

Abstract
Motivation: Secondary structure underpins the folding pattern and architecture of most proteins. Accurate assignment of the secondary structure elements is therefore an important problem. Although many approximate solutions of the secondary structure assignment problem exist, the statement of the problem has resisted a consistent and mathematically rigorous definition. A variety of comparative studies have highlighted major disagreements in the way the available methods define and assign secondary structure to coordinate data.
Results: We report a new method to infer secondary structure based on the Bayesian method of minimum message length inference. It treats assignments of secondary structure as hypotheses that explain the given coordinate data. The method seeks to maximize the joint probability of a hypothesis and the data. There is a natural null hypothesis and any assignment that cannot better it is unacceptable. We developed a program SST based on this approach and compared it with popular programs, such as DSSP and STRIDE among others. Our evaluation suggests that SST gives reliable assignments even on low-resolution structures.
Availability: http://www.csse.monash.edu.au/~karun/sst
Contact: arun.konagurthu@monash.edu (or lloyd.allison@monash.edu)
1 INTRODUCTION
Periodic hydrogen-bonding patterns in globular proteins give rise to elements of secondary structure—helices and sheets. The α-helix and β-sheets were among the first structural motifs predicted from first principles of stereochemistry by Pauling and Corey (1951). We now know these specific motifs are almost ubiquitous across the corpus of known structures. Eventually, other repetitive motifs were also identified, and the alphabet of secondary structures was expanded to include 310-helix, π-helix, β-turn, γ-turn, Ω-turn and β-bulges, among other minor elements. In what follows, we use the term secondary structure to include both the classical helices and sheets, and other common substructural elements.
Accurate assignment of secondary structure of proteins from coordinate data is an important and a challenging problem (Andersen and Rost, 2009). Secondary structure underpins the architectural organization in proteins. It simplifies the complex atom-level description of proteins and is therefore the key to generation of schematic diagrams of their three dimensional (3D) folding patterns (Lesk and Hardman, 1982; Richardson, 1981). They are central in training methods geared for predicting secondary structure from amino acid sequence (Andersen and Rost, 2009). They form a linchpin to efficient methods for structural comparison and analysis (Kamat and Lesk, 2007; Konagurthu et al., 2008).
Over the last 30 years, many programs were developed to address the problem of assigning secondary structure to protein coordinate data. A broad classification can be made of the assignment strategies: (i) methods that use distance and angle profiles of local fragments; (ii) methods that detect hydrogen bonds between backbone atoms; (iii) methods that use 3D geometry of local fragments; and (iv) methods that approximate the backbone trace with a set of straight lines.
The following reviews some of the major earlier contributions to the literature of this problem. Levitt and Greer (1977) were the first to generate an automatic method for secondary structure assignment, based on distance and dihedral angle profiles of Cα atoms over a sliding window of four residues. P-SEA (Labesse et al., 1997) is another method in this category which assigns secondary structural states using a short Cα distance mask and two Cα dihedral angle criteria. PROSS (Srinivasan and Rose, 1999) proposes an assignment based solely on backbone dihedral angles. Xtlsstr (King and Johnson, 1999) calculates backbone dihedral angles and distances and assigns secondary structural types that would be consistent with interactions of amide-amide groups observed from circular dichroism of a protein in ultraviolet range (Andersen and Rost, 2009). More recently, PALSSE (Majumdar et al., 2005) was designed to delineate protein structure into helices and strands, mainly using distance and torsion angle constraints to identify core elements which are later extended to longer segments. KAKSI (Martin et al., 2005) is based on Cα distances and backbone dihedral angles and designed primarily to show concordance with the manual assignments found in the protein data bank (PDB).
The most popular method in this space is ‘Dictionary of Secondary Structure of Proteins’ (DSSP) developed by Kabsch and Sander (1983). DSSP is based on detecting hydrogen bonds between nitrogen and carbonyl groups along the protein polypeptide chain using a Coulomb approximation of the hydrogen-bond energy function (Andersen and Rost, 2009). Many now consider this method a standard for secondary structural assignment (Martin et al., 2005). Since DSSP was published, several methods have been designed that rely on computing the hydrogen-bond energy between backbone atoms. STRIDE (Frishman and Argos, 1995) is among the successful variants of DSSP which uses a modified hydrogen-bond energy function as well as backbone dihedral angles to compute its assignment. SECSTR (Fodje and Al-Karadaghi, 2002) is another variant which improves the detection and assignment of π-helices which both DSSP and STRIDE have difficulty characterizing (Martin et al., 2005).
There are other methods which assign secondary structure using 3D features in a protein structure. Richards and Kundrot (1988) describe a method, DEFINE-S, to assign secondary structure using local geometry of ideal secondary structures. The P-CURVE (Sklenar et al., 1989) algorithm uses an helicoidal axis approach derived from a series of peptide planes to assign secondary structure. The VoTAP (Dupuis et al., 2004) algorithm relies on Voronoi tessellation of a residue contact map and then matching the contact map profiles to a consensus assignment of secondary structures by methods like DSSP and STRIDE.
In the last category are indirect methods, such as STICK (Taylor, 2001) and PMML (Konagurthu et al., 2011) which work by approximating the Cα spatial trace using a set of lines. These methods seek the best approximation of the protein backbone using piecewise lines. Only as a post-process to this approximation, each line segment is indirectly attributed a secondary structural type based on criteria such as the average rise and pitch of the Cα atoms within the segment. These approaches solve a related yet different problem, namely ‘the best line approximation of the protein chain’.
Consistent with this large number of proposed methods, assignment of secondary structure has been recognized to be an ‘inexact process’ (Cuff and Barton, 1999). Previous comparative studies have highlighted the difficulties of existing programs to assign secondary structure consistently (Andersen and Rost, 2009; Colloc'h et al., 1993; Cuff and Barton, 1999; Martin et al., 2005; Zhang et al., 2008). These disagreements can be major as shown by Colloc'h et al. (1993) where the percentage of agreement between DSSP, DEFINE-S and P-CURVE was only 63% on a residue basis. It has been observed that most disagreements arise in the terminal regions of the assigned secondary structural elements. Reflecting on this problem Robson and Garnier (1986) comment [as quoted by Martin et al. (2005)]: ‘In looking at a model of a protein, it is often easy to recognize helix and to a lesser extent sheet strands, but it is not easy to say whether the residues at the ends of these features be included in them or not. In addition, there are many distortions within such structures so that it is difficult to assess whether this represents merely a distortion, or a break in the structure. In fact, the problem is essentially that helices and sheets in globular proteins lack the regularity and clear definition found in the Pauling and Corey models.’
Given the complexity of the details of individual protein structures, it is not surprising that the secondary structure assignment problem has resisted a mathematically rigorous definition. The effect can be seen in the use of a variety of definitions by the existing tools, although all of them are reasonable. In this study, we describe an approach, SST, to the secondary structure assignment problem using minimum message length (MML) inference (Wallace and Boulton, 1968). Linking statistical inference with data compression, the goal is to communicate losslessly the coordinates of a protein using a two-part message. The first part transmits the secondary structure assignment as a hypothesis about the coordinates. The second-part transmits the details of coordinates not explained by the hypothesis. This gives rise to statistically robust objective function to optimize: find the best hypothesis on the coordinate data that minimizes the total two-part message length.
SST assigns secondary structure segments of the following types: α, 310 and π-helix (including left-handed versions of all these helices when they occur), sharp turns, β-strands and others (coil). SST in a post-processing step merges consecutive structures where appropriate, and groups all strands of a sheet, identifies β-bulges, to convert the results to a molecular biologist's conventional secondary structure description, and produces a PyMol script to visualize the secondary structural assignments. (Fig. 1.)
Fig. 1.
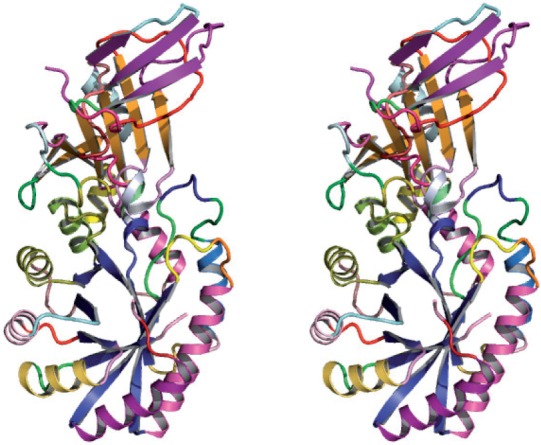
SST assigned secondary structure to coordinates of a 1.6Å crystal structure, Ornithine decarboxylase from mouse.
2 OVERVIEW OF MML CRITERION
The MML criterion provides an information–theoretic objective for problems of inference where the goal is to find the best explanation (or theory, hypothesis, model) for a set of observed data (Wallace and Boulton, 1968). MML relies on quantifying the amount of information required to convey losslessly the observed data in an explanation message. The best hypothesis is the one which can convey the entire data set in the shortest possible explanation message.
More formally, for some observed data D and a hypothesis H that offers an explanation of the data D, Bayes's theorem (Bayes and Price, 1763) gives
where P(H) is the prior probability of hypothesis H, P(D) is the prior probability of data D, P(H|D) is the posterior probability of H given D, and P(D|H) is the likelihood.
Using Shannon's mathematical theory of communication (Shannon, 1948), the amount of information for an explanation of the data D with the hypothesis H is given by
where I(x)=−log2(P(x)) gives the optimal code length to convey some event x whose probability is P(x).
This immediately gives an objective means to compare competing hypotheses. For hypotheses H1 and H2 on the same data D, we have
It follows that the best hypothesis H* over all competing hypotheses is the one where the expression I(H*)+I(D|H*) is minimized.
A concrete realization of the MML framework comes from describing it as a communication process between an imaginary transmitter (Alice) and receiver (Bob) connected over a Shannon channel. Alice's objective is to send the observed data D using an explanation message in a form such that Bob can receive and decode the data D precisely as Alice sees it. Alice and Bob agree on a codebook containing the general rules of communication composed solely of common knowledge about typical, hypothetical data. Anything that is not a part of the codebook must be strictly transmitted as a part of the message. If Alice can find the best hypothesis H* on the data, Bob will receive a decodable explanation message most economically: The best inference about the data is the hypothesis that minimizes the total message length.
Alice sends the explanation message of D in two parts. In the first part, she transmits the best hypothesis, H*, she could find on the data D taking I(H*) bits. In the second, she transmits the details of the observed data D not explained by H*, taking I(D|H*) bits (i.e. the deviations from H*). Notice that MML inference gives a natural trade-off between hypothesis complexity (I(H*)), and its goodness of fit to the data (I(D|H*)).
For a comprehensive resource on MML see Wallace (2005).
3 THE DESIGN OF THE COMMUNICATION FRAMEWORK
Protein coordinates for a single-polypeptide chain are represented as an ordered set of 3D points of the form 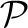 ={p1,···, pn}, where any pi corresponds to the ith Cα coordinate along N- to C-terminus of the protein chain. Each pi defines a 3D real-valued vector (pix, piy, piz) in Angstrom (Å) units, where each component of the vector comes specified (in the PDB) to three positions after the decimal place. Therefore, in this work, we treat the accuracy of measurement of the data as ϵ=0.001 Å (independent of the actual accuracy of the experimental structure determination). The transmitter (Alice) has to send a message to the receiver (Bob) who will then be able to reconstruct the original data from the encoded explanation message exactly. For coordinate data from the PDB, Bob will reconstruct each coordinate of each atom to the original precision of three digits after the decimal point.
={p1,···, pn}, where any pi corresponds to the ith Cα coordinate along N- to C-terminus of the protein chain. Each pi defines a 3D real-valued vector (pix, piy, piz) in Angstrom (Å) units, where each component of the vector comes specified (in the PDB) to three positions after the decimal place. Therefore, in this work, we treat the accuracy of measurement of the data as ϵ=0.001 Å (independent of the actual accuracy of the experimental structure determination). The transmitter (Alice) has to send a message to the receiver (Bob) who will then be able to reconstruct the original data from the encoded explanation message exactly. For coordinate data from the PDB, Bob will reconstruct each coordinate of each atom to the original precision of three digits after the decimal point.
3.1 Null model description of a protein coordinate data
MML gives a natural hypothesis test: The null-model corresponds to transmitting the data raw. If any hypothesis H on the data takes longer than the null model, then clearly H is unacceptable. However, the statement of the raw null model message (without any hypothesis) has to be economical; it must not be willfully inefficient.
The construction of an efficient null model for protein coordinates relies on the observation that the distance between successive Cα atoms in a protein chain is highly constrained at about 3.8Å with only small deviations from this value. The method starts with the transmission of the first Cα coordinate p1 in any choice of encoding that both transmitter and receiver agree on. (Stating p1 simply adds a constant overhead to the message length, whether transmitted via a null model message or an explanation using a hypothesis. A simple way to do away with this overhead is for Alice to translate such that p1 becomes the origin. p1 then need not be transmitted explicitly in the message and can be treated implicitly a part of the codebook.) Alice then computes the observed distance r between the successive Cα coordinates p1 and p2. This distance r can be communicated efficiently using an encoding over a normal distribution 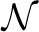 (μ, σ) with a certain fixed mean (μ) and a small standard deviation (σ) around it. Based on the prior knowledge of Cα–Cα distances between successive atoms, these values are set to μ=3.8Å and σ=0.4Å and are considered to be part of the codebook.
(μ, σ) with a certain fixed mean (μ) and a small standard deviation (σ) around it. Based on the prior knowledge of Cα–Cα distances between successive atoms, these values are set to μ=3.8Å and σ=0.4Å and are considered to be part of the codebook.
The probability density of a random variable x over a normal distribution with mean μ and a standard deviation σ is given by:
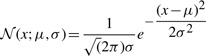 |
Therefore, the probability of stating any distance r to an accuracy of ϵ (given ϵ≪σ) using the above normal distribution is P(r)=ϵ×(x=r; μ, σ). This implies
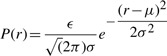 |
The optimal code length to transmit r is given by −log2(P(r)) bits:
| (1) |
Note that Bob will not be able to recover p2 simply from the transmitted information of p1 and the distance r between p1 to p2. p2 can lie anywhere on the surface of a sphere of radius r centered on p1. The precise location of p2 stated to ϵ can be transmitted by first dividing the surface area of this sphere into cells each of area ϵ2. This results in 4πr2/ϵ2 such cells distributed uniformly on the surface. These cells can be numbered using a convention that Alice and Bob both agree upon (as a part of the codebook). On the basis of this discretization of the sphere's surface area, Alice transmits the cell number c in which the observed p2 falls within. Assuming uniform probabilities, the probability that the point p2 falls in a cell number c is given by P(c)=ϵ2/4πr2. Following from this, the code length to state the cell number is:
| (2) |
Bob now has all the information to reconstruct p2 to the precision of ϵ using the information he has received.
With p2 known at Bob's end, Alice can proceed to encode in the same fashion p3 with respect to p2, then p4 with respect to p3 and so on until all the points in are transmitted.
Let ri (∀1≤i<n) denote the observed distance between any two successive Cα coordinates pi and pi+1. Let ci (∀1≤i<n) denote the cell number on the surface of the sphere of radius ri centered on the point pi in which the point pi+1 falls. The message length to transmit the entire Cα coordinate data in is therefore
| (3) |
where O(1) denotes the constant number of bits to state p1 (0 bits if Alice translates the coordinates such that p1 lies on the origin).
3.2 Models to describe segments of proteins
The secondary structure elements are used as a hypothesis to explain the coordinates. Here, we consider eight models to describe any contiguous stretch of Cα atoms (of arbitrary length) along the protein chain: (i) a right-handed α-helix; (ii) a left-handed α-helix; (iii) a right-handed 310-helix; (iv) a left-handed 310-helix; (v) a righ-handed π-helix; (vi) a left-handed π-helix; (vii) an extended β-strand; and (8) coil.
The Helical (1–6) and strand (7) models follow ideal Pauling–Corey geometry (Pauling and Corey, 1951) and are of arbitrary length. We term these seven collectively ideal models. (Pauling–Corey models are common knowledge and taken to be in the codebook.)
The coil model (8) is treated simply as a model that describes a segment of a protein raw, using the null model approach described above in Section 3.1.
3.3 Describing a protein segment using an Ideal model
Assume that at some stage of the transmission Bob has received Cα coordinates up to an intermediate point pi, that is he has received coordinates (p1, p2,···, pi) (i<n). Alice now will transmit a contiguous segment of coordinates pi to pj (1≤i<j≤n) using one of the ideal (helical or strand) models. If it is a good model, then the coordinates can be transmitted cheaply. (The discussion of the optimal choice is given in Section 4.)
The number of points to be transmitted in this segment is j−i since the start point of the segment pi is already known at the receiver's end. The remaining points pi+1,···, pj are transmitted as follows:
3.3.1 Transmitting the end point of the segment
The end point of the segment (pj) is transmitted using the sphere approach similar to the one described in Section 3.1. Instead of the distance between successive Cα coordinates, Alice transmits the distance dij between the start (pi) and end (pj) points. This is encoded using a normal distribution where the mean (μ) is taken as the distance (d*) between the start and end points from the ideal model containing j−i+1 points. The standard deviation σ of the end point is set to min((j−i)×0.2Å, 3Å) based on the length of the segment being transmitted and this rule is taken to be a part of the codebook.
On the basis of Equation 1, the code length to state dij to the accuracy of ϵ is given by
| (4) |
On the basis of equation 2, given the start point of the segment pi and distance dij of the end point pj, the end point can lie anywhere on a sphere with radius dij. pj can therefore be stated by specifying the cell number cij on the surface of this sphere in
| (5) |
3.3.2 Encoding the interior points
With the start and end points already known, there are j−i−1 interior points of the segment, pi+1,···, pj−1, yet to be transmitted. These points can be transmitted cheaply if the chosen ideal model agrees with the observed points in the segment. Alice uses the following procedure to transmit the interior points given a chosen ideal model. (Details of how the optimal choice is made appear in Section 4.)
Consider an ideal model containing l=j−i+1 points, denoted formally as 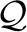 ={q1, q1,···, ql}. The coordinates in are orthogonally transformed to ′={q′1, q′2,···, q′l} such that:
={q1, q1,···, ql}. The coordinates in are orthogonally transformed to ′={q′1, q′2,···, q′l} such that:
q′1 is same as the start point pi of the segment;
the direction cosines of the vector connecting the start and end points of the ideal model q′l−q′1 and the direction cosines of the vector connecting the start and end points of the observed segment pj−pi are the same; and
the sum of the squared error of the (l−2) interior points of the segment with the corresponding interior points of the ideal model is minimized. That is, ∑1≤k≤l−2 |pi+k−q′1+k|2 is minimized, where |.| denotes the Euclidean vector norm.
Such a spatial transformation is related to the more general superposition problem that minimizes the sum of the squared distance between two corresponding vector sets (Kearsley, 1989). However, the transformation is further constrained such that the first points of the two sets are the same (Constraint 1) and the rotational axis for the ideal model is the vector between the start (pi) and end (pj) points of the segment (Constraint 2). The first two constraints can be achieved using elementary translation and rotation of the ideal coordinates.
Once is transformed such that the first two constraints are realized, the best rotation θ* of about the pj−pi axis has to be found so that Constraint 3 is realized. With an approach similar to the generalized superposition problem between two vector sets (Kearsley, 1989), this minimization problem can be solved analytically as an eigenvalue decomposition of a 2×2 square symmetric matrix in quaternion parameters of the corresponding points. (The detailed proof of the analytical method is too long for the main text and hence is provided as Supplementary Material.)
Once the transformation of to ′ is achieved as described above, Alice can transmit the interior points of the segment, pi+1,···, pj−1 by:
transmitting the best rotation about the pj−pi axis of the ideal model. (Note, Bob has already received the start and end points, pi and pj.);
transmitting the interior points pi+1··· pj−1 as spatial deviations from their corresponding transformed interior points of the ideal model. (Bob already knows pi and the coordinates of
of the ideal model from the codebook. After he receives the end point of the segment pj (using the sphere approach described above), the ideal coordinates can be transformed such that Constraints 1 and 2 of the transformation discussed above are realized. After Bob receives the rotation θ*, the ideal coordinates are rotated by that angle around the axis pj−pi whose information he already has. Once Alice sends the spatial deviations of interior points pi+1,···, pj−1 with respect to the transformed ideal coordinates, Bob can reconstruct the observed interior points of the segment.)
3.3.3 Transmitting the rotation
Rotation θ* is transmitted using a uniform distribution over a circle whose radius rθ* is the farthest distance of an interior point of the ideal model from the axis of rotation. Note that rθ* need not be transmitted because it is a property of the coordinates of the ideal model which the receiver already knows as a part of the codebook.
The rotation is transmitted by dividing the circumference of a circle of radius rθ* into arc segments of length ϵ and stating the segment number in which the rotated coordinate with the farthest radius to the axis falls. Thus, the code length of stating θ* is
| (6) |
3.3.4 Transmitting the interior points as spatial deviations
Let any error vector of an interior point of the segment with respect to the corresponding transformed interior point of the ideal model, ek≡(pi+k−q′1+k), 1≤k≤l−2 have the vector components (Δxk, Δyk, Δzk).
Each Δx, Δy and Δz of an interior point is transmitted using a normal distribution with a μ of 0 and a standard deviation σ set to the sample standard deviation computed from these error components.
Wallace (2005) gives the MML estimate of code length to transmit a set of independent data (Δx1, Δy1, Δz1), (Δx2, Δy2, Δz2), ··· (Δxl−2, Δyl−2, Δzl−1) using a normal distribution as:
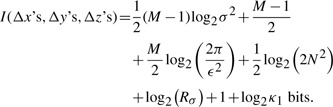 |
(7) |
where M=3×(l−2) is the total number of components of the error vectors ek being transmitted, Rσ gives the prior knowledge of the limits to log2 σ and κ1≈1/12 denotes the constant corresponding to quantizing lattices proposed by Conway and Sloane (1984). In this study, we assume that σ is bounded by 3Å because this is consistent with the limits of utility of root-mean-squared-deviation (RMSD) in superposition as a measure to estimate protein structural similarity.
Therefore, combining code lengths from equations 4–7, the code length required to transmit coordinates of a segment of a protein using any ideal model is give by
| (8) |
3.4 Describing a protein segment using the coil model
When transmitting a segment of a protein pi,···, pj as a coil, the coordinates are stated raw in that range using a null model (Section 3.1). Therefore, the code length of stating a segment pi,··· pj as a coil is
| (9) |
where I(rk) and I(ck) are code lengths given in equations 1 and 2.
3.5 Describing the protein as a collection of segments
Having laid the foundations of encoding segments of a protein using one of the ideal models or the coil model (Sections 3.3 and 3.4), this section deals with describing the entire protein coordinates as a collection of segments of a model type.
The main idea here is to find the best decomposition of points of a protein into segments where each segment is described using exactly one of eight potential models. Note that the decomposition, with the associated model descriptors, gives a secondary structural hypothesis of a protein.
Formally, a segmentation of ={p1,···, pn} gives an ordered subset of points ′={p′1≡pi1, p′2≡pi2,···, p′m≡pim} where 1=i1<i2<···<im=n. Each successive pair of points in ′, 〈p′1, p′2〉, 〈p′2, p′3〉,··· 〈p′m−1, p′m〉, defines the start and end points of a segment. (Notice that ′ gives m−1 segments of P, where end point of one segment is same as the start point of the next.) Associated with each segment 〈p′k, p′k+1〉 of length lk=ik+1−ik+1 is a model type tk, 1≤k≤m−1. A secondary structural assignment of is given by the segmentation {p′1,···, p′m} and its corresponding model assignment {t1,···, tm−1}.
We note that for n points in , there are 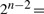 possible segmentations – the first and last points of ′ are the same as those in . Since each segment of any segmentation can be assigned to any of the eight possible model types, the total possible secondary structural assignments is given by the formula:
possible segmentations – the first and last points of ′ are the same as those in . Since each segment of any segmentation can be assigned to any of the eight possible model types, the total possible secondary structural assignments is given by the formula: 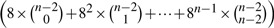 . For an average protein, this gives a massive search space. (An efficient dynamic programming method to find the best secondary structural assignment is detailed in Section 4.)
. For an average protein, this gives a massive search space. (An efficient dynamic programming method to find the best secondary structural assignment is detailed in Section 4.)
Any given segmentation ′ of and its associated model types acts as a secondary structural hypothesis of the given coordinate data. Alice can describe and transmit the coordinates in using this hypothesis over a two-part message.
3.5.1 First part of the explanation message
In the first part of the message, Alice communicates the segmentation ′={p′1,···, p′m} and its corresponding model assignments {t1,···, tm−1} as the hypothesis on the observed coordinate data in . This part of the message will be composed of the following:
the number of segments (m−1) in
′ and- for each segment p′k (1≤k≤m−1), communicate
- the length of the segment lk=ik+1−ik+1 and
- the model type tk to encode the points in that segment.
The number of segments (m−1) is an integer transmitted using a log* distribution assuming a universal prior on the distribution of numbers. Rissanen (1983) gives the code length of transmitting any integer n>0 as
| (10) |
where log2*(n)=log2 n+log2 log2 n+··· (over all +ve terms).
Next, the lengths of the segments are positive integers. Although these integers can also be transmitted using a log* distribution, it is rather inefficient because in practice the lengths of helices, strands and coils are constrained. Therefore, in this work, we, encode the lengths of the segments using a Poisson distribution with a predefined mean of λ for each model type: (The parameters λ for each of the eight types of models are treated to be a part of the codebook. In this work we empirically set λ=4 for coil and λ=5 for strands. The lengths of helices are transmitted using a mixture of two Poisson distributions with means 4 and 8.)
The code length to state any integer n>0 using this distribution is:
| (11) |
Finally, each model type t (encoded as an integer 0≤t≤7) of any segment is stated using a uniform distribution (uniform is the simplest choice. Since some models as more probable than others (e.g. α-helices and strands are significantly more probable than other models), a more elaborate coding scheme can also be considered taking into account the empirical distribution of various models.) in
| (12) |
In summary, combining equations 10–12, the code length of the first part of the message proposing the secondary structural hypothesis on is
| (13) |
3.5.2 Second part of the explanation message
In the second part of the message, Alice sends the actual details of the coordinates in economically given the hypothesis ′.
The procedure to transmit coordinate data of a segment of a protein using the ideal and coil models has been discussed in Sections 3.3 and 3.4.
Using the notations in Section 3.5, the hypothesis received in the first part of the message is of the form 〈p′k, p′k+1〉, with each segment of some length lk and type tk. The message length to transmit the coordinates given the above segmentation is:
| (14) |
where O(1) is the constant number of bits to state the first Cα coordinate p1 (Section 3.1), Imodel(p′k,···, p′k+1)=Iideal(p′k,···, p′k+1) if 0≤tk≤6 and Imodel=Icoil(p′k,···, p′k+1) if tk=7.
The total message length of communicating with ′ comes from combining equations 13 and 14
| (15) |
3.6 Problem Statement
From the earlier section, the problem of inferring the best secondary structural assignment can now be stated formally as follows: given containing n points, find the secondary structural segmentation ′ and its corresponding model assignment such that the total message length to transmit losslessly, Itotal (&′)=Ifirst(′)+Isecond(|′) is minimized.
4 INFERENCE OF SECONDARY STRUCTURE
This section describes the search method to find the best MML secondary structural segmentation from given coordinate data.
4.1 Constructing the code length matrices
Equation 15 gives the total message length of communicating coordinates in using a segmentation ′:
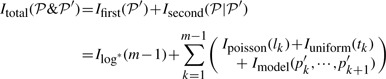 |
For a given protein, any pair of points can potentially be the start and end points of a segment. At the same time, a segment can be described using any of the eight models considered here. Therefore, the procedure to assign secondary structure to Cα coordinates {p1,···, pn} in a given protein begins by constructing a set of eight code length matrices, one for each model type t (0≤t≤7):
| (16) |
where any cell (i, j)1≤i<j≤n of the matrix for type t gives the code length of stating the segment pi to pj using the model t.
4.2 Finding the best secondary structural assigning
The segmentation of using various model types enforces a strict ordering constraint, satisfying the requirements for a solution by dynamic programming, even though the search space is huge as discussed in Section 3.5. Let any D(i) store the optimal message length of transmitting points p1,···, pi, for all 1≤i≤n. With the boundary condition of D(1)=0, the dynamic programming recurrence to find the optimal assignment is given by
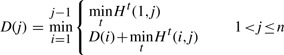 |
The above recurrence is used to fill the array D iteratively from 1 to n. On completion, the best secondary structure assignment can be derived by remembering the index i and type t from which the optimal D(j) is computed.
5 POST-PROCESSING
In the post-processing step, the above-defined successive segments of same type (helical or strand) from the MML inference are examined for moderate curvature. Further, sharp turns are identified and distinguished from coil assignments. Finally, β-sheets are identified by grouping together the assigned strands.
Checking for moderate curvature in helix and strand: The MML inference automatically gives the best (in the information–theoretic sense) piecewise approximation of a curved helix or strand. In a single pass through the secondary structural assignment generated by our method, we check for such curvatures and merge successive segments to form a larger segment.
Each helix (1≤t≤6) and strand (t=7) is represented by a vector. For a helical segment, the vector is the axis of the helix. For the strand segment, the vector is the least-square line fitting its Cα atoms. Two successive segments (of the same model type) are joined to form a single segment if the orientation angle of their vectors is within 30○. [For a detailed description of the finding the axes and orientation angle, see Konagurthu et al. (2008)]
Identifying sharp turns: sharp turns often have geometries that are conformationally similar to a turn of ideal helical models considered in this work. Any assignment of a helical segment of length less than or equal to four residues that is preceded and succeeded by other assigned segments is relabeled as a sharp turn.
However, α-helices often fray or tighten at their N- or C-terminal ends giving a short stretch of π or 310 helical segment. In order not to incorrectly assign these ends as sharp turns, the orientation of a candidate segment is checked against the preceding and succeeding segments and only assigned a sharp turn if the segment's orientation exceeds 45○ relative to its neighbouring segments.
Grouping strands forming β-sheet: all strand segments are extracted from a given assignment. A strand adjacency matrix is computed where two strands are treated to be adjacent if and only if they are in parallel (with orientation angle in the range ± 45○) or anti-parallel (with orientation angle in the range [135○ to 180○] or [−135○ to −180○]) orientation and there exist Van der Waals interactions between at least two pairs of atoms from the segment.
Strands from a β-sheet are then identified and grouped using a complete depth-first search on the strand adjacency matrix.
6 RESULTS
Implementation: a program (SST) implementing the method described in the previous section has been developed in C++ programming language. The program accepts protein coordinates in the Brookhaven PDB format and outputs the secondary structure assignment both at a segment level (stating the start and end point of each secondary structural segment) as well as at a residue level. The program also generates a PyMol script, which allows users to visualize the secondary stucture assignment. (Fig. 1).
Datasets and comparison methods: To study the performance of SST, we consider a dataset of 1737 PDB structures. These structures are the same as the dataset considered by Martin et al. (2005), excluding the structures which have been deprecated or those structures that failed running on any one of the considered methods. (See below.) These structures are divided into four datasets: high-resolution (HRes) dataset with 631 crystal structures solved to 1.7 Å or better; medium resolution (MRes) dataset with 582 crystal structures with resolution between 1.7 and 3 Å resolution; low-resolution (LRes) dataset with 306 structures with resolution >3 Å and finally, a dataset of 218 NMR structures.
In this work, we mainly compare SST with DSSP (Kabsch and Sander, 1983) and STRIDE (Frishman and Argos, 1995) exhaustively on the large structural dataset described earlier. Although there are other programs for secondary structure assignment, we had difficulty finding distributions that we could download. For those we managed to download, we could not install the programs due to unresolvable dependencies in their source code. However, we use a web server 2Struct (Klose et al., 2010), which allows manual submission of queries with a single point of access to a variety of secondary structure assignment methods beyond DSSP and STRIDE such as KAKSI (Martin et al., 2005), PALSSE (Majumdar et al., 2005), STICK (Taylor, 2001), XTLSSTR (King and Johnson, 1999) and P-SEA (Labesse et al., 1997). To facilitate comparisons with other methods, we randomly selected 30 low-resolution structures from the LRes dataset and manually collected the secondary structure assignments from the server. The list of structures used in this experiment can be download from http://www.csse.monash.edu.au/~karun/sst.
Comparison: we first assess the composition of the assigned regular secondary structures (helices and strands) using DSSP, STRIDE and SST over the four datasets described earlier. Overall DSSP assigns 34.5% of residues to helices and 19.9 of residues to strands. STRIDE assigns 37.3 and 27.4%, and SST assigns 40.4 and 25.3% of residues to helices and strands, respectively. In general, DSSP is conservative in assigning regular secondary structures resulting in shorter elements compared with output from STRIDE and SST. Examining the lengths of various secondary structural segments, we observe that the average length of a helix and strand segment assigned by SST is 12.1 and 6.1 residues, respectively. Unlike SST, which states the residue start and end points of each segment, computing the lengths of secondary structural elements from DSSP and STRIDE's output is problematic and error-prone (Majumdar et al., 2005).
Table 1 shows the comparison between SST, DSSP and STRIDE over high, medium, low resolution and NMR structures. To undertake this comparison, the assignments of the three programs are grouped into three classes: helix (of all types), strand and other. DSSP and STRIDE use similar methods for assignment based on detecting hydrogen bonds. Therefore, as one would expect, their assignments are highly similar. SST largely agrees with DSSP and STRIDE when assigning helices. Strands, however, show some disagreement. Visually examining several instances, we find that in many cases SST assigns longer strands than the other two methods. Agreement between strand assignments on NMR structures among the three methods is rather poor. Surprisingly, even DSSP and STRIDE differ enormously in this class even though their assignment methods are quite similar.
Table 1.
Performance of SST compared with DSSP and STRIDE on four datasets: HRes, MRes, LRes and NMR
| HRes (% Agreement) |
MRes (% Agreement) |
|||||
|---|---|---|---|---|---|---|
| Helix | Strand | Total | Helix | Strand | Total | |
| SST versus DSSP | 97.6 | 81.9 | 84.1 | 97.6 | 83.4 | 83.9 |
| SST versus STRIDE | 97.1 | 80.8 | 84.3 | 97.2 | 82.3 | 84.3 |
| STRIDE versus DSSP | 99.4 | 98.5 | 96.7 | 99.4 | 98.9 | 96.9 |
| LRes (% Agreement) |
NMR (% Agreement) |
|||||
| Helix | Strand | Total | Helix | Strand | Total | |
| SST versus DSSP | 97.7 | 84.3 | 82.7 | 98.4 | 53.8 | 51.5 |
| SST versus STRIDE | 97.2 | 82.3 | 83.8 | 94.4 | 78.9 | 83.9 |
| STRIDE versus DSSP | 99.3 | 98.3 | 96.0 | 99.6 | 64.6 | 68.7 |
Columns labelled ‘Helix’ and ‘Strand’ give the percentage agreement of residues assigned as helix and strand, respectively, between the two methods. Column ‘Total’ gives the percentage agreement over three classes: helix, strand and others.
Table 2 extends the comparison of SST with other popular methods for assignment on low-resolution structures. The table shows the percentage agreement of secondary structural assignments between the methods. Consistent with previous comparative studies (Colloc'h et al., 1993; Martin et al., 2005), we see considerable differences in the assignments. In the absence of a universally acknowledged gold standard for assignment, it becomes very difficult (if not impossible) to objectively validate one method to be truly better than the other. The observed differences mainly arise from the different criteria used by the methods. However, manually examining many cases where the methods differ, we find that most disagreements appear at the ends of various (helical or strand) segments. We will use a simple example to highlight the most common type of differences. Figure 2 gives the overall residue-level secondary structure assignment across different methods for a flavodoxin structure from Clostridium beijerinckii (wwPDB ID:5NLL). DSSP and STRIDE assignments are nearly identical to each other. From the figure, small disagreements between methods can be seen around the start and end points of various segments demarcated by SSTs segment view (labelled SST(SEG)). A major difference between DSSP and SST is the region Lys28…Asn34, which SST assigns as a strand. DSSP starts the segment three residues further at Asn31. Inspecting the structure, we find a backbone hydrogen bond between Asp29 and Met1. This might suggest the start of the strand at either Asp29 or one residue upstream at Lys28 as identified by SST. Also, in the region Glu62… Ile73, only SST correctly assigns a π-helical cap (Glu62… Phe66) leading into a α-helix.
Table 2.
Pairwise comparison between secondary structure assignment methods
| DSSP (%) | STRIDE (%) | KAKSI (%) | PALSSE (%) | P-SEA (%) | STICK (%) | XtlSStr (%) | |
|---|---|---|---|---|---|---|---|
| SST | 77.4 | 75.3 | 74.3 | 80.7 | 53.9 | 69.0 | 74.9 |
| DSSP | 86.1 | 76.9 | 76.0 | 57.1 | 71.8 | 77.6 | |
| STRIDE | 67.8 | 74.8 | 48.8 | 64.5 | 73.8 | ||
| KAKSI | 75.4 | 67.4 | 79.4 | 72.9 | |||
| PALSSE | 50.9 | 69.9 | 70.8 | ||||
| P-SEA | 66.6 | 54.1 | |||||
| STICK | 66.7 |
Each cell in the upper-triangular matrix gives the percentage agreement of the residue-level assignments between a pair of methods indicated in the first row and column. The agreement is measured over all three classes: helix, strand and other.
Fig. 2.

Residue-level secondary structure assignment of a 1.75 Å flavodoxin structure from Clostridium beijerinckii. SST residue-level assignment and the segment boundaries are shown in addition to the assignments across multiple methods. For details of the secondary structure codes, see http://www.csse.monash.edu.au/karun/sst/codes.html
To evaluate the consistency of SSTs secondary structural assignments on coordinates solved at different resolutions, we randomly selected 15 protein structures for which both the superseded low-resolution coordinates and the new high-resolution coordinates were available. Table 3 gives the list of considered structures along with the percentage agreement between SSTs assignment at different resolutions. The results indicate that SST produces consistent results on structures determined at different resolutions. The <10% differences (Table 3, last column) in agreement on the chosen structures may well represent genuine structural differences rather than shortcomings of the algorithm.
Table 3.
SST assignment sensitivity to changes in coordinate resolution. Resolution numbers marked with * are taken from the original papers
| Structure name | LRes PDB ID | HRes PDB ID | %Agree |
|---|---|---|---|
| Lysozyme | 2LZH (6.0 Å) | 2ZQ3 (1.6 Å) | 95.3 |
| Ferrochelatase | 1LD3 (2.6 Å) | 1DOZ (1.8 Å) | 97.4 |
| Glutamate Dehydrogenase | 1AUP (2.5 Å) | 1BGV (1.9 Å) | 90.6 |
| Pseudomonas Cytochrome | 151C (2.0 Å) | 351C (1.6 Å) | 93.9 |
| Bence-Jones Protein | 1BJL (2.9 Å)* | 3BJL (2.3 Å) | 90.2 |
| Concanavalin A | 4CNA (2.9 Å)* | 5CNA (2.0 Å) | 91.8 |
| Endochitinase | 1BAA (2.8 Å) | 2BAA (1.8 Å) | 95.5 |
| Ferredoxin Reductase | 1FNR (2.6 Å) | 1FND (1.7 Å) | 95.9 |
| Endonuclease III | 1ABK (2.0 Å) | 2ABK (1.6 Å) | 97.6 |
| Myohemerythrin | 1MHR (2.9 Å)* | 2MHR (1.3 Å) | 92.4 |
| Phosphofructokinase | 5PFK (7.0 Å)* | 6PFK (2.6 Å) | 95.3 |
| Serine Protease Inhibitor | 1QLP (2.9 Å) | 2PSI (2.0 Å) | 95.4 |
| Dimeric Hemoglobin | 1SDH (2.4 Å)* | 3SDH (1.4 Å) | 98.4 |
| Glutathione Reductase | 1GRS (3.0 Å)* | 3GRS (1.5 Å) | 94.4 |
| Calmodulin Fragment TR2C | 1TRC (3.6 Å) | 1FW4 (1.7 Å) | 93.9 |
Further, to illustrate the reliable segmentation produced by SST on structures with long, curved helices and strands, we chose two structures: Leucine zipper protein (wwPDB: 1NKP) composed of very long helices and Sucrose-specific porin protein (wwPDB: 1A0S) composed of long, curved strands forming β-barrels. Although SST initially breaks the curved segments into smaller pieces, the post-processing step explained in Section 5 reconstitutes these pieces back correctly into fuller segments. [Fig. 3 gives SSTs assignment on the porin protein (1A0S). The figure shows that the curved strands of the β-barrels have been reconstituted and grouped reasonably well in the post-processing step.]
Fig. 3.
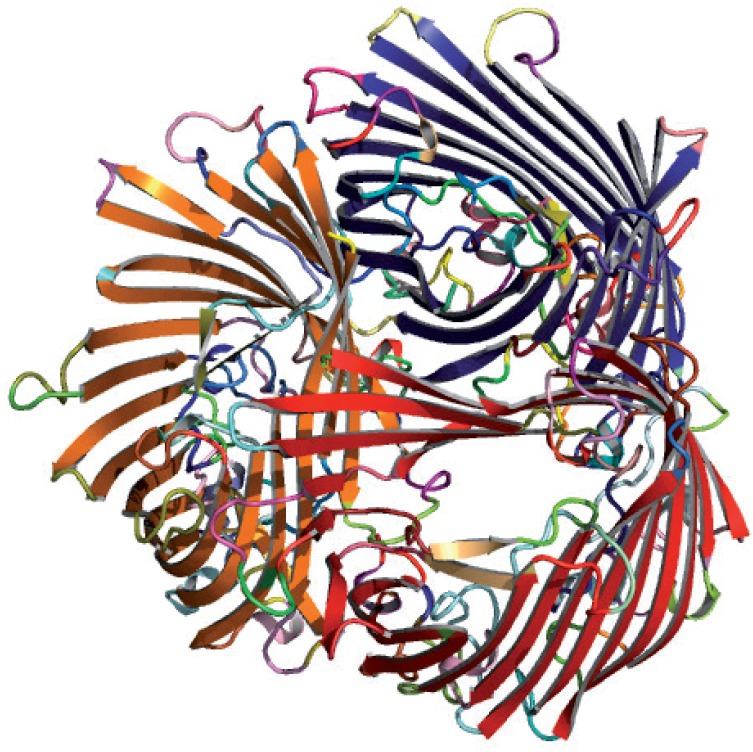
Automatically generated PyMol image of SSTs secondary structural assignment on sucrose-specific porin (ScrY) from Salmonella typhimurium (wwPDB: 1A0S)
Finally, as a difficult case we consider the 10 Å resolution protein coordinates of Elongation Factor Tu (GDB.Kirromycin) from Escherichia coli (wwPDB: 1qzd) solved using Cryo-Electron Microscopy. Its wwPDB file contains only Cα coordinate information. DSSP, STRIDE and P-SEA fail to process such information as the coordinates of other atoms are needed to decipher Hydrogen bonds. KAKSI and XTLSSTR are able to process this structure but assign all residues in the chain to coil. Of the considered methods, only SST, PALSSE and STICK assigned any secondary structure. For lack of space, the overall residue-level assignment across these three methods are presented in Figure 1 of the supplementary text. Examining the structure, PALSSE consistently overestimates the regular secondary structural regions by a large margin. STICK performs well, especially in identifying β-strands. However, it miscalculates several secondary structural elements. In comparison, SST produced the most reasonable segmentation of the three methods on visual inspection of the structure.
7 CONCLUSION
Reliable secondary structure assignment is an important problem. We have developed a novel information theoretic method to address this problem using the Bayesian framework of MML inference. Careful examination of the results over a large number of structures suggests that our method gives consistent assignments even on low-resolution data. We note that our method uses a dictionary of models composed of ideal secondary structural elements. The details of the models are explicit and open to scrutiny. It is likely that these models can be improved. (‘Essentially, all models are wrong, but some are useful.’—George Box.) However, modification to the models is an improvement if, and only if, it yields extra compression.
8 ACKNOWLEDGEMENTS
LA thanks Sally P. Allison for support. ASK acknowledges helpful discussions with Peter J. Stuckey during the development of this work, and thanks to Rekha Amar for proof reading this manuscript.
Funding: ASK's research is supported by Monash Larkins Fellowship.
Conflict of Interest: none declared.
REFERENCES
- Andersen C.A., Rost B. Secondary structure assignment. In: Gu J., Bourne P.E., editors. Structural Bioinformatics. Wiley-Blackwell; 2009. pp. 459–484. [Google Scholar]
- Bayes T., Price R. An essay towards solving a problem in the doctrine of chance. Philos. Trans. Roy. Soc. Lond. 1763;53:370–418. [Google Scholar]
- Colloc'h N., et al. Comparison of three algorithms for the assignment of secondary structure in proteins. Protein Eng. 1993;6:377–382. doi: 10.1093/protein/6.4.377. [DOI] [PubMed] [Google Scholar]
- Conway J.H., Sloane N.J.A. On the Voronoi regions of certain lattices. SIAM Journal on Algebraic and Discrete Methods. 1984;5:294–305. [Google Scholar]
- Cuff J.A., Barton G.J. Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. Proteins. 1999;34:508–519. doi: 10.1002/(sici)1097-0134(19990301)34:4<508::aid-prot10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Dupuis F., et al. Protein secondary structure assignment through Voronoi tessellation. Proteins. 2004;55:519–528. doi: 10.1002/prot.10566. [DOI] [PubMed] [Google Scholar]
- Fodje M., Al-Karadaghi S. Occurrence, conformational features and amino acid propensities for the π-helix. Protein Eng. 2002;15:353–358. doi: 10.1093/protein/15.5.353. [DOI] [PubMed] [Google Scholar]
- Frishman D., Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kamat A.P., Lesk A.M. Contact patterns between helices and strands of sheet define protein folding patterns. Proteins: Struct. Funct. Bioinformatics. 2007;66:869–876. doi: 10.1002/prot.21241. [DOI] [PubMed] [Google Scholar]
- Kearsley S.K. On the orthogonal transformation used for structural comparisons. Acta. Cryst. 1989;A45:208–210. [Google Scholar]
- King S., Johnson W. Assigning secondary structure from protein coordinate data. Proteins. 1999;35:313–320. [PubMed] [Google Scholar]
- Klose D.P., et al. 2Struct: the secondary structure server. Bioinformatics. 2010;20:2624–2625. doi: 10.1093/bioinformatics/btq480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konagurthu A.S., et al. Structural search and retrieval using tableau representation of protein folding patterns. Bioinformatics. 2008;24:645–651. doi: 10.1093/bioinformatics/btm641. [DOI] [PubMed] [Google Scholar]
- Konagurthu A.S., et al. Piecewise linear approximation of protein structures using the principle of minimum message length. 2011;27:i43i51. doi: 10.1093/bioinformatics/btr240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labesse G., et al. P-SEA: a new efficient assignment of secondary structure from C alpha trace of proteins. Comput Appl Bio Sci. 1997;13:291–295. doi: 10.1093/bioinformatics/13.3.291. [DOI] [PubMed] [Google Scholar]
- Lesk A.M., Hardman K.D. Computer-generated schematic diagrams of protein structures. Science. 1982;216:539–540. doi: 10.1126/science.7071602. [DOI] [PubMed] [Google Scholar]
- Levitt M., Greer J. Automatic identification of secondary structure in globular proteins. J. Mol. Biol. 1977;114:181–239. doi: 10.1016/0022-2836(77)90207-8. [DOI] [PubMed] [Google Scholar]
- Majumdar I., et al. PALSSE: A program to delineate linear secondary structural elements from protein structures. BMC Bioinformatics. 2005;6:202. doi: 10.1186/1471-2105-6-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J., et al. Protein secondary structure assignment revisited: a detailed analysis of different assignment methods. BMC Struct. Biol. 2005;5:17. doi: 10.1186/1472-6807-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L., Corey R. Configurations of polypeptide chains with favored orientations around single bonds: Two new pleated sheets. Proc. Natl. Acad. Sci. USA. 1951;37:729–740. doi: 10.1073/pnas.37.11.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson J.S. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- Richards F.M., Kundrot C.E. Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure. Proteins. 1988;3:71–78. doi: 10.1002/prot.340030202. [DOI] [PubMed] [Google Scholar]
- Rissanen J. A universal prior for integers and estimation by minimum description length. Ann. Stat. 1983;11:416–431. [Google Scholar]
- Robson B., Garnier J. Introduction to Proteins and Protein Engineering. Amsterdam: Elsevier Science Ltd; 1986. [Google Scholar]
- Shannon C.E. A mathematical theory of communication. Bell Syst. Technical Jrnl. 1948;27:379–423. [Google Scholar]
- Sklenar H., et al. Describing protein structure: a general algorithm yielding complete helicoidal parameters and unique overall axis. Proteins. 1989;6:46–60. doi: 10.1002/prot.340060105. [DOI] [PubMed] [Google Scholar]
- Srinivasan R., Rose G.D. A physical basis for protein secondary structure. Proc. Natl. Acad. Sci. USA. 1999;96:14258–14263. doi: 10.1073/pnas.96.25.14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor W.R. Defining linear segments in protein structures. J. Mol. Biol. 2001;310:1135–1150. doi: 10.1006/jmbi.2001.4817. [DOI] [PubMed] [Google Scholar]
- Wallace C.S., Boulton D.M. An information measure for classification. Comput. J. 1968;11:185–194. [Google Scholar]
- Wallace C.S. Statistical and Inductive Inference using Minimum Message Length. Information Science and Statistics. SpringerVerlag; 2005. [Google Scholar]
- Zhang W., et al. Assessing secondary structure assignment of protein structures by using pairwise sequence-alignment benchmarks. Proteins. 2008;71:61–67. doi: 10.1002/prot.21654. [DOI] [PubMed] [Google Scholar]