

Abstract
Objective
To determine the role of Notch signaling in mediating alcohol’s inhibition of smooth muscle cell (SMC) proliferation.
Methods and Results
Treatment of human coronary artery SMCs with ethanol (EtOH) decreased Notch 1 mRNA and Notch 1 intracellular domain protein levels, in the absence of any effect on Notch 3. EtOH treatment also decreased C-promoter binding factor-1 (CBF-1)/recombination signal-binding protein (RBP)-jk promoter activity and Notch target gene (hairy related transcription factor [HRT-1] or HRT-2) expression. These effects were concomitant with an inhibitory effect of EtOH on SMC proliferation. Overexpression of constitutively active Notch 1 intracellular domain or human hairy related transcription factor-1 (hHRT-1) prevented the EtOH-induced inhibition of SMC proliferation. In vivo, Notch 1 and HRT-1 mRNA expression was increased after ligation-induced carotid artery remodeling. The vessel remodeling response was inhibited in mice that received “moderate” amounts of alcohol by gavage daily; intimal-medial thickening was markedly reduced, and medial and neointimal SMC proliferating cell nuclear antigen expression was decreased. Moreover, Notch 1 and HRT-1 expression, induced after ligation injury, was inhibited by moderate alcohol consumption.
Conclusion
EtOH inhibits Notch signaling and, subsequently, SMC proliferation, in vitro and in vivo. The modulation of Notch signaling in SMCs by EtOH may be relevant to the cardiovascular protective effects of moderate alcohol consumption purported by epidemiological studies.
Keywords: alcohol, Notch, SMC growth, vascular remodeling, intimal-medial thickening, athersclerosis, restenosis
Although the probability of cardiovascular disease and morbidity increases with heavy alcohol (ethanol [EtOH]) intake,1–3 studies indicate that regular moderate alcohol consumption is associated with a reduced incidence and severity of atherosclerosis and its clinical sequelae, including myocardial infarction and ischemic stroke.4,5 “Moderate” alcohol consumption is generally considered to be in the range of 1 to 3 drinks per day, giving rise to blood alcohol concentrations (BACs) of 5 to 25 mmol/L.5 Mortality and morbidity attributable to coronary heart disease are reportedly 20% to 40% lower in light-to-moderate drinkers compared with abstainers.6,7 Beneficial effects of alcohol at the level of plasma lipoproteins and on platelets, endothelial cells, and smooth muscle cells (SMCs) have been reported. However, the precise signaling and molecular mechanisms by which EtOH elicits its putative cardioprotective effects are not yet fully understood.
One potentially novel target for alcohol is the Notch signaling pathway. It is apparent that Notch plays a pivotal role in the differentiation and function of adult vascular SMCs (VSMCs), whose growth and migration are key processes in the pathophysiological features of atherosclerosis and in vessel remodeling in general. Several groups have described a role for Notch signaling in vitro and in vivo in repressing VSMC differentiation, an effect that is mediated via the induction of its target genes (hairy enhancer of split [HES] and HRT).8–10 Studies in animal models of vascular injury demonstrate that the expression of several components of the Notch pathway, including Notch 1 and 3, Jagged 1 and 2, and Notch target genes, are altered after experimentally induced vascular injury; and that Notch is a critical factor in determining the SMC proliferative response.9,11–13 Recent evidence points to a preferential role for Notch 1, rather than Notch 3, in mediating neointimal formation after vascular injury.14 Researchers have previously reported an inhibitory effect of EtOH on SMC proliferation15–17 and an EtOH-induced reduction in phenotype conversion of SMCs and in neointimal formation after balloon injury has been reported in animal models.18–20 The current study examined whether modulation of Notch signaling may mediate alcohol’s inhibitory effect on VSMC proliferation in vitro and in vivo.
Methods
Cell Culture and EtOH Treatment
Human coronary artery SMCs (HCASMCs) were commercially obtained and cultured in optimized SMC medium (Clonetics), supplemented with human (h) epidermal growth factor, insulin, h fibroblast growth factor, and 5% FCS. Cells were characterized by staining positively for SMC α-actin and calponin and weakly for myosin and smoothelin.
EtOH, 200 proof, was diluted in media to achieve the desired concentration before being added to SMC cultures (in multiwell plates or Petri dishes, wrapped in parafilm). For animal studies, EtOH (0.8 g/kg) in a total volume of 200 μL was delivered directly into the stomach of mice by oral gavage using a bulb-tipped gastric gavage needle attached to a syringe. This treatment resulted in a peak BAC of approximately 15 mmol/L. The control group was gavaged with a calorically matched water-cornstarch mixture.
Western Blot Analysis
Proteins from cell lysates (12 to 15 μg) were resolved on SDS-PAGE (12% resolving and 5% stacking) before transfer onto nitrocellulose membrane. Membranes were stained with Ponceau S and probed for β-actin or GAPDH, to ensure equal protein loading and transfer, and rinsed in wash buffer (PBS-containing 0.05% Tween-20) before being probed as previously described.21
Quantitative Real-Time RT-PCR
Total RNA (1 to 2 μg), isolated from cells using a kit (RNeasy), was reverse transcribed using another kit (iscript cDNA Synthesis kit). The gene-specific oligonucleotide sequences were as previously described.22 Real-time RT-PCR was performed using a machine (Stratagene MX3005) and a kit (SYBER green jumpstart PCR kit), as described by the manufacturer.
Cell Proliferation and Apoptosis
Changes in SMC number were determined by cell counting. Cells were serum starved for 24 hours and then seeded at 1×104 cells per well onto 6-well plates in 5% growth media, with or without EtOH (25 mmol/L). Cells were counted each day, and the average of 3 wells was quantified by using a hemocytometer. In parallel studies, proliferating cell nuclear antigen (PCNA) protein expression was determined by Western blot after treatment with EtOH. Staining using a kit (Annexin V: PE Apoptosis Detection Kit I) was performed according to the manufacturer’s instructions to quantitatively determine the percentage of cells within a population that was actively undergoing apoptosis.
Plasmid Preparation, Transient Transfection, and Luciferase and β-Galactosidase Assays
Plasmids were prepared for transfection using a plasmid midi kit. Cells were plated onto 6-well plates 2 days before transfection, at a density of 1×105 cells per well, and were transfected at 70% confluence. Plasmid transfection was performed using a commercially available system (Gene Pulser Xcell). The cells were transfected with various expression constructs, with the addition of 5.0 μg of DNA per well. Transfection efficiency was confirmed and normalized to β-galactosidase activity after cotransfection with cytomegalovirus (CMV)- β-galactosidase (LacZ), a plasmid-encoding β-galactosidase activity. Western blot analysis was also performed, whenever possible, to confirm overexpression of effector proteins. In luciferase reporter studies, cells were harvested 16 to 24 hours after transfection, using ×1 reporter lysis buffer. Transactivation of reporter genes was evaluated by the luciferase assay and normalized to β-galactosidase activity. The latter was performed according to the manufacturer’s instructions (high-sensitivity β-galactosidase assay).
Small and Interfering RNA Transfection
For gene-silencing studies, a system (Gene Pulser Xcell) was used for transient transfection of HCASMCs with gene-specific small and interfering RNA (siRNA). Briefly, 2×105 cells were transfected with 2.5 μg of siRNA targeting Notch 1 or a scrambled negative control siRNA (catalogue No. 4611) in 75 μL of siRNA electroporation buffer.
Mouse Carotid Artery Partial Ligation
The carotid artery ligation model of vascular injury and remodeling was performed essentially as described by Korshunov and Berk.23 All procedures were approved by the University of Rochester Animal Care Committee. After buprenorphine analgesia and induction of anesthesia using inhalational isoflurane, the mouse was positioned on a clean operating table, with a warming pad to maintain body temperature. The animal was clipped, and the surgical site was prepared using betadine solution and alcohol. A midline cervical incision was made. With the aid of a dissecting microscope, the left external and internal carotid arterial branches were isolated and ligated with a 6-0 silk suture, reducing left carotid blood flow to flow via the patent occipital artery. The neck incision (2 layers, muscle and skin) was closed with a running suture using 4-0 coated Vicryl. Ligation of the carotid artery in this manner results in a decrease in blood flow in the left carotid artery, concomitant with an increase in the right carotid artery, with an intact endothelial monolayer, when compared with a sham-operated control.
Preparation of Carotid Artery RNA
Mice were perfused with heparin-saline solution. Carotid arteries were collected in Trizol and homogenized using a tissue disperser (Ultra-Turrax), and RNA was prepared according to the manufacturer’s specifications.
Immunohistochemistry and Histomorphometry
Mice were perfusion fixed with 10% paraformaldehyde in sodium phosphate buffer (pH 7.0), 14 days after ligation; and the carotids were harvested and embedded in paraffin. Starting 500 μm below the carotid bifurcation landmark, a series of cross-sections (10×5 μm) was made, every 200 μm through 2 mm length of carotid artery. Cross-sections were stained with Verhoeff–van Gieson stain for elastic laminae, and sections were imaged using a microscope (Nikon TE300) equipped with a digital camera (Spot RT). Digitized images were analyzed using microcomputer imaging device software. Assuming a circular structure in vivo, the circumference of the lumen was used to calculate the lumen area; the intimal area was defined by the luminal surface and internal elastic lamina; the medial area was defined by the internal elastic lamina and the external elastic lamina; and the adventitial area was the area between the external elastic lamina and the outer edge, essentially as previously described by Korshunov and Berk.23 For immunohistochemistry, sections were stained by α-actin (Abcam, 1:100 dilution), PCNA (using a kit), or isotype control IgG, followed by biotinylated secondary antibody and streptavidin–horseradish peroxidase. Specimens were then developed with diaminobenzadine/water substrate and counterstained with Harris hematoxylin. Carotid sections were imaged using a microscope (Nikon TE300) equipped with a digital camera (Spot RT).
Blood and Media EtOH Concentration
Whole blood samples, collected by submandibular bleeding, were diluted 1:5 with water (100 μL of blood plus 400 μL of water) and analyzed for EtOH using head-space gas chromatography on an instrument by the Strong Health Clinical Laboratories (University of Rochester Medical Center, Rochester, NY). The method uses N-propanol as the internal standard and is sensitive to 10 mg/dL of EtOH. The media EtOH concentration over time was determined, in the absence or presence of SMCs, using an EtOH assay kit.
Data Analysis
Results are expressed as mean±SEM. Experimental points were performed in triplicate, with a minimum of 3 independent experiments (SMCs) or a minimum of 5 animals per group. An ANOVA was performed on cell count data, and a Wilcoxon signed-rank test was used for comparison of 2 groups when compared with normalized control. P≤0.05 was considered significant.
Results
EtOH Inhibits SMC Proliferation
We confirmed the inhibitory effect of EtOH on the proliferation of SMCs by measuring HCASMC counts and PCNA expression as an index of cell growth. Daily exposure of HCASMCs to EtOH (25 mmol/L) significantly decreased their proliferation over 7 days (Figure 1A). In addition, PCNA expression was significantly decreased (50% to 75%) after EtOH treatment (25 mmol/L for 24 hours) (Figure 1B). The effect of EtOH on HCASMC apoptosis was determined by flow cytometry detection using a stain (Annexin V/7-amino-actinomycin D [AAD]). EtOH treatment caused a small increase (approximately 3% to 4%) in the percentage of apoptotic SMCs (Figure 1C).
Figure 1.

EtOH inhibits SMC proliferation. A, HCASMCs were treated with or without EtOH (25 mmol/L). Cell counts of parallel triplicate wells were performed daily. Representative growth curves are shown. The experiment was repeated with similar results. B, Representative Western blot of PCNA protein expression in HCASMCs treated with or without EtOH (25 mmol/L for 24 hours). C, Flow cytometric analysis of stained HCASMCs (Annexin V/7-AAD). Percentages of apoptotic cells are shown in the top left quadrant for each condition. PE indicates phycoerythrin.
EtOH Inhibits Notch Signaling in HCASMCs
HCASMCs were treated with EtOH, and Notch receptor and target gene mRNA and protein levels were determined. EtOH treatment (25 mmol/L for 24 hours) significantly decreased (approximately 65%) Notch 1 mRNA expression and Notch 1 intracellular domain (IC) protein expression in these cells (Figure 2A and 2C). EtOH also significantly decreased the mRNA expression of the Notch target genes HRT-1 and HRT-2 (Figure 2B). No significant effects on Notch 3 mRNA or N3 IC protein levels were observed after treatment with EtOH (Figure 2). In contrast to EtOH, methanol and butanol had no significant effect on either Notch expression or PCNA levels in HCASMCs (supplemental Figure I; available online at http://atvb.ahajournals.org). In parallel cultures, EtOH treatment decreased baseline transactivation of CBF-1/RBP-JK–dependent promoter activity (57.0±7.0% and 64.0±6.0% decrease for 10- and 25-mmol/L EtOH, respectively) (Figure 2D).
Figure 2.

EtOH inhibits Notch signaling in HCASMCs. A and B, Quantitative RT-PCR analysis of Notch 1 and Notch 3 receptors (A) and Notch target gene HRT-1 and HRT-2 mRNA (B) after EtOH (25 mmol/L) treatment for 24 hours. Data are normalized to GAPDH and represent the mean±SEM (n=4). C, Representative Western blots, together with cumulative data showing a decrease in Notch 1 IC but no effect on Notch 3 IC with EtOH (25 mmol/L) treatment. (Blots were reprobed for β-actin as a loading control.) Data are given as the mean±SEM (n=4). D, CBF-1/RBP-JK–dependent promoter activity, evaluated by luciferase assay and normalized to β-galactosidase activity, after EtOH treatment for 24 hours. Data are given as the mean±SEM (n=3). *P≤0.05 vs control.
Overexpression of Notch 1 IC or HRT-1 Blocks Inhibition of SMC Proliferation by EtOH
HCASMCs were transfected with constitutively active Notch 1 IC, then treated with EtOH for 24 hours. Their PCNA protein levels were determined by Western blotting. Overexpression of Notch 1 IC in these cells was confirmed by Western blotting (data not shown). EtOH treatment (25 and 50 mmol/L) decreased (approximately 50%) SMC PCNA levels (Figure 3A). Notch 1 IC overexpression significantly increased PCNA, compared with mock transfected cells, and blocked the inhibition of PCNA by EtOH (Figure 3A). Furthermore, overexpression of HRT-1 also increased SMC PCNA levels and cell proliferation and blocked the EtOH inhibitory effect (Figure 3B).
Figure 3.

Notch 1 IC or HRT-1 overex-pression blocks inhibition of SMC proliferation by EtOH. A and B, SMCs were transfected without (mock) or with constitutively active Notch 1 IC (A) or hHRT-1 (B) and treated with EtOH for 24 hours; PCNA protein levels were measured by Western blot as an index of proliferation. Blots were reprobed for β-actin or GAPDH as a loading control. A, *P<0.05 vs mock control (0-mmol/L EtOH). A representative Western blot is shown, with cumulative data (n=3). B, Top, A representative Western blot of 3 is shown; bottom, growth curves for control and EtOH-treated cells, with or without HRT-1 overexpression.
Effect of Notch 1 Knockdown on SMC Proliferation and the EtOH Effect
Gene silencing of Notch 1 with specific targeted siRNA duplexes was confirmed at the mRNA and protein level. Inhibition of Notch 1 with siRNA, or treatment with EtOH, inhibited SMC growth (supplemental Figure II) to a similar extent. There was no significant additive or synergistic effect of using Notch 1 siRNA and EtOH together, suggesting that EtOH does not have antiproliferative effects independent of Notch 1.
Effect of EtOH Feeding on BACs
Male C57Bl/6J wild-type mice, aged 8 to 10 weeks, each weighing approximately 24 g, received either 1.0-g/kg alcohol (EtOH) or 0.8 g/kg in 200 μL of water by oral gavage. Samples of blood (200 μL) were obtained 30, 60, and 120 minutes after administration; and BAC was determined by head-space gas chromatography. The BACs recorded were approximately 23 and 15 mmol/L, respectively, after 30 minutes, with levels becoming undetectable (<2.2 mmol/L) after only 2 hours (supplemental Figure IIIA). We decided to use 0.8-g/kg EtOH as our experimental dosing regimen. The effect of daily alcohol consumption on mouse body weight was also determined. There was a small, but significant, decrease in body weight in both groups after carotid ligation that was regained by the end of the experimental period; however, there was no difference in body weight over time between the alcohol and control groups (supplemental Figure IIIB).
EtOH Feeding Inhibits Vessel Remodeling and Notch 1 mRNA Induction After Carotid Ligation
Partial ligation of the carotid artery in a wild-type C57BL/6J mouse induced a reproducible remodeling response, assessed 2 weeks after ligation, that included neointimal lesion formation and an increase in medial and adventitial volume compared with sham-operated vessels (Figure 4, Figure 5, and supplemental Figure IV). The remodeling response was inhibited in mice that received moderate levels of alcohol (EtOH, 0.8 g/kg; peak BAC, approximately 15 mmol/L) daily by gavage. Specifically, the neointimal, medial, and adventitial volumes were significantly decreased in alcohol-fed ligated mice compared with control ligated mice such that they approached sham vessel values (Figures 4 and 5). The intima:media ratio was 1.30±0.29 versus 0.14±0.08 for control versus alcohol-fed animals (n=6 in each group), respectively (supplemental Figure IV). PCNA expression, determined by immunohistochemistry, was increased in medial and neointimal SMCs in ligated controls; this was markedly inhibited in alcohol-fed mice (Figure 5). Interestingly, in sham-operated animals, daily alcohol feeding caused an increase in carotid artery lumen volume, compared with control mice. However, there was no difference in the lumen volume of remodeled vessels in the control and alcohol-fed animals (Figure 4).
Figure 4.

The effect of daily moderate alcohol feeding on vessel remodeling after carotid artery ligation. Carotid artery vessel compartment volumes (evaluated over a 1-mm carotid length) for control and EtOH-fed, sham-operated, and ligated groups. Data are given as the mean±SEM, 4 sections analyzed per animal (n=3 animals [sham], and n=6 animals [ligated]). *P≤0.05 vs control ligated.
Figure 5.
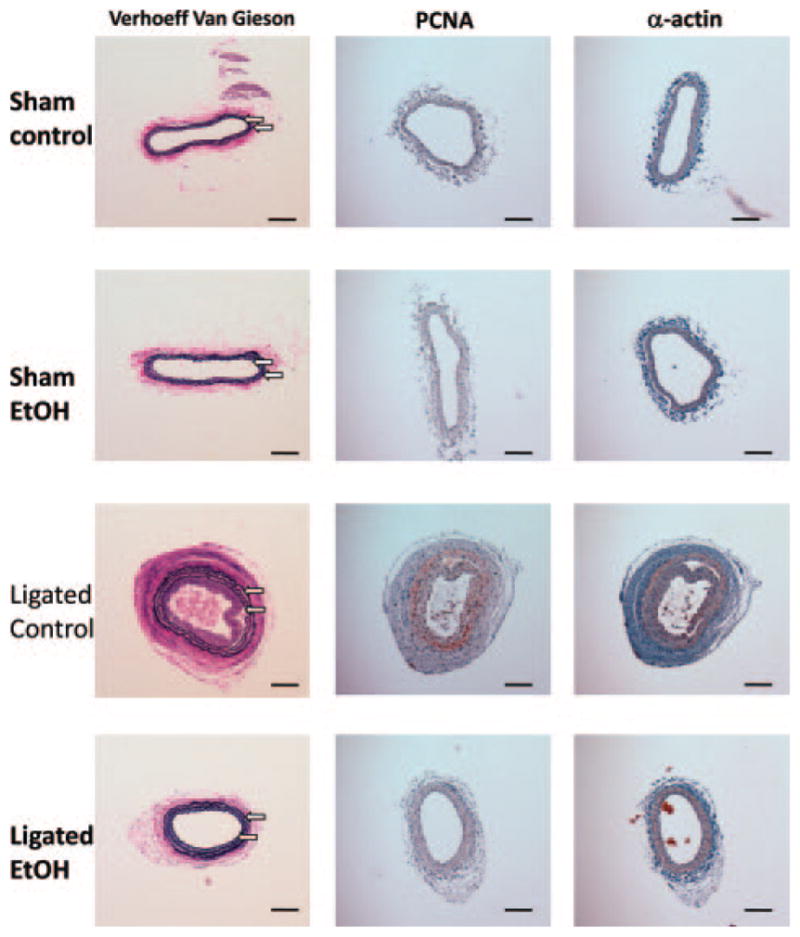
Immunohistochemical staining for PCNA and α-actin in representative sections of carotid arteries of C57BL/6J mice, 14 days after sham operation (Sham) or partial ligation (Ligated) in control or alcohol-fed animals. Verhoeff–van Gieson–stained sections are also shown. The arrows denote internal and external elastic laminae (original magnification ×10). The scale bar indicates 100 μm.
Notch 1 and Notch 3 mRNA levels were determined in the carotid arteries of control and alcohol-fed mice. In control animals, compared with sham vessels, there was a significant increase in Notch 1, but not Notch 3, mRNA expression in ligated remodeled vessels. This injury-induced increase in Notch 1 was completely inhibited by alcohol feeding (Figure 6A). Similarly, HRT-1 mRNA induced after injury was inhibited in alcohol-fed mice, compared with controls (Figure 6B).
Figure 6.

A and B, Quantitative RT-PCR analysis of Notch 1 and Notch 3 mRNA (A) and HRT-1 mRNA (B) isolated from whole carotid arteries of control or alcohol (EtOH)–fed C57Bl/6J mice, 2 weeks after partial ligation or sham operation. Data are normalized to GAPDH (n=5). *P≤0.05 vs control ligated.
Discussion
We demonstrated that alcohol (EtOH) inhibits VSMC proliferation in vitro by inhibiting Notch signaling in these cells. Moreover, daily alcohol feeding markedly reduced intima-media thickening concomitant with inhibition of induced Notch 1 and HRT expression after carotid ligation injury in the mouse. These findings provide new mechanistic data that may be relevant to how moderate alcohol consumption affects SMC proliferation and vascular remodeling and, consequently, cardiovascular mortality.
In the adult, SMCs proliferate at an extremely low rate and their principal function is contraction and regulation of blood vessel diameter. Unlike either skeletal or cardiac muscle, which is terminally differentiated, SMCs within adult animals retain plasticity and can undergo reversible changes in phenotype in response to a variety of stimuli, including growth factors and hemodynamic forces.24,25 An example of this plasticity is seen in response to vascular injury when SMCs dramatically increase their rate of cell growth and synthetic capacity (including the production of extracellular matrix components). VSMC proliferation plays a critical role in atherosclerosis and during restenosis after angioplasty and its regulation; therefore, it is of considerable clinical interest.
Previous studies have investigated the effect of alcohol on SMC growth and on injury-induced vascular remodeling. Alcohol inhibited SMC proliferation and migration in vitro,15,16,26 inhibited neointimal hyperplasia after balloon injury in animals,18–20 reduced restenosis after percutaneous transluminal coronary angioplasty and stent implantation in patients,27 and inhibited the progression and initiation of atherosclerotic lesions in hyperlipidemic mice.28 Herein, we demonstrate that alcohol treatment inhibits HCASMC proliferation in vitro and that daily feeding of alcohol, in amounts considered equivalent to those found in moderate drinkers, inhibited carotid artery vessel remodeling (ie, intimal-medial thickening) in response to flow reduction. Because SMCs are the predominant cell type responsible for intimal-medial thickening in this model23,29 and given our in vitro findings, our data are supportive of the concept that moderate alcohol consumption inhibits SMC proliferation and, thus, vascular remodeling. EtOH feeding also strongly inhibited the increase in adventitial volume seen in this model. The adventitia is predominately composed of fibroblasts and connective tissue cells; thus, the inhibitory effect of alcohol on the adventitia cannot be ascribed to inhibition of VSMC growth and warrants separate investigation.
Notch signaling is an important regulator of cell fate decisions during embryogenesis and also in adults in certain circumstances.30 SMCs preferentially express the receptors Notch 1 and Notch 3. Engagement of Notch by ligand results in 2 cleavage events that allow release of the IC of Notch (also referred to as notch intracellular domain [NICD]), thereby allowing translocation to the nucleus.31 Therefore, enforced expression of Notch IC provides a constitutively active signaling form of the receptor. The major signaling pathway evoked by Notch IC translocation into the nucleus is relayed by the transcriptional repressor, RBP-Jk, which is also (CBF1/(Su[H]Lag-1)-type transcription factor (CSL). Notch IC/CSL signaling leads to cell type–dependent activation of Notch target genes, including the HES gene and HES-related transcription factors (HRTs).32,33 Researchers have previously shown that Notch receptors and downstream target genes (HES and HRT) are crucial in controlling adult SMC growth, migration, and apoptosis in vitro and in vivo.8–10 Herein, overexpression of HRT-1 increased SMC proliferation and inhibited the EtOH antiproliferative effect, supporting a role for Notch 1–regulated gene expression in the regulation of SMC growth. It was also demonstrated that hemodynamic forces, such as cyclic strain, can modulate Notch receptor signaling and proliferation in VSMCs.34 Moreover, previous studies suggest that the expression of several Notch components, including receptors (Notch 1 and 3), ligands (Jagged 1 and Jagged 2), and target genes (HRT-1 and HRT-2) are altered after experimentally induced vascular injury (balloon catheter denudation)9,11,14,35 in human atherosclerotic lesions36 and after flow-induced injury.37 Intimal hyperplasia after vascular injury (wire injury) was significantly decreased in HRT-2−/− mice38 and SMCs from HRT-2−/− mice revealed that these mutant cells proliferate at a reduced rate compared with wild-type cells. The overexpression of HRT-1 or HRT-2 in VSMCs led to increased VSMC proliferation associated with reduced levels of the cyclin-dependent kinase inhibitors p21waf1/cip1 (Cdkn1a) or p27kip1 (Cdkn1b).39
However, despite the evidence supporting an important role for Notch in regulating SMC proliferation/vascular remodeling, and the growth inhibitory effects of EtOH on SMCs, to our knowledge, there have been no previous studies investigating the effect of alcohol on the Notch pathway in SMCs. Therefore, our study represents the first evidence of an alcohol-induced modulation of Notch signaling in SMCs. EtOH, at the dose studied, inhibited Notch 1 in HCASMCs, in the absence of any effect on Notch 3. Moreover, Notch 1 mRNA expression was exclusively induced in the carotid artery after ligation injury, an induction that was inhibited by alcohol feeding. Our data support the important role of Notch 1 IC (but not Notch 3 IC) in contributing to the intimal hyperplasia typical of this animal model. This fits well with the recent report by Li et al,14 indicating a role for Notch 1, but not Notch 3, in SMCs in mediating neointimal formation in a carotid ligation vascular injury model. Notch signaling is a tightly controlled pathway with several potential regulatory points. Studies investigating precisely how EtOH inhibits Notch signaling in SMCs are under way in our laboratory. Interestingly, it was recently reported that EtOH promotes Notch activity and proliferation in endothelial cells,22 the opposite of the effect seen herein in SMCs. Taken together, our studies point to an intriguing vascular cell-specific effect of EtOH with respect to Notch that merits further investigation.
In conclusion, alcohol, at levels consistent with moderate consumption, inhibits Notch signaling and, thus, proliferation in VSMCs in vitro and during vessel remodeling in response to flow reduction in vivo. Modulation of Notch by alcohol in SMCs may be an important mechanism involved in alcohol’s purported cardiovascular protective effects.
Supplementary Material
Acknowledgments
We thank Eric Olson, PhD, UT Southwestern, Dallas, for providing the HRT-1 expression vector (pcDNA3.1-N-MYC-hHRT-1).
Sources of Funding
This study was supported in part by grants K99HL095650 (Dr Morrow) and AA-12610 (Dr Redmond) from the National Institutes of Health; and Grant-in-Aid 0855865D from the American Heart Association (Dr Cullen).
Footnotes
Disclosures
None.
References
- 1.Klatsky AL, Armstrong MA, Friedman GD. Alcohol and mortality. Ann Intern Med. 1992;117:646–654. doi: 10.7326/0003-4819-117-8-646. [DOI] [PubMed] [Google Scholar]
- 2.Klatsky AL. Alcohol and cardiovascular health. Physiol Behav. 2010;100:76–81. doi: 10.1016/j.physbeh.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 3.Thun MJ, Peto R, Lopez AD, Monaco JH, Henley SJ, Heath CW, Jr, Doll R. Alcohol consumption and mortality among middle-aged and elderly US adults. N Engl J Med. 1997;337:1705–1714. doi: 10.1056/NEJM199712113372401. [DOI] [PubMed] [Google Scholar]
- 4.Corrao G, Rubbiati L, Bagnardi V, Zambon A, Poikolainen K. Alcohol and coronary heart disease: a meta-analysis. Addiction. 2000;95:1505–1523. doi: 10.1046/j.1360-0443.2000.951015056.x. [DOI] [PubMed] [Google Scholar]
- 5.Mukamal KJ, Conigrave KM, Mittleman MA, Camargo CA, Jr, Stampfer MJ, Willett WC, Rimm EB. Roles of drinking pattern and type of alcohol consumed in coronary heart disease in men. N Engl J Med. 2003;348:109–118. doi: 10.1056/NEJMoa022095. [DOI] [PubMed] [Google Scholar]
- 6.Costanzo S, Di Castelnuovo A, Donati MB, Iacoviello L, de Gaetano G. Alcohol consumption and mortality in patients with cardiovascular disease: a meta-analysis. J Am Coll Cardiol. 2010;55:1339–1347. doi: 10.1016/j.jacc.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Mukamal KJ, Chen CM, Rao SR, Breslow RA. Alcohol consumption and cardiovascular mortality among U.S. adults, 1987 to 2002. J Am Coll Cardiol. 2010;55:1328–1335. doi: 10.1016/j.jacc.2009.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sweeney C, Morrow D, Birney YA, Coyle S, Hennessy C, Scheller A, Cummins PM, Walls D, Redmond EM, Cahill PA. Notch 1 and 3 receptor signaling modulates vascular smooth muscle cell growth, apoptosis, and migration via a CBF-1/RBP-Jk dependent pathway. FASEB J. 2004;18:1421–1423. doi: 10.1096/fj.04-1700fje. [DOI] [PubMed] [Google Scholar]
- 9.Wang W, Campos AH, Prince CZ, Mou Y, Pollman MJ. Coordinate Notch3-hairy-related transcription factor pathway regulation in response to arterial injury: mediator role of platelet-derived growth factor and ERK. J Biol Chem. 2002;277:23165–23171. doi: 10.1074/jbc.M201409200. [DOI] [PubMed] [Google Scholar]
- 10.Wang W, Prince CZ, Hu X, Pollman MJ. HRT1 modulates vascular smooth muscle cell proliferation and apoptosis. Biochem Biophys Res Commun. 2003;308:596–601. doi: 10.1016/s0006-291x(03)01453-0. [DOI] [PubMed] [Google Scholar]
- 11.Lindner V, Booth C, Prudovsky I, Small D, Maciag T, Liaw L. Members of the Jagged/Notch gene families are expressed in injured arteries and regulate cell phenotype via alterations in cell matrix and cell-cell interaction. Am J Pathol. 2001;159:875–883. doi: 10.1016/S0002-9440(10)61763-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrow D, Scheller A, Birney YA, Sweeney C, Guha S, Cummins PM, Murphy R, Walls D, Redmond EM, Cahill PA. Notch-mediated CBF-1/RBP-J{kappa}-dependent regulation of human vascular smooth muscle cell phenotype in vitro. Am J Physiol Cell Physiol. 2005;289:C1188–C1196. doi: 10.1152/ajpcell.00198.2005. [DOI] [PubMed] [Google Scholar]
- 13.Morrow D, Guha S, Sweeney C, Birney Y, Walshe T, O’Brien C, Walls D, Redmond EM, Cahill PA. Notch and vascular smooth muscle cell phenotype. Circ Res. 2008;103:1370–1382. doi: 10.1161/CIRCRESAHA.108.187534. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Takeshita K, Liu PY, Satoh M, Oyama N, Mukai Y, Chin MT, Krebs L, Kotlikoff MI, Radtke F, Gridley T, Liao JK. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation. 2009;119:2686–2692. doi: 10.1161/CIRCULATIONAHA.108.790485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hendrickson RJ, Cahill PA, McKillop IH, Sitzmann JV, Redmond EM. Ethanol inhibits mitogen activated protein kinase activity and growth of vascular smooth muscle cells in vitro. Eur J Pharmacol. 1998;362:251–259. doi: 10.1016/s0014-2999(98)00771-7. [DOI] [PubMed] [Google Scholar]
- 16.Sayeed S, Cullen JP, Coppage M, Sitzmann JV, Redmond EM. Ethanol differentially modulates the expression and activity of cell cycle regulatory proteins in rat aortic smooth muscle cells. Eur J Pharmacol. 2002;445:163–170. doi: 10.1016/s0014-2999(02)01761-2. [DOI] [PubMed] [Google Scholar]
- 17.Ghiselli G, Chen J, Kaou M, Hallak H, Rubin R. Ethanol inhibits fibroblast growth factor-induced proliferation of aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:1808–1813. doi: 10.1161/01.ATV.0000090140.20291.CE. [DOI] [PubMed] [Google Scholar]
- 18.Liu MW, Anderson PG, Luo JF, Roubin GS. Local delivery of ethanol inhibits intimal hyperplasia in pig coronary arteries after balloon injury. Circulation. 1997;96:2295–2301. doi: 10.1161/01.cir.96.7.2295. [DOI] [PubMed] [Google Scholar]
- 19.Merritt R, Guruge BL, Miller DD, Chaitman BR, Bora PS. Moderate alcohol feeding attenuates postinjury vascular cell proliferation in rabbit angioplasty model. J Cardiovasc Pharmacol. 1997;30:19–25. doi: 10.1097/00005344-199707000-00004. [DOI] [PubMed] [Google Scholar]
- 20.Liu MW, Lin SJ, Chen YL. Local alcohol delivery may reduce phenotype conversion of smooth muscle cells and neointimal formation in rabbit iliac arteries after balloon injury. Atherosclerosis. 1996;127:221–227. doi: 10.1016/s0021-9150(96)05959-x. [DOI] [PubMed] [Google Scholar]
- 21.Morrow D, Cullen JP, Cahill PA, Redmond EM. Cyclic strain regulates the Notch/CBF-1 signaling pathway in endothelial cells: role in angiogenic activity. Arterioscler Thromb Vasc Biol. 2007;27:1289–1296. doi: 10.1161/ATVBAHA.107.142778. [DOI] [PubMed] [Google Scholar]
- 22.Morrow D, Cullen JP, Cahill PA, Redmond EM. Ethanol stimulates endothelial cell angiogenic activity via a Notch- and angiopoietin-1-dependent pathway. Cardiovasc Res. 2008;79:313–321. doi: 10.1093/cvr/cvn108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korshunov VA, Berk BC. Flow-induced vascular remodeling in the mouse: a model for carotid intima-media thickening. Arterioscler Thromb Vasc Biol. 2003;23:2185–2191. doi: 10.1161/01.ATV.0000103120.06092.14. [DOI] [PubMed] [Google Scholar]
- 24.Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 25.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 26.Hendrickson RJ, Cahill PA, Sitzmann JV, Redmond EM. Ethanol enhances basal and flow-stimulated nitric oxide synthase activity in vitro by activating an inhibitory guanine nucleotide binding protein. J Pharmacol Exp Ther. 1999;289:1293–1300. [PubMed] [Google Scholar]
- 27.Niroomand F, Hauer O, Tiefenbacher CP, Katus HA, Kuebler W. Influence of alcohol consumption on restenosis rate after percutaneous transluminal coronary angioplasty and stent implantation. Heart. 2004;90:1189–1193. doi: 10.1136/hrt.2003.025627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emeson EE, Manaves V, Emeson BS, Chen L, Jovanovic I. Alcohol inhibits the progression as well as the initiation of atherosclerotic lesions in C57Bl/6 hyperlipidemic mice. Alcohol Clin Exp Res. 2000;24:1456–1466. [PubMed] [Google Scholar]
- 29.Davies MG. Intimal hyperplasia. In: Johnston CA, editor. Rutherford’s Vascular Surgery. Philadelphia, PA: Saunders Elsevier; 2010. [Google Scholar]
- 30.Shawber CJ, Kitajewski J. Notch function in the vasculature: insights from zebrafish, mouse and man. Bioessays. 2004;26:225–234. doi: 10.1002/bies.20004. [DOI] [PubMed] [Google Scholar]
- 31.Schweisguth F. Regulation of notch signaling activity. Curr Biol. 2004;14:R129–R138. [PubMed] [Google Scholar]
- 32.Lai EC. Keeping a good pathway down: transcriptional repression of Notch pathway target genes by CSL proteins. EMBO Rep. 2002;3:840–845. doi: 10.1093/embo-reports/kvf170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iso T, Hamamori Y, Kedes L. Notch signaling in vascular development. Arterioscler Thromb Vasc Biol. 2003;23:543–553. doi: 10.1161/01.ATV.0000060892.81529.8F. [DOI] [PubMed] [Google Scholar]
- 34.Morrow D, Sweeney C, Birney YA, Cummins PM, Walls D, Redmond EM, Cahill PA. Cyclic strain inhibits Notch receptor signaling in vascular smooth muscle cells in vitro. Circ Res. 2005;96:567–575. doi: 10.1161/01.RES.0000159182.98874.43. [DOI] [PubMed] [Google Scholar]
- 35.Campos AH, Wang W, Pollman MJ, Gibbons GH. Determinants of Notch-3 receptor expression and signaling in vascular smooth muscle cells: implications in cell-cycle regulation. Circ Res. 2002;91:999–1006. doi: 10.1161/01.res.0000044944.99984.25. [DOI] [PubMed] [Google Scholar]
- 36.Doi H, Iso T, Yamazaki M, Akiyama H, Kanai H, Sato H, Kawai-Kowase K, Tanaka T, Maeno T, Okamoto E, Arai M, Kedes L, Kurabayashi M. HERP1 inhibits myocardin-induced vascular smooth muscle cell differentiation by interfering with SRF binding to CArG box. Arterioscler Thromb Vasc Biol. 2005;25:2328–2334. doi: 10.1161/01.ATV.0000185829.47163.32. [DOI] [PubMed] [Google Scholar]
- 37.Morrow D, Cullen JP, Liu W, Guha S, Sweeney C, Birney YA, Collins N, Walls D, Redmond EM, Cahill PA. Sonic Hedgehog induces Notch target gene expression in vascular smooth muscle cells via VEGF-A. Arterioscler Thromb Vasc Biol. 2009;29:1112–1118. doi: 10.1161/ATVBAHA.109.186890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakata Y, Xiang F, Chen Z, Kiriyama Y, Kamei CN, Simon DI, Chin MT. Transcription factor CHF1/Hey2 regulates neointimal formation in vivo and vascular smooth muscle proliferation and migration in vitro. Arterioscler Thromb Vasc Biol. 2004;24:2069–2074. doi: 10.1161/01.ATV.0000143936.77094.a4. [DOI] [PubMed] [Google Scholar]
- 39.Havrda MC, Johnson MJ, O’Neill CF, Liaw L. A novel mechanism of transcriptional repression of p27kip1 through Notch/HRT2 signaling in vascular smooth muscle cells. Thromb Haemost. 2006;96:361–370. doi: 10.1160/TH06-04-0224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.