

Abstract
Motivation: The number of completely sequenced genomes is continuously rising, allowing for comparative analyses of genomic variation. Such analyses are often based on whole-genome alignments to elucidate structural differences arising from insertions, deletions or from rearrangement events. Computational tools that can visualize genome alignments in a meaningful manner are needed to help researchers gain new insights into the underlying data. Such visualizations typically are either realized in a linear fashion as in genome browsers or by using a circular approach, where relationships between genomic regions are indicated by arcs. Both methods allow for the integration of additional information such as experimental data or annotations. However, providing a visualization that still allows for a quick and comprehensive interpretation of all important genomic variations together with various supplemental data, which may be highly heterogeneous, remains a challenge.
Results: Here, we present two complementary approaches to tackle this problem. First, we propose the SuperGenome concept for the computation of a common coordinate system for all genomes in a multiple alignment. This coordinate system allows for the consistent placement of genome annotations in the presence of insertions, deletions and rearrangements. Second, we present the GenomeRing visualization that, based on the SuperGenome, creates an interactive overview visualization of the multiple genome alignment in a circular layout. We demonstrate our methods by applying them to an alignment of Campylobacter jejuni strains for the discovery of genomic islands as well as to an alignment of Helicobacter pylori, which we visualize in combination with gene expression data.
Availability: GenomeRing and example data is available at http://it.inf.uni-tuebingen.de/software/genomering/
Contact: kay.nieselt@uni-tuebingen.de
1 INTRODUCTION
Advances in high-throughput sequencing technologies have dramatically increased the speed at which genomes are sequenced (Bennet, 2004; Droege and Hill, 2008; Porreca et al., 2006; Eid et al., 2009; Rothberg et al., 2011). This led to the establishment of large-scale genome sequencing projects such as the 1000 genomes project (Durbin et al., 2010), the 1001 genomes project in Arabidopsis thaliana (Weigel and Mott, 2009), the 10K genomes project (Haussler et al., 2009), which aims at sequencing vertebrate genomes, the insect genomes initiative i5k (Robinson et al., 2011) as well as many projects sequencing prokaryotic species, often at the level of individual strains. The genome sequencing projects are conducted with different long-term goals. While the 10K and i5k initiatives aim at collecting genomes across a large part of the tree of vertebrates and insects, respectively, the 1000 and 1001 genome projects focus on genetic variation within one species. Regarding the prokaryotic species, genome projects often focus on this latter aspect, comparing different strains of bacteria with the goal of understanding the genetic basis of pathogenicity and drug resistance, the adaptability to environments, the extent of horizontal gene transfer as well as to elucidate the architectural diversity of bacterial genomes.
Parallel to the increase in genomic data with the development of new sequencing technologies, powerful visualization tools have been developed and continue being developed. An excellent review on methods as well as the challenges of visualizing genomes has recently been published by Nielsen et al. (2010). Generally, one can distinguish two approaches to the visualization of genomes: A single genome is visualized (often in comparison to a reference genome), or multiple genomes are compared. In the first case, genome browsers are typically utilized, which represent the genome linearly and can display multiple variables in parallel ‘tracks’ aligned to the genomic coordinates. Such tracks can contain annotation, experimental, or statistical data.
For the comparison of multiple genomes, the same linear approach as applied by genome browsers can be used. A typical example is the viewer integrated into Mauve (Darling et al., 2004). However, large changes such as inversions can quickly lead to visual clutter, and it can be difficult to deduce similarities and differences between genomes from the visualization. Some genome viewers employ a circular approach to visualize one genome with annotation and experimental data, or to present an alignment of several genomes. Circos (Krzywinski et al., 2009) is one of the most often used circular genome visualization tools. It displays genomic data as a circular plot, in which the relationships of genomic elements are displayed using arcs. It is, therefore, particularly useful for visualizing variation within one genome, but it can also be applied to visualize the relationship of several genomes. While Circos without doubt produces aesthetically very attractive figures, it has the major disadvantage of only presenting a static, non-interactive view of the data.
Further circular genome viewers are MEDEA (Broad Institute, 2009) and MizBee (Meyer et al., 2009). Another circular approach is taken by the BLAST Ring Image Generator (BRIG) that visualizes multiple prokaryotic genomes (Alikhan et al., 2011). Each genome is compared to a reference genome using BLAST. The hits between each genome and the reference are then visualized as concentric rings using different colors for each genome. Additional rings, representing meta information, such as GC content, can be added. Its main focus is to accompany sequencing projects, in particular to handle and visualize assembly data.
Though a number of excellent visualization and analysis tools are already available for researchers working with multiple genome sequences [see Nielsen et al. (2010) for a review], an important obstacle remains to be overcome. While genomes in studies of strain diversity are usually highly similar, sharing long stretches of conserved sequence, they still show differences due to larger events such as inversions, translocations and insertions/deletions. In the context of such differences, researchers are faced with the problem of mapping existing annotations as well as experimental data to each of the aligned genomes in a consistent manner. The coordinate transformations necessary to visualize annotations in the context of alignments are implemented implicitly in programs that deal with the analysis and visualization of alignments, such as Mauve, tools in the VISTA suite (Frazer et al., 2004), or others. However, as the basis for our proposed visualization, we need an approach that allows us to explicitly generate a joint coordinate system that can be used to consistently specify coordinates of annotations (and also of experimental data such as mapped sequencing reads or RNAseq expression graphs), which can be used independently from a specific analysis or visualization software. In addition, many existing methods require the specification of one ‘reference’ genome that is afforded a special status (e.g. coordinates within insertions with regard to that reference can not be expressed), which we consider an artifact of the method rather than a choice based on biological facts.
Here, we present two complementary approaches to solve this problem. First, we present the SuperGenome algorithm, which computes a common coordinate system for all genomes in a multiple alignment. Using this coordinate system, genome annotations can be placed consistently in the presence of insertions, deletions and rearrangements between the different genomes. Second, we present the GenomeRing visualization which, based on the SuperGenome coordinate system, visualizes the multiple genome alignment in a circular layout. Its main advantages are a much more appealing and clearer visual presentation of deletion, insertion and rearrangement events compared to linear alignment viewers, as well as more interactivity than existing circular visualizations. We designed GenomeRing to be a fast, interactive overview tool for alignments of several (ideally less than 10) genomes with high similarity (less than 25 genomic events for optimal visual clarity). The general idea and proof-of-concept visualizations of our methods were submitted to the Illumina iDEA challenge 2011, where our submission was selected as the most creative algorithm.
We have now integrated the GenomeRing visualization with Mayday (Battke et al., 2010), our visual analysis platform for ‘omics’ data. As a result, GenomeRing can be linked with all other visualizations offered by Mayday, including a traditional, linear genome browser.
2 METHODS
2.1 SuperGenome construction
The construction of the SuperGenome is based on whole-genome alignments. In the case of genomic rearrangements, these can be viewed as a collection of local alignments, also called blocks. In this context, we define our concept of a ‘SuperGenome’ as a representation of the multiple sequence alignment with an additional common coordinate system, and mappings between this coordinate system and the aligned sequences.
To achieve this, we process the set of blocks as follows: for each block, the alignment information is used to calculate a bidirectional mapping between the coordinate system of the SuperGenome and the original coordinates of each input genome contained in the block. The SuperGenome coordinate system is based on the alignment coordinates of all concatenated blocks, whose ordering is derived from the reference genome of the alignment. Note that the chosen order of the blocks is not crucial for the functionality of the SuperGenome concept.
In comparison to the traditional alignment concept, which defines pairwise mappings between the coordinates of the involved sequences, the SuperGenome has the advantage that independent alignment blocks are combined into a global coordinate system. This makes it also possible to assign coordinates to unaligned regions.
For the generation of whole-genome alignments for prokaryotic organisms, we decided to use the progressiveMauve algorithm (Darling et al., 2010) of the genome alignment software Mauve [Darling et al. (2004); version 2.3.1], since besides insertions and deletions, Mauve is also able to discover genomic rearrangements, i.e. translocations and inversions. If such events occur, the alignment is provided as a set of blocks, where each block represents a region in two or more genomes that can be collinearly aligned.
In GenomeRing, however, we do not only want to visualize rearrangements but also large-scale insertions and deletions. This requires further processing. For this, each aligned sequence in a block is scanned for gaps that are longer than a user-defined threshold. The start and end coordinates of these gaps are stored as break points. Using the break points of all sequences, the block is split up into subblocks, which represent insertions or deletions in one or more of the aligned genomes. Subblocks that are smaller than a user-defined threshold are discarded and neighboring subblocks are merged if their conservation pattern, i.e. the set of contained genomes, is the same.
The set of all remaining subblocks is the basis for the GenomeRing visualization. By adjusting the parameter for the minimal block length, the user can choose whether only large events will be displayed, which is especially useful for more diverse genomes, or whether smaller insertions and deletions should also be visualized.
2.2 Layout of GenomeRing
To visualize a SuperGenome alignment, we created GenomeRing, an interactive circular visualization and integrated it into our visual analytics software Mayday (Battke et al., 2010).
The blocks computed by the SuperGenome algorithm give rise to circle segments sized according to their length and ordered as defined by the SuperGenome ordering. Each block results in two segments of identical angular extent, one on the outer (‘forward’) ring, one on the inner (‘backward’) ring (Fig. 1). Assume the alignment contains n genomes. We split each of the two rings into n ‘lanes’, each of which is assigned to exactly one genome.
Fig. 1.
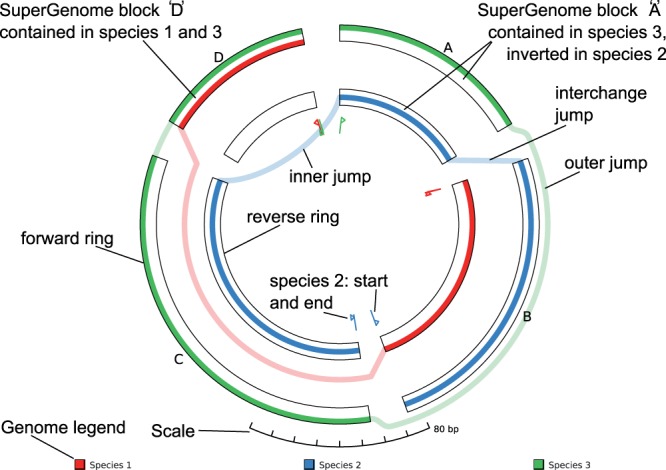
GenomeRing and its view elements. The rings of the SuperGenome are laid out in two concentric circles representing the forward resp. the reverse direction. Each genome is represented by a colored path connecting the blocks according to their order in the genome. Small flags indicate each genome's start and end position. Deletions, inversions and translocations result in jumps, either outside of the outer or inside of the inner circle (representing deletions, translocations), or between circles (representing inversions, inverted translocations). The synthetic example shown here comprises three genomes: The genome of species ‘1’ contains the blocks D and B (inverted); Species ‘2’ contains B, A (inverted), and C (inverted); Species ‘3’ contains A, C and D
With these preliminaries, we can visualize genomes as follows: Each aligned genome G is a concatenation of blocks. This is visualized as a directed path connecting the SuperGenome's blocks in the order that they have within G. The path is drawn with a unique, distinct color assigned to G, based on the ‘quantitative’ color scheme suggested by ColorBrewer (Harrower and Brewer, 2003). Within each block, the path uses the lane assigned to G. Blocks that are not in G are not part of the path. If a block appears in its native direction in the respective genome, the path includes the respective segment on the outer ring. If the block is inverted in G, the segment on the inner ring is used. The start and end of G are represented by small flags drawn inside of the inner ring, which also indicate where the path is heading (from the start) and where it is coming from (towards the end).
Several types of connections have to be visualized: Direct connections exist if two consecutive SuperGenome blocks are also consecutive in G and appear in the same direction. The genome's path simply connects the two consecutive blocks, staying on the same radius. Second, a deletion in G results in a jump connection. This can either be an outer jump (both blocks are on the outer ring), or an inner jump (both blocks are on the inner ring), which are visualized by introducing a curved connection outside the outer ring, or inside the inner ring, respectively. Third, inversions lead to interchange connections that link blocks on the outer circle with blocks on the inner circle, or vice versa. Interchange connections can also jump over deleted blocks, as described above.
The visual clarity of the presentation is greatly influenced by two factors: The total number of jumps and interchange connections, and the number of jump edges that overlay each other.
First, jump and interchange connections should be avoided because these lead to visual clutter. To minimize the number of indirect block connections, as well as to highlight different aspects of the alignment, users can reorder the SuperGenome blocks in a number of ways, either based on their ‘native’ order in one of the aligned genomes, or reflecting the order chosen by the SuperGenome algorithm (see Section 2.1), or by using one of the block sorting algorithms (described in Section 2.3). Finding an optimal block order is of high importance, as exemplified by Figure 2 which shows the same alignment as Figure 1, but requires only two short connections instead of three long arcs and one short connection.
Fig. 2.
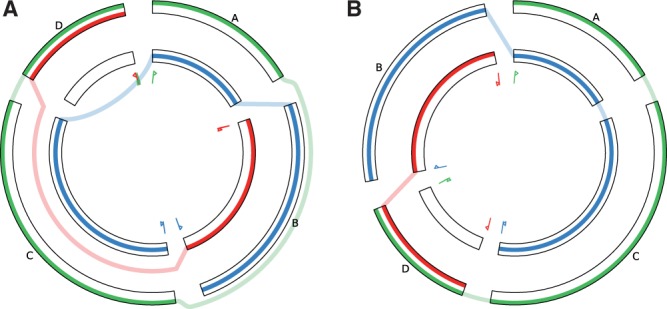
The influence of block sorting. The alignment of (A) is shown sorted according to the green genome (B). This optimal sort order reduces the number of jumps from four to two. The long jumps over a total of three blocks which added up to over 300○ in length have become unnecessary
Second, we use different rings to differentiate between blocks present in forward and backward direction, respectively, in each genome. Thus the direction in which a genome's path traverses each one of the SuperGenome blocks can be freely chosen by the layout algorithm, instead of always traversing segments in a clockwise (or counter-clockwise) direction depending on their direction of incorporation in the genome. This allows us to reduce the number of jump edges as well as their length (in degrees) by finding sets of consecutive blocks that have the same direction in a genome G and, for each such set, choosing to traverse all consecutive blocks in the direction which maximizes the number of direct connections between sets of consecutive blocks. This leads to a much more appealing visualization as it allows limiting the maximal angular length of jump edges to <180○.
Finally, our path layout algorithm minimizes the overlap between jump edges by placing each edge such that the length of overlap (in degrees) is minimal.
In addition to the SuperGenome blocks and the genomes' paths, the view contains a scale indicating the number of bases displayed per degree, and a legend which maps each color to the respective genome's identifier. Blocks in the SuperGenome are either labeled numerically by the SuperGenome algorithm, or with user-defined names [e.g. with the name of a well-known pathogenicity island (Hacker et al., 1990)].
Interactivity is an important factor to allow users to understand the presented view, and to create figures for dissemination to collaborators as well as for publication. The GenomeRing view allows for free rotation, zooming and panning using direct mouse interaction. When zooming in, additional detail, such as annotated genes, can be presented (see Section 2.4). Individual genomes can be hidden from view and the automatically assigned color can be changed. The SuperGenome block labels can also be hidden. Clicking on any position inside a lane of a block shows a tooltip window with a description of the position in terms of its coordinate (base pairs from the start), the size of the block, the index of the block and the offset in base pairs from the block's start. All of these numbers are given both for the SuperGenome coordinate system and for the coordinate system of the respective genome of the lane.
Three fundamental parameters of the view can be interactively adjusted: The radial spacing between jump connections, the size of the gaps between SuperGenome blocks and the width of the paths representing the genomes. Adjusting these, users can create very different visualizations of the same alignment. For example, reducing the inter-block gaps to zero presents a view focusing on the presence and absence of blocks in the particular genomes as well as which genomes share each block. Increasing the inter-block gap until each block is drawn with zero angular extent, on the other hand, creates a view highlighting shared evolutionary events, such as inversions and translocations. Note that all inter-block gaps are drawn with the same extent, irrespective of the actual number of bases that were removed during filtering (see Section 2.1).
To facilitate understanding of even very complex views with larger numbers of events, path animation can be added: A dash pattern is moved along each genome's path to visualize the direction of the path, traveling from the genome's start to its end position. We propose that this can help users in understanding the displayed alignment.
2.3 Block sorting
A clear visualization of the different genomes in GenomeRing strongly depends on the ordering of the blocks. The default ordering as provided by the SuperGenome may not in every case be a good choice to visualize the multiple genome alignment since very long connecting arcs can be the result. Therefore, we allow users to rearrange the blocks by three different approaches.
First, each of the aligned genomes can be chosen as a basis for an ordering. This results in a consecutive ordering of the blocks necessary to display the chosen genome, while leaving the order of the other blocks with respect to each other unchanged. This strategy is useful when one wants to focus on a single genome.
The second approach is to find an ordering that optimizes an objective function. This approach can find an ordering of the blocks such that a clear visualization of all genomes in GenomeRing is obtained. Here, we present three criteria for minimization in order to find an optimal arrangement of the blocks in the SuperGenome. These are:
-
Minimization of the number of jumps
If two blocks A, B are consecutive in one genome G, but not consecutive in the SuperGenome, the result is a jump connection in G's path. By minimizing the total number of jumps found for all genomes, this method minimizes the number of non-consecutive blocks.
-
Minimization of the number of skipped blocks
A jump connection (as defined above) gives rise to one or more skipped blocks, as several blocks can lie between A and B. This strategy minimizes the total number of skipped blocks regarding all genomes displayed in GenomeRing.
-
Minimization of the total jump length
This method is related to the previous strategy. However, instead of using the number of skipped blocks for each jump, here a jump is weighted by the length of the resulting connecting arc. The total jump length is the sum of the absolute magnitudes of the angles between each pair of connected blocks, computed for all genomes.
For each of these strategies an iterative process is applied in order to minimize the cost function f. This cost function f determines the costs for visualizing the alignment with the currently defined block ordering given one of the abovementioned minimization criteria. The minimization process then operates as follows:
The cost function f is applied to the initial arrangement to determine the cost c.
A genome G is chosen and the blocks in the SuperGenome are sorted according to G. This changes only the order of the blocks contained in G, leaving all other blocks in their original ordering with respect to each other. If this new arrangement results in a smaller cost c′, it is chosen as the new best arrangement and c is updated to c=c′.
-
New orderings are calculated by swapping pairs of blocks in GenomeRing. This is done for all possible pairs.
The cost function f is applied to evaluate the new arrangement.
New arrangements of the blocks are calculated by moving each block through the SuperGenome, i.e. removing it from its original position and inserting it at another position. This is done for each block and each possible insertion position. Again the cost function f is applied.
Steps 2–4 are performed for each genome in GenomeRing, always using the optimal ordering found in the previous rounds.
To guarantee that a minimum is reached, the process is repeated until the cost function converges, and no smaller costs c′<c can be found.
Our approach has a runtime in the order of O(n2·b2) for n genomes and b blocks in the SuperGenome, as b2 possibilities exist for swapping blocks, as well as for moving blocks. A naive approach enumerating all possible block orderings would result in a runtime of O(b!), which is clearly infeasible even for a moderate number of blocks. For example, 15 blocks result in about 1012 different arrangements that have to be evaluated.
Finally, our third approach allows users to interactively change the arrangement of the blocks after visual inspection. All three strategies can be used in combination.
2.4 Linked visualizations
GenomeRing is integrated into our visual analytics platform Mayday (Battke et al., 2010) as a visualization which can display data from multiple per-species data sets. Using Mayday's facilities for data and meta-information management, we can for example add information about gene expression in the GenomeRing visualization. For instance, genes that have been found to be coregulated (using statistical methods) can be mapped to the SuperGenome blocks to allow users to quickly identify genomic colocation. Another interesting application is to map differentially expressed genes to the SuperGenome and then to find out whether some of these map to ‘regions of interest’, such as pathogenicity islands.
Within Mayday, all visualizations are implemented as linked views. As a result, users can select genes of interest from any of the available visualizations and directly see them highlighted in the GenomeRing view. Furthermore, double-clicking on any position within a genome displayed in GenomeRing will center a linked instance of Mayday's genome browser (Symons et al., 2010) on the respective genomic position. The genome browser can thus be used to investigate detailed information including gene annotations, expression data, mapped reads from RNA-seq experiments, sequence information and meta-data such as P-values from statistical tests.
All visualizations, including the GenomeRing view, can be exported as publication-quality figures in various bitmap (PNG, TIFF, JPG) and vector formats (SVG, PDF) in arbitrary resolution.
3 RESULTS
3.1 Discovery of genomic islands in Campylobacter jejuni strains
One example for the application of our method is the discovery of large-scale deletions or insertions. One reason for long insertions can be genomic islands. These are regions in a genome which are usually acquired as a result of horizontal gene transfer. They are of great interest because they often contain genes encoding proteins related to pathogenicity or drug resistance.
To demonstrate the ability of our concept to identify such regions, we applied it to an alignment of four Campylobacter jejuni strains (RM1221, NCTC11168, 81-176, 81116). Campylobacter jejuni is a Gram-negative microaerophilic bacterium, which is one of the major causes of gastroenteritis (Snelling et al., 2005).
For the SuperGenome generation, the minimal block size was set to 10 kb. This resulted in a SuperGenome consisting of 14 blocks, which we visualized with GenomeRing (Fig. 3). The majority of the blocks contain all four strains. There are four large insertion blocks for C. jejuni RM1221, which are apparent at first glance. They correspond to genomic islands, which are referred to as C. jejuni-integrated elements (CJIEs) (Fouts et al., 2005; Parker et al., 2006). CJIE1, which is also known as CMLP1, is a Campylobacter Mu-like phage. CJIE3 is a putative integrated plasmid while CJIE2 and CJIE4 also contain phage-related proteins.
Fig. 3.
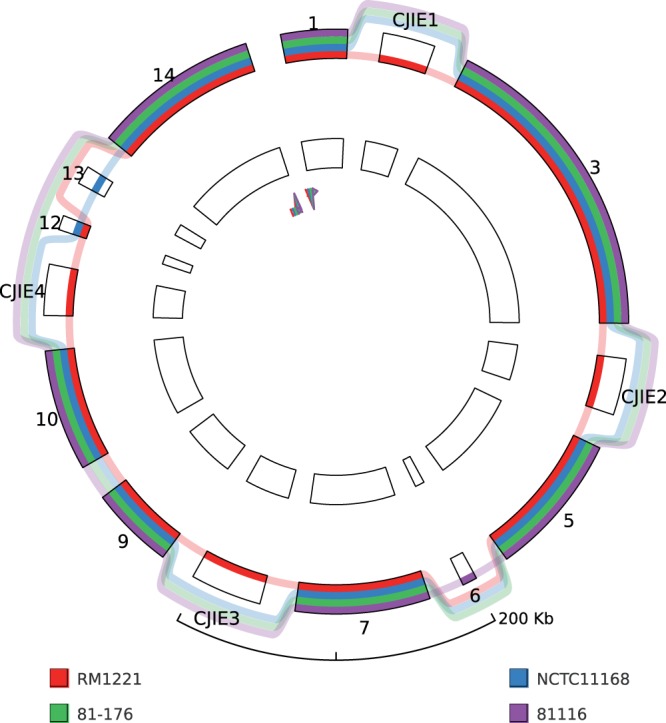
GenomeRing visualization of an alignment of four C. jejuni strains (RM1221, NCTC11168, 81-176, 81116). The four genomic islands in RM1221 (CJIE1–4) appear as insertions in the view and the blocks are labeled accordingly. For this view, the minimal block length was set to 10 kb. None of the genomes contains an inversion, thus the reverse ring is empty
This example nicely shows our visualization concept. While a linear viewer defines blocks as a collinear alignment, we add blocks also for insertions and deletions so that organisms that contain or lack the respective regions in the alignment can be quickly visually identified.
3.2 Environment-specific gene expression in Helicobacter pylori
The integration of GenomeRing with Mayday allows for linking the view to other visualizers. This includes Mayday's genome browser, where genomic annotations, such as the location of genes, as well as related expression values can be visualized. In addition, genomic annotation loaded in the genome browser can be mapped into the lane of the respective genome in the GenomeRing visualization.
We demonstrate this for an alignment of three Helicobacter pylori strains (26695, J99, P12) and gene expression data for H. pylori 26695. Helicobacter pylori is a Gram-negative, microaerophilic bacterium that populates the human stomach causing gastritis and even gastric cancer (Cover and Blaser, 2009). Because of its role as a major human pathogen, genetic factors responsible for its pathogenicity are of great interest. (Sharma et al., 2010) published a comprehensive transcriptomic study of H. pylori strain 26695 under various experimental conditions. The organism was grown to mid-logarithmic phase (ML), under acid stress (AS) as well as in contact with responsive gastric epithelial cells (AG) and non-responsive liver cells (HU). In addition, the transcriptome was measured when the organism was grown in cell culture medium (PL).
When applying the SuperGenome construction to the alignment of the three H. pylori strains, we set the threshold for the minimal block size to 50 kb, which only preserves very large events. This results in a SuperGenome consisting of eight blocks, of which two represent inversions between strain 26695 and J99/P12 (Fig. 4).
Fig. 4.
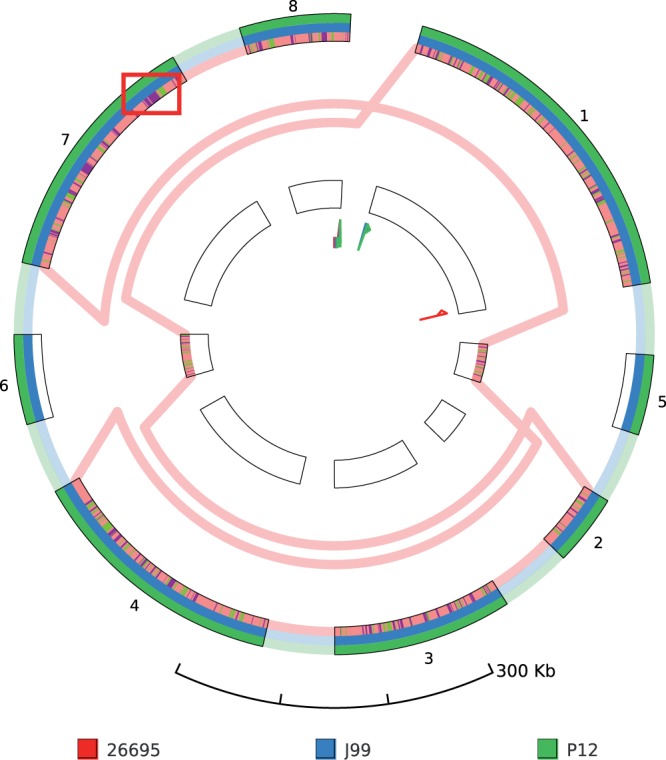
GenomeRing visualization of an alignment of three H. pylori strains (26695, J99, P12). For this view, the minimal block length was set to 50 kb. Two large-scale inversions between 26695 and J99/P12 are represented by blocks 5 and 6. Gene expression data for 26695 has been mapped into the corresponding lane (red) of all blocks. Genes upregulated in condition HU or AS are shown on the red lane, colored purple and green, respectively. The region that is shown in more detail in Fig. 5 is highlighted by a red rectangle (not part of GenomeRing visualization)
We integrated the expression data of the study by Sharma et al. (2010) into GenomeRing as follows: After loading the expression data into Mayday we performed a z-score normalization and a k-means clustering. By this, we were able to identify groups of genes that are differentially regulated under specific experimental conditions. We selected two large expression profile clusters of which one contains genes which are upregulated during acid stress (AS) and another one which contains genes upregulated when the organism is in contact with liver cells (HU). Using different colors, both groups were mapped into the lane of H. pylori strain 26695 in the GenomeRing visualization. By doing so, it is possible to get an instant overview about where in the genome the genes are located that specifically react to a certain condition.
It appears that genes reacting to the same condition tend to be organized in chromosomal clusters in many cases, which can be seen by stretches of visualized gene loci with the same color. The investigation of chromosomal clusters of co-expressed genes is of interest because it allows researchers to generate hypotheses about the function of these genes as they are often involved in similar biological processes.
An example of such a locus is highlighted in Figure 4. By double-clicking on that region in the GenomeRing visualization, the Mayday genome browser instance, which is linked to the view, jumps to that locus, thus allowing a more detailed inspection (Fig. 5). Here, we combined the locus information of the genes with a heatmap track showing the expression for all experimental conditions. In addition, wiggle tracks show the expression level in single-nucleotide resolution for the two conditions (HU, AS), as calculated from the RNAseq data.
Fig. 5.

Visualization of the genomic region in H. pylori 26695 highlighted in Figure 4 using Mayday's track-based genome browser. The five tracks shown here from top to bottom are as follows: (A) genomic coordinates in the H. pylori 26695 genome; (B) locus-specific expression value heatmap of genes upregulated under HU or AS condition (forward strand: above the baseline, reverse strand: below the baseline). The heatmap shows the expression for all five experimental conditions (from top to bottom: AG, AS, HU, ML, PL); (C) visualization of protein-coding genes located in that region. The chromosomal gene clusters upregulated under the HU or AS condition are labeled by horizontal braces (not part of the genome browser visualization); (D) wiggle track for RNAseq data from the HU condition (reverse strand); (E) wiggle track for RNAseq data from the AS condition (forward strand)
Two chromosomal clusters of coexpressed genes can be found in this region. One larger cluster contains genes upregulated under the HU condition. Another smaller cluster that consists of genes upregulated under acid stress (AS) is located further downstream. An inspection of the functional annotation of these genes revealed that the larger cluster primarily consists of ribosomal proteins while the smaller cluster contains only four genes, two of which encode cation efflux system proteins (czcA), which have been shown to be required for growth at low pH (Bijlsma et al., 2000). The other two genes are annotated as hypothetical. However, these hypothetical proteins might be related to the same system as indicated by their coexpression.
This brief analysis demonstrates how our concept allows for the generation of hypotheses by an iterative visual inspection of data at several levels. Starting with GenomeRing at a global overview level, which shows structural differences between genomes but which in addition can incorporate locus-specific data, users can step down to a level of analyzing single gene loci or even more fine-grained information such as RNAseq data in single nucleotide resolution, as also illustrated in Fig. 5.
4 DISCUSSION
In this work, we presented two complementary approaches to the multiple genome alignment problem.
Our SuperGenome method computes a consistent coordinate system for a multiple genome alignment. As the SuperGenome mapping is performed on single nucleotide level, high-resolution expression height graphs resulting from RNAseq or tiling array experiments can also be investigated within a common coordinate system, which is especially useful for comparative analyses. The comparison of gene expression, for example, is possible even if an ortholog mapping between the organisms is not available. In addition, the SuperGenome allows for the inspection of conserved intergenic regions, e.g. to discover yet unknown coding or non-coding transcripts.
Genomic annotations can also be mapped into the SuperGenome enabling users to compare the gene content of a region between several organisms independently from the location of that region in the respective genomes. Even if translocations and inversions occur, the regions still map to the same coordinates in the SuperGenome.
The SuperGenome is complemented by GenomeRing to visualize completely aligned genomes within a common coordinate system to get a quick and broad overview of the structural differences between these genomes. Thus for each region that appears in one of the aligned genomes, it is immediately apparent which of the other genomes also contain or lack that region.
The SuperGenome approach in general is not limited to visualization with GenomeRing. Conventional genome browsers can be applied in parallel to obtain more detailed information on a specific locus in a chosen genome. We have therefore linked GenomeRing with Mayday's genome browser. Furthermore, linear visualizations of the whole genome alignment based on the SuperGenome is of course feasible. Linear representations of genome alignments certainly have their strong points, especially when larger number of genomes are compared. However, the circular approach can be an effective means to minimize visual clutter (Nielsen et al., 2010), as it allows connecting edges to be, on average, much shorter than in a linear view (see Fig. 6 for a comparison). For example, if in one genome the last block of the SuperGenome is to be connected with the first block, the circular layout can represent this by a very short edge, while the linear layout requires an edge traversing the whole length of the alignment. Furthermore, the circular layout results in two possible directions for each edge, clockwise or counter-clockwise, allowing us to limit the maximal length of each edge to <180○ while at the same time offering the possibility to choose a longer edge route to avoid overlapping edges.
Fig. 6.
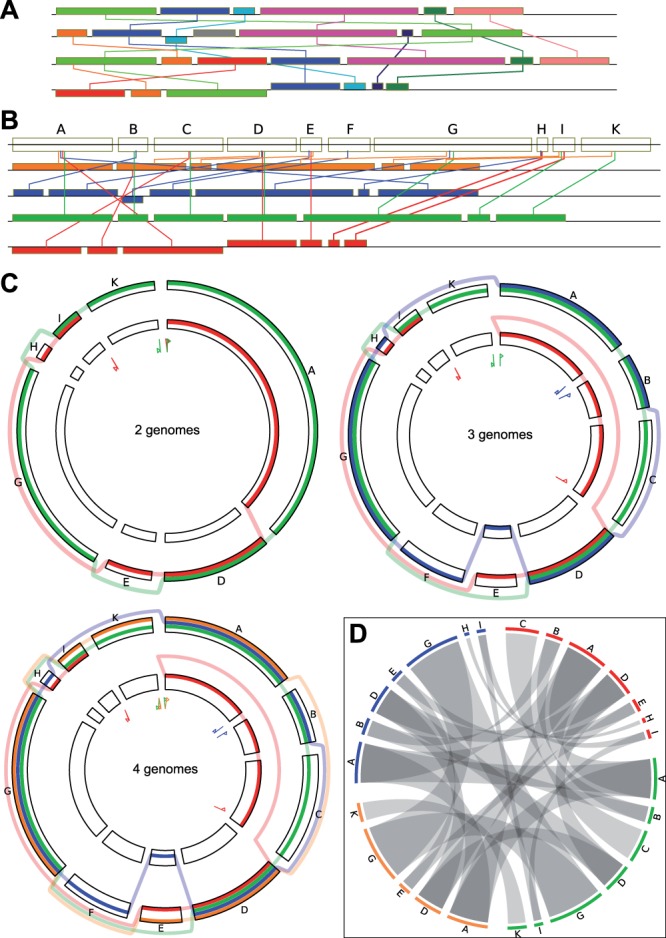
Linear versus circular view. An alignment of four genomes is shown. (A) block-based display in the style of Mauve (Darling et al., 2004) using colors to distinguish blocks and edges to link identical blocks in the aligned genomes. (B) linear representation of the SuperGenome for this example, using one color for each genome. (C) circular GenomeRing visualization of the same example, built step-by-step from the alignment, starting with two genomes. The inversion and deletion events accounting for the differences between the two genomes are very clearly visible. The third genome gives rise to new blocks by its new start/end coordinates, by a deletion (new blocks A–C), and by an insertion (new block F). The addition of the fourth, orange, genome does not induce the formation of any more blocks, but simply adds a new path. The increase in visual complexity is quite small when compared with the linear views. (D) visualization of the same alignment using Circos. Aligned blocks are connected
It is important to note that the GenomeRing approach to MSA visualization presents a different angle than traditional visualizations. Most linear viewers, such as the one contained in Mauve, as well as the traditional Circos plot for genome comparison, show the order of the alignment blocks for each genome. This allows users to quickly identify the composition of each genome, but results in a possibly large number of edges indicating block identities. Our approach, on the other hand, focuses on the identification of differences and similarities between genomes: By using one circle segment for each alignment block, and one color for each genome's path, users can immediately identify the composition of each block. Determining, for example, which genomes contain a certain genomic island is straightforward in GenomeRing. If blocks are ordered independently for each genome in order to achieve collinearity within each genome, identifying a genomic island requires the recognition of the corresponding block by a common attribute. As colors are often used to identify blocks, the human visual system, which is only capable of distinguishing a small number of distinct colors (Ware, 2008), becomes the limiting factor, and users are forced to use mouse interaction to check whether their assumption about block identities are correct. Based on these considerations, we present GenomeRing as an addition to the researcher's toolbox, complementing existing (linear) viewers, and not as a replacement for existing tools.
The GenomeRing visualization can not only be used to display multiple alignments of whole genomes resulting from the SuperGenome algorithm. Because of its flexible implementation, it can also be used to display other types of alignments, for example of a cluster of genes (each block representing one gene), to allow for the investigation of synteny. Another application could be the analysis of splice variants, where each block represents one exon.
Currently, GenomeRing is designed to display the alignment of a single chromosome. To display alignments spanning multiple chromosomes (or on bacterial genomes spanning several plasmids), we envision two strategies. First, the alignment could be presented as the concatenation of several SuperGenomes, one computed for each chromosome (Fig. 7 left). This is already possible with the current GenomeRing version. Second, several circular views (one per chromosome) could be displayed in a common visualization with an additional type of jump edge connecting blocks from different chromosomes to represent inter-chromosomal translocations (Fig. 7 right).
Fig. 7.
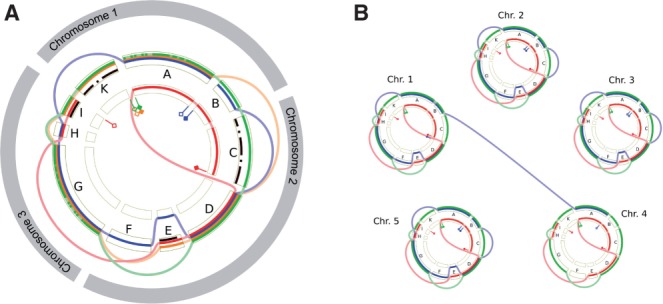
Visualization of multiple chromosomes in an alignment. (A) concatenating chromosomal alignments. (B) representing each chromosome by a circular view and adding inter-circle jumps to represent evolutionary events involving several chromosomes
An important feature of GenomeRing is the possibility to rearrange blocks in the SuperGenome in order to enhance visual clarity, as well as to highlight different aspects of the multiple genome alignment. Clearly, our strategy of finding a good ordering for the visualization of the genomes in GenomeRing only results in a local optimum and thus does not guarantee to find an optimal solution for a specified optimization criterion. However, a full evaluation of all possible arrangements of blocks in the SuperGenome is infeasible. We approach this problem with a quadratic-time heuristic which allows us to find a nearly optimal solution in acceptable time.
An optimal sorting of the blocks according to some objective function might on the other hand not be optimal for the user. In the future, we, therefore, plan to develop further strategies for the rearrangement of blocks in the SuperGenome that incorporates information gained from a user study.
A user study for GenomeRing could provide highly valuable information on several aspects. Regarding the block ordering methods, such a study could provide information on two different questions. First, it could allow us to elucidate how the blocks of the SuperGenome have to be arranged such that individual blocks contained in a specific genome can easily be spotted by the user. Second, one could gain information on the interpretability of the multiple genome alignment and how this correlates with different block arrangements. Including such information in the design of a sorting algorithm could strongly improve the visualization.
In the course of such a user study, we could also explore the effectiveness of the provided interaction methods, and perhaps also include semi-automatic methods to highlight features of possible interest, or to zoom and pan to such features. Furthermore, different strategies for the edge layout could be evaluated. By allowing paths to traverse blocks in clockwise or counter-clockwise direction, we reduce visual clutter. An important question to address would be, whether users experience difficulties interpreting the alignment, i.e. whether minimizing the number or length of jump edges leads to a visualization which is not optimal from the user's point of view. Users might prefer a visualization using only a single direction of block traversal, at the expense of increased visual complexity. Another central feature that we would like to investigate is whether users understand that the genome paths in GenomeRing only show the order of the blocks, and not the direction of the actual bases within each block.
Another route for further development lies in the summarization of events to display different views of the SuperGenome depending on the current zoom level. As too many blocks lead to visual clutter due to the possible increase in the number of connecting edges, high-level views could summarize small blocks depending on some measure of similarity, for instance. Clearly, every visual approach has its limitations due to the limited resolution and/or the limits of screen area, and also due to the limits of the human visual system. We have conducted preliminary tests that show that visualization of more than 10 genomes is, in most cases, infeasible. Likewise, large numbers of ‘long-range’ events such as translocated inversions result in increasingly complex visualizations. GenomeRing is not designed to visualize hundreds of genomes, blocks or events. In our view, the challenge of adequately visualizing such very large alignments, or alignments with a large divergence between species lies not in finding a visualization that shows every detail at maximum resolution, but rather in appropriate summarization and a (semi-automatic) focusing on the important events.
5 CONCLUSION
GenomeRing is a highly interactive tool for multiple alignment visualization based on SuperGenome coordinates. The concept of the SuperGenome together with GenomeRing provides a quick and broad overview through the display of genomic events from completely aligned genomes and allows for a detailed analysis of specific genes through the linkage of GenomeRing to several other visualizations incorporated into Mayday, such as Mayday's genome browser. These aspects and the applicability of GenomeRing to other fields of research make it a highly usable visualization strategy complementing already existing visualization techniques. In addition, the SuperGenome coordinate system can be widely applied beyond the field of visualization, as it provides a generic solution to the problem of consistently specifying coordinates in multiple genome alignments without attributing a special status to an arbitrarily chosen reference sequence.
ACKNOWLEDGEMENTS
The authors wish to thank Dr Cynthia Sharma for kindly providing the H. pylori expression data. Funding from the DFG Priority Program 1335 ‘Scalable Visual Analytics’ is gratefully acknowledged.
Funding: DFG priority program 1335 ‘Scalable Visual Analytics’.
Conflict of Interest: none declared.
REFERENCES
- Alikhan N., et al. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battke F., et al. Mayday – Integrative analytics for expression data. BMC Bioinformatics. 2010;11:121. doi: 10.1186/1471-2105-11-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet S. Solexa ltd. Pharmacogenomics. 2004;5:433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- Bijlsma J.J., et al. Identification of loci essential for the growth ofHelicobacter pyloriunder acidic conditions. J. Infect. Dis. 2000;182:1566–1569. doi: 10.1086/315855. [DOI] [PubMed] [Google Scholar]
- Broad Institute. MEDEA comparative genomic visualization with Adobe Flash. 2009 http://www.broadinstitute.org/annotation/medeaaccessed (December 21, 2011) [Google Scholar]
- Cover T.L., Blaser M.J. Helicobacter pyloriin health and disease. Gastroenterology. 2009;136:1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A., et al. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A.E., et al. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5 doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droege M., Hill B. The Genome Sequencer FLX System–longer reads, more applications, straight forward bioinformatics and more complete data sets. J. Biotechnol. 2008;136:3–10. doi: 10.1016/j.jbiotec.2008.03.021. [DOI] [PubMed] [Google Scholar]
- Durbin R., et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J., et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Fouts D.E., et al. Major structural differences and novel potential virulence mechanisms from the genomes of multipleCampylobacterspecies. PLoS Biol. 2005;3 doi: 10.1371/journal.pbio.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer K.A., et al. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32(Suppl. 2):W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker J., et al. Deletions of chromosomal regions coding for fimbriae and hemolysins occur in vitro and in vivo in various extra intestinalEscherichia coliisolates. Microbial Pathogenesis. 1990;8:213–225. doi: 10.1016/0882-4010(90)90048-u. [DOI] [PubMed] [Google Scholar]
- Harrower M., Brewer C. ColorBrewer.org: an online tool for selecting colour schemes for maps. The Map Reader. 2003:261–268. [Google Scholar]
- Haussler D., et al. Genome 10K: a proposal to obtain whole-genome sequence for 10 000 vertebrate species. J. Hered. 2009;100:659–674. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M., et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M., et al. MizBee: A multiscale synteny browser. Visualization Comput. Graph. IEEE Trans. 2009;15:897–904. doi: 10.1109/TVCG.2009.167. [DOI] [PubMed] [Google Scholar]
- Nielsen C., et al. Visualizing genomes: techniques and challenges. Nat. Methods. 2010;7:S5–S15. doi: 10.1038/nmeth.1422. [DOI] [PubMed] [Google Scholar]
- Parker C.T., et al. Comparative genomic analysis ofCampylobacter jejunistrains reveals diversity due to genomic elements similar to those present inC. jejunistrain RM1221. J. Clin. Microbiol. 2006;44:4125–4135. doi: 10.1128/JCM.01231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca G.J., et al. Polony DNA sequencing. Curr. Protocols Mol. Biol. 2006;76:7.8.1–7.8.22. doi: 10.1002/0471142727.mb0708s76. [DOI] [PubMed] [Google Scholar]
- Robinson G., et al. Creating a buzz about insect genomes. Science. 2011;331:1386. doi: 10.1126/science.331.6023.1386. [DOI] [PubMed] [Google Scholar]
- Rothberg J., et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- Sharma C.M., et al. The primary transcriptome of the major human pathogenHelicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- Snelling W.J., et al. Campylobacter jejuni. Lett. Appl. Microbiol. 2005;41:297–302. doi: 10.1111/j.1472-765X.2005.01788.x. [DOI] [PubMed] [Google Scholar]
- Symons S., et al. Integrative systems biology visualization with MAYDAY. J. Integrative Bioinformatics. 2010;7:115. doi: 10.2390/biecoll-jib-2010-115. [DOI] [PubMed] [Google Scholar]
- Ware C. Visual Thinking: for Design, (Morgan Kaufmann Series in Interactive Technologies). 1st. Waltham, Massachusetts, USA: Morgan Kaufmann; 2008. [Google Scholar]
- Weigel D., Mott R. The 1001 genomes project forArabidopsis thaliana. Genome Biol. 2009;10:107. doi: 10.1186/gb-2009-10-5-107. [DOI] [PMC free article] [PubMed] [Google Scholar]