly more active species.
No definite trend between redox behavior and antitumor activity of [Rh2(carboxylato)4(H2O)2] complexes was observed (182). In an attempt to sensitize cells to irradiation, RhII carboxylates were compared to cisplatin and metronidazole. The lower redox potential of the RhII compounds as compared to metronidazole led to the conclusion that they do not undergo electron transfer reactions upon interaction with DNA-derived radicals. The increase in radiation sensitivity with RhII carboxylates, but not cisplatin, was attributed to the ability of the rhodium compounds to deplete intracellular thiols (71).
Additionally, photoactivation of [Rh2(carboxylate)4(H2O)2] with visible light in the presence of electron acceptors was analyzed. This process causes formation of one-electron-oxidized complexes of the general formula [Rh2(carboxylate)4(H2O2)+], capable of cleaving plasmid DNA (119). However, to the best of our knowledge no detailed studies on the role of redox processes or ROS formation in the modes of action of rhodium compounds have been reported. Only recently, the [Rh2(PheAla)2(acetato)2] complex was shown to exhibit its anticancer activity by an ROS-independent mechanism (114) and the activity of several monosubstituted dirhodiumII,II complexes was not affected by changes in GSH levels (2).
H. Cobalt
Cobalt (Co) has two naturally occurring oxidation states, CoII and CoIII. In general, cobalt is a very rare metal but a biologically important cofactor in vitamin B12-dependent enzymes. Vitamin B12 (cobalamin) represents a relatively inert CoIII ion in a substituted corrin macrocycle (Fig. 20). In addition to the four nitrogens of the corrin macrocycle, the CoIII of the B12 coenzyme possesses an axial 5-deoxyadenosine or methyl group. In the biological context, vitamin B12 acts as a coenzyme in a wide spectrum of metabolic processes, including methylmalonyl CoA mutase and type II RR (found in bacteria and archea). However, the actual number of known B12-dependent enzymes remains comparatively small and, therefore, most organisms need cobalamin in vanishingly small quantities. Humans require between 1 and 2 μg per day, which is ingested from our diet and is taken up by an elaborate absorption mechanism (305). Exclusively members of the Archea and certain eubacteria are able to synthesize cobalamin via a complex biosynthetic pathway (310). Further, only a few proteins containing cobalt not coordinated to the corrin macrocyclic system have been characterized.
FIG. 20.

Vitamin B12 (Cobalamin).
Regarding health risks, uptake of cobalt at larger quantities has been demonstrated to be carcinogenic at least in rodents. The underlying mechanisms involve genotoxicity by both radical-mediated mechanisms as well as direct interference of cobalt with DNA repair (35). CoII catalyzes the generation of hydroxyl radicals from H2O2 in a Fenton-like reaction (compare Section II.C.). After intraperitoneal injection in rats, CoII evoked the formation of oxidative DNA base damage in kidney, liver, and lung (187). In case of DNA repair, CoII interferes with nucleotide excision repair probably by substituting for zinc ions in zinc finger proteins, for example, XPA (200). Moreover, cobalt enhances the effects of other carcinogens like benzo[a]pyrene (362).
Despite these limitations due to adverse effects on normal cells and tissues, cobalt-containing compounds recently attracted considerable interest as systemic anticancer agents. First, cobalamin is substituted together with folic acid in chemotherapy regimens involving antimetabolites to reduce unwanted side effects. Additionally, since fast proliferating cells require higher amount of cobalamin than normal cells, cobalamin-conjugates with radioisotopes or cytotoxic compounds like, for example, nitrosylcobalamin or a cisplatincobalamin have been developed to achieve enhanced tumor accumulation via the respective receptor-mediated uptake system (29, 133, 325). The studies on cytotoxic/cytostatic cobalt complexes as anticancer therapeutics have more or less focused on the following types of cobalt compounds (Fig. 21): (i) hexacarbonyldicobalt complexes with alkyne ligands (cobalt alkyne complexes) containing two covalently linked Co0 atoms (285), (ii) [CoIII(NH3)6]Cl3, (iii) CoIII complexes with Schiff base ligands (282), including salen (135), and (iv) CoII and/or CoIII complexes with cytotoxic mustamine (398), mithramycin (163), and thiouracil (184) ligands. Regarding the cobalt alkyne complexes, potent anticancer activity was shown in vitro and in vivo of complexes containing the propargylic ester of acetylsalicylic acid (Co-Ass, Fig. 21) especially against breast cancer cells (181). With regard to their activity, minor modifications on the molecule resulted in distinct variations, whereby profound intracellular accumulation and the higher lipophilicity of the complex as compared to the free ligand might be of great importance (285). Mode of action studies indicated that—even though binding to DNA—cobalt alkyne complexes do not substantially target DNA in living cells and several observations suggest that the activity of Co-Ass (Fig. 21) might be based on the interaction of the ligand acetylsalicylic acid (aspirin) with cyclooxygenase enzymes (COX-1 and COX-2). This would fit well with the often observed hypersensitivity of breast cancer cells against COX inhibitors. The preferential accumulation in malignant cells indicates that Co-Ass might represent a “tumor-targeted aspirin.” Indeed, its anticancer activity was distinctly higher as compared to aspirin (286). However, redox processes were not discussed to be involved significantly in the mode of action of cobalt alkyne complexes.
FIG. 21.
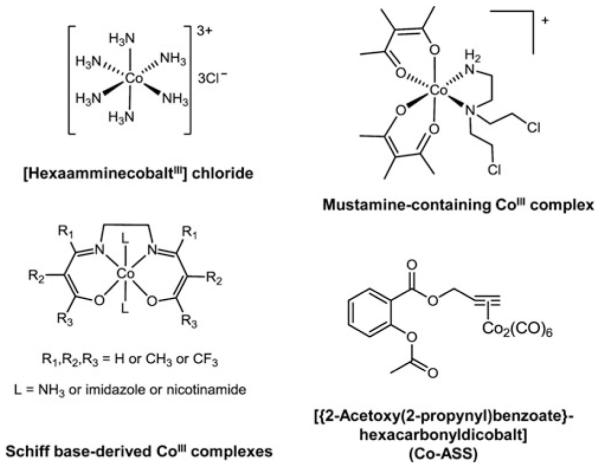
Anticancer cobalt compounds.
With regard to redox processes in the anticancer activity of cobalt complexes, two aspects are of central interest: (i) activation of CoIII complexes in hypoxic environment by reduction to CoII and release of the ligand, and (ii) generation of ROS by a catalytic autooxidation process especially by Schiff base complexes but also [CoIII(NH3)6]Cl3 (265, 266). Regarding the principal hypothesis of activation by reduction/hypoxia (similar to PtIV and RuIII compounds compare Section IV.C.,V.A.2., and V.D.), the drug must be able to exist in an inactivated higher oxidation state (the prodrug) and an activated lower oxidation state (the effective drug). As reductants are present throughout the body, it has been assumed for CoIII drugs that not the reduction but the delayed reoxidation of such compounds is responsible for the hyper-activation in the hypoxic tissues (89). Several CoIII complexes have been demonstrated to be reduced to CoII within the hypoxic tumor tissue. As CoII complexes are more labile, the cytotoxic ligands may be released from the “metal chaperon” and exert their anticancer activity. The reactions that are taking place are shown in Figure 22 (285). At least in some cases this activation step was proven to be tumor specific as detected for example by X-ray absorption near edge structure (XANES) (41) and efficient activation can be further promoted by ionizing radiation (252). Such, CoIII complexes containing nitrogen mustard ligands were demonstrated to be active under hypoxic conditions (399, 400) and the cytotoxic ligand 8-hydroxyquinoline (5) or the potent DNA minor groove alkylator azachloromethylbenzindoline (4) were released from the CoIII complex in hypoxic solutions by ionizing radiation. However, other studies based on cobalt complexes of bi- and tridentate cytotoxic or fluorescent ligands (397, 411) as well as pulse radiolysis experiments (10) have indicated that hypoxia selectivity of CoIII complexes might not completely be based on redox cycling. Instead other mechanisms like ligand exchange without prior reduction of CoIII or competition with O2 for biological reductants could be involved. Indeed, ascorbate and cysteine can reduce but also coordinate to CoIII (368). Yamamoto et al. showed that both cysteine and ascorbate were able to release fluorescent ligands from complexes even though they are—based on their reduction potentials— unlikely to be reduced by these cellular reductants (411).
FIG. 22.
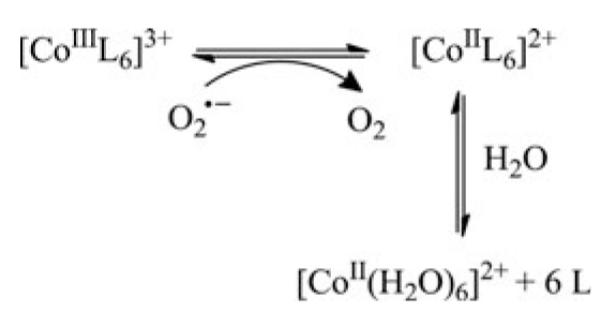
Ligand release after reduction of CoIII complexes (285). In the case of CoIII complexes it is assumed that in the hypoxic tumor tissue the CoIII metal center can be reduced to CoII, for example, by superoxide radicals. Due to the lower stability of the CoII complexes the cytotoxic ligands are released under formation of [CoII(H2O2)6]2+.
The reduction step of CoIII complexes might not only lead to the release of cytotoxic ligands but also to generation of ROS based on a catalytic autooxidation process (281). As mentioned above, even CoII ions themselves induce generation of ROS in vivo and in vitro by catalyzing the generation of hydroxyl radicals from H2O2 in a Fenton-like reaction (compare Section II.C.) (35). After exposure to [CoIII(NH3)6]Cl3, enhanced lipid peroxidation and upregulation of other oxidative stress parameters were found in the kidneys of mice. With EPR spin-trapping it was demonstrated that several CoII complexes are able to generate oxygen-derived free radicals under physiological conditions which were inhibited by addition of 5′-diphosphate or citrate (145). In the presence of peroxides, a nitrilotriacetate CoII complex formed hydroxyl radicals, whereas in case of an EDTA–CoII complex only oxidation to CoIII but no ROS generation was observed. Also, cobalt metal particles in suspension and in the presence of SOD generated OH•. Chelators like anserine enhance but 1,10-phenanthroline and desferioxamine reduced OH• generation from H2O2 by CoII. Interestingly, in a series of cobalt(3,4-diarylsalen) complexes the oxidizing potency did not reflect the anticancer activity against human cancer cell lines, suggesting that in case of these compounds superoxide radical-mediated active species are not the major effectors. Thus, other mechanisms might be important including DNA intercalation (135). Moreover, it has been recently demonstrated that CoII ions can replace Mg2+ in enzymatic physiological enzyme reactions, which strongly enhance DNA cleavage by topoisomerase IIα (20).
In several rodent tumor models comparable antitumor activity and DNA damage have been described as a consequence of treatment with redox-active CoII/III complexes with tetradentate Schiff base ligands derived from acetylacetone and ethylenediamine or biogenous and/or syntheticnitrogen-containing ligands, like phthalocyanines and vitamin B12 derivatives (281, 283, 391). Such redox-active complexes may act, in addition to the already mentioned cytotoxic ligand release, by other mechanisms, including binding of the histidine units of polypeptide chains like in case of [Co(acetylacetonate-ethylenediimine)(NH3)2]+ with metmyoglobin (40). Moreover, these complexes may catalyze auto-oxidation of ascorbic acid involving generation of O2• −,OH•, and H2O2 (391). Thus, cobalt complexes accumulated in malignant tissues should exhibit enhanced antitumor activity in cooperation with ascorbic acid as shown for the cobalt phthalocyanine complexes and Co compounds of the B12 series (281, 391).
In summary, cobalt complexes are mainly in the focus of interest in experimental cancer therapy research because of their ability to redox-dependent targeting the malignant tissue of solid tumors. It is surprising that despite the intense research efforts during the last decades none of these compounds has reached clinical evaluation as anticancer drug so far.
I. Manganese
Manganese (Mn) is an essential trace metal. Several enzymes have Mn cofactors, including oxidoreductases, transferases, and hydrolases. One of the best investigated enzymes is the Mn-containing SOD (compare Section II.A.). As oxidative stress is important in numerous diseases, including cancer, synthetic antioxidants have been extensively investigated especially in cancer chemoprevention and antiaging research. Within these, especially Mn-containing complexes as SOD mimics exhibited high antioxidative potential. From the chemical view, Mn complexes exhibit rich redox chemistry. Important examples are Mn-porphyrin compounds that have accessible oxidation states ranging from + 2 to + 5 under physiological conditions (27). Their redox potentials are similar to those of several RuIII anticancer agents (compare Section V.D.). The primarily developed Mn-containing SOD mimics are based on corroles, porphyrins, salens, and cyclic polyamine ligand systems (Fig. 23). These SOD mimics possess tumor growth-inhibiting (27) as well as cancer-preventing properties (279) [for a recent comprehensive review see (27)].
FIG. 23.

Manganese drugs under preclinical development as SOD mimics.
Within the Mn compounds, Mn-porphyrin complexes are the best investigated, which appear particularly advantageous due to their low toxicity and their ability to cross cell membranes. The most potent complexes have MnIII/MnII reduction potentials between the potential of O2• − reduction (E1/2 + 0.89 V vs. NHE pH 7.0) and oxidation (E1/2-0.16V vs. NHE pH 7.0), similar to endogenous SOD (E1/2 ~ + 0.3 V vs. NHE) (27, 28). Further, the catalytical rate constant kcat for O2• − dismutation equals nearly the kcat of SOD enzymes. These properties enable Mn-porphyrins to easily donate and accept electrons from redox active compounds, such as cellular reductants (28, 110).
In the context of cancer, particularly the highly positive charged Mn-porphyrin complex Mn(III) meso-tetrakis(N-ethylpyridinum-2-yl)porphyrin (MnTE-2-PyP5+) (Fig. 23) was investigated. Even though as single agent only low anticancer activity against cancer cell lines was observed, MnTE-2-PyP5+ had antiangiogenic properties in vivo, especially in combination with hyperthermia and radiation (32, 306, 418). The mode of action is thought to be related on the one hand to its anti-oxidative properties by downregulation of cellular levels of reactive nitrogen species and ROS on the other hand, to its prooxidative properties. The latter leads to oxidation of biological targets such as cysteines, for example, in signaling proteins by increased generation of H2O2 particularly occurring in cells with insufficient peroxide metabolism (407). Consequently, several biological functions are altered by the anti- and prooxidative properties of MnTE-2-PyP5+, including inhibition of AP-1 and NF-κB activity and downregulation of HIF1α,VEGF, and TGF-β (28, 110, 385). Accumulation studies showed that MnTE-2-PyP5+ was able to accumulate in vivo in heart mitochondria to levels sufficient to exert its antioxidant activity. In vitro accumulation studies with macrophages and lipopolysaccharide-stimulated macrophages demonstrated that the positively charged porphyrins favor the nucleus with its anionic nucleic acids in contrast to the cytosol (27, 28, 361). Moreover, it has been shown that treatment of cancer cells in a combination regimen consisting of MnTE-2-PyP5+ and glucocorticoids, cyclophosphamide, or doxorubicin sensitized cells in some cases to these chemotherapeutics (174).
Beside MnTE-2-PyP5+, the macrocyclic Mn(II) polyamine M40403 (Fig. 23) has been investigated in combination with chemotherapy, radiotherapy, and immune-stimulating inter-leukin-2 treatment. Prevention of side effects by the manganese compounds became obvious, and therefore M40403 has been granted orphan drug designation for prevention of radiation- or chemotherapy-induced oral mucositis in cancer patients in 2008. However, recent studies suggest that combination of M40403 with cytotoxic agents not only prevents side effects but also increases the anticancer activity by enhanced pro-oxidative effects. The M40403 produced H2O2 may especially target rapidly dividing cancer cells with impaired peroxide metabolism and high levels of endogenous oxidative stress (7, 208, 407). Interestingly, also mangafodipir (Fig. 24), a paramagnetic MnII-containing contrast agent for magnetic resonance imaging of the liver, enhanced cytotoxicity of anticancer agents and decreased hematotoxicity. Besides mimicking SOD, mangafodipir has also catalase- and glutathione reductase-like properties allowing interaction with several points of cellular redox homeostasis (7, 407).
FIG. 24.
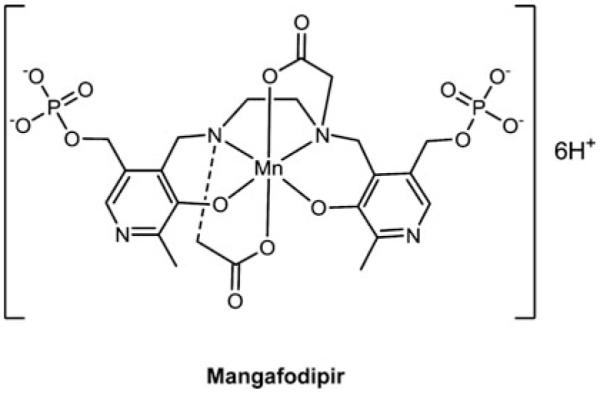
Structure of Mangafodipir. This compound is in clinical use as contrast agent.
In summary, Mn compounds exert a number of interesting properties that might be useful in the development of metal-based anticancer agents. However, none of them are in clinical trials for the application as cancer chemotherapeutics so far (326, 407).
J. Complexes with redox silent metal centers in clinical trials
There are currently several strategies in (pre)clinical development that use metal-containing drugs to interfere with the redox balance of cancer cells, where the central metal core is not directly involved in this “redox activity.” Among these, two promising compounds (Fig. 25), namely, motexafin gadolinium (228) and tetrathiomolybdate (47, 134, 290), have been tested in several clinical trials. Gadolinium motexafin (MGd) is a texaphyrin coordinated to a nonredox-active gadolinium(III) cation. However, the aromatic texaphyrin ring system of MGd is easily reduced (first reduction potential of −0.041 V vs. NHE in dimethylformamide), for example, O2• − . In the presence of oxygen, this is supposed to result in redox cycling, oxidative stress, and disruption of the cellular redox homeostasis (228, 378). Indeed, ROS formation after MGd treatment has been shown in cell culture and animal experiments initiating the clinical testing to exploit these redox properties to sensitize cancer cells to radiation therapy (324). Several clinical studies were published especially on the combination therapy of whole-brain radiation and MGd in patients with brain metastases reporting in some cases encouraging response rates (54). Unfortunately, no survival benefit by addition of MGd to whole-brain radiation in patients with brain metastases from lung cancer was found in a large-scale phase III study despite an improvement in neurocognitive functions and a prolonged time to neurologic progression (247). Consequently, MGd has not been approved for clinical use as anticancer therapeutic so far.
FIG. 25.
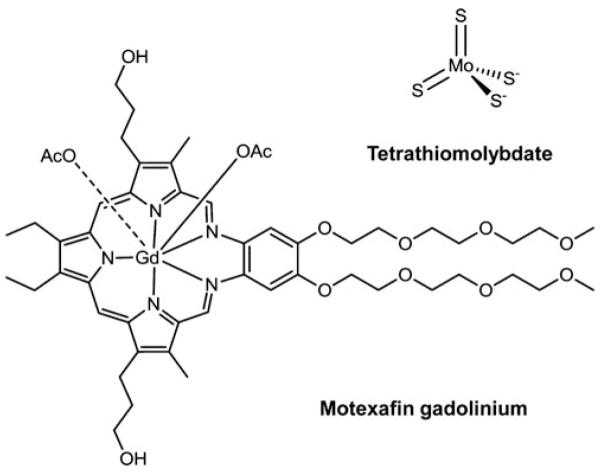
Anticancer complexes with redox-silent metal centers under clinical investigation.
Another clinically evaluated compound is tetrathiomolybdate (TM), which has been developed as copper chelating agent. Numerous reports describe elevated copper levels in serum and malignant tissues of cancer patients, which directly correlate with cancer progression (134). TM has been shown to interfere with the cellular redox balance by inhibition of copper-containing enzymes, such as ceruloplasmin, ascorbate oxidase, cytochrome oxidase, or Cu/Zn SODs (compare Section II.A.) (9). However, the binding of copper by TM does not involve any redox reaction but is based on the formation of stable ternary adducts with copper-containing proteins (9). Based on its antiangiogenic activity, TM has been evaluated against several cancer types in clinical studies indicated some clinical efficacy (244). Recently, chelation of copper by TM was demonstrated to enhanced sensitivity of tumor models against cisplatin based on augmented cisplatin uptake via the copper transporter CTR1 (169).
VI. Conclusion
Based on the availability of the human genome and the development of high-throughput omics methods, experimental cancer therapy research was dominated by rational drug development and molecular targeted approaches during the last decades. In parallel, studies on classical chemotherapeutics uncovered that also for such old-fashioned cytotoxic drug (including anticancer metal complexes)-specific molecular targets in addition to DNA might exist. Moreover, it was emerging that several tumor-specific biochemical/biophysical conditions, like altered pH, redox milieu, and hypoxia distinctly impact on the activity of more or less all applied anticancer drugs. Interactions with such tumor-specific conditions/mechanisms are now increasingly utilized in (pre)clinical anticancer drug development by novel and creative approaches focusing on (i) enhanced drug transport (in)to the malignant tissue, (ii) activation of prodrugs in the malignant compartment, (iii) increase of cytotoxicity against cancer cells by drug metabolism in the malignant cells, and (iv) circumvention of resistance development. All these approaches offer chances to develop better tumor-specific cancer therapeutics with enhanced activity in molecularly defined tumor entities and patient subgroups.
With regard to metal-based anticancer drugs, the great success of cisplatin but also its limitations based on side effects and resistance development were strong stimuli for the development of novel and more tumor-specific metal complexes. In that context, it has to be admitted, that—considering the enormous number of compounds synthesized—the count of clinically approved substances remains comparably low. Nevertheless, the above-mentioned developments toward an in-depth understanding of the molecular changes affecting cancer cells or the tumor microenvironment also offer novel avenues for the development of smart, tumor-targeting metal compounds. Recent success stories like the profound activity of ATO against APL based on a highly specific targeting of the oncogenic fusion protein (compare Section V.C.), give hope in that respect.
Alterations of the redox status and, consequently, upregulation of oxidative stress and its molecular consequences are well known for malignant tumors and now more and more recognized as platform for the development of novel cancer-targeting drugs (381). Moreover, these profound alterations in the redox status of malignant tissues might not only be a consequence of misbalance between cell proliferation, mitochondrial activity, and blood supply, but also a direct result of tumor-specific gene mutations. For example, the metabolic enzymes isocitrate dehydrogenase-1 and −2 (IDH1, IDH2) were found mutated in an extended subgroup of glioma and AML patients. The mutations were shown to alter the redox status of cancer cells with enhanced radical stress, and to activate the hypoxia-inducible factor 1 alpha (1, 412, 430).
Based on their chemical characteristics, redox-active metal-drugs are naturally in the focus of interest in this research field. This review summarizes the impact of altered redox conditions on the anticancer activity of clinically approved and innovative redox-active metal compounds. From this overview it becomes obvious that redox processes are important players both in the mode of action as well as in metabolism, transport, and distribution of anticancer metal complexes. Although for many promising drug candidates an extended array of data exist also several limitations are obvious. For example, a mode of action comparable to cisplatin is frequently anticipated for all metal drugs and, consequently, multiple studies have focused exclusively on the interaction of metal complexes with DNA. However, during the last decades increasing evidence is accumulating that such a view is too short-sighted and obviously more integrated approaches are needed. Such especially for gold, arsenic, and ruthenium compounds, important cytosolic targets are emerging (compare Section V.B. to V.D.). Moreover even in case of clinically approved platinum compounds, the modes of action might severely differ. For example, recent data suggest an important contribution of immunogenic cell death to the anticancer activity of oxali- but not cisplatin (371, 432). Moreover, the literature on redox processes in the activity of anticancer metal complexes has often focused on cell-free in vitro systems. Although highly informative, the translation of the gained knowledge to the in vivo situation—both at the level of the living (tumor) cell and the whole organism—is extremely complicated and challenging. However, the availability of modern analytical techniques as well as sophisticated, transgenic cell and animal models should severely support such integrated attempts. These considerations suggest that the molecular mechanisms underlying the anticancer activity of metal complexes need to be re-evaluated and, based on the gained knowledge, the development of more tumor-specific and less toxic anticancer metal compounds has to be further promoted by multidisciplinary research teams. Then a revival of metal-compounds to successfully fight human cancer seems not only feasible but even inevitable.
Acknowledgments
Many thanks to Dr. Leonilla Elbling and Michael Micksche for inspiring discussions. This work was supported by the Austrian Science Fund grants #L568, #P22072, and #I496.
Abbreviations Used
- AML
acute myeloid leukemia
- ANT
adenine nucleotide translocator
- APL
acute promyelocytic leukemia
- ASK-1
apoptosis signal-regulating kinase
- Ass
propargylic ester of acetylsalicylic acid
- ATO
arsenic trioxide
- ATP7A/B
P-type ATPase
- Auoxo6
[Au2(6,6′-dimethyl-2,2′-bipyridine)(μ-O)2]PF6
- Auranofin
[tetra-O-acetyl-β-D-(glucopyranosyl) thio](triethylphosphine)gold(I)
- BHA
buthylated hydroxyanisole
- BPYTA
2,2′-bipyridyl-6-carbothioamide
- BSO
L-buthionine-(S,R)-sulfoximine
- COX
cyclooxygenase
- COX17
cyclooxygenase 17
- CTR1
copper transporter 1
- CuNG
N-(2-hydroxyacetophenone)glycinato copper(II)
- D2PYPP
bis(di-2-pyridylphosphino)propane
- DACH
(1R,2R)-cyclohexanediamine
- DMT1
divalent metal transporter
- DMTU
dimethylthiourea
- DNCB
dinitrochlorobenzene
- DPPE
bis(diphenylphosphine)ethane
- EDTA
ethylenediaminetetraacetato
- GPx
glutathione peroxidase
- GR
glutathione reductase
- GSAO
4-(N-(S-glutathionylacetyl) amino)phenylarsonous acid
- GSH
glutathione
- GST
glutathione-S-transferases
- H2O2
hydrogen peroxide
- HO-1
heme oxigenase-1
- HSAB
hard and soft acids and bases
- Iproplatin
cis,trans,cis-[PtCl2(OH)2(isopropylamine)2]
- JM118
cis-amminedichlorido-(cyclohexylamine)platinum(II)
- LIP
labile iron pool
- MER1
S-dimethylarsino-thiosuccinic acid
- METVAN
[Bis(4,7-dimethyl-1,10-phenanthroline)sulfatooxovanadium(IV)]
- MGd
gadolinium motexafin
- MnTE-2-PyP5+
Mn(III) meso-tetrakis(N-ethylpyridinum-2-yl)porphyrin
- MOA
mode of action
- MRP
multi-drug resistance
- NAC
N-acetylcysteine
- NHE
normal hydrogen electrode
- O2• −
superoxide radical
- OH•
hydroxyl radical
- Phen
1,10-phenanthroline
- ROS
reactive oxygen species
- RR
ribonucleotide reductase
- Satraplatin
cis,trans-[PtCl2(OAc)2(NH3)(cyclohexylamine)]
- SOD
superoxide dismutases
- Tetraplatin
[PtCl4(d,l-cyclohexane-1,2-diamine)]
- Tf
transferrin
- TfR1
transferrin receptor
- TGR
thioredoxin glutathione reductase
- TM
tetrathiomolybdate
- TrxR
thioredoxin reductase
- TSC
α-N-heterocyclic carboxaldehyde thiosemicarbazonates
- UPR
unfolded protein response
- XANES
X-ray absorption near edge structure
- ZIO-101
darinaparsin
- γ-GT
γ-glutamyl transpeptidase
References
- 1.Abdel-Wahab O, Levine RL. Metabolism and the leukemic stem cell. J Exp Med. 2010;207:677–680. doi: 10.1084/jem.20100523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguirre JD, Angeles-Boza AM, Chouai A, Pellois JP, Turro C, Dunbar KR. Live cell cytotoxicity studies: documentation of the interactions of antitumor active dirhodium compounds with nuclear DNA. J Am Chem Soc. 2009;131:11353–11360. doi: 10.1021/ja9021717. [DOI] [PubMed] [Google Scholar]
- 3.Ahmadi R, Urig S, Hartmann M, Helmke BM, Koncarevic S, Allenberger B, Kienhoefer C, Neher M, Steiner HH, Unterberg A, Herold-Mende C, Becker K. Antiglioma activity of 2,2′:6′,2″-terpyridineplatinum(II) complexes in a rat model—effects on cellular redox metabolism. Free Radic Biol Med. 2006;40:763–778. doi: 10.1016/j.freeradbiomed.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 4.Ahn GO, Botting KJ, Patterson AV, Ware DC, Tercel M, Wilson WR. Radiolytic and cellular reduction of a novel hypoxia-activated cobalt(III) prodrug of a chloromethylbenzindoline DNA minor groove alkylator. Biochem Pharmacol. 2006;71:1683–1694. doi: 10.1016/j.bcp.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Ahn GO, Ware DC, Denny WA, Wilson WR. Optimization of the auxiliary ligand shell of Cobalt(III)(8-hydro-xyquinoline) complexes as model hypoxia-selective radiation-activated prodrugs. Radiat Res. 2004;162:315–325. doi: 10.1667/rr3229. [DOI] [PubMed] [Google Scholar]
- 6.Aird RE, Cummings J, Ritchie AA, Muir M, Morris RE, Chen H, Sadler PJ, Jodrell DI. In vitro and in vivo activity and cross resistance profiles of novel ruthenium (II) organometallic arene complexes in human ovarian cancer. Br J Cancer. 2002;86:1652–1657. doi: 10.1038/sj.bjc.6600290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alexandre J, Nicco C, Chereau C, Laurent A, Weill B, Goldwasser F, Batteux F. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J Natl Cancer Inst. 2006;98:236–244. doi: 10.1093/jnci/djj049. [DOI] [PubMed] [Google Scholar]
- 8.Altamura C, Squitti R, Pasqualetti P, Gaudino C, Palazzo P, Tibuzzi F, Lupoi D, Cortesi M, Rossini PM, Vernieri F. Ceruloplasmin/Transferrin system is related to clinical status in acute stroke. Stroke. 2009;40:1282–1288. doi: 10.1161/STROKEAHA.108.536714. [DOI] [PubMed] [Google Scholar]
- 9.Alvarez HM, Xue Y, Robinson CD, Canalizo-Hernandez MA, Marvin RG, Kelly RA, Mondragon A, Penner-Hahn JE, O’Halloran TV. Tetrathiomolybdate inhibits copper trafficking proteins through metal cluster formation. Science. 2010;327:331–334. doi: 10.1126/science.1179907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson RF, Denny WA, Ware DC, Wilson WR. Pulse radiolysis studies on the hypoxia-selective toxicity of a colbalt-mustard complex. Br J Cancer. 1996;(Suppl 27):S48–S51. [PMC free article] [PubMed] [Google Scholar]
- 11.Anestal K, Arner ES. Rapid induction of cell death by selenium-compromised thioredoxin reductase 1 but not by the fully active enzyme containing selenocysteine. J Biol Chem. 2003;278:15966–15972. doi: 10.1074/jbc.M210733200. [DOI] [PubMed] [Google Scholar]
- 12.Antholine WE, Taketa F. Effects of 2-formylpyridine monothiosemicarbazonato copper II on red cell components. J Inorg Biochem. 1984;20:69–78. doi: 10.1016/0162-0134(84)80007-0. [DOI] [PubMed] [Google Scholar]
- 13.Aposhian HV, Zakharyan RA, Avram MD, Sampayo-Reyes A, Wollenberg ML. A review of the enzymology of arsenic metabolism and a new potential role of hydrogen peroxide in the detoxication of the trivalent arsenic species. Toxicol Appl Pharmacol. 2004;198:327–335. doi: 10.1016/j.taap.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 14.Arion VB, Reisner E, Fremuth M, Jakupec MA, Keppler BK, Kukushkin VY, Pombeiro AJL. Synthesis, X-ray diffraction structures, spectroscopic properties, and in vitro antitumor activity of isomeric (1H-1,2,4-triazole)Ru(III) complexes. Inorg Chem. 2003;42:6024–6031. doi: 10.1021/ic034605i. [DOI] [PubMed] [Google Scholar]
- 15.Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol. 2006;16:420–426. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 16.Arner ES, Nakamura H, Sasada T, Yodoi J, Holmgren A, Spyrou G. Analysis of the inhibition of mammalian thioredoxin, thioredoxin reductase, and glutaredoxin by cis-diamminedichloroplatinum (II) and its major metabolite, the glutathione-platinum complex. Free Radic Biol Med. 2001;31:1170–1178. doi: 10.1016/s0891-5849(01)00698-0. [DOI] [PubMed] [Google Scholar]
- 17.Arredondo M, Nunez MT. Iron and copper metabolism. Mol Aspects Med. 2005;26:313–327. doi: 10.1016/j.mam.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Aubrecht J, Narla RK, Ghosh P, Stanek J, Uckun FM. Molecular genotoxicity profiles of apoptosis-inducing vanadocene complexes. Toxicol Appl Pharmacol. 1999;154:228–235. doi: 10.1006/taap.1998.8592. [DOI] [PubMed] [Google Scholar]
- 19.Balamurugan K, Schaffner W. Copper homeostasis in eukaryotes: teetering on a tightrope. Biochim Biophys Acta. 2006;1763:737–746. doi: 10.1016/j.bbamcr.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Baldwin EL, Byl JA, Osheroff N. Cobalt enhances DNA cleavage mediated by human topoisomerase II alpha in vitro and in cultured cells. Biochemistry. 2004;43:728–735. doi: 10.1021/bi035472f. [DOI] [PubMed] [Google Scholar]
- 21.Bales BC, Pitie M, Meunier B, Greenberg MM. A minor groove binding copper-phenanthroline conjugate produces direct strand breaks via beta-elimination of 2-deoxyribonolactone. J Am Chem Soc. 2002;124:9062–9063. doi: 10.1021/ja026970z. [DOI] [PubMed] [Google Scholar]
- 22.Ballatori N, Krance SM, Marchan R, Hammond CL. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol Aspects Med. 2009;30:13–28. doi: 10.1016/j.mam.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bard AJ, Faulkner LR. Electrochemical Methods Fundamentals and Applications. John Wiley & Sons; New York: 2001. [Google Scholar]
- 24.Bard AJ, Parsons R, Jordan J, editors. Standard Potentials in Aqueous Solution. Dekker; New York: 1985. p. 834. [Google Scholar]
- 25.Barrette WC, Jr., Johnson HW, Jr., Sawyer DT. Voltam-metric evaluation of the effective acidities (pKa’) for Broensted acids in aprotic solvents. Anal Chem. 1984;56:1890–1898. doi: 10.1021/ac00275a030. [DOI] [PubMed] [Google Scholar]
- 26.Basu S, Majumder S, Chatterjee S, Ganguly A, Efferth T, Choudhuri SK. Detection and characterization of a glutathione conjugate of a novel copper complex. In Vivo. 2009;23:401–408. [PubMed] [Google Scholar]
- 27.Batinic-Haberle I, Reboucas JS, Spasojevic I. Superoxide dismutase mimics: chemistry, pharmacology, and therapeutic potential. Antioxid Redox Signal. 2010;13:877–918. doi: 10.1089/ars.2009.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Batinic-Haberle I, Spasojevic I, Tse H, Tovmasyan A, Rajic Z, St. Clair D, Vujaskovic Z, Dewhirst M, Piganelli J. Design of Mn porphyrins for treating oxidative stress injuries and their redox-based regulation of cellular transcriptional activities. Amino Acids. 2010 doi: 10.1007/s00726-010-0603-6. [Epub ahead of print]; DOI: 10.1007/s00726-010-0636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauer JA, Lupica JA, Schmidt H, Morrison BH, Haney RM, Masci RK, Lee RM, Didonato JA, Lindner DJ. Nitrosylcobalamin potentiates the anti-neoplastic effects of chemotherapeutic agents via suppression of survival signaling. PLoS One. 2007;2:e1313. doi: 10.1371/journal.pone.0001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becker K, Herold-Mende C, Park JJ, Lowe G, Schirmer RH. Human thioredoxin reductase is efficiently inhibited by (2,2′:6′,2″-terpyridine)platinum(II) complexes. Possible implications for a novel antitumor strategy. J Med Chem. 2001;44:2784–2792. doi: 10.1021/jm001014i. [DOI] [PubMed] [Google Scholar]
- 31.Bencini A, Failli P, Valtancoli B, Bani D. Low molecular weight compounds with transition metals as free radical scavengers and novel therapeutic agents. Cardiovasc Hematol Agents Med Chem. 2010;8:128–146. doi: 10.2174/187152510791698389. [DOI] [PubMed] [Google Scholar]
- 32.Benjamin JM, Ines B-H, Ivan S, Zahid NR, Mitchell SA, Zeljko V, Mark WD. A manganese porphyrin super-oxide dismutase mimetic enhances tumor radioresponsiveness. Int J Radiat Oncol Biol Phys. 2005;63:545–552. doi: 10.1016/j.ijrobp.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 33.Bernhardt PV, Sharpe PC, Islam M, Lovejoy DB, Kalinowski DS, Richardson DR. Iron chelators of the dipyridylketone thiosemicarbazone class: precomplexation and transmetalation effects on anticancer activity. J Med Chem. 2009;52:407–415. doi: 10.1021/jm801012z. [DOI] [PubMed] [Google Scholar]
- 34.Bertolaso L, Martini A, Bindini D, Lanzoni I, Parmeggiani A, Vitali C, Kalinec G, Kalinec F, Capitani S, Previati M. Apoptosis in the OC-k3 immortalized cell line treated with different agents. Audiology. 2001;40:327–335. doi: 10.3109/00206090109073130. [DOI] [PubMed] [Google Scholar]
- 35.Beyersmann D, Hartwig A. Carcinogenic metal compounds: recent insight into molecular and cellular mechanisms. Arch Toxicol. 2008;82:493–512. doi: 10.1007/s00204-008-0313-y. [DOI] [PubMed] [Google Scholar]
- 36.Bhuiyan MS, Fukunaga K. Cardioprotection by vanadium compounds targeting Akt-mediated signaling. J Pharmacol Sci. 2009;110:1–13. doi: 10.1254/jphs.09r01cr. [DOI] [PubMed] [Google Scholar]
- 37.Biaglow JE, Miller RA. The thioredoxin reductase/thioredoxin system: novel redox targets for cancer therapy. Cancer Biol Ther. 2005;4:6–13. doi: 10.4161/cbt.4.1.1434. [DOI] [PubMed] [Google Scholar]
- 38.Binet F, Chiasson S, Girard D. Arsenic trioxide induces endoplasmic reticulum stress-related events in neutrophils. Int Immunopharmacol. 2010;10:508–512. doi: 10.1016/j.intimp.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 39.Bishayee A, Waghray A, Patel MA, Chatterjee M. Vanadium in the detection, prevention and treatment of cancer: the in vivo evidence. Cancer Lett. 2010;294:1–12. doi: 10.1016/j.canlet.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 40.Blum O, Haiek A, Cwikel D, Dori Z, Meade TJ, Gray HB. Isolation of a myoglobin molten globule by selective cobalt(III)-induced unfolding. Proc Natl Acad Sci U S A. 1998;95:6659–6662. doi: 10.1073/pnas.95.12.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonnitcha PD, Hall MD, Underwood CK, Foran GJ, Zhang M, Beale PJ, Hambley TW. XANES investigation of the Co oxidation state in solution and in cancer cells treated with Co(III) complexes. J Inorg Biochem. 2006;100:963–971. doi: 10.1016/j.jinorgbio.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 42.Bose RN, Weaver EL. A long-lived ascorbate radical in the platinum(II) catalysed reductions of platinum(IV) antitumor drugs. J Chem Soc Dalton Trans. 1997;11:1797–1799. [Google Scholar]
- 43.Bower JJ, Leonard SS, Chen F, Shi X. As(III) transcriptionally activates the gadd45a gene via the formation of H2O2. Free Radic Biol Med. 2006;41:285–294. doi: 10.1016/j.freeradbiomed.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 44.Bowling BD, Doudican N, Manga P, Orlow SJ. Inhibition of mitochondrial protein translation sensitizes melanoma cells to arsenic trioxide cytotoxicity via a reactive oxygen species dependent mechanism. Cancer Chemother Pharmacol. 2008;63:37–43. doi: 10.1007/s00280-008-0705-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brabec V, Novakova O. DNA binding mode of ruthenium complexes and relationship to tumor cell toxicity. Drug Resist Updat. 2006;9:111–122. doi: 10.1016/j.drup.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 46.Breuer W, Shvartsman M, Cabantchik ZI. Intracellular labile iron. Int J Biochem Cell Biol. 2008;40:350–354. doi: 10.1016/j.biocel.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 47.Brewer GJ, Dick RD, Grover DK, LeClaire V, Tseng M, Wicha M, Pienta K, Redman BG, Jahan T, Sondak VK, Strawderman M, LeCarpentier G, Merajver SD. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: Phase I study. Clin Cancer Res. 2000;6:1–10. [PubMed] [Google Scholar]
- 48.Brigelius-Flohe R, Kipp A. Glutathione peroxidases in different stages of carcinogenesis. Biochim Biophys Acta. 2009;1790:1555–1568. doi: 10.1016/j.bbagen.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 49.Buettner GR. The pecking order of free radicals and anti-oxidants: lipid peroxidation, alpha-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 50.Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat Res. 1996;145:532–541. [PubMed] [Google Scholar]
- 51.Byrnes RW, Antholine WE, Petering DH. Oxidation-reduction reactions in Ehrlich cells treated with copper-neocuproine. Free Radic Biol Med. 1992;13:469–478. doi: 10.1016/0891-5849(92)90141-3. [DOI] [PubMed] [Google Scholar]
- 52.Byrnes RW, Mohan M, Antholine WE, Xu RX, Petering DH. Oxidative stress induced by a copper-thiosemicarba-zone complex. Biochemistry. 1990;29:7046–7053. doi: 10.1021/bi00482a014. [DOI] [PubMed] [Google Scholar]
- 53.Canumalla AJ, Al-Zamil N, Phillips M, Isab AA, Shaw CF., 3rd Redox and ligand exchange reactions of potential gold(I) and gold(III)-cyanide metabolites under biomimetic conditions. J Inorg Biochem. 2001;85:67–76. doi: 10.1016/s0162-0134(00)00224-5. [DOI] [PubMed] [Google Scholar]
- 54.Carde P, Timmerman R, Mehta MP, Koprowski CD, Ford J, Tishler RB, Miles D, Miller RA, Renschler MF. Multi-center phase Ib/II trial of the radiation enhancer motexafin gadolinium in patients with brain metastases. J Clin Oncol. 2001;19:2074–2083. doi: 10.1200/JCO.2001.19.7.2074. [DOI] [PubMed] [Google Scholar]
- 55.Carr JL, Tingle MD, McKeage MJ. Satraplatin activation by haemoglobin, cytochrome C and liver micro-somes in vitro. Cancer Chemother Pharmacol. 2006;57:483–490. doi: 10.1007/s00280-005-0069-5. [DOI] [PubMed] [Google Scholar]
- 56.Cascales M, Martin-Sanz P, Craciunescu DG, Mayo I, Aguilar A, Robles-Chillida EM, Cascales C. Alterations in hepatic peroxidation mechanisms in thioacetamide-induced tumors in rats. Effect of a rhodium(III) complex. Carcinogenesis (London) 1991;12:233–240. doi: 10.1093/carcin/12.2.233. [DOI] [PubMed] [Google Scholar]
- 57.Casini A, Cinellu MA, Minghetti G, Gabbiani C, Coronnello M, Mini E, Messori L. Structural and solution chemistry, antiproliferative effects, and DNA and protein binding properties of a series of dinuclear gold(III) compounds with bipyridyl ligands. J Med Chem. 2006;49:5524–5531. doi: 10.1021/jm060436a. [DOI] [PubMed] [Google Scholar]
- 58.Casini A, Gabbiani C, Sorrentino F, Rigobello MP, Bindoli A, Geldbach TJ, Marrone A, Re N, Hartinger CG, Dyson PJ, Messori L. Emerging protein targets for anticancer metallodrugs: inhibition of thioredoxin reductase and cathepsin B by antitumor ruthenium(II)-arene compounds. J Med Chem. 2008;51:6773–6781. doi: 10.1021/jm8006678. [DOI] [PubMed] [Google Scholar]
- 59.Chakraborty A, Ghosh R, Roy K, Ghosh S, Chowdhury P, Chatterjee M. Vanadium: a modifier of drug-metabolizing enzyme patterns and its critical role in cellular proliferation in transplantable murine lymphoma. Oncology. 1995;52:310–314. doi: 10.1159/000227480. [DOI] [PubMed] [Google Scholar]
- 60.Chaney SG, Campbell SL, Bassett E, Wu Y. Recognition and processing of cisplatin- and oxaliplatin-DNA adducts. Crit Rev Oncol Hematol. 2005;53:3–11. doi: 10.1016/j.critrevonc.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 61.Chaney SG, Gibbons GR, Wyrick SD, Podhasky P. An unexpected biotransformation pathway for tetrachloro-(d,ltrans)-1,2-diaminocyclohexaneplatinum(IV) (tetraplatin) in the L1210 cell line. Cancer Res. 1991;51:969–973. [PubMed] [Google Scholar]
- 62.Chaney SG, Wyrick S, Till GK. Invitro biotransformations of tetrachloro(D,L-trans)-1,2-diaminocyclohexaneplatinum (IV) (tetraplatin) in rat plasma. Cancer Res. 1990;50:4539–4545. [PubMed] [Google Scholar]
- 63.Chatterjee D, Ward MS, Shepherd RE. Detection of N-3 and N-7-coordinated [RuII(edta)(5′-GMP)]4- complexes and the N-1 protonation equilibrium of the RuIII derivative. Inorg Chim Acta. 1999;285:170–177. [Google Scholar]
- 64.Chatterjee S, Mookerjee A, Basu JM, Chakraborty P, Ganguly A, Adhikary A, Mukhopadhyay D, Ganguli S, Banerjee R, Ashraf M, Biswas J, Das PK, Sa G, Chatterjee M, Das T, Choudhuri SK. A novel copper chelate modulates tumor associated macrophages to promote anti-tumor response of T cells. PLoS One. 2009;4:e7048. doi: 10.1371/journal.pone.0007048. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Chen D, Milacic V, Frezza M, Dou QP. Metal complexes, their cellular targets and potential for cancer therapy. Curr Pharm Des. 2009;15:777–791. doi: 10.2174/138161209787582183. [DOI] [PubMed] [Google Scholar]
- 66.Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, Han ZG, Ni JH, Shi GY, Jia PM, Liu MM, He KL, Niu C, Ma J, Zhang P, Zhang TD, Paul P, Naoe T, Kitamura K, Miller W, Waxman S, Wang ZY, de The H, Chen SJ, Chen Z. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89:3345–3353. [PubMed] [Google Scholar]
- 67.Chen HH, Kuo MT. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met Based Drugs. 2010 doi: 10.1155/2010/430939. [Epub ahead of print]; DOI: 10.1155/2010/430939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen L, Lee PF, Ranford JD, Vittal JJ, Wong SY. Reduction of the anti-cancer drug analogue cis,trans, cis[PtCl2(OCOCH3)(2)(NH3)(2)] by L-cysteine and L-methionine and its crystal structure. J Chem Soc Dalton Trans. 1999;8:1209–1212. [Google Scholar]
- 69.Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116:565–576. doi: 10.1016/s0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- 70.Chiaverini N, De Ley M. Protective effect of metallothionein on oxidative stress-induced DNA damage. Free Radic Res. 2010;44:605–613. doi: 10.3109/10715761003692511. [DOI] [PubMed] [Google Scholar]
- 71.Chibber R, Stratford IJ, O Neill P, Sheldon PW, Ahmed I, Lee B. The interaction between radiation and complexes of cis-Pt(II) and Rh(II): studies at the molecular and cellular level. Int J Radiat Biol. 1985;48:513–524. doi: 10.1080/09553008514551581. [DOI] [PubMed] [Google Scholar]
- 72.Chikira M, Tomizawa Y, Fukita D, Sugizaki T, Sugawara N, Yamazaki T, Sasano A, Shindo H, Palaniandavar M, Antholine WE. DNA-fiber EPR study of the orientation of Cu(II) complexes of 1,10-phenanthroline and its derivatives bound to DNA: mono(phenanthroline)-copper(II) and its ternary complexes with amino acids. J Inorg Biochem. 2002;89:163–173. doi: 10.1016/s0162-0134(02)00378-1. [DOI] [PubMed] [Google Scholar]
- 73.Chirino YI, Pedraza-Chaverri J. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp Toxicol Pathol. 2009;61:223–242. doi: 10.1016/j.etp.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 74.Choi S, Filotto C, Bisanzo M, Delaney S, Lagasee D, Whitworth JL, Jusko A, Li CR, Wood NA, Willingham J, Schwenker A, Spaulding K. Reduction and anticancer activity of platinum(IV) complexes. Inorg Chem. 1998;37:2500–2504. [Google Scholar]
- 75.Chowdhury UK, Zakharyan RA, Hernandez A, Avram MD, Kopplin MJ, Aposhian HV. Glutathione-S-transferase-omega [MMA(V) reductase] knockout mice: enzyme and arsenic species concentrations in tissues after arsenate administration. Toxicol Appl Pharmacol. 2006;216:446–457. doi: 10.1016/j.taap.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 76.Christodoulou J, Sadler PJ, Tucker A. H NMR of albumin in human blood plasma: drug binding and redox reactions at Cys34. FEBS Lett. 1995;376:1–5. doi: 10.1016/0014-5793(95)01231-2. [DOI] [PubMed] [Google Scholar]
- 77.Clarke MJ. Ruthenium metallopharmaceuticals. Coordination Chem Rev. 2003;236:209–233. [Google Scholar]
- 78.Clarke MJ, Jansen B, Marx KA, Kruger R. Biochemical effects of binding pentaammineaquaruthenium(2 +) to DNA and oxidation to [(NH3)5RuIII]n-DNA. Inorg Chim Acta. 1986;124:13–28. [Google Scholar]
- 79.Clarke MJ, Zhu F, Frasca DR. Non-Platinum Chemotherapeutic Metallopharmaceuticals. Chem Rev. 1999;99:2511–2533. doi: 10.1021/cr9804238. [DOI] [PubMed] [Google Scholar]
- 80.Corazza A, Harvey I, Sadler PJ. H,13C-NMR and X-ray absorption studies of copper(I) glutathione complexes. Eur J Biochem. 1996;236:697–705. doi: 10.1111/j.1432-1033.1996.0697d.x. [DOI] [PubMed] [Google Scholar]
- 81.Corti A, Casini AF, Pompella A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch Biochem Biophys. 2010;500:107–115. doi: 10.1016/j.abb.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 82.Cox AG, Winterbourn CC, Hampton MB. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem J. 2010;425:313–325. doi: 10.1042/BJ20091541. [DOI] [PubMed] [Google Scholar]
- 83.Crans DC, Zhang B, Gaidamauskas E, Keramidas AD, Willsky GR, Roberts CR. Is vanadate reduced by thiols under biological conditions? changing the redox potential of V(V)/V(IV) by complexation in aqueous solution. Inorg Chem. 2010;49:4245–4256. doi: 10.1021/ic100080k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cullen KJ, Newkirk KA, Schumaker LM, Aldosari N, Rone JD, Haddad BR. Glutathione S-transferase pi amplification is associated with cisplatin resistance in head and neck squamous cell carcinoma cell lines and primary tumors. Cancer Res. 2003;63:8097–8102. [PubMed] [Google Scholar]
- 85.Custodio JB, Cardoso CM, Santos MS, Almeida LM, Vicente JA, Fernandes MA. Cisplatin impairs rat liver mitochondrial functions by inducing changes on membrane ion permeability: prevention by thiol group protecting agents. Toxicology. 2009;259:18–24. doi: 10.1016/j.tox.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 86.D’Cruz OJ, Uckun FM. Metvan: a novel oxovanadium(IV) complex with broad spectrum anticancer activity. Expert Opin Investig Drugs. 2002;11:1829–1836. doi: 10.1517/13543784.11.12.1829. [DOI] [PubMed] [Google Scholar]
- 87.Dai J, Weinberg RS, Waxman S, Jing Y. Malignant cells can be sensitized to undergo growth inhibition and apoptosis by arsenic trioxide through modulation of the glutathione redox system. Blood. 1999;93:268–277. [PubMed] [Google Scholar]
- 88.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 89.Denny WA. Prodrug strategies in cancer therapy. Eur J Med Chem. 2001;36:577–595. doi: 10.1016/s0223-5234(01)01253-3. [DOI] [PubMed] [Google Scholar]
- 90.Denny WA. Hypoxia-activated prodrugs in cancer therapy: progress to the clinic. Future Oncol. 2010;6:419–428. doi: 10.2217/fon.10.1. [DOI] [PubMed] [Google Scholar]
- 91.Desoize B. Metals and metal compounds in cancer treatment. Anticancer Res. 2004;24:1529–1544. [PubMed] [Google Scholar]
- 92.Diaz Z, Colombo M, Mann KK, Su H, Smith KN, Bohle DS, Schipper HM, Miller WH., Jr Trolox selectively enhances arsenic-mediated oxidative stress and apoptosis in APL and other malignant cell lines. Blood. 2005;105:1237–1245. doi: 10.1182/blood-2004-05-1772. [DOI] [PubMed] [Google Scholar]
- 93.Dilda PJ, Hogg PJ. Arsenical-based cancer drugs. Cancer Treat Rev. 2007;33:542–564. doi: 10.1016/j.ctrv.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 94.Dilda PJ, Ramsay EE, Corti A, Pompella A, Hogg PJ. Metabolism of the tumor angiogenesis inhibitor 4-(N-(SGlutathionylacetyl)amino)phenylarsonous acid. J Biol Chem. 2008;283:35428–35434. doi: 10.1074/jbc.M804470200. [DOI] [PubMed] [Google Scholar]
- 95.Dittrich C, Scheulen ME, Jaehde U, Kynast B, Gneist M, Richly H, Schaad S, Arion VB, Keppler BK. Phase I and pharmacokinetic study of sodium trans- [tetrachlorobis(1H-indazole)ruthenate(III)]/indazolehydrochloride (1:1.1) (FFC14A, KP1019) in patients with solid tumors–a study of the CESAR Central European Society for Anticancer Drug Research—EWIV. Proceedings of the American Association for Cancer Research. 2005;46:P472. [Google Scholar]
- 96.Djordjevic C, Wampler GL. Antitumor activity and toxicity of peroxo heteroligand vanadates(V) in relation to biochemistry of vanadium. J Inorg Biochem. 1985;25:51–55. doi: 10.1016/0162-0134(85)83007-5. [DOI] [PubMed] [Google Scholar]
- 97.Don AS, Kisker O, Dilda P, Donoghue N, Zhao X, Decollogne S, Creighton B, Flynn E, Folkman J, Hogg PJ. A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell. 2003;3:497–509. doi: 10.1016/s1535-6108(03)00109-0. [DOI] [PubMed] [Google Scholar]
- 98.dos Santos NA, Martins NM, Curti C, Pires Bianchi, Mde L, dos Santos AC. Dimethylthiourea protects against mitochondrial oxidative damage induced by cisplatin in liver of rats. Chem Biol Interact. 2007;170:177–186. doi: 10.1016/j.cbi.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 99.Dougan SJ, Habtemariam A, McHale SE, Parsons S, Sadler PJ. Catalytic organometallic anticancer complexes. Proc Natl Acad Sci. 2008;105:11628–11633. doi: 10.1073/pnas.0800076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eastman A. Cross-linking of glutathione to DNA by cancer chemotherapeutic platinum coordination complexes. Chem Biol Interact. 1987;61:241–248. doi: 10.1016/0009-2797(87)90004-4. [DOI] [PubMed] [Google Scholar]
- 101.Eastman A. Glutathione-mediated activation of anticancer platinum(IV) complexes. Biochem Pharmacol. 1987;36:4177–4178. doi: 10.1016/0006-2952(87)90581-8. [DOI] [PubMed] [Google Scholar]
- 102.Ejima K, Layne MD, Carvajal IM, Nanri H, Ith B, Yet SF, Perrella MA. Modulation of the thioredoxin system during inflammatory responses and its effect on heme oxygenase-1 expression. Antioxid Redox Signal. 2002;4:569–575. doi: 10.1089/15230860260220067. [DOI] [PubMed] [Google Scholar]
- 103.El-Naggar MM, El-Waseef AM, El-Halafawy KM, El-Sayed IH. Antitumor activities of vanadium(IV), manganese(IV), iron(III), cobalt(II) and copper(II) complexes of 2-methylaminopyridine. Cancer Lett. 1998;133:71–76. doi: 10.1016/s0304-3835(98)00213-4. [DOI] [PubMed] [Google Scholar]
- 104.Elder RC, Zhao Z, Zhang Y, Dorsey JG, Hess EV, Tepperman K. Dicyanogold (I) is a common human metabolite of different gold drugs. J Rheumatol. 1993;20:268–272. [PubMed] [Google Scholar]
- 105.Ellis LT, Er HM, Hambley TW. The influence of the axial ligands of a series of platinum(IV) anticancer complexes on their reduction to platinum(II) and reaction with DNA. Aust J Chem. 1995;48:793–806. [Google Scholar]
- 106.Evangelou AM. Vanadium in cancer treatment. Crit Rev Oncol Hematol. 2002;42:249–265. doi: 10.1016/s1040-8428(01)00221-9. [DOI] [PubMed] [Google Scholar]
- 107.Evens AM, Tallman MS, Gartenhaus RB. The potential of arsenic trioxide in the treatment of malignant disease: past, present, and future. Leuk Res. 2004;28:891–900. doi: 10.1016/j.leukres.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 108.Faure R, Vincent M, Dufour M, Shaver A, Posner BI. Arrest at the G2/M transition of the cell cycle by protein-tyrosine phosphatase inhibition: studies on a neuronal and a glial cell line. J Cell Biochem. 1995;59:389–401. doi: 10.1002/jcb.240590310. [DOI] [PubMed] [Google Scholar]
- 109.Fenton HJH. On a new reaction of tartaric acid. Chem News. 1876;33:190. [Google Scholar]
- 110.Ferrer-Sueta G, Batinic-Haberle I, Spasojevic I, Fridovich I, Radi R. Catalytic scavenging of peroxynitrite by isomeric Mn(III) N-methylpyridylporphyrins in the presence of reductants. Chem Res Toxicol. 1999;12:442–449. doi: 10.1021/tx980245d. [DOI] [PubMed] [Google Scholar]
- 111.Finch RA, Liu MC, Cory AH, Cory JG, Sartorelli AC. Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone; 3-AP): an inhibitor of ribonucleotide reductase with antineoplastic activity. Adv Enzyme Regul. 1999;39:3–12. doi: 10.1016/s0065-2571(98)00017-x. [DOI] [PubMed] [Google Scholar]
- 112.Fokkema E, Groen HJ, Helder MN, de Vries EG, Meijer C. JM216-, JM118-, and cisplatin-induced cytotoxicity in relation to platinum-DNA adduct formation, glutathione levels and p53 status in human tumour cell lines with different sensitivities to cisplatin. Biochem Pharmacol. 2002;63:1989–1996. doi: 10.1016/s0006-2952(02)00983-8. [DOI] [PubMed] [Google Scholar]
- 113.Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 2009;30:1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Frade RFM, Candeias NR, Duarte CMM, André V, Duarte M Teresa, Gois PMP, Afonso CAM. New dirhodium complex with activity towards colorectal cancer. Bioorg Med Chem Lett. 2010;20:3413–3415. doi: 10.1016/j.bmcl.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 115.Franco R, Cidlowski JA. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009;16:1303–1314. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- 116.Frasca DR, Clarke MJ. Alterations in the binding of [Cl(NH3)5RuIII]2 + to DNA by glutathione: reduction, autoxidation, coordination, and decomposition. J Am Chem Soc. 1999;121:8523–8532. [Google Scholar]
- 117.Fries JF, Bloch D, Spitz P, Mitchell DM. Cancer in rheumatoid arthritis: a prospective long-term study of mortality. Am J Med. 1985;78:56–59. doi: 10.1016/0002-9343(85)90247-5. [DOI] [PubMed] [Google Scholar]
- 118.Fruhauf S, Zeller WJ. In vitro evaluation of platinum, titanium and ruthenium metal complexes in cisplatin-sensitive and -resistant rat ovarian tumors. Cancer Chemother Pharmacol. 1991;27:301–307. doi: 10.1007/BF00685116. [DOI] [PubMed] [Google Scholar]
- 119.Fu PKL, Bradley PM, Turro C. DNA Cleavage by Photogenerated Rh2(O2CCH3)4(H2O)2+ Inorg Chem. 2001;40:2476–2477. doi: 10.1021/ic010118w. [DOI] [PubMed] [Google Scholar]
- 120.Galanski M, Jakupec MA, Keppler BK. Update of the preclinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches. Curr Med Chem. 2005;12:2075–2094. doi: 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- 121.Gallagher J, Chen CH, Pan CQ, Perrin DM, Cho YM, Sigman DS. Optimizing the targeted chemical nuclease activity of 1,10-phenanthroline-copper by ligand modification. Bioconjug Chem. 1996;7:413–420. doi: 10.1021/bc960028t. [DOI] [PubMed] [Google Scholar]
- 122.Gandin V, Fernandes AP, Rigobello MP, Dani B, Sorrentino F, Tisato F, Bjornstedt M, Bindoli A, Sturaro A, Rella R, Marzano C. Cancer cell death induced by phosphine gold(I) compounds targeting thioredoxin reductase. Biochem Pharmacol. 2010;79:90–101. doi: 10.1016/j.bcp.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 123.Ganyc D, Talbot S, Konate F, Jackson S, Schanen B, Cullen W, Self WT. Impact of trivalent arsenicals on seleno-protein synthesis. Environ Health Perspect. 2007;115:346–353. doi: 10.1289/ehp.9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gibson D. The mechanism of action of platinum anticancer agents—what do we really know about it? Dalton Trans. 2009;48:10681–10689. doi: 10.1039/b918871c. [DOI] [PubMed] [Google Scholar]
- 125.Giommarelli C, Corti A, Supino R, Favini E, Paolicchi A, Pompella A, Zunino F. Gamma-glutamyltransferase-dependent resistance to arsenic trioxide in melanoma cells and cellular sensitization by ascorbic acid. Free Radic Biol Med. 2009;46:1516–1526. doi: 10.1016/j.freeradbiomed.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 126.Glickstein H, El RB, Shvartsman M, Cabantchik ZI. Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood. 2005;106:3242–3250. doi: 10.1182/blood-2005-02-0460. [DOI] [PubMed] [Google Scholar]
- 127.Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780:1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Golemovic M, Quintas-Cardama A, Manshouri T, Orsolic N, Duzkale H, Johansen M, Freireich EJ, Kantarjian H, Zingaro RA, Verstovsek S. MER1, a novel organic arsenic derivative, has potent PML-RARalpha- independent cytotoxic activity against leukemia cells. Invest New Drugs. 2010;28:402–412. doi: 10.1007/s10637-009-9267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 130.Gourdier I, Crabbe L, Andreau K, Pau B, Kroemer G. Oxaliplatin-induced mitochondrial apoptotic response of colon carcinoma cells does not require nuclear DNA. Oncogene. 2004;23:7449–7457. doi: 10.1038/sj.onc.1208047. [DOI] [PubMed] [Google Scholar]
- 131.Grad JM, Bahlis NJ, Reis I, Oshiro MM, Dalton WS, Boise LH. Ascorbic acid enhances arsenic trioxide-induced cytotoxicity in multiple myeloma cells. Blood. 2001;98:805–813. doi: 10.1182/blood.v98.3.805. [DOI] [PubMed] [Google Scholar]
- 132.Gregus Z, Nemeti B. The glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase works as an arsenate reductase in human red blood cells and rat liver cytosol. Toxicol Sci. 2005;85:859–869. doi: 10.1093/toxsci/kfi158. [DOI] [PubMed] [Google Scholar]
- 133.Gupta Y, Kohli DV, Jain SK. Vitamin B12-mediated transport: a potential tool for tumor targeting of antineo-plastic drugs and imaging agents. Crit Rev Ther Drug Carrier Syst. 2008;25:347–379. doi: 10.1615/critrevtherdrugcarriersyst.v25.i4.20. [DOI] [PubMed] [Google Scholar]
- 134.Gupte A, Mumper RJ. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat Rev. 2009;35:32–46. doi: 10.1016/j.ctrv.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 135.Gust R, Ott I, Posselt D, Sommer K. Development of cobalt(3,4-diarylsalen) complexes as tumor therapeutics. J Med Chem. 2004;47:5837–5846. doi: 10.1021/jm040763n. [DOI] [PubMed] [Google Scholar]
- 136.Haber F, Weiss J. The catalytic decomposition of hydrogen peroxide by iron salts. Proc R Soc Lond Ser A. 1934;147:332–351. [Google Scholar]
- 137.Hall MD, Amjadi S, Zhang M, Beale PJ, Hambley TW. The mechanism of action of platinum(IV) complexes in ovarian cancer cell lines. J Inorg Biochem. 2004;98:1614–1624. doi: 10.1016/j.jinorgbio.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 138.Hall MD, Hambley TW. Platinum(IV) antitumour compounds: their bioinorganic chemistry. Coordination Chem Rev. 2002;232:49–67. [Google Scholar]
- 139.Hall MD, Mellor HR, Callaghan R, Hambley TW. Basis for design and development of platinum(IV) anticancer complexes. J Med Chem. 2007;50:3403–3411. doi: 10.1021/jm070280u. [DOI] [PubMed] [Google Scholar]
- 140.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford University Press; New York: 1988. p. 374. [Google Scholar]
- 141.Hambley TW, Battle AR, Deacon GB, Lawrenz ET, Fallon GD, Gatehouse BM, Webster LK, Rainone S. Modifying the properties of platinum (IV) complexes in order to increase biological effectiveness. J Inorg Biochem. 1999;77:3–12. doi: 10.1016/s0162-0134(99)00133-6. [DOI] [PubMed] [Google Scholar]
- 142.Han YH, Kim SZ, Kim SH, Park WH. Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a G2 arrest of the cell cycle and apoptosis accompanied with the depletion of GSH. Cancer Lett. 2008;270:40–55. doi: 10.1016/j.canlet.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 143.Hancock CN, Stockwin LH, Han B, Divelbiss RD, Jun JH, Malhotra SV, Hollingshead MG, Newton DL. A copper chelate of thiosemicarbazone NSC 689534 induces oxidative/ER stress and inhibits tumor growth in vitro and in vivo. Free Radic Biol Med. 2011;50:110–121. doi: 10.1016/j.freeradbiomed.2010.10.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hanigan MH, Devarajan P. Cisplatin nephrotoxicity: molecular mechanisms. Cancer Ther. 2003;1:47–61. [PMC free article] [PubMed] [Google Scholar]
- 145.Hanna PM, Kadiiska MB, Mason RP. Oxygen-derived free radical and active oxygen complex formation from cobalt(II) chelates in vitro. Chem Res Toxicol. 1992;5:109–115. doi: 10.1021/tx00025a019. [DOI] [PubMed] [Google Scholar]
- 146.Hannemann J, Baumann K. Cisplatin-induced lipid peroxidation and decrease of gluconeogenesis in rat kidney cortex: different effects of antioxidants and radical scavengers. Toxicology. 1988;51:119–132. doi: 10.1016/0300-483x(88)90143-6. [DOI] [PubMed] [Google Scholar]
- 147.Harding MM, Mokdsi G. Antitumour metallocenes: structure-activity studies and interactions with biomolecules. Curr Med Chem. 2000;7:1289–1303. doi: 10.2174/0929867003374066. [DOI] [PubMed] [Google Scholar]
- 148.Hartinger CG, Dyson PJ. Bioorganometallic chemistry-from teaching paradigms to medicinal applications. Chem Soc Rev. 2009;38:391–401. doi: 10.1039/b707077m. [DOI] [PubMed] [Google Scholar]
- 149.Hartinger CG, Zorbas-Seifried S, Jakupec MA, Kynast B, Zorbas H, Keppler BK. From bench to bedside—preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate (III)] (KP1019 or FFC14A) JInorg Biochem. 2006;100:891–904. doi: 10.1016/j.jinorgbio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 150.Hayakawa T, Kobayashi Y, Cui X, Hirano S. A new metabolic pathway of arsenite: arsenic-glutathione complexes are substrates for human arsenic methyltransferase Cyt19. Arch Toxicol. 2005;79:183–191. doi: 10.1007/s00204-004-0620-x. [DOI] [PubMed] [Google Scholar]
- 151.Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. Cancer chemoprevention mechanismsmediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal. 2010;13:1713–1748. doi: 10.1089/ars.2010.3221. [DOI] [PubMed] [Google Scholar]
- 152.Healy J, Tipton K. Ceruloplasmin and what it might do. J Neural Transm. 2007;114:777–781. doi: 10.1007/s00702-007-0687-7. [DOI] [PubMed] [Google Scholar]
- 153.Hedstrom E, Eriksson S, Zawacka-Pankau J, Arner ES, Selivanova G. p53-dependent inhibition of TrxR1 contributes to the tumor-specific induction of apoptosis by RITA. Cell Cycle. 2009;8:3576–3583. doi: 10.4161/cc.8.21.9977. [DOI] [PubMed] [Google Scholar]
- 154.Heffeter P, Bock K, Atil B, Hoda MA Reza, Korner W, Bartel C, Jungwirth U, Keppler BK, Micksche M, Berger W, Koellensperger G. Intracellular protein binding patterns of the anticancer ruthenium drugs KP1019 and KP1339. J Biol Inorg Chem. 2010;15:737–748. doi: 10.1007/s00775-010-0642-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Heffeter P, Jungwirth U, Jakupec M, Hartinger C, Galanski M, Elbling L, Micksche M, Keppler B, Berger W. Resistance against novel anticancer metal compounds: differences and similarities. Drug Resist Updat. 2008;11:1–16. doi: 10.1016/j.drup.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 156.Heffeter P, Popovic-Bijelic A, Saiko P, Dornetshuber R, Jungwirth U, Voevodskaya N, Biglino D, Jakupec MA, Elbling L, Micksche M, Szekeres T, Keppler BK, Graslund A, Berger W. Ribonucleotide reductase as one important target of [tris(1,10-phenanthroline)lanthanum(III)] trithiocyanate (KP772) Curr Cancer Drug Targets. 2009;9:595–607. doi: 10.2174/156800909789056962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hellberg V, Wallin I, Eriksson S, Hernlund E, Jerremalm E, Berndtsson M, Eksborg S, Arner ES, Shoshan M, Ehrsson H, Laurell G. Cisplatin and oxaliplatin toxicity: importance of cochlear kinetics as a determinant for ototoxicity. J Natl Cancer Inst. 2009;101:37–47. doi: 10.1093/jnci/djn418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 159.Hoke GD, McCabe FL, Faucette LF, Bartus JO, Sung CM, Jensen BD, Heys JR, Rush GF, Alberts DW, Johnson RK, et al. In vivo development and in vitro characterization of a subclone of murine P388 leukemia resistant to bis (diphenylphosphine)ethane. Mol Pharmacol. 1991;39:90–97. [PubMed] [Google Scholar]
- 160.Hoke GD, Rush GF, Bossard GF, McArdle JV, Jensen BD, Mirabelli CK. Mechanism of alterations in isolated rat liver mitochondrial function induced by gold complexes of bidentate phosphines. J Biol Chem. 1988;263:11203–11210. [PubMed] [Google Scholar]
- 161.Hoke GD, Rush GF, Mirabelli CK. The mechanism of acute cytotoxicity of triethylphosphine gold(I) complexes. III. Chlorotriethylphosphine gold(I)-induced alterations in isolated rat liver mitochondrial function. Toxicol Appl Pharmacol. 1989;99:50–60. doi: 10.1016/0041-008x(89)90110-5. [DOI] [PubMed] [Google Scholar]
- 162.Holleman AF, Wiberg E, Wiberg N. Inorganic Chemistry. de Gryter; Berlin: 2001. p. 1924. [Google Scholar]
- 163.Hou MH, Lu WJ, Huang CY, Fan RJ, Yuann JM. Effects of polyamines on the DNA-reactive properties of dimeric mithramycin complexed with cobalt(II): implications for anticancer therapy. Biochemistry. 2009;48:4691–4698. doi: 10.1021/bi900092w. [DOI] [PubMed] [Google Scholar]
- 164.Housecroft CE, Sharpe AG. Inorganic Chemistry. Pearson Education Limited; Harlow: 2005. p. 949. [Google Scholar]
- 165.Howard RA, Spring TG, Bear JL. The interaction of rhodium(II) carboxylates with enzymes. Cancer Res. 1976;36:4402–4405. [PubMed] [Google Scholar]
- 166.Howell SB, Safaei R, Larson CA, Sailor MJ. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol Pharmacol. 2010;77:887–894. doi: 10.1124/mol.109.063172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hu J, Liu YF, Wu CF, Xu F, Shen ZX, Zhu YM, Li JM, Tang W, Zhao WL, Wu W, Sun HP, Chen QS, Chen B, Zhou GB, Zelent A, Waxman S, Wang ZY, Chen SJ, Chen Z. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 2009;106:3342–3347. doi: 10.1073/pnas.0813280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J Biol Chem. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- 169.Ishida S, McCormick F, Smith-McCune K, Hanahan D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell. 2010;17:574–583. doi: 10.1016/j.ccr.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jakimowicz P, Ostropolska L, Pruchnik FP. Interaction of [Rh2(O2CCH3)4(H2O)2] and [Rh2(O2CCH(OH)Ph)2(phen)2 (H2O)2](O2CCH(OH)Ph)2 with sulfhydryl compounds and ceruloplasmin. Met-Based Drugs. 2000;7:201–209. doi: 10.1155/MBD.2000.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jakupec MA, Galanski M, Keppler BK. Tumour-inhibiting platinum complexes—state of the art and future perspectives. Rev Physiol Biochem Pharmacol. 2003;146:1–54. doi: 10.1007/s10254-002-0001-x. [DOI] [PubMed] [Google Scholar]
- 173.Jakupec MA, Reisner E, Eichinger A, Pongratz M, Arion VB, Galanski M, Hartinger CG, Keppler BK. Redox-active antineoplastic ruthenium complexes with indazole: correlation of in vitro potency and reduction potential. J Med Chem. 2005;48:2831–2837. doi: 10.1021/jm0490742. [DOI] [PubMed] [Google Scholar]
- 174.Jaramillo MC, Frye JB, Crapo JD, Briehl MM, Tome ME. Increased manganese superoxide dismutase expression or treatment with manganese porphyrin potentiates dexamethasone-induced apoptosis in lymphoma cells. Cancer Res. 2009;69:5450–5457. doi: 10.1158/0008-5472.CAN-08-4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jaspers I, Samet JM, Erzurum S, Reed W. Vanadium-induced kappaB-dependent transcription depends upon peroxide-induced activation of the p38 mitogen-activated protein kinase. Am J Respir Cell Mol Biol. 2000;23:95–102. doi: 10.1165/ajrcmb.23.1.3989. [DOI] [PubMed] [Google Scholar]
- 176.Jennette KW, Lippard SJ, Vassiliades GA, Bauer WR. Metallointercalation reagents. 2-hydroxyethanethiolato (2,2′,2′-terpyridine)-platinum(II) monocation binds strongly to DNA by intercalation. Proc Natl Acad Sci U S A. 1974;71:3839–3843. doi: 10.1073/pnas.71.10.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jeon KI, Byun MS, Jue DM. Gold compound auranofin inhibits IkappaB kinase (IKK) by modifying Cys-179 of IKKbeta subunit. Exp Mol Med. 2003;35:61–66. doi: 10.1038/emm.2003.9. [DOI] [PubMed] [Google Scholar]
- 178.Jiang H, Ma Y, Chen X, Pan S, Sun B, Krissansen GW, Sun X. Genistein synergizes with arsenic trioxide to suppress human hepatocellular carcinoma. Cancer Sci. 2010;101:975–983. doi: 10.1111/j.1349-7006.2009.01464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jiang M, Dong Z. Regulation and pathological role of p53 in cisplatin nephrotoxicity. J Pharmacol Exp Ther. 2008;327:300–307. doi: 10.1124/jpet.108.139162. [DOI] [PubMed] [Google Scholar]
- 180.Jing Y, Dai J, Chalmers-Redman RM, Tatton WG, Waxman S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood. 1999;94:2102–2111. [PubMed] [Google Scholar]
- 181.Jung M, Kerr DE, Senter PD. Bioorganometallic chemistry—synthesis and antitumor activity of cobalt carbonyl complexes. Arch Pharm (Weinheim) 1997;330:173–176. doi: 10.1002/ardp.19973300604. [DOI] [PubMed] [Google Scholar]
- 182.Kadishi KM, Das K, Howard R, Dennis A, Bear JL. 264- Redox Reactions and Antitumor Activity of Tetra-[mu]-Carboxylatodirhodium(II) Bioelectrochem Bioenerg. 1978;5:741–753. [Google Scholar]
- 183.Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, measurement, and participation in cellular processes(1) Free Radic Biol Med. 2002;33:1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 184.Kamalakannan P, Venkappayya D. Synthesis and characterization of cobalt and nickel chelates of 5-dimethylaminomethyl-2-thiouracil and their evaluation as antimicrobial and anticancer agents. J Inorg Biochem. 2002;90:22–37. doi: 10.1016/s0162-0134(02)00413-0. [DOI] [PubMed] [Google Scholar]
- 185.Kapitza S, Jakupec MA, Uhl M, Keppler BK, Marian B. The heterocyclic ruthenium(III) complex KP1019 (FFC14A) causes DNA damage and oxidative stress in colorectal tumor cells. Cancer Lett. 2005;226:115–121. doi: 10.1016/j.canlet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 186.Kapitza S, Pongratz M, Jakupec MA, Heffeter P, Berger W, Lackinger L, Keppler BK, Marian B. Heterocyclic complexes of ruthenium(III) induce apoptosis in colorectal carcinoma cells. J Cancer Res Clin Oncol. 2005;131:101–110. doi: 10.1007/s00432-004-0617-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kasprzak KS, Zastawny TH, North SL, Riggs CW, Diwan BA, Rice JM, Dizdaroglu M. Oxidative DNA base damage in renal, hepatic, and pulmonary chromatin of rats after intraperitoneal injection of cobalt(II) acetate. Chem Res Toxicol. 1994;7:329–35. doi: 10.1021/tx00039a009. [DOI] [PubMed] [Google Scholar]
- 188.Katsaros N, Anagnostopoulou A. Rhodium and its compounds as potential agents in cancer treatment. Crit Rev Oncol Hematol. 2002;42:297–308. doi: 10.1016/s1040-8428(01)00222-0. [DOI] [PubMed] [Google Scholar]
- 189.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 190.Khan MF, Ohno Y, Takanaka A. Effect of tetrakismu-3,5-diisopropylsalicylatodiaquodicopper(II) on the status of reduced glutathione in freshly isolated hepatocytes. Arch Toxicol. 1992;66:587–591. doi: 10.1007/BF01973390. [DOI] [PubMed] [Google Scholar]
- 191.Kido Y, Khokhar AR, Siddik ZH. Glutathione-mediated modulation of tetraplatin activity against sensitive and resistant tumor cells. Biochem Pharmacol. 1994;47:1635–1642. doi: 10.1016/0006-2952(94)90542-8. [DOI] [PubMed] [Google Scholar]
- 192.Kido Y, Khokhar AR, Yoshida M, Thai GW, Siddik ZH. Pharmacokinetics of tetraplatin administered intraperitoneally with reduced glutathione in mice. Drug Metab Dispos. 1994;22:312–317. [PubMed] [Google Scholar]
- 193.Kim H, Son HY, Bailey SM, Lee J. Deletion of hepatic Ctr1 reveals its function in copper acquisition and compensatory mechanisms for copper homeostasis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G356–G364. doi: 10.1152/ajpgi.90632.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 195.Kim NH, Lee MY, Park SJ, Choi JS, Oh MK, Kim IS. Auranofin blocks interleukin-6 signalling by inhibiting phosphorylation of JAK1 and STAT3. Immunology. 2007;122:607–614. doi: 10.1111/j.1365-2567.2007.02679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kim NH, Oh MK, Park HJ, Kim IS. Auranofin, a gold(I)-containing antirheumatic compound, activates Keap1/Nrf2 signaling via Rac1/iNOS signal and mitogen-activated protein kinase activation. J Pharmacol Sci. 2010;113:246–254. doi: 10.1254/jphs.09330fp. [DOI] [PubMed] [Google Scholar]
- 197.Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radical Biol Med. 1999;27:1208–1218. doi: 10.1016/s0891-5849(99)00145-8. [DOI] [PubMed] [Google Scholar]
- 198.Knox RJ, Friedlos F, Lydall DA, Roberts JJ. Mechanism of cytotoxicity of anticancer platinum drugs: evidence that cis-diamminedichloroplatinum(II) and cis-diammine-(1,1-cyclobutanedicarboxylato)platinum(II) differ only in the kinetics of their interaction with DNA. Cancer Res. 1986;46:1972–1979. [PubMed] [Google Scholar]
- 199.Kolberg M, Strand KR, Graff P, Andersson KK. Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta. 2004;1699:1–34. doi: 10.1016/j.bbapap.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 200.Kopera E, Schwerdtle T, Hartwig A, Bal W. Co(II) and Cd(II) substitute for Zn(II) in the zinc finger derived from the DNA repair protein XPA, demonstrating a variety of potential mechanisms of toxicity. Chem Res Toxicol. 2004;17:1452–1458. doi: 10.1021/tx049842s. [DOI] [PubMed] [Google Scholar]
- 201.Kopf-Maier P, Krahl D. Tumor inhibition by metallocenes: ultrastructural localization of titanium and vanadium in treated tumor cells by electron energy loss spectroscopy. Chem Biol Interact. 1983;44:317–328. doi: 10.1016/0009-2797(83)90059-5. [DOI] [PubMed] [Google Scholar]
- 202.Kostova I. Gold coordination complexes as anticancer agents. Anticancer Agents Med Chem. 2006;6:19–32. doi: 10.2174/187152006774755500. [DOI] [PubMed] [Google Scholar]
- 203.Kostova I. Titanium and vanadium complexes as anticancer agents. Anticancer Agents Med Chem. 2009;9:827–842. doi: 10.2174/187152009789124646. [DOI] [PubMed] [Google Scholar]
- 204.Kowol CR, Trondl R, Heffeter P, Arion VB, Jakupec MA, Roller A, Galanski M, Berger W, Keppler BK. Impact of metal coordination on cytotoxicity of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (triapine) and novel insights into terminal dimethylation. J Med Chem. 2009;52:5032–5043. doi: 10.1021/jm900528d. [DOI] [PubMed] [Google Scholar]
- 205.Kruszewski M. Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat Res Fundam Mol Mech Mutagen. 2003;531:81–92. doi: 10.1016/j.mrfmmm.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 206.Kuwabara M, Yoon C, Goyne T, Thederahn T, Sigman DS. Nuclease activity of 1,10-phenanthroline-copper ion: reaction with CGCGAATTCGCG and its complexes with netropsin and EcoRI. Biochemistry. 1986;25:7401–7408. doi: 10.1021/bi00371a023. [DOI] [PubMed] [Google Scholar]
- 207.Laib JE, Shaw CF, 3rd, Petering DH, Eidsness MK, Elder RC, Garvey JS. Formation and characterization of aurothioneins: Au,Zn,Cd-thionein, Au,Cd-thionein, and (thiomalato-Au)chi-thionein. Biochemistry. 1985;24:1977–1986. doi: 10.1021/bi00329a027. [DOI] [PubMed] [Google Scholar]
- 208.Laurent A, Nicco C, Chereau C, Goulvestre C, Alexandre J, Alves A, Levy E, Goldwasser F, Panis Y, Soubrane O, Weill B, Batteux F. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005;65:948–956. [PubMed] [Google Scholar]
- 209.Lee MT, Ahmed T, Haddad R, Friedman ME. Inhibition of several enzymes by gold compounds. II. Beta-glucuronidase, acid phosphatase and L-malate dehydrogenase by sodium thiomalatoraurate (I), sodium thiosulfatoaurate (I) and thioglucosoaurate (I) J Enzyme Inhib. 1989;3:35–47. doi: 10.3109/14756368909030362. [DOI] [PubMed] [Google Scholar]
- 210.Lemma K, Berglund J, Farrell N, Elding LI. Kinetics and mechanism for reduction of anticancer-active tetrachloroam(m)ine platinum(IV) compounds by glutathione. J Biol Inorg Chem. 2000;5:300–306. doi: 10.1007/pl00010658. [DOI] [PubMed] [Google Scholar]
- 211.Lemma K, House DA, Retta N, Elding LI. Kinetics and mechanism for reduction of halo- and haloam(m)ine platinum(IV) complexes by L-ascorbate. Inorganica Chimica Acta. 2002;331:98–108. [Google Scholar]
- 212.Lemma K, Sargeson AM, Elding LI. Kinetics and mechanism for reduction of oral anticancer platinum(IV) dicarboxylate compounds by L-ascorbate ions. J Chem Society-Dalton Transactions. 2000;7:1167–1172. [Google Scholar]
- 213.Lever ABP. Electrochemical parametrization of metal complex redox potentials, using the ruthenium(III)/ruthenium(II) couple to generate a ligand electrochemical series. Inorg Chem. 1990;29:1271–1285. [Google Scholar]
- 214.Li H, Qian ZM. Transferrin/transferrin receptor-mediated drug delivery. Med Res Rev. 2002;22:225–250. doi: 10.1002/med.10008. [DOI] [PubMed] [Google Scholar]
- 215.Li H, Sun H, Qian ZM. The role of the transferrin-transferrin-receptor system in drug delivery and targeting. Trends Pharmacol Sci. 2002;23:206–209. doi: 10.1016/s0165-6147(02)01989-2. [DOI] [PubMed] [Google Scholar]
- 216.Li J, Waters SB, Drobna Z, Devesa V, Styblo M, Thomas DJ. Arsenic (+ 3 oxidation state) methyltransferase andthe inorganic arsenic methylation phenotype. Toxicol Appl Pharmacol. 2005;204:164–169. doi: 10.1016/j.taap.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 217.Li Y, Schellhorn HE. New developments and novel therapeutic perspectives for vitamin C. J Nutr. 2007;137:2171–2184. doi: 10.1093/jn/137.10.2171. [DOI] [PubMed] [Google Scholar]
- 218.Lide DR. CRC Handbook of Chemistry and Physics. 83rd Edition 2002. p. 2664. [Google Scholar]
- 219.Lin S, Del Razo LM, Styblo M, Wang C, Cullen WR, Thomas DJ. Arsenicals inhibit thioredoxin reductase in cultured rat hepatocytes. Chem Res Toxicol. 2001;14:305–311. doi: 10.1021/tx0001878. [DOI] [PubMed] [Google Scholar]
- 220.Liochev SI. The mechanism of “Fenton-like” reactions and their importance for biological systems. A biologist’s view. Met Ions Biol Syst. 1999;36:1–39. [PubMed] [Google Scholar]
- 221.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 222.Liu ZQ. Chemical methods to evaluate antioxidant ability. Chem Rev. 2010;110:5675–5691. doi: 10.1021/cr900302x. [DOI] [PubMed] [Google Scholar]
- 223.Lo YC, Ko TP, Su WC, Su TL, Wang AH. Terpyridine-platinum(II) complexes are effective inhibitors of mammalian topoisomerases and human thioredoxin reductase 1. J Inorg Biochem. 2009;103:1082–1092. doi: 10.1016/j.jinorgbio.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 224.Lowe G, Droz AS, Vilaivan T, Weaver GW, Park JJ, Pratt JM, Tweedale L, Kelland LR. Cytotoxicity of 2,2′:6′,2″-terpyridineplatinum(II) complexes against human ovarian carcinoma. J Med Chem. 1999;42:3167–3174. doi: 10.1021/jm991053y. [DOI] [PubMed] [Google Scholar]
- 225.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Luo S, Levine RL. Methionine in proteins defends against oxidative stress. Faseb J. 2009;23:464–72. doi: 10.1096/fj.08-118414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10:997–1030. doi: 10.1089/ars.2007.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Magda D, Miller RA. Motexafin gadolinium: a novel redox active drug for cancer therapy. Semin Cancer Biol. 2006;16:466–476. doi: 10.1016/j.semcancer.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 229.Magherini F, Modesti A, Bini L, Puglia M, Landini I, Nobili S, Mini E, Cinellu MA, Gabbiani C, Messori L. Exploring the biochemical mechanisms of cytotoxic gold compounds: a proteomic study. J Biol Inorg Chem. 2009;15:573–582. doi: 10.1007/s00775-010-0624-3. [DOI] [PubMed] [Google Scholar]
- 230.Majumder S, Dutta P, Mookerjee A, Choudhuri SK. The role of a novel copper complex in overcoming doxorubicin resistance in Ehrlich ascites carcinoma cells in vivo. Chem Biol Interact. 2006;159:90–103. doi: 10.1016/j.cbi.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 231.Majumder S, Panda GS, Choudhuri S Kumar. Synthesis, characterization and biological properties of a novel copper complex. Eur J Med Chem. 2003;38:893–898. doi: 10.1016/j.ejmech.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 232.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 233.Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003;278:9100–9106. doi: 10.1074/jbc.M210284200. [DOI] [PubMed] [Google Scholar]
- 234.Mandl J, Szarka A, Banhegyi G. Vitamin C: update on physiology and pharmacology. Br J Pharmacol. 2009;157:1097–1110. doi: 10.1111/j.1476-5381.2009.00282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Mann KK, Wallner B, Lossos IS, Miller WH., Jr. Darinaparsin: a novel organic arsenical with promising anti-cancer activity. Expert Opin Investig Drugs. 2009;18:1727–1734. doi: 10.1517/13543780903282759. [DOI] [PubMed] [Google Scholar]
- 236.Marcon G, Carotti S, Coronnello M, Messori L, Mini E, Orioli P, Mazzei T, Cinellu MA, Minghetti G. Gold(III) complexes with bipyridyl ligands: solution chemistry, cytotoxicity, and DNA binding properties. J Med Chem. 2002;45:1672–1677. doi: 10.1021/jm010997w. [DOI] [PubMed] [Google Scholar]
- 237.Marshall LE, Graham DR, Reich KA, Sigman DS. Cleavage of deoxyribonucleic acid by the 1,10-phenanthroline-cuprous complex. Hydrogen peroxide requirement and primary and secondary structure specificity. Biochemistry. 1981;20:244–250. doi: 10.1021/bi00505a003. [DOI] [PubMed] [Google Scholar]
- 238.Martins NM, Santos NA, Curti C, Bianchi ML, Santos AC. Cisplatin induces mitochondrial oxidative stress with resultant energetic metabolism impairment, membrane rigidification and apoptosis in rat liver. J Appl Toxicol. 2008;28:337–344. doi: 10.1002/jat.1284. [DOI] [PubMed] [Google Scholar]
- 239.Masuda H, Tanaka T, Takahama U. Cisplatin generates superoxide anion by interaction with DNA in a cell-free system. Biochem Biophys Res Commun. 1994;203:1175–1180. doi: 10.1006/bbrc.1994.2306. [DOI] [PubMed] [Google Scholar]
- 240.Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. 1996;178:179–185. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 241.Matulis SM, Morales AA, Yehiayan L, Croutch C, Gutman D, Cai Y, Lee KP, Boise LH. Darinaparsin induces a unique cellular response and is active in an arsenic trioxide-resistant myeloma cell line. Mol Cancer Ther. 2009;8:1197–1206. doi: 10.1158/1535-7163.MCT-08-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.McCann M, Geraghty M, Devereux M, O’Shea D, Mason J, O’Sullivan L. Insights into the mode of action of the anti-Candida activity of 1,10-phenanthroline and its metal chelates. Met Based Drugs. 2000;7:185–193. doi: 10.1155/MBD.2000.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.McCoubrey A, Latham HC, Cook PR, Rodger A, Lowe G. Picoline-2,2′:6′,2″-terpyridine-platinum(II)—a potent intercalator of DNA. FEBS Lett. 1996;380:73–78. doi: 10.1016/0014-5793(95)01537-x. [DOI] [PubMed] [Google Scholar]
- 244.Medici V, Sturniolo GC. Tetrathiomolybdate, a copper chelator for the treatment of Wilson disease, pulmonary fibrosis and other indications. IDrugs. 2008;11:592–606. [PubMed] [Google Scholar]
- 245.Meijer C, Mulder NH, Timmer-Bosscha H, Sluiter WJ, Meersma GJ, de Vries EG. Relationship of cellular glutathione to the cytotoxicity and resistance of seven platinum compounds. Cancer Res. 1992;52:6885–6889. [PubMed] [Google Scholar]
- 246.Mellish KJ, Kelland LR, Harrap KR. In vitro platinum drug chemosensitivity of human cervical squamous cell carcinoma cell lines with intrinsic and acquired resistance to cisplatin. Br J Cancer. 1993;68:240–250. doi: 10.1038/bjc.1993.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Meyers CA, Smith JA, Bezjak A, Mehta MP, Liebmann J, Illidge T, Kunkler I, Caudrelier JM, Eisenberg PD, Meerwaldt J, Siemers R, Carrie C, Gaspar LE, Curran W, Phan SC, Miller RA, Renschler MF. Neurocognitive function and progression in patients with brain metastases treated with whole-brain radiation and motexafin gadolinium: results of a randomized phase III trial. J Clin Oncol. 2004;22:157–165. doi: 10.1200/JCO.2004.05.128. [DOI] [PubMed] [Google Scholar]
- 248.Miao L, St Clair DK. Regulation of superoxide dismutase genes: implications in disease. Free Radic Biol Med. 2009;47:344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal. 2008;10:1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Milacic V, Dou QP. The tumor proteasome as a novel target for gold(III) complexes: implications for breast cancer therapy. Coord Chem Rev. 2009;253:1649–1660. doi: 10.1016/j.ccr.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Milano G, Caldani C, Khater R, Launay JM, Soummer AM, Namer M, Schneider M. Time- and dose-dependentinhibition of erythrocyte glutathione peroxidase by cisplatin. Biochem Pharmacol. 1988;37:981–982. doi: 10.1016/0006-2952(88)90196-7. [DOI] [PubMed] [Google Scholar]
- 252.Milbank JB, Stevenson RJ, Ware DC, Chang JY, Tercel M, Ahn GO, Wilson WR, Denny WA. Synthesis and evaluation of stable bidentate transition metal complexes of 1-(chloromethyl)-5-hydroxy-3-(5,6,7-trimethoxyindol-2-ylcarbonyl)-2,3-dihy dro-1H-pyrrolo[3,2-f]quinoline (seco-6-azaCBI-TMI) as hypoxia selective cytotoxins. J Med Chem. 2009;52:6822–6834. doi: 10.1021/jm9008746. [DOI] [PubMed] [Google Scholar]
- 253.Mistry P, Lee C, McBrien DC. Intracellular metabolites of cisplatin in the rat kidney. Cancer Chemother Pharmacol. 1989;24:73–79. doi: 10.1007/BF00263124. [DOI] [PubMed] [Google Scholar]
- 254.Mookerjee A, Basu JM, Majumder S, Chatterjee S, Panda GS, Dutta P, Pal S, Mukherjee P, Efferth T, Roy S, Choudhuri SK. A novel copper complex induces ROS generation in doxorubicin resistant Ehrlich ascitis carcinoma cells and increases activity of antioxidant enzymes in vital organs in vivo. BMC Cancer. 2006;6:267. doi: 10.1186/1471-2407-6-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Morales AA, Gutman D, Cejas PJ, Lee KP, Boise LH. Reactive oxygen species are not required for an arsenic trioxide-induced antioxidant response or apoptosis. J Biol Chem. 2009;284:12886–12895. doi: 10.1074/jbc.M806546200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Morbidelli L, Donnini S, Filippi S, Messori L, Piccioli F, Orioli P, Sava G, Ziche M. Antiangiogenic properties of selected ruthenium(III) complexes that are nitric oxide scavengers. Br J Cancer. 2003;88:1484–1491. doi: 10.1038/sj.bjc.6600906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Morinville A, Maysinger D, Shaver A. From Vanadis to Atropos: vanadium compounds as pharmacological tools in cell death signalling. Trends Pharmacol Sci. 1998;19:452–460. doi: 10.1016/s0165-6147(98)01257-7. [DOI] [PubMed] [Google Scholar]
- 258.Moriya M, Ho YH, Grana A, Nguyen L, Alvarez A, Jamil R, Ackland ML, Michalczyk A, Hamer P, Ramos D, Kim S, Mercer JF, Linder MC. Copper is taken up efficiently from albumin and alpha2-macroglobulin by cultured human cells by more than one mechanism. Am J Physiol Cell Physiol. 2008;295:C708–C721. doi: 10.1152/ajpcell.00029.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Mostert V, Hill KE, Ferris CD, Burk RF. Selective induction of liver parenchymal cell heme oxygenase-1 in selenium-deficient rats. Biol Chem. 2003;384:681–687. doi: 10.1515/BC.2003.076. [DOI] [PubMed] [Google Scholar]
- 260.Mukherjee B, Patra B, Mahapatra S, Banerjee P, Tiwari A, Chatterjee M. Vanadium—an element of atypical biological significance. Toxicol Lett. 2004;150:135–143. doi: 10.1016/j.toxlet.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 261.Mura P, Camalli M, Bindoli A, Sorrentino F, Casini A, Gabbiani C, Corsini M, Zanello P, Rigobello MP, Messori L. Activity of rat cytosolic thioredoxin reductase is strongly decreased by trans-[bis(2-amino-5- methylthiazole)tetrachlororuthenate(III)]: first report of relevant thioredoxin reductase inhibition for a ruthenium compound. J Med Chem. 2007;50:5871–5874. doi: 10.1021/jm0708578. [DOI] [PubMed] [Google Scholar]
- 262.Na HK, Surh YJ. Transcriptional regulation via cysteine thiol modification: a novel molecular strategy for chemo-prevention and cytoprotection. Mol Carcinog. 2006;45:368–380. doi: 10.1002/mc.20225. [DOI] [PubMed] [Google Scholar]
- 263.Nagahara N. Intermolecular disulfide bond to modulate protein function as a redox-sensing switch. Amino Acids. 2010 doi: 10.1007/s00726-010-0508-4. [Epub ahead of print]; DOI: 10.1007/s00726-010-0508-4. [DOI] [PubMed] [Google Scholar]
- 264.Narasimhan J, Antholine WE, Chitambar CR, Petering DH. Inhibition of iron uptake in HL60 cells by 2-formylpyridine monothiosemicarbazonato Cu(II) Arch Biochem Biophys. 1991;289:393–398. doi: 10.1016/0003-9861(91)90429-m. [DOI] [PubMed] [Google Scholar]
- 265.Naura AS, Kalla NR, Sharma RP, Sharma R. Antic-arcinogenic effects of hexaamminecobalt(III) chloride in mice initiated with diethylnitrosamine. Biol Trace Elem Res. 2007;119:147–165. doi: 10.1007/s12011-007-0051-7. [DOI] [PubMed] [Google Scholar]
- 266.Naura AS, Sharma R. Toxic effects of hexaammine cobalt(III) chloride on liver and kidney in mice: implication of oxidative stress. Drug Chem Toxicol. 2009;32:293–299. doi: 10.1080/01480540902882234. [DOI] [PubMed] [Google Scholar]
- 267.Nemirovski A, Kasherman Y, Tzaraf Y, Gibson D. Reduction of cis,trans,cis-[PtCl2(OCOCH3)2(NH3)2] by aqueous extracts of cancer cells. J Med Chem. 2007;50:5554–5556. doi: 10.1021/jm070740j. [DOI] [PubMed] [Google Scholar]
- 268.Nemirovski A, Vinograd I, Takrouri K, Mijovilovich A, Rompel A, Gibson D. New reduction pathways for ctc-[PtCl2(CH3CO2)2(NH3)(Am)] anticancer prodrugs. Chem Commun (Camb) 2010;46:1842–1844. doi: 10.1039/b925721g. [DOI] [PubMed] [Google Scholar]
- 269.Neumann CA, Cao J, Manevich Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle. 2009;8:4072–4078. doi: 10.4161/cc.8.24.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Neumann CA, Fang Q. Are peroxiredoxins tumor suppressors? Curr Opin Pharmacol. 2007;7:375–380. doi: 10.1016/j.coph.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 271.Nicolis I, Curis E, Deschamps P, Benazeth S. Arsenite medicinal use, metabolism, pharmacokinetics and monitoring in human hair. Biochimie. 2009;91:1260–1267. doi: 10.1016/j.biochi.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 272.Nobili S, Mini E, Landini I, Gabbiani C, Casini A, Messori L. Gold compounds as anticancer agents: chemistry, cellular pharmacology, and preclinical studies. Med Res Rev. 2009;30:550–580. doi: 10.1002/med.20168. [DOI] [PubMed] [Google Scholar]
- 273.Nocentini G, Barzi A. The 2,2′-bipyridyl-6-carbothioamide copper(II) complex differs from the iron(II) complex in its biochemical effects in tumor cells, suggesting possible differences in the mechanism leading to cytotoxicity. Biochem Pharmacol. 1996;52:65–71. doi: 10.1016/0006-2952(96)00139-6. [DOI] [PubMed] [Google Scholar]
- 274.Nocentini G, Barzi A. Antitumor activity of 2,2′-bipyridyl-6-carbothioamide: a ribonucleotide reductase inhibitor. Gen Pharmacol. 1997;29:701–706. doi: 10.1016/s0306-3623(97)00011-6. [DOI] [PubMed] [Google Scholar]
- 275.Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol. 2003;23:916–922. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.O’Rourke TJ, Weiss GR, New P, Burris HA, 3rd, Rodriguez G, Eckhardt J, Hardy J, Kuhn JG, Fields S, Clark GM, et al. Phase I clinical trial of ormaplatin (tetraplatin, NSC 363812) Anticancer Drugs. 1994;5:520–526. doi: 10.1097/00001813-199410000-00002. [DOI] [PubMed] [Google Scholar]
- 277.Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. 2005;37:1264–1269. doi: 10.1038/ng1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood. 2006;108:1388–1394. doi: 10.1182/blood-2006-02-003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Okun Z, Kupershmidt L, Amit T, Mandel S, Bar-Am O, Youdim MBH, Gross Z. Manganese corroles prevent intracellular nitration and subsequent death of insulin-producing cells. ACS Chem Biol. 2009;4:910–914. doi: 10.1021/cb900159n. [DOI] [PubMed] [Google Scholar]
- 280.Orino K, Lehman L, Tsuji Y, Ayaki H, Torti SV, Torti FM. Ferritin and the response to oxidative stress. Biochem J. 2001;357:241–247. doi: 10.1042/0264-6021:3570241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Osinsky S, Levitin I, Bubnovskaya L, Sigan A, Ganusevich I, Kovelskaya A, Valkovskaya N, Campanella L, Wardman P. Selectivity of effects of redox-active cobalt(III) complexes on tumor tissue. Exp Oncol. 2004;26:140–144. [PubMed] [Google Scholar]
- 282.Osinsky SP, Levitin IY, Bubnovskaya LN, Ganusevich II, Tsikalova MV, Istomin YP, Zhavrid EA, Volpin ME. Modifying effect of organocobalt complexes on the tumour response to anticancer treatments. Anticancer Res. 1997;17:3457–3462. [PubMed] [Google Scholar]
- 283.Osinsky SP, Levitin Y, Sigan AL, Bubnovskaya LN, Ganusevich II, Campanella L, Wardman P. Redox-active cobalt complexes as promising antitumor agents. Russ Chem Bull Int Ed. 2003;52:2636–2645. [Google Scholar]
- 284.Otiko G, Razi MT, Sadler PJ, Isab AA, Rabenstein DL. A 1H nmr study of the interaction of aurothiomalate (“Myocrisin”) with human red blood cells in vitro. J Inorg Biochem. 1983;19:227–235. doi: 10.1016/0162-0134(83)85027-2. [DOI] [PubMed] [Google Scholar]
- 285.Ott I, Gust R. Non platinum metal complexes as anti-cancer drugs. Arch Pharm (Weinheim) 2007;340:117–126. doi: 10.1002/ardp.200600151. [DOI] [PubMed] [Google Scholar]
- 286.Ott I, Schmidt K, Kircher B, Schumacher P, Wiglenda T, Gust R. Antitumor-active cobalt-alkyne complexes derived from acetylsalicylic acid: studies on the mode of drug action. J Med Chem. 2005;48:622–629. doi: 10.1021/jm049326z. [DOI] [PubMed] [Google Scholar]
- 287.Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80:1895–1903. doi: 10.1016/j.bcp.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 288.Palmiter RD. Protection against zinc toxicity by metallothionein and zinc transporter 1. Proc Natl Acad Sci U S A. 2004;101:4918–4923. doi: 10.1073/pnas.0401022101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol. 2005;202:199–211. doi: 10.1016/j.taap.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 290.Pass HI, Brewer GJ, Dick R, Carbone M, Merajver S. A phase II trial of tetrathiomolybdate after surgery for malignant mesothelioma: final results. Ann Thorac Surg. 2008;86:383–389. doi: 10.1016/j.athoracsur.2008.03.016. discussion 390. [DOI] [PubMed] [Google Scholar]
- 291.Pastore A, Federici G, Bertini E, Piemonte F. Analysis of glutathione: implication in redox and detoxification. Clin Chim Acta. 2003;333:19–39. doi: 10.1016/s0009-8981(03)00200-6. [DOI] [PubMed] [Google Scholar]
- 292.Paul MK, Kumar R, Mukhopadhyay AK. Dithiothreitol abrogates the effect of arsenic trioxide on normal rat liver mitochondria and human hepatocellular carcinoma cells. Toxicol Appl Pharmacol. 2008;226:140–152. doi: 10.1016/j.taap.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 293.Pavlishchuk VV, Addison AW. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 DegC. Inorg Chim Acta. 2000;298:97–102. [Google Scholar]
- 294.Pearson RG. Hard and soft acids and bases. J Am Chem Soc. 1963;85:3533–3539. [Google Scholar]
- 295.Pedersen MO, Larsen A, Stoltenberg M, Penkowa M. The role of metallothionein in oncogenesis and cancer prognosis. Prog Histochem Cytochem. 2009;44:29–64. doi: 10.1016/j.proghi.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 296.Pendyala L, Cowens JW, Chheda GB, Dutta SP, Creaven PJ. Identification of cis-dichloro-bis-isopropylamine platinum(II) as a major metabolite of iproplatin in humans. Cancer Res. 1988;48:3533–3536. [PubMed] [Google Scholar]
- 297.Pendyala L, Creaven PJ, Perez R, Zdanowicz JR, Raghavan D. Intracellular glutathione and cytotoxicity of platinum complexes. Cancer Chemother Pharmacol. 1995;36:271–278. doi: 10.1007/BF00689042. [DOI] [PubMed] [Google Scholar]
- 298.Petrick JS, Ayala-Fierro F, Cullen WR, Carter DE, Aposhian H Vasken. Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Chang human hepatocytes. Toxicol Appl Pharmacol. 2000;163:203–207. doi: 10.1006/taap.1999.8872. [DOI] [PubMed] [Google Scholar]
- 299.Petzold H, Xu J, Sadler PJ. Metal and ligand control of sulfenate reactivity: arene ruthenium thiolato-mono-S-oxides. Angewandte Chem Int Ed Engl. 2008;47:3008–3011. doi: 10.1002/anie.200705342. [DOI] [PubMed] [Google Scholar]
- 300.Piccioli F, Sabatini S, Messori L, Orioli P, Hartinger CG, Keppler BK. A comparative study of adduct formation between the anticancer ruthenium(III) compound HInd trans-[RuCl4(Ind)2] and serum proteins. J Inorg Biochem. 2004;98:1135–1142. doi: 10.1016/j.jinorgbio.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 301.Pizarro AM, Sadler PJ. Unusual DNA binding modes for metal anticancer complexes. Biochimie. 2009;91:1198–1211. doi: 10.1016/j.biochi.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Pope LM, Reich KA, Graham DR, Sigman DS. Products of DNA cleavage by the 1,10-phenanthroline-copper complex. Inhibitors of Escherichia coli DNA polymerase I. J Biol Chem. 1982;257:12121–12128. [PubMed] [Google Scholar]
- 303.Porter JB, Rafique R, Srichairatanakool S, Davis BA, Shah FT, Hair T, Evans P. Recent insights into interactions of deferoxamine with cellular and plasma iron pools: implications for clinical use. Ann N Y Acad Sci. 2005;1054:155–168. doi: 10.1196/annals.1345.018. [DOI] [PubMed] [Google Scholar]
- 304.Qian ZM, Li H, Sun H, Ho K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev. 2002;54:561–587. doi: 10.1124/pr.54.4.561. [DOI] [PubMed] [Google Scholar]
- 305.Qureshi AA, Rosenblatt DS, Cooper BA. Inherited disorders of cobalamin metabolism. Crit Rev Oncol Hematol. 1994;17:133–151. doi: 10.1016/1040-8428(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 306.Rabbani ZN, Spasojevic I, Zhang X, Moeller BJ, Haberle S, Vasquez-Vivar J, Dewhirst MW, Vujaskovic Z, Batinic-Haberle I. Antiangiogenic action of redox-modulating Mn(III) meso-tetrakis(N-ethylpyridinium-2-yl)porphyrin, MnTE-2-PyP5 + , via suppression of oxidative stress in a mouse model of breast tumor. Free Radic Biol Med. 2009;47:992–1004. doi: 10.1016/j.freeradbiomed.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Rackham O, Nichols SJ, Leedman PJ, Berners-Price SJ, Filipovska A. A gold(I) phosphine complex selectively induces apoptosis in breast cancer cells: implications for anticancer therapeutics targeted to mitochondria. Biochem Pharmacol. 2007;74:992–1002. doi: 10.1016/j.bcp.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 308.Rademaker-Lakhai JM, van den Bongard D, Pluim D, Beijnen JH, Schellens JH. A phase I and pharmacological study with imidazolium-trans-DMSO-imidazoletetrachlororuthenate, a novel ruthenium anticancer agent. Clin Cancer Res. 2004;10:3717–3727. doi: 10.1158/1078-0432.CCR-03-0746. [DOI] [PubMed] [Google Scholar]
- 309.Ramirez-Ramirez N, Mendoza-Diaz G, Pedraza-Reyes M. Degradation of single stranded nucleic acids by the chemical nuclease activity of the metal complex [Cu(-phen)(nal)] Bioinorg Chem Appl. 2003;1:25–34. doi: 10.1155/S1565363303000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Raux E, Schubert HL, Warren MJ. Biosynthesis of cobalamin (vitamin B12): a bacterial conundrum. Cell Mol Life Sci. 2000;57:1880–1893. doi: 10.1007/PL00000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Ray RS, Ghosh B, Rana A, Chatterjee M. Suppression of cell proliferation, induction of apoptosis and cell cycle arrest: chemopreventive activity of vanadium in vivo and in vitro. Int J Cancer. 2007;120:13–23. doi: 10.1002/ijc.22277. [DOI] [PubMed] [Google Scholar]
- 312.Raynaud FI, Mistry P, Donaghue A, Poon GK, Kelland LR, Barnard CF, Murrer BA, Harrap KR. Biotransformation of the platinum drug JM216 following oral administration to cancer patients. Cancer Chemother Pharmacol. 1996;38:155–162. doi: 10.1007/s002800050464. [DOI] [PubMed] [Google Scholar]
- 313.Raynaud FI, Odell DE, Kelland LR. Intracellular metabolism of the orally active platinum drug JM216: influence of glutathione levels. Br J Cancer. 1996;74:380–386. doi: 10.1038/bjc.1996.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Rebillard A, Jouan-Lanhouet S, Jouan E, Legembre P, Pizon M, Sergent O, Gilot D, Tekpli X, Lagadic-Gossmann D, Dimanche-Boitrel MT. Cisplatin-induced apoptosis involves a Fas-ROCK-ezrin-dependent actin remodelling in human colon cancer cells. Eur J Cancer. 2010;46:1445–1455. doi: 10.1016/j.ejca.2010.01.034. [DOI] [PubMed] [Google Scholar]
- 315.Rebillard A, Lagadic-Gossmann D, Dimanche-Boitrel MT. Cisplatin cytotoxicity: DNA and plasma membrane targets. Curr Med Chem. 2008;15:2656–2663. doi: 10.2174/092986708786242903. [DOI] [PubMed] [Google Scholar]
- 316.Rebouche CJ. Ascorbic acid and carnitine biosynthesis. Am J Clin Nutr. 1991;54:1147S–1152S. doi: 10.1093/ajcn/54.6.1147s. [DOI] [PubMed] [Google Scholar]
- 317.Reisner E, Arion VB, Guedes da Silva MFC, Lichtenecker R, Eichinger A, Keppler BK, Kukushkin VY, Pombeiro AJL. Tuning of redox potentials for the design of rutheniumanticancer drugs—an electrochemical study of [trans-RuCl4L(DMSO)]- and [trans-RuCl4L2]- complexes, where L = imidazole, 1,2,4-triazole, indazole. Inorg Chem. 2004;43:7083–7093. doi: 10.1021/ic049479c. [DOI] [PubMed] [Google Scholar]
- 318.Reisner E, Arion VB, Keppler BK, Pombeiro AJL. Electron-transfer activated metal-based anticancer drugs. Inorg Chim Acta. 2008;361:1569–1583. [Google Scholar]
- 319.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 320.Rigobello MP, Folda A, Dani B, Menabo R, Scutari G, Bindoli A. Gold(I) complexes determine apoptosis with limited oxidative stress in Jurkat T cells. Eur J Pharmacol. 2008;582:26–34. doi: 10.1016/j.ejphar.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 321.Rigobello MP, Messori L, Marcon G, Cinellu MA, Bragadin M, Folda A, Scutari G, Bindoli A. Gold complexes inhibit mitochondrial thioredoxin reductase: consequences on mitochondrial functions. J Inorg Biochem. 2004;98:1634–1641. doi: 10.1016/j.jinorgbio.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 322.Ronconi L, Marzano C, Zanello P, Corsini M, Miolo G, Macca C, Trevisan A, Fregona D. Gold(III) dithiocarbamate derivatives for the treatment of cancer: solution chemistry, DNA binding, and hemolytic properties. J Med Chem. 2006;49:1648–1657. doi: 10.1021/jm0509288. [DOI] [PubMed] [Google Scholar]
- 323.Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Platinum compounds: a new class of potent antitumour agents. Nature. 1969;222:385–386. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 324.Rosenthal DI, Nurenberg P, Becerra CR, Frenkel EP, Carbone DP, Lum BL, Miller R, Engel J, Young S, Miles D, Renschler MF. A phase I single-dose trial of gadolinium texaphyrin (Gd-Tex), a tumor selective radiation sensitizer detectable by magnetic resonance imaging. Clin Cancer Res. 1999;5:739–745. [PubMed] [Google Scholar]
- 325.Ruiz-Sanchez P, Konig C, Ferrari S, Alberto R. Vitamin B(12) as a carrier for targeted platinum delivery: in vitro cytotoxicity and mechanistic studies. J Biol Inorg Chem. 2011;16:33–44. doi: 10.1007/s00775-010-0697-z. [DOI] [PubMed] [Google Scholar]
- 326.Salvemini D, Doyle TM, Cuzzocrea S. Superoxide, peroxynitrite and oxidative/nitrative stress in inflammation. Biochem Soc Trans. 2006;34:965–970. doi: 10.1042/BST0340965. [DOI] [PubMed] [Google Scholar]
- 327.Samimi G, Kishimoto S, Manorek G, Breaux JK, Howell SB. Novel mechanisms of platinum drug resistance identified in cells selected for resistance to JM118 the active metabolite of satraplatin. Cancer Chemother Pharmacol. 2007;59:301–312. doi: 10.1007/s00280-006-0271-0. [DOI] [PubMed] [Google Scholar]
- 328.Sammes PG, Yahioglu G. 1,10-Phenanthroline—a versatile ligand. Chem Soc Rev. 1994;23:327–334. [Google Scholar]
- 329.Sanchez Y, Amran D, Fernandez C, de Blas E, Aller P. Genistein selectively potentiates arsenic trioxide-induced apoptosis in human leukemia cells via reactive oxygen species generation and activation of reactive oxygen species-inducible protein kinases (p38-MAPK, AMPK) Int J Cancer. 2008;123:1205–1214. doi: 10.1002/ijc.23639. [DOI] [PubMed] [Google Scholar]
- 330.Santos NA, Bezerra CS, Martins NM, Curti C, Bianchi ML, Santos AC. Hydroxyl radical scavenger ameliorates cisplatin-induced nephrotoxicity by preventing oxidative stress, redox state unbalance, impairment of energetic metabolism and apoptosis in rat kidney mitochondria. Cancer Chemother Pharmacol. 2008;61:145–155. doi: 10.1007/s00280-007-0459-y. [DOI] [PubMed] [Google Scholar]
- 331.Saryan LA, Ankel E, Krishnamurti C, Petering DH, Elford H. Comparative cytotoxic and biochemical effects of ligands and metal complexes of alpha-N-heterocyclic carboxaldehyde thiosemicarbazones. J Med Chem. 1979;22:1218–1221. doi: 10.1021/jm00196a013. [DOI] [PubMed] [Google Scholar]
- 332.Saryan LA, Mailer K, Krishnamurti C, Antholine W, Petering DH. Interaction of 2-formylpyridine thiosemicarbazonato copper(II) with Ehrlich ascites tumor cells. Biochem Pharmacol. 1981;30:1595–1604. doi: 10.1016/0006-2952(81)90386-5. [DOI] [PubMed] [Google Scholar]
- 333.Sasada T, Nakamura H, Ueda S, Sato N, Kitaoka Y, Gon Y, Takabayashi A, Spyrou G, Holmgren A, Yodoi J. Possible involvement of thioredoxin reductase as well as thioredoxin in cellular sensitivity to cis-diamminedichloroplatinum (II) Free Radic Biol Med. 1999;27:504–514. doi: 10.1016/s0891-5849(99)00101-x. [DOI] [PubMed] [Google Scholar]
- 334.Sava G, Bergamo A, Zorzet S, Gava B, Casarsa C, Cocchietto M, Furlani A, Scarcia V, Serli B, Iengo E, Alessio E, Mestroni G. Influence of chemical stability on the activity of the antimetastasis ruthenium compound NAMI-A. Eur J Cancer. 2002;38:427–435. doi: 10.1016/s0959-8049(01)00389-6. [DOI] [PubMed] [Google Scholar]
- 335.Sava G, Zorzet S, Mestroni G, Zassinovich G. Anti-neoplastic activity of planar rhodium(I) complexes in mice bearing Lewis lung carcinoma and P388 leukemia. Anticancer Res. 1985;5:249–252. [PubMed] [Google Scholar]
- 336.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 337.Schilder RJ, LaCreta FP, Perez RP, Johnson SW, Brennan JM, Rogatko A, Nash S, McAleer C, Hamilton TC, Roby D, et al. Phase I and pharmacokinetic study of ormaplatin (tetraplatin, NSC 363812) administered on a day 1 and day 8 schedule. Cancer Res. 1994;54:709–717. [PubMed] [Google Scholar]
- 338.Schluga P, Hartinger CG, Egger A, Reisner E, Galanski M, Jakupec MA, Keppler BK. Redox behavior of tumor-inhibiting ruthenium(III) complexes and effects of physiological reductants on their binding to GMP. Dalton Trans. 2006;14:1796–1802. doi: 10.1039/b511792e. [DOI] [PubMed] [Google Scholar]
- 339.Schuhmacher-Wolz U, Dieter HH, Klein D, Schneider K. Oral exposure to inorganic arsenic: evaluation of its carcinogenic and non-carcinogenic effects. Crit Rev Toxicol. 2009;39:271–298. doi: 10.1080/10408440802291505. [DOI] [PubMed] [Google Scholar]
- 340.Scrivens PJ, Alaoui-Jamali MA, Giannini G, Wang T, Loignon M, Batist G, Sandor VA. Cdc25A-inhibitory properties and antineoplastic activity of bisperoxovanadium analogues. Mol Cancer Ther. 2003;2:1053–1059. [PubMed] [Google Scholar]
- 341.Shao J, Zhou B, Chu B, Yen Y. Ribonucleotide reductase inhibitors and future drug design. Curr Cancer Drug Targets. 2006;6:409–431. doi: 10.2174/156800906777723949. [DOI] [PubMed] [Google Scholar]
- 342.Shao J, Zhou B, Di Bilio AJ, Zhu L, Wang T, Qi C, Shih J, Yen Y. A Ferrous-Triapine complex mediates formation of reactive oxygen species that inactivate human ribonucleotide reductase. Mol Cancer Ther. 2006;5:586–592. doi: 10.1158/1535-7163.MCT-05-0384. [DOI] [PubMed] [Google Scholar]
- 343.Shaw IC. Gold-based therapeutic agents. Chem Rev. 1999;99:2589–2600. doi: 10.1021/cr980431o. [DOI] [PubMed] [Google Scholar]
- 344.Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW, Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z, Wang ZY. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89:3354–3360. [PubMed] [Google Scholar]
- 345.Shi TS, Berglund J, Elding LI. Kinetics and mechanism for reduction of trans-dichlorotetracyanoplatinate(IV) by thioglycolic acid, L-cysteine, DL-penicillamine, and glutathione in aqueous solution. Inorg Chem. 1996;35:3498–3503. [Google Scholar]
- 346.Shi TS, Berglund J, Elding LI. Reduction of transdichloro- and trans-dibromo-tetracyano-platinate(IV) by L-methionine. J Chem Soc Dalton Trans. 1997;12:2073–2077. [Google Scholar]
- 347.Shi X, Dalal NS. Hydroxyl radical generation in the NADH/microsomal reduction of vanadate. Free Radic Res Commun. 1992;17:369–376. doi: 10.3109/10715769209083141. [DOI] [PubMed] [Google Scholar]
- 348.Shi X, Flynn DC, Liu K, Dalal N. Vanadium (IV) formation in the reduction of vanadate by glutathione reductase/NADPH and the role of molecular oxygen. Ann Clin Lab Sci. 1997;27:422–427. [PubMed] [Google Scholar]
- 349.Shi X, Jiang H, Mao Y, Ye J, Saffiotti U. Vanadium(IV)-mediated free radical generation and related 2′-deoxyguanosine hydroxylation and DNA damage. Toxicology. 1996;106:27–38. doi: 10.1016/0300-483x(95)03151-5. [DOI] [PubMed] [Google Scholar]
- 350.Shi XL, Dalal NS. Flavoenzymes reduce vanadium(V) and molecular oxygen and generate hydroxyl radical. Arch Biochem Biophys. 1991;289:355–361. doi: 10.1016/0003-9861(91)90423-g. [DOI] [PubMed] [Google Scholar]
- 351.Shi Y, Amin K, Sato BG, Samuelsson SJ, Sambucetti L, Haroon ZA, Laderoute K, Murphy BJ. The metal-responsive transcription factor-1 protein is elevated in human tumors. Cancer Biol Ther. 2010;9:469–476. doi: 10.4161/cbt.9.6.10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Shimizu Y, Hendershot LM. Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid Redox Signal. 2009;11:2317–2331. doi: 10.1089/ars.2009.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Sigman DS, Graham DR, D’Aurora V, Stern AM. Oxygen-dependent cleavage of DNA by the 1,10-phenanthroline. cuprous complex. Inhibition of Escherichia coli DNA polymerase I. J Biol Chem. 1979;254:12269–12272. [PubMed] [Google Scholar]
- 354.Sigman DS, Mazumder A, Perrin DM. Chemical Nucleases. Chem Rev. 1993;93:2295–2316. [Google Scholar]
- 355.Singh S, Vrishni S, Singh BK, Rahman I, Kakkar P. Nrf2-ARE stress response mechanism: a control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free Radic Res. 2010;44:1267–1288. doi: 10.3109/10715762.2010.507670. [DOI] [PubMed] [Google Scholar]
- 356.Smith PF, Hoke GD, Alberts DW, Bugelski PJ, Lupo S, Mirabelli CK, Rush GF. Mechanism of toxicity of an experimental bidentate phosphine gold complexed anti-neoplastic agent in isolated rat hepatocytes. J Pharmacol Exp Ther. 1989;249:944–50. [PubMed] [Google Scholar]
- 357.Snyder RM, Mirabelli CK, Crooke ST. Cellular association, intracellular distribution, and efflux of auranofin via sequential ligand exchange reactions. Biochem Pharmacol. 1986;35:923–932. doi: 10.1016/0006-2952(86)90078-x. [DOI] [PubMed] [Google Scholar]
- 358.Sodhi A, Gupta P. Increased release of hydrogen peroxide (H2O2) and superoxide anion (O-2) by murine macrophages in vitro after cis-platin treatment. Int J Immunopharmacol. 1986;8:709–714. doi: 10.1016/0192-0561(86)90006-8. [DOI] [PubMed] [Google Scholar]
- 359.Soignet SL, Frankel SR, Douer D, Tallman MS, Kantarjian H, Calleja E, Stone RM, Kalaycio M, Scheinberg DA, Steinherz P, Sievers EL, Coutre S, Dahlberg S, Ellison R, Warrell RP., Jr. United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol. 2001;19:3852–3860. doi: 10.1200/JCO.2001.19.18.3852. [DOI] [PubMed] [Google Scholar]
- 360.Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J, Scheinberg DA, Pandolfi PP, Warrell RP., Jr. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med. 1998;339:1341–1348. doi: 10.1056/NEJM199811053391901. [DOI] [PubMed] [Google Scholar]
- 361.Spasojevic I, Sheng H, Warner DS, Batinic-Haberle I. Metalloporphyrins are versatile and powerful therapeutics: biomimetics of SOD, peroxyredoxin, and cyt P450. 2nd World Conference on Magic Bullets (Ehrlich II); Nürnberg, Germany. 2008. [Google Scholar]
- 362.Steinhoff D, Mohr U. On the question of a carcinogenic action of cobalt-containing compounds. Exp Pathol. 1991;41:169–174. doi: 10.1016/s0232-1513(11)80084-8. [DOI] [PubMed] [Google Scholar]
- 363.Stewart DJ. Mechanisms of resistance to cisplatin and carboplatin. Crit Rev Oncol Hematol. 2007;63:12–31. doi: 10.1016/j.critrevonc.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 364.Stordal B, Pavlakis N, Davey R. Oxaliplatin for the treatment of cisplatin-resistant cancer: a systematic review. Cancer Treat Rev. 2007;33:347–357. doi: 10.1016/j.ctrv.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 365.Sulyok M, Hann S, Hartinger CG, Keppler BK, Stingeder G, Koellensperger G. Two dimensional separation schemes for investigation of the interaction of an anticancer ruthenium(III) compound with plasma proteins. Journal of Analytical Atomic Spectrometry. 2005;20:856–863. [Google Scholar]
- 366.Surowiak P, Materna V, Kaplenko I, Spaczynski M, Dietel M, Lage H, Zabel M. Augmented expression of metallothionein and glutathione S-transferase pi as unfavourable prognostic factors in cisplatin-treated ovarian cancer patients. Virchows Arch. 2005;447:626–633. doi: 10.1007/s00428-005-1228-0. [DOI] [PubMed] [Google Scholar]
- 367.Sweeney CJ, Takimoto C, Wood L, Porter JM, Tracewell WG, Darwish M, D’Andrea DM, Remick SC. A pharmacokinetic and safety study of intravenous arsenic trioxide in adult cancer patients with renal impairment. Cancer Chemother Pharmacol. 2010;66:345–356. doi: 10.1007/s00280-009-1169-4. [DOI] [PubMed] [Google Scholar]
- 368.Tajmir-Riahi HA. Vitamin C interaction with cobalt-ammine cations. Synthesis, spectroscopic and structural characterization of cobalt-pentammine and cobalt-tetrammine sugar complexes containing L-ascorbate anion. Biophys Chem. 1986;25:37–41. doi: 10.1016/0301-4622(86)85065-7. [DOI] [PubMed] [Google Scholar]
- 369.Talbot S, Nelson R, Self WT. Arsenic trioxide and auranofin inhibit selenoprotein synthesis: implications for chemotherapy for acute promyelocytic leukaemia. Br J Pharmacol. 2008;154:940–948. doi: 10.1038/bjp.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Tapio S, Grosche B. Arsenic in the aetiology of cancer. Mutat Res. 2006;612:215–246. doi: 10.1016/j.mrrev.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 371.Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, Mendiboure J, Pignon JP, Jooste V, van Endert P, Ducreux M, Zitvogel L, Piard F, Kroemer G. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–491. doi: 10.1038/onc.2009.356. [DOI] [PubMed] [Google Scholar]
- 372.Theil EC. Coordinating responses to iron and oxygen stress with DNA and mRNA promoters: the ferritin story. Biometals. 2007;20:513–521. doi: 10.1007/s10534-006-9063-6. [DOI] [PubMed] [Google Scholar]
- 373.Thomas DJ. Molecular processes in cellular arsenic metabolism. Toxicol Appl Pharmacol. 2007;222:365–373. doi: 10.1016/j.taap.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 374.Thompson DC, Vaisman A, Sakata MK, Wyrick SD, Holbrook DJ, Chaney SG. Organ-specific biotransformation of ormaplatin in the Fischer 344 rat. Cancer Chemother Pharmacol. 1995;36:439–447. doi: 10.1007/BF00686194. [DOI] [PubMed] [Google Scholar]
- 375.Tian C, Gao P, Zheng Y, Yue W, Wang X, Jin H, Chen Q. Redox status of thioredoxin-1 (TRX1) determines the sensitivity of human liver carcinoma cells (HepG2) to arsenic trioxide-induced cell death. Cell Res. 2008;18:458–471. doi: 10.1038/cr.2007.112. [DOI] [PubMed] [Google Scholar]
- 376.Timerbaev AR, Foteeva LS, Rudnev AV, Abramski JK, Polec-Pawlak K, Hartinger CG, Jarosz M, Keppler BK. Probing the stability of serum protein-ruthenium(III) drug adducts in the presence of extracellular reductants using CE. Electrophoresis. 2007;28:2235–2240. doi: 10.1002/elps.200600707. [DOI] [PubMed] [Google Scholar]
- 377.Timerbaev AR, Hartinger CG, Aleksenko SS, Keppler BK. Interactions of Antitumor Metallodrugs with Serum Proteins: Advances in Characterization Using Modern Analytical Methodology. Chem Rev. 2006;106:2224–2248. doi: 10.1021/cr040704h. [DOI] [PubMed] [Google Scholar]
- 378.Tonissen KF, Di Trapani G. Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy. Mol Nutr Food Res. 2009;53:87–103. doi: 10.1002/mnfr.200700492. [DOI] [PubMed] [Google Scholar]
- 379.Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol. 2004;122:339–352. doi: 10.1007/s00418-004-0696-7. [DOI] [PubMed] [Google Scholar]
- 380.Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-glutathionylation following oxidative and nitrosative stress. J Biol Chem. 2009;284:436–445. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 382.Trask C, Silverstone A, Ash CM, Earl H, Irwin C, Bakker A, Tobias JS, Souhami RL. A randomized trial of carboplatin versus iproplatin in untreated advanced ovarian cancer. J Clin Oncol. 1991;9:1131–1137. doi: 10.1200/JCO.1991.9.7.1131. [DOI] [PubMed] [Google Scholar]
- 383.Trigona WL, Mullarky IK, Cao Y, Sordillo LM. Thioredoxin reductase regulates the induction of haem oxygenase-1 expression in aortic endothelial cells. Biochem J. 2006;394:207–216. doi: 10.1042/BJ20050712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Tsang SY, Tam SC, Bremner I, Burkitt MJ. Research communication copper-1,10-phenanthroline induces inter-nucleosomal DNA fragmentation in HepG2 cells, resulting from direct oxidation by the hydroxyl radical. Biochem J. 1996;317:13–16. doi: 10.1042/bj3170013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Tse HM, Milton MJ, Piganelli JD. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen-presenting cells: implication for their use in targeting oxidation-reduction reactions in innate immunity. Free Radical Biology and Medicine. 2004;36:233–247. doi: 10.1016/j.freeradbiomed.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 386.Tseng CH. Arsenic methylation, urinary arsenic metabolites and human diseases: current perspective. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2007;25:1–22. doi: 10.1080/10590500701201695. [DOI] [PubMed] [Google Scholar]
- 387.Tsimberidou AM, Camacho LH, Verstovsek S, Ng C, Hong DS, Uehara CK, Gutierrez C, Daring S, Stevens J, Komarnitsky PB, Schwartz B, Kurzrock R. A phase I clinical trial of darinaparsin in patients with refractory solid tumors. Clin Cancer Res. 2009;15:4769–4776. doi: 10.1158/1078-0432.CCR-08-2984. [DOI] [PubMed] [Google Scholar]
- 388.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 389.Vergara E, Casini A, Sorrentino F, Zava O, Cerrada E, Rigobello MP, Bindoli A, Laguna M, Dyson PJ. Anticancer therapeutics that target selenoenzymes: synthesis, characterization, in vitro cytotoxicity, and thioredoxin reductase inhibition of a series of gold(I) complexes containing hydrophilic phosphine ligands. ChemMedChem. 2010;5:96–102. doi: 10.1002/cmdc.200900370. [DOI] [PubMed] [Google Scholar]
- 390.Villeneuve NF, Lau A, Zhang DD. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin protea-some system: an insight into cullin-ring ubiquitin ligases. Antioxid Redox Signal. 2010;13:1699–1712. doi: 10.1089/ars.2010.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Vol’pin ME, Levitin I, Osinsky SP. pH-dependent organocobalt sources for active radical species: a new type of anticancer agents. Met Ions Biol Syst. 1999;36:485–519. [PubMed] [Google Scholar]
- 392.Volckova E, Weaver E, Bose RN. Insight into the reactive form of the anticancer agent iproplatin. Eur J Med Chem. 2008;43:1081–1084. doi: 10.1016/j.ejmech.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 393.Wang F, Xu J, Habtemariam A, Bella J, Sadler PJ. Competition between glutathione and guanine for a ruthenium(II) arene anticancer complex: detection of a sulfenato intermediate. J Am Chem Soc. 2005;127:17734–17743. doi: 10.1021/ja053387k. [DOI] [PubMed] [Google Scholar]
- 394.Wang T, Zhang X, Li JJ. The role of NF-kappaB in the regulation of cell stress responses. Int Immunopharmacol. 2002;2:1509–1520. doi: 10.1016/s1567-5769(02)00058-9. [DOI] [PubMed] [Google Scholar]
- 395.Wang Y, He QY, Sun RW, Che CM, Chiu JF. GoldIII porphyrin 1a induced apoptosis by mitochondrial death pathways related to reactive oxygen species. Cancer Res. 2005;65:11553–11564. doi: 10.1158/0008-5472.CAN-05-2867. [DOI] [PubMed] [Google Scholar]
- 396.Wardman P, Candeias LP. Fenton chemistry: an introduction. Radiat Res. 1996;145:523–531. [PubMed] [Google Scholar]
- 397.Ware DC, Brothers PJ, Clark GR, Denny WA, Palmer BD, Wilson WR. Synthesis, structures and hypoxia-selective cytotoxicity of cobalt(III) complexes containing tridentate amine and nitrogen mustard ligands. Dalton Trans. 2000;6:925–932. [Google Scholar]
- 398.Ware DC, Palmer BD, Wilson WR, Denny WA. Hypoxia-selective antitumor agents. 7. Metal complexes of aliphatic mustards as a new class of hypoxia-selective cytotoxins. Synthesis and evaluation of cobalt(III) complexes of bidentate mustards. J Med Chem. 1993;36:1839–1846. doi: 10.1021/jm00065a006. [DOI] [PubMed] [Google Scholar]
- 399.Ware DC, Palmer HR, Brothers PJ, Rickard CE, Wilson WR, Denny WA. Bis-tropolonato derivatives of cobalt(III) complexes of bidentate aliphatic nitrogen mustards as potential hypoxia-selective cytotoxins. J Inorg Biochem. 1997;68:215–224. doi: 10.1016/s0162-0134(97)00090-1. [DOI] [PubMed] [Google Scholar]
- 400.Ware DC, Palmer HR, Pruijn FB, Anderson RF, Brothers PJ, Denny WA, Wilson WR. Bis(dialkyl)dithiocarbamato cobalt(III) complexes of bidentate nitrogen mustards: synthesis, reduction chemistry and biological evaluation as hypoxia-selective cytotoxins. Anticancer Drug Des. 1998;13:81–103. [PubMed] [Google Scholar]
- 401.Waters SB, Devesa V, Del Razo LM, Styblo M, Thomas DJ. Endogenous reductants support the catalytic function of recombinant rat cyt19, an arsenic methyltransferase. Chem Res Toxicol. 2004;17:404–409. doi: 10.1021/tx0342161. [DOI] [PubMed] [Google Scholar]
- 402.Weaver EL, Bose RN. Platinum(II) catalysis and radical intervention in reductions of platinum(IV) antitumor drugs by ascorbic acid. J Inorg Biochem. 2003;95:231–239. doi: 10.1016/s0162-0134(03)00136-3. [DOI] [PubMed] [Google Scholar]
- 403.Weissbach H, Resnick L, Brot N. Methionine sulfoxide reductases: history and cellular role in protecting against oxidative damage. Biochim Biophys Acta. 2005;1703:203–212. doi: 10.1016/j.bbapap.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 404.Wells WW, Rocque PA, Xu DP, Yang Y, Deits TL. Interactions of platinum complexes with thioltransferase(glutaredoxin), in vitro. Biochem Biophys Res Commun. 1991;180:735–741. doi: 10.1016/s0006-291x(05)81127-1. [DOI] [PubMed] [Google Scholar]
- 405.Witte AB, Anestal K, Jerremalm E, Ehrsson H, Arner ES. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic Biol Med. 2005;39:696–703. doi: 10.1016/j.freeradbiomed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 406.Wolf CR, Hayward IP, Lawrie SS, Buckton K, McIntyre MA, Adams DJ, Lewis AD, Scott AR, Smyth JF. Cellular heterogeneity and drug resistance in two ovarian adenocarcinoma cell lines derived from a single patient. Int J Cancer. 1987;39:695–702. doi: 10.1002/ijc.2910390607. [DOI] [PubMed] [Google Scholar]
- 407.Wondrak GT. Redox-Directed Cancer Therapeutics: Molecular Mechanisms and Opportunities. Antioxid Redox Signal. 2009;11:3013–3069. doi: 10.1089/ars.2009.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Woynarowski JM, Chapman WG, Napier C, Herzig MC, Juniewicz P. Sequence- and region-specificity of oxaliplatin adducts in naked and cellular DNA. Mol Pharmacol. 1998;54:770–777. doi: 10.1124/mol.54.5.770. [DOI] [PubMed] [Google Scholar]
- 409.Wu J, Henderson C, Feun L, Van Veldhuizen P, Gold P, Zheng H, Ryan T, Blaszkowsky LS, Chen H, Costa M, Rosenzweig B, Nierodzik M, Hochster H, Muggia F, Abbadessa G, Lewis J, Zhu AX. Phase II study of darinaparsin in patients with advanced hepatocellular carcinoma. Invest New Drugs. 2010;28:670–676. doi: 10.1007/s10637-009-9286-9. [DOI] [PubMed] [Google Scholar]
- 410.Wu XX, Ogawa O, Kakehi Y. Enhancement of arsenic trioxide-induced apoptosis in renal cell carcinoma cells by L-buthionine sulfoximine. Int J Oncol. 2004;24:1489–1497. [PubMed] [Google Scholar]
- 411.Yamamoto N, Danos S, Bonnitcha PD, Failes TW, New EJ, Hambley TW. Cellular uptake and distribution of cobalt complexes of fluorescent ligands. J Biol Inorg Chem. 2008;13:861–871. doi: 10.1007/s00775-008-0374-7. [DOI] [PubMed] [Google Scholar]
- 412.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Yan YK, Melchart M, Habtemariam A, Sadler PJ. Organometallic chemistry, biology and medicine: ruthenium arene anticancer complexes. Chem Commun (Camb): 2005;4764:4776. doi: 10.1039/b508531b. [DOI] [PubMed] [Google Scholar]
- 414.Yang CH, Kuo ML, Chen JC, Chen YC. Arsenic trioxide sensitivity is associated with low level of glutathione in cancer cells. Br J Cancer. 1999;81:796–799. doi: 10.1038/sj.bjc.6690766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Yang M, Kroft SH, Chitambar CR. Gene expression analysis of gallium-resistant and gallium-sensitive lymphoma cells reveals a role for metal-responsive transcription factor-1, metallothionein-2A, and zinc transporter-1 in modulating the antineoplastic activity of gallium nitrate. Mol Cancer Ther. 2007;6:633–643. doi: 10.1158/1535-7163.MCT-06-0557. [DOI] [PubMed] [Google Scholar]
- 416.Yang X, Chen-Barrett Y, Arosio P, Chasteen ND. Reaction paths of iron oxidation and hydrolysis in horse spleen and recombinant human ferritins. Biochemistry. 1998;37:9743–9750. doi: 10.1021/bi973128a. [DOI] [PubMed] [Google Scholar]
- 417.Yangyuoru PM, Webb JW, Shaw CF., 3rd. Glutathionato-S-Gold(III) complexes formed as intermediates in the reduction of auricyanide by glutathione. J Inorg Biochem. 2008;102:584–593. doi: 10.1016/j.jinorgbio.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 418.Ye X, Fels D, Dedeugd C, Dewhirst MW, Leong K, Batinic-Haberle1 I. Antioxidants and Novel Therapeutics. Free Radical Biology and Medicine. 2009;47:S137. [Google Scholar]
- 419.Yi J, Gao F, Shi G, Li H, Shi X, Tang X. Apoptosis susceptibility of tumor cells to arsenic trioxide and the inherent cellular level of reactive oxygen species. Chin Med J (Engl) 2002;115:603–606. [PubMed] [Google Scholar]
- 420.Youn HS, Lee JY, Saitoh SI, Miyake K, Hwang DH. Auranofin, as an anti-rheumatic gold compound, suppresses LPS-induced homodimerization of TLR4. Biochem Biophys Res Commun. 2006;350:866–871. doi: 10.1016/j.bbrc.2006.09.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Yu Y, Kalinowski DS, Kovacevic Z, Siafakas AR, Jansson PJ, Stefani C, Lovejoy DB, Sharpe PC, Bernhardt PV, Richardson DR. Thiosemicarbazones from the old to new: iron chelators that are more than just ribonucleotide reductase inhibitors. J Med Chem. 2009;52:5271–5294. doi: 10.1021/jm900552r. [DOI] [PubMed] [Google Scholar]
- 422.Zakharyan RA, Sampayo-Reyes A, Healy SM, Tsaprailis G, Board PG, Liebler DC, Aposhian HV. Human monomethylarsonic acid (MMA(V)) reductase is a member of the glutathione-S-transferase superfamily. Chem Res Toxicol. 2001;14:1051–1057. doi: 10.1021/tx010052h. [DOI] [PubMed] [Google Scholar]
- 423.Zakharyan RA, Tsaprailis G, Chowdhury UK, Hernandez A, Aposhian HV. Interactions of sodium selenite, glutathione, arsenic species, and omega class human glutathione transferase. Chem Res Toxicol. 2005;18:1287–1295. doi: 10.1021/tx0500530. [DOI] [PubMed] [Google Scholar]
- 424.Zassinovich G, Mestroni G, Camus A. Diolefinic complexes of rhodium(I) and iridium(I) with nitrogen-containing ligands. J Organomet Chem. 1975;91:379–88. [Google Scholar]
- 425.Zelenko O, Gallagher J, Xu Y, Sigman DS. Chemical nuclease activity of 1,10-phenanthroline-copper. Isotopic probes of mechanism. Inorg Chem. 1998;37:2198–2204. doi: 10.1021/ic971154r. [DOI] [PubMed] [Google Scholar]
- 426.Zeller WJ, Fruhauf S, Chen G, Keppler BK, Frei E, Kaufmann M. Chemoresistance in rat ovarian tumours. Eur J Cancer. 1991;27:62–67. doi: 10.1016/0277-5379(91)90063-j. [DOI] [PubMed] [Google Scholar]
- 427.Zhang N, Wu ZM, McGowan E, Shi J, Hong ZB, Ding CW, Xia P, Di W. Arsenic trioxide and cisplatin synergism increase cytotoxicity in human ovarian cancer cells: Therapeutic potential for ovarian cancer. Cancer Sci. 2009;100:2459–2464. doi: 10.1111/j.1349-7006.2009.01340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Zhang X, Frezza M, Milacic V, Ronconi L, Fan YH, Bi CF, Fregona D, Dou QP. Inhibition of Tumor Proteasome Activity by Gold-Dithiocarbamato Complexes via Both Redox-Dependent and -Independent Processes. J Cell Biochem. 2010;109:162–172. doi: 10.1002/jcb.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lallemand-Breitenbach V, Jeanne M, Zhang QY, Yang HY, Huang QH, Zhou GB, Tong JH, Zhang Y, Wu JH, Hu HY, de The H, Chen SJ, Chen Z. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 2010;328:240–243. doi: 10.1126/science.1183424. [DOI] [PubMed] [Google Scholar]
- 430.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Zhu XH, Shen YL, Jing YK, Cai X, Jia PM, Huang Y, Tang W, Shi GY, Sun YP, Dai J, Wang ZY, Chen SJ, Zhang TD, Waxman S, Chen Z, Chen GQ. Apoptosis and growth inhibition in malignant lymphocytes after treatment with arsenic trioxide at clinically achievable concentrations. J Natl Cancer Inst. 1999;91:772–778. doi: 10.1093/jnci/91.9.772. [DOI] [PubMed] [Google Scholar]
- 432.Zitvogel L, Kepp O, Senovilla L, Menger L, Chaput N, Kroemer G. Immunogenic tumor cell death for optimal anticancer therapy: the calreticulin exposure pathway. Clin Cancer Res. 2010;16:3100–3104. doi: 10.1158/1078-0432.CCR-09-2891. [DOI] [PubMed] [Google Scholar]