

Abstract
Motivation: Computational identification of genomic structural variants via high-throughput sequencing is an important problem for which a number of highly sophisticated solutions have been recently developed. With the advent of high-throughput transcriptome sequencing (RNA-Seq), the problem of identifying structural alterations in the transcriptome is now attracting significant attention.
In this article, we introduce two novel algorithmic formulations for identifying transcriptomic structural variants through aligning transcripts to the reference genome under the consideration of such variation. The first formulation is based on a nucleotide-level alignment model; a second, potentially faster formulation is based on chaining fragments shared between each transcript and the reference genome. Based on these formulations, we introduce a novel transcriptome-to-genome alignment tool, Dissect (DIScovery of Structural Alteration Event Containing Transcripts), which can identify and characterize transcriptomic events such as duplications, inversions, rearrangements and fusions. Dissect is suitable for whole transcriptome structural variation discovery problems involving sufficiently long reads or accurately assembled contigs.
Results: We tested Dissect on simulated transcripts altered via structural events, as well as assembled RNA-Seq contigs from human prostate cancer cell line C4-2. Our results indicate that Dissect has high sensitivity and specificity in identifying structural alteration events in simulated transcripts as well as uncovering novel structural alterations in cancer transcriptomes.
Availability: Dissect is available for public use at: http://dissect-trans.sourceforge.net
Contact: denizy@mit.edu; fhach@cs.sfu.ca; cenk@cs.sfu.ca
1 INTRODUCTION
The transcriptome refers to the complete collection of RNA sequences transcribed from portions of the genome; these include not only mRNAs but also non-coding RNAs. Genomic structural alterations involving transcribed regions of the genome will appear in the associated transcript sequences. Although the whole transcriptome is much smaller than the whole genome, in the context of structural alterations, RNA-Seq data can be more difficult to analyze, partially due to splicing, which can produce several transcripts from the same gene. In comparison to the wild-type transcripts, post-transcriptional processes can also introduce structural alterations into these sequences.
To analyze structural variation within transcriptomic high-throughput sequencing (HTS) data (a.k.a. RNA-Seq) one typically needs to find the most likely transcript-to-genome alignment under the possibility of structural alteration events such as: (i) internal duplications, which result in two separate segments in the transcript sequence aligning to the same segment of the genomic sequence; (ii) inversions, which result in a segment of the transcript sequence aligning to the opposite strand of the genome than the rest of the transcript in an inverted fashion; (iii) rearrangements, which result in a change of ordering of the aligned segments; and (iv) fusions, which result in the transcript sequence aligning to two genes that are on two different chromosomes or far apart on the same chromosome (Figure 1). Note that an inversion can be of the type (i) suffix-inversion (or prefix-inversion), which involve a single breakpoint, where a suffix of the transcript sequence aligns to the strand opposite of that of the corresponding prefix; and (ii) internal-inversion, which involves a pair of breakpoints, where the portion of the inverted transcript sequence aligns to the strand opposite to that of the flanking portions.
Fig. 1.
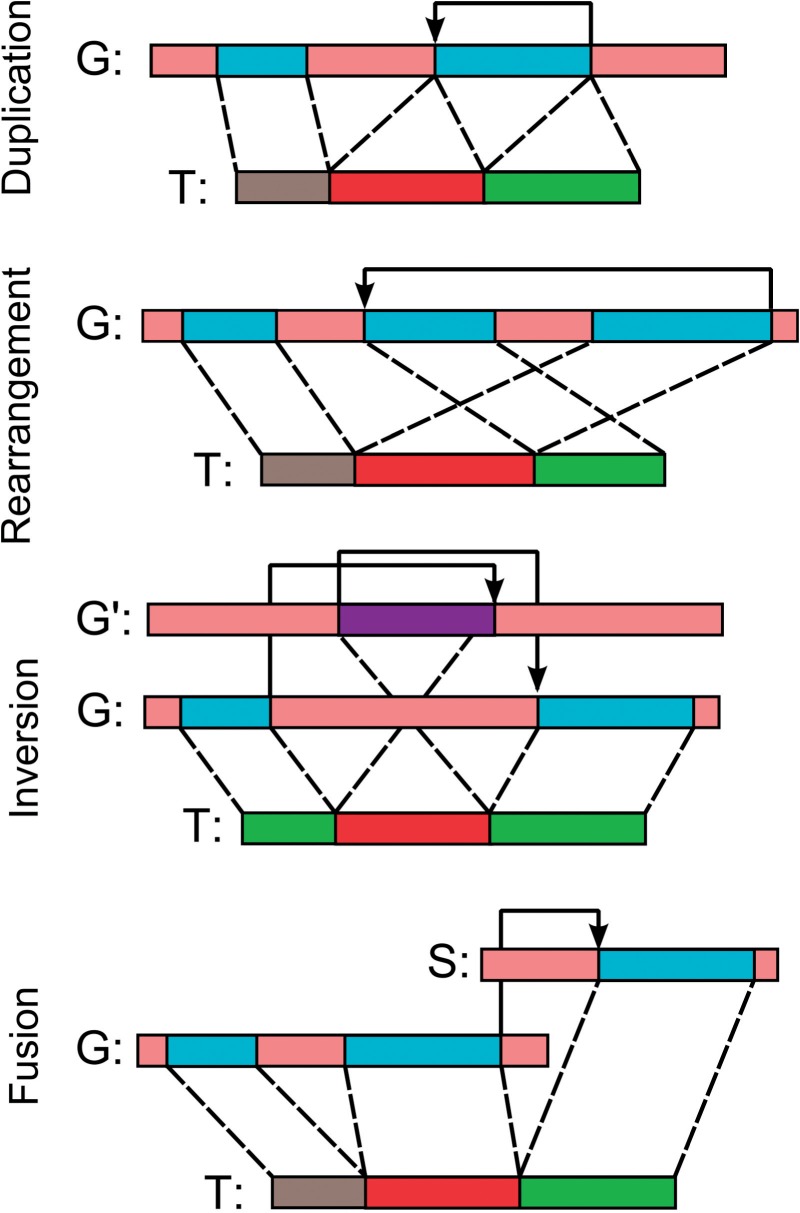
Structural alteration events considered in this article. T represent the transcript, G and S represent two genomic regions. G′ is the complementary strand for G. Boundaries between red and green blocks indicate event breakpoints; arrows represent corresponding genomic transitions in the alignment. Apart from the event types shown in the figure, duplication events can appear as non-tandem and fusions can be between two different strands
Several studies have detected such transcriptomic structural alterations in eukaryotic species (Anderson and Staley, 2008; Gingeras, 2009; Horiuchi and Aigaki, 2006; Horiuchi et al., 2003; Labrador et al., 2001), including mice (Hirano and Noda, 2004), rats (Caudevilla et al., 1998; Frantz et al., 1999) and humans (Akiva et al., 2006; Bäsecke et al., 2002; Brassesco, 2008; Herai and Yamagishi, 2010; Kannan et al., 2011; Rickman et al., 2009). These structural alterations are rarely observed in healthy human tissues and their normal roles have not been determined. However, specific instances of transcriptomic structural alterations (which can be a result of genomic alterations or transcriptomic processing) have been correlated with disease states, especially in cancer (De Braekeleer et al., 2011; Dorrance et al., 2008; Takahashi, 2011).
Several high-throughput methods for detecting structural alterations by analyzing mappings of RNA-seq reads have been developed (Asmann et al., 2011; Ge et al., 2011; Inaki et al., 2011; Levin et al., 2009; Li et al., 2011; McPherson et al., 2011a; McPherson et al., 2011b; McPherson et al., 2012; Nacu et al., 2011; Sboner et al., 2010), mostly focusing on fusions, partially due to their abundance in cancer, but also due to the relative ease of identifying them computationally. As HTS technologies progress, the length of the read-sequences they produce grows dramatically, and is expected to continue growing to over 1000 bp per read. A longer read has a greater possibility of containing segments from more than two exons, complicating the process of mapping such a read. Existing structural alteration detection tools are limited by their reliance on read-mapping tools designed for the short read-sequences produced by the original HTS technologies; they cannot take advantage of increasing read length. Furthermore, the increase in read length improves the accuracy of de novo transcriptome assembly tools, leading to opportunities for the analysis of full transcript sequences.
Existing methods: Many methods have been proposed for spliced transcriptome-to-genome sequence alignment, including Gapped BLAST (Altschul et al., 1997), BLAT (Kent, 2002), Exonerate (Slater and Birney, 2005), EST_GENOME (Mott, 1997) and GMAP (Wu and Watanabe, 2005). These methods implicitly assume the transcript sequences are devoid of structural alterations and use a seed-and-extend alignment strategy combined with a fragment chaining method. In the context of genomic sequences, alignment under structural alterations has been considered since the early 2000s. From a theoretical perspective, Cormode et al. introduced block edit distance (Cormode et al., 2000) as the minimum number segment deletions, duplications and translocations in addition to single-nucleotide insertions, deletions to transform (and align) one sequence to another. Block edit distance in its most general setting is NP-hard to compute; Ergün et al. (2003) investigated many variants of the block edit distance under several restrictions that make the alignment problem computationally tractable. One such variant involving 1-monotonous alignments was implemented in the context of genome-to-genome alignments (Brudno et al., 2003). Unfortunately no such method exists for transcript-to-genome alignments.
Contributions: In this study, we introduce novel algorithmic formulations of the transcript-to-genome alignment under structural alterations problem and describe solutions for several gap penalty models. Our first formulation is a nucleotide-level alignment model that assumes the transcript sequence is a chain of unidirectional copies of segments taken from the genome—as investigated in Ergün et al. (2003). We show how to sparsify the alignment table using a convex gap penalty model; we also show how to incorporate splice signal scores to the model. Our second formulation aims to reduce the running time and memory cost of the initial nucleotide-level alignment problem by sacrificing the sensitivity upon structures shorter than a user-defined threshold value. In this formulation, we assume each alignment unit is a short segment shared between the transcript and the genome, possibly containing some mismatches. We then aim to find the fragment chain that gives the best overall alignment score based on the penalties described in the first formulation.
This study also introduces a novel computational tool, Dissect (DIScovery of Structural alteration Event Containing Transcripts), suitable for high-throughput transcriptome studies. To the best of our knowledge, Dissect is the first comprehensive stand-alone software for detecting and characterizing novel structural alterations in RNA-Seq data, and capable of direct global alignment of long transcript sequences to a genome. We report experimental results obtained by Dissect on a simulated mouse transcriptome database containing nucleotide-level and structural noise, as well as assembled RNA-Seq reads from the human prostate cancer cell line C4-2.
2 METHODS
In this article, we introduce a generalization of the transcriptome to genome spliced alignment problem, which allows the detection of transcriptional aberrations such as duplications, rearrangements and inversions. The model we use for our formulation corresponds to the restricted asymmetric variant of the block edit distance (Ergün et al., 2003), in which the transcript sequence is represented by substrings extracted from the genome sequence, which are stitched together in various formations. Even though such an alignment model ignores the intermediate evolutionary steps of genomic modifications, it can still act as a realistic model of the transcription process with structural alterations, allowing the alignment of chimeric transcripts containing introns/deletions, novel insertions, duplications, rearrangements and inversions. One major caveat of aligning two genomic sequences using this approach is the omission of duplications in one of the aligned sequences (Brudno et al., 2003), whereas the same approach does not necessarily pose a limitation for transcriptome to genome alignment since transcriptional structural alterations involve duplications on the transcript side, yet genomic duplications that appear in the transcript are not crucial for our analysis.
We further generalize our formulation for handling the special case of fusions that correspond to the alignment of a single transcript to two independent genomic sequences such that there can only be a single transition from the first sequence to the second and no transition from the second to the first. Finally, we incorporate additional score models for canonical splice signals into our formulation to represent a realistic model of the transcriptional machinery biased on canonical splice sites.
Below we describe a nucleotide-level transcriptome to genome alignment formulation for detection of structural alterations within the transcript. Based upon this approach, we also describe a number of algorithmic formulations for ‘chaining’ fragments shared between the transcript and the genome sequences (within some small number of mismatches), considering alternative structural formations of the resulting fragment chain. Our formulation considers a number of genomic gap penalty models, in the form of general, convex and log-scale cost functions and transcriptional insertion penalties with a constant gap penalty model.
2.1 Nucleotide-level transcriptome to genome alignment under structural alterations
Given T=t1 t2… tN, a transcript sequence and G=g1 g2… gM, a genomic sequence (including a gene), let the complementary genomic sequence to G be G′=g′1 g′2… g′M, where g′i represents the complement of gi. Also let S=s1 s2… sL be a secondary genomic sequence (e.g. another gene) independent from G, and let S′=s′1s′2… s′L be its complement. S represents the potential fusion partner for G in the context of T.
We define an alignment of T to {G, G′, S, S′} under specific set of structural alterations As (which will be clarified later in the text) to be a mapping f from the nucleotides of T to those of G, G′, S, S′, as well as ϕ, representing a single-nucleotide gap on the genome side of the alignment (note that f is not an invertible function). To prevent multiple fusions within the alignment, we restrict As such that if a nucleotide in T is aligned to S or S′, none of the subsequent nucleotides in T can be aligned to G or G′—ensuring that T can be obtained by fusing at most two genes and there can be at most one ‘transition’ from G/G′ to S/S′.
We now define the score of an alignment with structural alterations As as
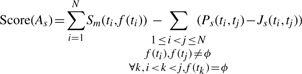 |
Here Sm denotes the alignment score of each ti to f(ti)—which is higher if ti=f(ti). The second contribution to the alignment score is due to the penalties for all genomic transitions in the alignment, i.e. segments in T which are all mapped to ϕ, indicating a gap. Now we assign Ps as:
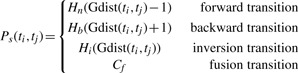 |
Here Gdist(a, b) denotes the genomic distance, namely, |f(a)−f(b)|. Note that forward transitions are genomic transitions between a pair of aligned positions that lie on the same sequence (i.e. G, G′, S or S′) such that the order of alignment is preserved with respect to the alignment direction (e.g. when aligned to complementary strand order will be reverse as well). The penalty function for forward transitions is a user-defined function Hn (see the note below). In the presence of duplication and rearrangement events, aligned positions will be in reverse order with respect to their alignment direction and these types of backwards transitions are penalized by the user-defined function Hb. Another type of transition considered is the inverted transitions penalized by the user-defined function Hi; here, one alignment lies on the original sequence and the other on the complementary sequence. Finally, a constant gap penalty Cf is applied to fusion transitions in which ti aligns to G/G′ and tj aligns to S/S′. Note again that although Hn, Hb and Hi are user-defined functions, Hb and Hi (which correspond to structural alterations in the transcript) should be costlier than Hn (which corresponds to the regular genomic gap, presumably spanning an intron).
The second component of the transition penalty, Js, is an additional score deducted from the penalty depending on the existence of canonical splice signals at the junction sites:
Here min_intron corresponds to the minimum considered length of an intron (anything shorter is treated as a deletion) and Jb, Je are binary functions that indicate whether the beginning and ending splice sites of the transition correspond to canonical splice signals. Note that the above formulation assumes a single-canonical splice signal pair (e.g. GT-AG) and the penalty is additive with respect to the beginning and ending sites.
Based on the definitions above, observe that in the special case of an insertion in T, such that ti+1… tj−1 are all aligned to ϕ and f(ti) and f(tj) are consecutive nucleotides in G, G′, S and S′, the penalty is zero.
An efficient algorithm for the nucleotide-level transcriptome to genome alignment with structural alterations problem. Given a limited variant of the alignment and the score function above, where only forward transitions are considered without splice signal scores on a single-genomic sequence G, there is a simple algorithm (for arbitrary Hn) with running time O(NM2). This algorithm constructs an alignment table, XG, of size N×M and initializes its first row as max(Sm(t1, gj), Sm(t1, ϕ)) for all j∈[1, M]. For each of the remaining rows, the transitions from the previous row are evaluated representing genomic gaps between valid transcript position pairs. The gaps in the transcript are calculated as vertical transitions between two adjacent rows in the alignment table. This allows the construction of each row in O(M2) time, yielding the above running time.
Galil and Giancarlo (1989) and Miller and Myers (1988) independently showed that when a restricted distance penalty scheme is assumed, the running time needed to construct a row can be reduced to O(M log(M)) using sparse dynamic programming. This restriction assumes a convex gap penalty function, h: ℤ+∪{0}→ℝ such that h(x)−h(x−1)≥h(x+1)−h(x)≥0. Galil and Giancarlo (1989) further reduced the running time to O(M) for simple convex gap penalty functions, with the condition that for every x1, x2∈ℤ, x1<x2 and r∈ℝ, the smallest integer value y that satisfies y>x2 and h(y−x1)−h(y−x2)−r≤0 can be calculated in constant time. Log-scale distance penalty functions [in the form a+b*log(distance)], which model gap penalties quite realistically [as the intron lengths are geometrically distributed (Burge and Karlin, 1997)], satisfy the simple convexity property. This allows the exact transcript to genome alignment without structural alterations to be performed in O(NM) time.
We now show how to extend the above algorithms to handle splice signal scores and transcriptional structural alterations in the form of duplications, rearrangements, inversions and fusions.
Even if genomic distance penalties can be chosen as convex functions, the contribution from Js may violate the convexity. To resolve this issue, we construct each row of the alignment table by the use of two independent processes; the first process calculates the genomic transitions from the previous row for the positions that constitute a canonical splice starting site, and the second process does the same for the positions that do not constitute a canonical splice starting site. For each of these processes, since Jb remains constant for a fixed position in the previous row, the set of forward transitions between two rows satisfy the convexity property. To obtain the optimal entries in a given row, we take the higher of the two values.
Even though the above formulation is for forward transitions only, we can perform all sparse dynamic programming operations in reverse order (with switched indices); as a result we can split each of the two processes described above into further two processes, one for forward and another one for backward transitions. This way we can capture duplication and rearrangement events that require a backwards transition in the alignment.
For handling inverted transitions we use a second alignment table, XG′ for aligning T with G′, which is initialized in a similar fashion to XG (naturally Sm values are obtained for the complementary nucleotide). Since the original sparse dynamic programming solution is designed for any arbitrary score values taken from the previous row, obtaining the row from table XG′ will still be valid. Therefore, we can further split the four processes described above for handling inverted and non-inverted transitions; each entry will then be assigned to the maximum valued result out of the eight computed. Computing a row in table XG′ can be carried out similarly: non-inverted transitions involving forward/backward and canonical/non-canonical processes will be computed using the previous row in table XG′, where the remaining inverted transitions will be computed using the table XG.
For fusion cases, we use two new alignment tables XS and XS′ to our formulation (for aligning T with both S and S′ ). Even though the processes for constructing the rows of XS/XS′ are identical to XG/XG′, the initialization step requires handling potential fusion transitions. Before starting the row construction process we first identify the optimal fusion transition to each row in XS/XS′. For k-th row, the highest scoring entry within the first k−1 rows in XG or XG′ constitutes the highest scoring fusion transition score, combined with the constant fusion transition penalty Cf. Since fusion transition penalties are independent from genomic position, the highest scoring table entry gives the optimal fusion transition for any of the cells in the row with the same transition score.
The above algorithmic formulation provides solutions for arbitrary, convex and simple convex penalty formulations for handling structural alterations and splice signal scores within respective running times of O(NM2), O(NM log(M)), and O(NM).
2.2 Fragment chaining for transcriptome to genome alignment under structural alterations
The problem of transcriptome to the genome alignment with structural alterations can be optimally solved in polynomial time under the assumption that the transcript sequence is composed of substrings copied from the genome sequence. For high-throughput transcriptome to genome alignment studies, however, running time and memory requirements of nucleotide-level alignment will be costly even for log-scale gap penalty functions.
In this section, we describe the algorithmic formulation for a ‘lower resolution’ solution to the transcript to genome alignment with structural alterations. Given a set of ‘fragments’ between the transcript and the genome sequences, this approach aims to find the optimal chain of fragments within certain constraints that will give the maximum alignment score with respect to the fragment ‘qualities’ and transition penalties (Figure 2). If the fragment length is one, this formulation reduces to the nucleotide-level formulation given.
Fig. 2.
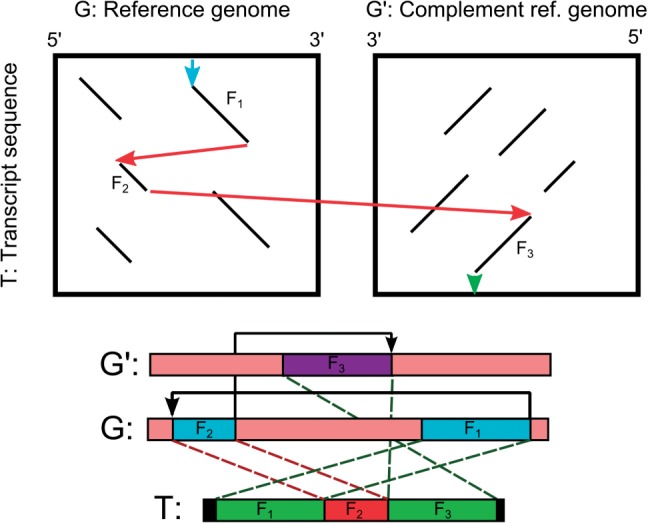
Fragment chaining in the presence of a rearrangement and an inversion. The fragments involved include two segments from T associated with segments from G and another segment from T associated with a segment from G′. The figure depicts how the fragments reveal themselves in the alignment tables and how they can be chained to get the overall alignment
Given a transcript sequence T and a pair of genomic sequences G and S (and their complementary sequences G′ and S′), a fragment F is a segment of G, S, G′ or S′ which is also present in T within a small number of mismatches. Associated with F, we have (i) the starting position in T; (ii) the starting position in one of the genome sequences G, G′, S and S′; (iii) the length of the fragment; and (iv) the similarity score for the fragment, respectively, denoted by F.ts, F.gs, F.len and F.score. Similarly F.ge and F.te will denote the ending position of the respective sequences; e.g. for forward alignments, F.ge=F.gs+F.len−1 and F.te=F.ts+F.len−1. Fragments from G or S are aligned to T in the forward direction; the fragments from G′ or S′ are aligned to T in the reverse direction with complementary nucleotides. The score of the fragment is a function of its length and the number of mismatches between itself and its occurrence in T.
In the algorithmic formulation below, we are given a set (Fset) of K fragments shared between T and genomic sequences, G, G′, S and S′, which are at least of a user specified length and have an alignment score higher than a user specified threshold (we describe how we obtain Fset later in the text). A pair of fragments can overlap in the transcript or in the genome sequence. However, for the description below, we do not consider a fragment in Fset, which is a sub-fragment of (fully included in, and in the same direction with) another fragment in the genome sequence and the transcript.
We define a valid disjoint fragment chain C as an ordered subset of Fset involving k≤K fragments, (F1, F2,…, Fk), such that (i) for each pair of subsequent fragments Fi, Fi+1 (subsequent fragments are said to be chained) we have Fi.te<Fi+1.ts; and (ii) if Fi is aligned to S/S′, no Fj for j>i is aligned to G/G′.
Our goal here is to find the valid disjoint fragment chain Cd (of length B≤K) over Fset with the highest possible ‘score’ with respect to the scoring function fscore and transition penalty function fpenalty given as
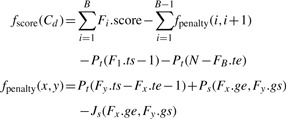 |
Here, the ‘transcript gap penalty function’ (according to the constant gap penalty scheme described in our nucleotide-level alignment formulation) is set to be Pt(dist)=Cgap*dist. The original genomic transition penalty function Ps and canonical splice signal scoring function Js are as per the nucleotide-level alignment formulation.
It is possible to solve the problem described above by going through the fragments in Fset according to their starting position in T, computing the best scoring chain ending with each fragment via dynamic programming (see the description of the Dissect method). For any pair of user-defined functions Ps and Js this algorithm can find the optimal disjoint fragment chain in Fset in O(K2) time. Although it may be possible to improve the running time for restricted genomic gap penalty models involving, e.g., convex and simple convex cost functions, this algorithm is easy to implement and has proven to be sufficiently fast on the datasets we experimented with.
Fragment chains with overlapping fragments. In a real-life experimental setting, a chain of fragments should be allowed to overlap to handle situations involving highly similar flanking sequences of a pair of chained fragments. Here, we develop a generalized version of the formulation given above which allows the chaining of a prefix of a fragment Fi to a suffix of another fragment Fj so that the chosen prefix and suffix do not overlap in the transcript. First we redefine the concept of a valid fragment chain and then investigate different overlap resolution schemes.
Let a valid fragment chain with overlaps, C, be a sequence of k≤K fragments, (F1, F2,…, Fk), such that (i) the starting and ending positions of the fragments in the transcript increase throughout the chain and (ii) if Fi the chain is aligned to S/S′, Fj cannot be aligned to G/G′ for any j>i.
Our goal is to find the optimal overlapping fragment chain Co (of length B≤K) with a modified score function that differs from the original score function in only the transition penalty function, fpenalty, described below:
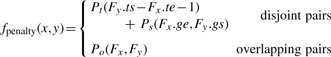 |
Here Po(x, y) represents the special transition penalty for overlapping fragment pairs. Notice that the splice signal score function, Js, is omitted from the penalty function. This is due to the complications that can be caused by the integration of splice signal scores and overlapping fragments.
Given a valid overlapping fragment chain, (F1,…, Fk), an overlap split position between chained fragments Fi and Fi+1, is a position r∈[Fi+1.ts−1, Fi.te] indicating the modified ending position of Fi, and r+1 indicating the modified starting position of Fi+1 in the transcript. Below we show how to obtain the overlap split positions, and effectively resolve overlaps between a pair of chained fragments.
In a simple overlap resolution model, the penalty Po can be set to the sum of the length of the overlapping interval and the penalty of the genomic gap: for an overlapping fragment pair Fi and Fi+1, we define Po=Ps(F′i.ge, F′i+1.gs)+(Fi.te−Fi+1.ts+1), where F′i and F′i+1 represent the updated (shortened) fragments. Since the overlap length is known, we simply have to find the overlap split position r that minimizes Ps.
Notice that for fragment pairs that are aligned in the same direction, the genomic distance between a pair of fragments increases with the length of the overlap—independent of the overlap split position; thus any overlap split position will do. For fragments that are aligned in different directions however, the overlap split position has an effect on the genomic distance between the two fragments. There are three different scenarios to consider in this case: (i) first fragment is located ‘earlier’ in the genome; (ii) first fragment is located ‘later’ in the genome; and (iii) the overlapping regions of both fragments overlap in the genome as well. For each of the above scenarios, respectively, selecting the ‘latest’ overlap split position, the ‘earliest’ overlap split position, and the overlap split position that makes the two updated fragments closest in the genome minimizes the genomic transition penalty—under the assumption that inversion transition penalty increases with the genomic distance. Since all these three cases can be handled in constant time, computing the optimal overlap split position for any pair of fragments can be performed in constant time. As a result, we can employ an algorithm quite similar to the one described for disjoint fragment chaining. The only difference is, when selecting the valid chains ending at each fragment Fi, the algorithm will need to also consider the fragments that overlap with Fi, but do not start earlier than Fi.ts in T or end earlier than Fi.te. As per the algorithm for disjoint fragment chaining, this variant of the fragment chaining with overlapping fragments method needs O(K2) time.
For further improving the accuracy, one needs to consider the (eliminated) mismatches within the overlapping region. The penalty function, Po, can now be defined as: Po(Fi, Fi+1)=Ps(F′i.ge, F′i+1.gs)+(Fi.score−F′i.score)+(Fi+1.score−F′i+1.score). The optimal split position of an overlapping fragment pair is that which minimizes the sum of the contributions from the updated genomic distance and the number of mismatches retained in the updated fragments. A naive method to handle this variant of the problem checks each position within the overlap to compute the minimum value for Po. However, if there are no mismatches in either fragment, the problem reduces to that described above, and thus will have an efficient solution. Furthermore, if each fragment in Fset can only have a constant number of mismatches (one can enforce this in the definition of a fragment), a simple brute force search will compute the optimal split position for each pair in constant time, preserving the running time of O(K2).
The algorithmic formulation described above provides the theoretical underpinnings of the computational method we devised for identifying structural alterations leading to a transcript. However, as will be described in the next section, we need to take further steps to make the fragment chaining solution efficient and its results close to those implied by nucleotide-level formulation towards a practical solution.
2.3 Whole genome analysis and discovery of novel transcriptional structural alterations with Dissect
In this section, we describe some of the details of our computational tool Dissect. Dissect has three main stages:
Genomic region inference. This stage begins by sampling anchors from the transcript sequence and mapping to reference genome within a user-defined mismatch threshold. Although there are a number of ‘spliced’ alignment methods in the literature, they either perform a local (Kent, 2002) or an anchor-specific (Wu and Watanabe, 2005) analysis of anchor mappings. Since we aim to detect structural alterations, the order or direction of anchors are not necessarily preserved within the alignment. Thus, a global region inference approach oblivious to order or direction is more suitable.
For our purposes, an anchor is a substring of constant length LA, of the transcript sequence T of length N. We generate the set of anchors from the transcript by sampling a user-defined number of equally distanced anchors of length LA, the first and last anchors corresponding to the beginning and the ending of the transcript, respectively. Then we find all possible mappings of an anchor in the genome through the cache-oblivious short read mapper, mrsFAST (Hach et al., 2010), eliminating the anchors that have more mappings than a user-specified threshold.
Within a set of anchor mappings Smap={m1, m2,…, mK} of size K, each mapping mi (to a genomic region) is represented as mi=(mi.t, mi.g, mi.score), which, respectively, correspond to the starting position of the anchor in the transcript, the starting position of the mapping in the genome, and mapping (similarity) score. Given the complete set of anchor mappings, we determine a ‘genomic region of interest’ by finding all intervals within the genome (or two disjoint intervals for the fusion cases) to which a high number of distinct anchors are mapped—with high-alignment scores. Since the region is preferred to be compact, our aligner only searches among the intervals that start at the starting position of an anchor mapping and end at the ending position of a mapping. This condition removes all intervals that have unnecessary extensions at each end and reduces the number of possible genomic regions to O(K2).
For our purposes, a genomic region R is an interval that locally maximizes the following score:
Here cN≥1, cα≥1 and cL>0 are user-defined normalization parameters for adjusting relative significance of the number of anchors contained within the region to the length of the inferred region. M, on the other hand, is the function defined to be the sum of best mapping scores of all anchors mapped to the region.
Our genomic region inference method initially sorts all anchor mappings according to their genomic position. It then goes over each mapping position and calculates the above score for all possible genomic intervals starting at that position and commits to the one with the highest score—in linear time via dynamic programming.
A second type of genomic region inference needs to find fusion regions that appear as intervals separated by long inter-genic regions on the same genomic sequence (e.g. intra-chromosomal fusions) or possible different genomic sequences (e.g. inter-chromosomal fusions). Since Dissect does not utilize gene annotation for region inference, we do not differentiate between single-gene alignments and inter-genic fusions between closely located genes. This step essentially corresponds to the inference of two separate regions and a transcript cut position that yield the highest double region inference score, which is the sum of scores of the two regions such that the first region score is only calculated over the anchor samples taken from upstream of the transcript cut position and the second region score is only calculated upon the samples taken from downstream.
Instead of looking for combinations of regions that give the optimal double region score, Dissect scans over all possible anchor split positions and search for single regions for both ends of the transcript independently. Optimal double region score for this anchor split is the sum of optimal single regions that cover anchors on each side. Given cA anchors sampled from the transcript, we find the optimal double region pair in O(cA*K2) time.
Even though the region inference methods described above search for the highest scoring genomic region/region pair, Dissect processes all regions whose scores are within a constant factor of the highest score (and within a user-defined maximum number of disjoint regions threshold). Each of these reported genomic regions/region pairs are analyzed separately within the downstream alignment pipeline.
An important issue to address is the relation between inferred single regions and double regions. Even though we can efficiently find the highest scoring single/double regions from a given anchor set and its mappings, it is not possible to objectively compare scores of inferred single/double regions. For instance, if there is a long intron in the alignment of the transcript, the double region that spans the exons on each side of the intron might score higher than a single region that encompasses the entire transcript alignment if cα is 1. Furthermore, a distance threshold would require prior knowledge of fusions within the genome that is analyzed. Dissect uses a double layer inference step as a workaround to this single/double region score comparison issue. In the first inference layer, we find a set of highest scoring single regions. If any of these regions cover a user determined percentage of the sampled anchors in the transcript, a double region is not searched for. If there is no such single region however, our method searches for high-scoring double regions. If there is no high-quality single or double region, our method does not report a region and considers the given transcript sequence as not represented within the genome with high similarity.
As the final step of this stage, regions that are overlapping or close to each other are combined into single regions (these include fusion regions that are relatively close to each other). In addition, the region boundaries are extended allowing flanking sequence from the beginning/ending anchors that are not represented in the set of mappings. The resulting genomic intervals are passed on to the second stage of Dissect which finds the optimal fragment chain with structural alterations between the transcript and inferred regions.
Fragment set construction and chaining. In the fragment set construction stage, we construct a set of fragments shared between the transcript and the genomic region(s) inferred in the previous stage. For that purpose we modified mrsFAST alignment method (Hach et al., 2010), to identify ‘seeds’ (of a user specified length—which can be overlapping) in the transcript and maps them to both strands of the genomic sequence. After obtaining all possible seed mappings, the modified mrsFAST extends each fragment on both ends under certain extension constraints. These constraints are defined in the form of thresholds that limit total number of errors, number of consecutive errors that can appear in the fragment and minimum sequence similarity required for each k-mer of the fragment. After the fragment set is constructed, we employ the overlapping fragment chaining algorithm once using forward and a second time using reverse splicing signals (GT-AG and CT-AC). In the case of a single region inferred in the previous stage, the chaining solution will only consist of G/G′ sequences. If a pair of regions are inferred, the chaining solution will consider all G/G′ and S/S′ sequences allowing fusion transitions.
A key difference between our original formulation and the chaining method used in Dissect is the use of splice signal scores. Even though splice signal scores were omitted in our original overlapping chaining formulation, in practice it would be useful to have a two layered chaining/post-processing approach that constructs the chain that does not attempt to determine the exact overlap split positions, but performs a more accurate overlap analysis together with splice signals as a post-processing step after the optimal chain is obtained. The optimality condition here is also modified in the sense that splice signal score is incorporated when two disjoint fragments are chained together, yet omitted for overlapping fragments.
At the end of fragment chaining step, a tentative chain is obtained that represents the general structure of the alignment, yet overlap split positions are not exactly specified in the resulting chain. Dissect detects the exact split position through a post-refinement method described below.
Post-refinement of the fragment chain. In the post-refinement stage, we adjust the boundaries of fragment pairs that potentially contain minor misalignments due to the limitations introduced in the fragment construction step. To resolve these, we implemented several post-refinement steps that (i) combine fragments which are separated by a single-nucleotide indel or a mismatch in their alignment; (ii) classify and modify short overlaps in the genome; (iii) fill in short gaps in the transcript between adjacent fragments in the chain; and (iv) find optimal split position for overlapping fragment pairs.
In the case of two adjacent fragments being separated by an indel or a mismatching transcript-genome nucleotide, the two fragments are simply combined into a single fragment that contain an error in between. Clearly, if the fragments are separated by an indel, the combined fragment will also contain indels. Even though this is against the original fragment construction constraints; as a post-processing method, it only affects the resulting chain and not the chaining formulation.
When there is an adjacent fragment pair with a short overlapping region in the genome, Dissect does not directly report the overlap region as a duplication as this can also be an inserted region that displays sequence similarity to one of the flanking sequences. Since it is difficult to differentiate between the two, we allow the user to define lower and upper thresholds on length. Overlaps that are shorter than the lower threshold are treated as insertions, whereas overlaps that are longer than the upper threshold are reported as regular duplicated regions in the transcript. The overlap regions that fall in between are reported as an ambiguous insertion/duplication region should be further analyzed by the use of gene annotations.
We have two additional refinement methods that require efficient implementations: (i) If two adjacent fragments have a gap in between them in the transcript, the formation might indicate a novel insertion of the size of the gap. (ii) Alternatively, the region of the gap might belong to one of the exons represented by adjacent fragments (or both), yet may have relatively low similarity to the corresponding genomic region. To test the latter case, we perform a double-sided semi-global alignment on the flanking sequences of the fragment pair in the genome and the gap sequence. This alignment scheme aims to optimize the sum of the semi-global (overlap-detection) alignment scores on each side of a fixed split position in the transcript gap region. The method we apply is analogous to the Sandwich DP method proposed for GMAP spliced alignment algorithm (Wu and Watanabe, 2005). The key difference in our application is the consideration of alignment with various structural alterations and their effect of fragment directionality. Within the alignment table, we also mark the GT-AG/CT-AC splice signals and take them into account for the computation of the new transition penalty between the updated fragments. This allows a fair alignment score comparison between the chains that go through this post-processing step and the chains that do not. Note that this gap refinement scheme has a running time of O(l2) per refinement (l is the length of the transcript gap).
As mentioned in the description of the fragment chaining stage; the exact overlap split position for overlapping fragments is not determined during the execution of the dynamic programming method but is left for a more detailed analysis in the post-refinement stage. In this stage, Dissect searches for the optimal overlap resolution according to an extended version of the overlap resolution scheme described in the previous section. In this version of the overlap resolution scheme, we combine splice signal scores with the original accurate overlap resolution that considers mismatch retention and updated genomic distance for the overlap penalty. Even though this extension was not feasible during fragment chaining, if the overlaps are resolved in an iterative fashion from the fragment pairs at the beginning of the chain towards the end, the number of splice sites that need to be checked in the genome stays within O(N). Incorporating splice site scores in this manner also allows us to have a fair comparison basis when comparing the relative scores of the fragment chains for various regions inferred in the first stage.
When two adjacent overlapping fragments have near perfect sequence similarity and no splice signals to identify the exact splicing position, there can be multiple overlap split positions with equal updated fragment scores. In such cases, our aligner reports the earliest split in the transcript but also provides an output field indicating equivalent split positions. This additional output can be used to reconstruct all optimal overlap splitting selections.
3 RESULTS
We first report the performance of Dissect on simulation datasets derived from NCBI RefSeq transcript sequences and Known Gene gene structure database (Hsu et al., 2006), which are subjected to nucleotide-level substitution/indel noise with varying frequencies, novel oligonucleotide sequence insertions, and structural alterations at different length distributions, including exon duplications, inversions, rearrangements and transcript–transcript fusions. To demonstrate the performance of Dissect on real human transcriptome data, we report on an RNA-Seq dataset comprising 50 bp reads from the prostate cancer cell line C4-2, assembled through Trans-AbySS transcriptome assembler (Birol et al., 2009).
Wild-type transcripts with novel insertions and nucleotide-level alterations. We first evaluate Dissect's false discovery rate through the use of a dataset comprised of wild-type transcripts. For that, we used NCBI RefSeq mRNA annotation dataset including the whole mouse transcriptome (build of July 18, 2011). This annotation dataset is (presumably) composed of wild-type transcripts that do not contain structural alterations. We evaluated Dissect's false event discovery rate by aligning all transcripts from this dataset to the mouse reference genome (build mm9). Since most of these sequences have very close matches to genes in the mouse genome, we also used this dataset to evaluate the accuracy of Dissect alignments (obtained through fragment chaining) at nucleotide-level resolution.
After the removal of poly-A tails, the entire dataset containing 28 060 RefSeq sequences of average length of 2848 nucleotides, were aligned to the mouse reference genome using the default parameters of Dissect. We report on the highest scoring alignment for each transcript sequence. Among them, 27 922 (99.51%) did not contain structural alterations, 49 (0.17%) were reported to contain a structural event and 38 (0.14%) were identified to contain a short ambiguous insertion or duplication event. The remaining 51 sequences had no high-similarity alignments. On a standard single-core processor, aligning the entire dataset of 28 060 sequences with Dissect took <80 min.
Next, we aimed to observe how Dissect's false event discovery rate varies as a function of sequence divergence (between the transcript and the genome)—and thus sequencing error rate. For that, we modified the original RefSeq sequences by adding nucleotide-level substitution/indel errors. These errors were added independently at random in each position of a transcript sequence: based on a recent study on error rates in Illumina sequencing (Minoche et al., 2011), single-nucleotide indel to substitution error ratio was set to 1/150 and insertion to deletion ratio was set to 1/10. When the sequencing error rate was set to 1%, 27 917 (99.4%) sequences out of 28 060 were aligned as wild-type transcripts without no alterations. For 52 sequences, a high-similarity alignment was not reported. Structural alterations were reported for only 57 transcripts and short ambiguous duplication/insertions were detected for 34 transcripts. When the sequencing error rate was set to 4%, the number of transcripts with a wild-type transcript alignment was reduced to 27 756 (98.9%). Among the remaining 304 sequences, 188 did not produce a high-similarity alignment.
Finally, we used the latest RefSeq mRNA annotation dataset for whole human transcriptome (build of July 18, 2011) for the purpose of evaluating Dissect's false event discovery rate in the presence of short-to-medium size novel insertions.
To simulate a realistic sample of novel human genome insertions, we sampled substrings of varying length from the set of insertion sequences reported in a novel insertion characterization study (Kidd et al., 2010), and inserted them to the transcript sequences at random exon breakpoints. Our dataset included 33 460 sequences that were devoid of structural alterations and had nucleotide-level accurate alignments (after the removal of poly-A tails). We equally partitioned this dataset into four subsets, each subject to a specific insertion size. To obtain realistic novel insertion sequences, we used 2363 known novel insertion sequences (Kidd et al., 2010), from which we randomly picked a position in each sequence and extracted the sequence of the required length.
Table 1 depicts the false event detection rate for novel insertions shorter than 35 nucleotides. The higher rate of false positives for longer insertion sizes is caused by Dissect's high sensitivity to sequence similarity. Since the insertion sequences are obtained from a real novel insertion study for the human genome, there might be sequences highly similar to the insertion nearby the aligned gene loci, which increase the risk of identifying false rearrangements.
Table 1.
Alignment results of Dissect for the simulated wild-type transcriptome dataset with novel insertions
| Insertion length | Total | WT | All events | A. D/I | N.A. |
|---|---|---|---|---|---|
| 6–20 bases | 8365 | 8335 | 12 | 16 | 2 |
| 21–35 bases | 8365 | 8284 | 52 | 23 | 6 |
| 36–50 bases | 8365 | 8223 | 106 | 24 | 13 |
| 51–65 bases | 8365 | 8117 | 204 | 20 | 24 |
Rows represent the length interval of the novel insertion distributions (e.g. insertions reported in the first row are uniformly distributed between 6 and 20 nucleotides). Columns indicate the output labels of Dissect: All events column represents the total number of transcripts Dissect has identified as a structural alteration A. D/I column represents the alignments that contain a short ambiguous interval that cannot be verified with certainty as an insertion or a duplication, and N.A. column indicates the number of transcript sequences for which Dissect did not return a valid high-similarity alignment.
Simulated transcriptional events. To estimate the sensitivity of Dissect, we initially prepared wild-type transcriptome datasets without any structural alterations using the Known Genes mouse gene structure annotation database (Hsu et al., 2006) and modified extracted wild-type transcript sequences according to various structural alteration scenarios. In this step, any transcript sequence shorter than 50 bp is removed, since structural modifications in such short transcript sequences often prevent reliable mapping of the anchor sequences used by Dissect.
The thirteen simulations described below aim to emulate the aberrant formations that can occur in transcripts due to structural alterations. These simulations involve: (i) tandem duplications of the full transcript; (ii) tandem duplication of the longest exon; (iii) tandem duplication of the shortest exon; (iv) internal-inversion of the longest exon; (v) internal-inversion of the shortest exon; (vi) suffix-inversion with a breakpoint close to middle (prefix to suffix ratio: 36–65%); (vii) suffix-inversion with a breakpoint close to the beginning/end, (prefix to suffix ratio: 16–35% or 66–85%); (viii) rearrangement of the full transcript sequence from a particular split position; (ix) rearrangement of adjacent exons; (x) rearrangement of non-adjacent exons; (xi) well-balanced fusions (shorter fused sequence is ≥%60 of the longer one); (xii) moderately-balanced fusions (30%≤ short to long ratio <60%); and (xiii) imbalanced fusion (short to long ratio: <%30). The distribution of transcript alignments according to these event type labels for various event simulations are given in Table 2.
Table 2.
The number of structural alterations detected by Dissect for the simulation datasets
| Tot. | Tot-E. | Fusion | Inv. | F. Dup. | F. Rea. | |
|---|---|---|---|---|---|---|
| Exp. 1 | 5234 | 5099 | 1 | 5 | 5092 | 1 |
| Exp. 2 | 5234 | 5172 | 0 | 1 | 5171 | 0 |
| Exp. 3 | 5234 | 5093 | 0 | 0 | 5093 | 0 |
| Exp. 4 | 4788 | 4762 | 0 | 4762 | 0 | 0 |
| Exp. 5 | 4788 | 4331 | 0 | 4331 | 0 | 0 |
| Exp. 6 | 3188 | 3125 | 0 | 3125 | 0 | 0 |
| Exp. 7 | 4654 | 4501 | 0 | 4501 | 0 | 0 |
| Exp. 8 | 4788 | 4512 | 2 | 8 | 3 | 4499 |
| Exp. 9 | 4788 | 4623 | 0 | 8 | 2 | 4613 |
| Exp. 10 | 4316 | 4255 | 0 | 14 | 4 | 4237 |
| Exp. 11 | 1312 | 1237 | 1232 | 5 | 0 | 0 |
| Exp. 12 | 1558 | 1433 | 1433 | 0 | 0 | 0 |
| Exp. 13 | 2363 | 562 | 562 | 0 | 0 | 0 |
Tot., total number of transcript sequences; Tot-E., total number of discovered structural event containing transcripts; Fusion, total number of fusions; Inv., inversion events including inverted duplications, inverted rearrangements, in-place inversions, and suffix-inversions; F. Dup., forward duplications; F. Rea., forward rearrangement events.
Transcriptomic structural alterations in prostate cancer cell line C4-2. We applied Dissect to high-coverage 50 bp RNA-Seq read data from human prostate cancer cell line C4-2. The reads were assembled using short-read transcriptome assembler Trans-Abyss (Birol et al., 2009) version 1.2.0, using k-mer sizes of 26 and 49—the minimum overlap length between two reads to be combined in a contig. For two contigs to be merged we required 10 pair-end mappings between the contigs.
Among a total of 576 381 contigs assembled, Dissect did not return a high-quality alignment for 167 187 of them. Among the remaining contigs, 391 293 of them were aligned with no structural alterations. In 4 309 contigs, Dissect detected an ambiguous short insertion/duplication region. In 13 583 contigs, Dissect discovered a structural alteration: 1 044 fusions, 1 331 duplications, 555 rearrangements and 10 653 inversion events. In total, 10 992 of 12 539 non-fusion event contigs displayed ≥90% overlap with a single-gene annotated in HG18 Known Genes dataset. Among 10 653 inversions, 69 are multiple breakpoint inversions and another 79 contain combined duplication/rearrangement events. Within the remaining 10 505 single-breakpoint suffix-inversion events, 2600 contain overlapping regions within the two strands and 7905 have non-overlapping suffix/prefix formation.
We compared Dissect alignment results on contigs from C4-2 with long range PCR validated fusions reported by the fusion discovery tool Comrad (McPherson et al., 2011a) on the same dataset. Among eight validated gene fusion pairs, RERE-PIK3CD, HPR-MRPS10, CCDC43-YBX2, TFDP1-GRK1, BMPR2-FAM117B, GPS2-MPP2, MIPOL1-DGKB and ITPKC-PPFIA3, Dissect correctly identified six of them: RERE-PIK3CD, HPR-MRPS10, CCDC43-YBX2, BMPR2-FAM117B, MIPOL1-DGKB and ITPKC-PPFIA3. Note that because the two genes involved in the fusion BMPR2-FAM117B are in close genomic proximity, Dissect returned a rearrangement, rather than a fusion as per it is set to do. Among the two fusions Dissect could not identify, there was no contig returned by the assembler that spanned the TFDP1-GRK1 fusion breakpoint and the assembled contig spanning the GPS2-MPP2 fusion breakpoint was highly imbalanced ( 10%:90%). Note that for three out of these four genes, GPS2, MPP2 and TFDP1, Dissect also reported wild-type alignments, without any evidence for a fusion event.
To better understand and differentiate the limitations of Dissect from that of the assembly process, we extracted breakpoint sequences of length 200 bp for each of the eight gene fusion events given above. Dissect produced alignments that correctly capture the fusion breakpoint for each of the eight fusions. Six of these breakpoint sequences were reported as straightforward fusions. Among the remaining two breakpoints transitions, BMPR2-FAM117B was identified as a rearrangement event and TFDP1-GRK was identified as a wild-type alignment due to close proximity of the fused genes: a comparison with gene annotation uncovered the inter-genic structure of the breakpoints discovered.
4 CONCLUSION
We introduce novel algorithmic formulations for the problem of aligning transcripts to a genome under structural alterations such as duplications, inversions, rearrangements and fusions.
Our first formulation involves nucleotide-level alignment that can detect structural alterations by a single-unified dynamic programming approach. The fastest algorithms we developed for this formulation require O(NM log(M)) time for convex genomic gap penalties and O(MN) time for simple convex (including logarithmic) gap penalties (M and N correspond to the lengths of transcript and genome sequences, respectively).
Our second formulation allows a faster but lower-sensitivity solution for a whole genome alignment setting. Given a set of shared fragments between the transcript and the genome, we show how to obtain an optimal chain of fragments in O(K2) time for disjoint or overlapping fragments (K being the total number of fragments).
We also present a novel computational tool, Dissect, which implements the fragment chaining formulation described above. Dissect achieves high sensitivity and specificity in identifying structural alterations in simulated datasets, as well as in uncovering gene fusions in a prostate cancer cell line.
ACKNOWLEDGEMENTS
We would like to thank Fereydoun Hormozdiari, Andrew McPherson and Trans-Abyss development team at Michael Smith Genome Sciences Centre for helpful discussions and Iman Hajirasouliha for proofreading this article.
Funding: PIMS Mathematical Biology Fellowship to D.Y.; NSERC and SFU-CTEF supported BCID project to S.C.S (in part). Canadian Cancer Foundation to S.C.S. and C.C.C (in part).
Conflict of Interest: none declared.
REFERENCES
- Akiva P., et al. Transcription-mediated gene fusion in the human genome. Genome Res. 2006;16:30–36. doi: 10.1101/gr.4137606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., et al. Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson A.M., Staley J.P. Long-distance splicing. Proc. Natl. Acad. Sci. USA. 2008;105:6793–6794. doi: 10.1073/pnas.0803068105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmann Y.W., et al. A novel bioinformatics pipeline for identification and characterization of fusion transcripts in breast cancer and normal cell lines. Nucleic Acids Res. 2011;39:e100. doi: 10.1093/nar/gkr362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäsecke J., et al. Leukemia- and lymphoma-associated genetic aberrations in healthy individuals. Ann. Hematol. 2002;81:64–75. doi: 10.1007/s00277-002-0427-x. [DOI] [PubMed] [Google Scholar]
- Birol I., et al. De novo transcriptome assembly with abyss. Bioinformatics. 2009;25:2872–2877. doi: 10.1093/bioinformatics/btp367. [DOI] [PubMed] [Google Scholar]
- Brassesco M.S. Leukemia/lymphoma-associated gene fusions in normal individuals. Genet. Mol. Res. 2008;7:782–790. doi: 10.4238/vol7-3gmr486. [DOI] [PubMed] [Google Scholar]
- Brudno M., et al. Glocal alignment: finding rearrangements during alignment. Bioinformatics. 2003;19(Suppl. 1):i54–i62. doi: 10.1093/bioinformatics/btg1005. [DOI] [PubMed] [Google Scholar]
- Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Caudevilla C., et al. Natural trans-splicing in carnitine octanoyltransferase pre-mRNAs in rat liver. Proc. Natl. Acad. Sci. USA. 1998;95:12185–12190. doi: 10.1073/pnas.95.21.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormode G., et al. Proceedings of the Eleventh Annual ACM-SIAM Symposium on Discrete Algorithms (SODA) USA: 2000. Communication complexity of document exchange; pp. 197–206. [Google Scholar]
- De Braekeleer E., et al. Abl1 fusion genes in hematological malignancies: a review. Eur. J. Haematol. 2011;86:361–371. doi: 10.1111/j.1600-0609.2011.01586.x. [DOI] [PubMed] [Google Scholar]
- Dorrance A.M., et al. The Mll partial tandem duplication: differential, tissue-specific activity in the presence or absence of the wild-type allele. Blood. 2008;112:2508–2511. doi: 10.1182/blood-2008-01-134338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergün F., et al. Proceedings of Foundations of Software Technology and Theoretical Computer Science (FSTTCS) India; 2003. Comparing sequences with segment rearrangements; pp. 183–194. [Google Scholar]
- Frantz S.A., et al. Exon repetition in mRNA. Proc. Natl. Acad. Sci. USA. 1999;96:5400–5405. doi: 10.1073/pnas.96.10.5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galil Z., Giancarlo R. Speeding up dynamic programming with applications to molecular biology. Theor. Comput. Sci. 1989;64:107–118. [Google Scholar]
- Ge H., et al. Fusionmap: detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics. 2011;27:1922–1928. doi: 10.1093/bioinformatics/btr310. [DOI] [PubMed] [Google Scholar]
- Gingeras T.R. Implications of chimaeric non-co-linear transcripts. Nature. 2009;461:206–211. doi: 10.1038/nature08452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hach F., et al. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods. 2010;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herai R.H., Yamagishi M.E.B. Detection of human interchromosomal trans-splicing in sequence databanks. Brief Bioinform. 2010;11:198–209. doi: 10.1093/bib/bbp041. [DOI] [PubMed] [Google Scholar]
- Hirano M., Noda T. Genomic organization of the mouse MSH4 gene producing bicistronic, chimeric and antisense mRNA. Gene. 2004;342:165–177. doi: 10.1016/j.gene.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Horiuchi T., Aigaki T. Alternative trans-splicing: a novel mode of pre-mRNA processing. Biol. Cell. 2006;98:135–140. doi: 10.1042/BC20050002. [DOI] [PubMed] [Google Scholar]
- Horiuchi T., et al. Alternative trans-splicing of constant and variable exons of a drosophila axon guidance gene, lola. Genes. Dev. 2003;17:2496–2501. doi: 10.1101/gad.1137303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F., et al. The UCSC known genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- Inaki K., et al. Transcriptional consequences of genomic structural aberrations in breast cancer. Genome Res. 2011;21:676–687. doi: 10.1101/gr.113225.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan K., et al. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proc. Natl. Acad. Sci. USA. 2011;108:9172–9177. doi: 10.1073/pnas.1100489108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J. Blat–the blast-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd J.M., et al. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat. Methods. 2010;7:365–371. doi: 10.1038/nmeth.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrador M., et al. Protein encoding by both DNA strands. Nature. 2001;409:1000. doi: 10.1038/35059000. [DOI] [PubMed] [Google Scholar]
- Levin J.Z., et al. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol. 2009;10:R115. doi: 10.1186/gb-2009-10-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., et al. Fusionhunter: identifying fusion transcripts in cancer using paired-end RNA-seq. Bioinformatics. 2011;27:1708–1710. doi: 10.1093/bioinformatics/btr265. [DOI] [PubMed] [Google Scholar]
- McPherson A., et al. Comrad: detection of expressed rearrangements by integrated analysis of RNA-seq and low coverage genome sequence data. Bioinformatics. 2011a;27:1481–1488. doi: 10.1093/bioinformatics/btr184. [DOI] [PubMed] [Google Scholar]
- McPherson A., et al. Defuse: an algorithm for gene fusion discovery in tumor RNA-seq data. PLoS Comput. Biol. 2011b;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson A., et al. Proceedings of Research in Computational Molecular Biology (RECOMB) Spain: 2012. Discovery of complex genomic rearrangements in cancer using high-throughput sequencing; pp. 181–182. [Google Scholar]
- Miller W., Myers E.W. Sequence comparison with concave weighting functions. Bull. Math. Biol. 1988;50:97–120. doi: 10.1007/BF02459948. [DOI] [PubMed] [Google Scholar]
- Minoche A.E., et al. Evaluation of genomic high-throughput sequencing data generated on illumina HISeq and genome analyzer systems. Genome Biol. 2011;12:R112. doi: 10.1186/gb-2011-12-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott R. Est_genome: a program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl. Biosci. 1997;13:477–478. doi: 10.1093/bioinformatics/13.4.477. [DOI] [PubMed] [Google Scholar]
- Nacu S., et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genomics. 2011;4:11. doi: 10.1186/1755-8794-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman D.S., et al. Slc45a3-elk4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009;69:2734–2738. doi: 10.1158/0008-5472.CAN-08-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sboner A., et al. Fusionseq: a modular framework for finding gene fusions by analyzing paired-end RNA-sequencing data. Genome Biol. 2010;11:R104. doi: 10.1186/gb-2010-11-10-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater G.S.C., Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J. Hematol. Oncol. 2011;4:13. doi: 10.1186/1756-8722-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.D., Watanabe C.K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]