

Abstract
The close phylogenetic relationship of the important pathogen Streptococcus pneumoniae and several species of commensal streptococci, particularly Streptococcus mitis and Streptococcus pseudopneumoniae, and the recently demonstrated sharing of genes and phenotypic traits previously considered specific for S. pneumoniae hamper the exact identification of S. pneumoniae. Based on sequence analysis of 16S rRNA genes of a collection of 634 streptococcal strains, identified by multilocus sequence analysis, we detected a cytosine at position 203 present in all 440 strains of S. pneumoniae but replaced by an adenosine residue in all strains representing other species of mitis group streptococci. The S. pneumoniae-specific sequence signature could be demonstrated by sequence analysis or indirectly by restriction endonuclease digestion of a PCR amplicon covering the site. The S. pneumoniae-specific signature offers an inexpensive means for validation of the identity of clinical isolates and should be used as an integrated marker in the annotation procedure employed in 16S rRNA-based molecular studies of complex human microbiotas. This may avoid frequent misidentifications such as those we demonstrate to have occurred in previous reports and in reference sequence databases.
INTRODUCTION
Exact differentiation of the important pathogen Streptococcus pneumoniae and its close commensal relatives is a prerequisite for full appreciation of their clinical significance, pathogenic properties, and epidemiology both in routine clinical microbiology and in research. Although a number of phenotypic traits were previously assumed to be specific markers of S. pneumoniae, recent reports demonstrated that such properties, including optochin susceptibility, capsule production, and bile solubility, are also present in a substantial proportion of isolates of the commensal Streptococcus mitis (2, 3, 6, 19, 23, 40). Moreover, the realization that clinical isolates of S. pneumoniae, e.g., many isolates from conjunctivitis, do not express a capsule (hence the term “atypical pneumococci”) further emphasizes this problem (4, 7). Similar problems apply to molecular identification methods. Because the full 16S rRNA gene sequences of S. pneumoniae, Streptococcus pseudopneumoniae, S. mitis, and Streptococcus oralis are more than 99% identical (21), attempts to identify clinical isolates by BLAST searches with an unidentified 16S rRNA gene sequence against sequences in recognized databases are often unsuccessful or may provide misleading results. This is becoming an increasing problem due to the frequent and automated use of this procedure in clinical microbiology and in the annotation process employed in attempts to map complex microbiotas, such as in the Human Microbiome Project. Likewise, the suggested identification of clinical isolates of S. pneumoniae by detection of genes encoding regulatory proteins of capsule biosynthesis, e.g., cpsA/wzg, or putative virulence proteins such as autolysin (lytA), pneumolysin (ply), the surface protein PspA (pspA) (1, 30) is bound to result in a number of misidentifications due to the presence of homologs of these genes in a proportion of commensal streptococci (19, 20, 23, 27, 38, 40).
The biological explanation for the described problems is the phylogenetic history of these streptococcal species. In spite of striking differences in pathogenic potential, S. pneumoniae and S. mitis, S. pseudopneumoniae, Streptococcus oralis, Streptococcus infantis, and Streptococcus oligofermentans share an immediate common ancestor. We have suggested that reductive evolution of the genomes of S. mitis and S. pseudopneumoniae from an S. pneumoniae-like ancestor during a process of adaptation to a commensal lifestyle explains the patchy occurrence of virulence genes and phenotypic properties usually associated with S. pneumoniae in these species (23). In addition, horizontal gene transfer and homologous recombination contribute to the blurring of species boundaries and the ensuing difficulties in unequivocal identification of clinical isolates (8, 14, 15, 17, 23, 40). Thus, evidence of exchange of homologous gene sequences between species invalidates the many suggested identification procedures based on sequence comparisons of single housekeeping genes such as sodA, gdh, rpoA, recA, recN, etc. (11, 12, 22, 28, 31, 34, 41).
One crucial problem in the validation of new identification procedures for streptococci has been the lack of a “gold standard.” As a result of improved sequencing technologies, identification based on sequence comparisons of multiple housekeeping genes has become technically and economically feasible. Multilocus sequence analysis (MLSA) eliminates the potential problems resulting from homologous recombination and has been applied with success in the identification of several bacterial species that are difficult to differentiate by simpler methods (5, 13, 17, 18, 23, 29, 25, 33). Schemes based on four to seven gene loci have proven valuable in differentiation of Streptococcus species (5, 17, 18). Specifically, an MLSA-based procedure for identification of S. pneumoniae was reported by Hanage et al. (16). Some of these identification procedures can now be performed online with reference to comprehensive reference databases of concatenated sequences of six or seven genes (http://spneumoniae.mlst.net/sql/concatenate/isitaddto.asp and http://viridans.emlsa.net/). While providing a gold standard for reference and research purposes, MLSA is not yet feasible in routine clinical microbiology due to the economics and the workload involved.
During analysis of 16S rRNA gene sequences of mitis group streptococci, we noticed a nucleotide position at which sequences of S. pneumoniae strains differed from those of all other mitis group streptococci. To test whether this distinct sequence signature can be used for identification of S. pneumoniae, we analyzed 646 strains that were unequivocally identified to species level by MLSA and confirmed the validity of the signature for differentiation of S. pneumoniae from other mitis group streptococci.
MATERIALS AND METHODS
Bacterial strains.
The study was based on DNA sequences of 16S rRNA genes and selected housekeeping genes of 634 strains of mitis group streptococci and included designated type strains of all 13 recognized species of the mitis group of the genus Streptococcus. All strains were identified by cluster analysis of concatenated housekeeping genes (multilocus sequence analysis [MLSA]) extracted from the respective databases or determined by us. Most of the strains were identified by MLSA analysis of concatenated partial sequences of the housekeeping genes map, pfl, ppaC, pyk, rpoB, sodA, and tuf according to procedures and principles described by Bishop et al. (5) (http://viridans.emlsa.net). A minor proportion of the strains (SK618, SK673, SK676, SK680, SK848, SK851, SK852, SK853, SK854, SK856, SK858, SK862, SK865, and SK867) were identified as S. pneumoniae based on phylogenetic analysis of concatemers of the housekeeping genes gdh, ddl, rpoB, and sodA as described by Kilian et al. (23).
Sequences of 16S rRNA genes and the housekeeping gene sequences used for the MLSA were retrieved by BLASTN on the NCBI database against complete genome sequences of strains of relevant Streptococcus species, using the sequence from S. pneumoniae strain TIGR4 (NCBI gene ID 929754) as the query sequence. All four copies of the 16S rRNA gene were retrieved from the genomes to enable detection of potential sequence polymorphism as previously demonstrated for mitis group streptococci (23). The Illumina sequence data files for 240 strains of S. pneumoniae from a study recently reported by Croucher et al. (9) were available only as unassembled short reads generated by Illumina sequencing. These sequences, available at http://www.ebi.ac.uk/ena/data/view/ERP000139, were downloaded as “.fastq” files. The sequences were extracted and placed in a multi-Fasta file for each strain, and a comparative assembly (32) using the “AMOScmp-shortReads-alignmentTrimmed” command in the AMOS assemble utilities version 3.1.0 (http://sourceforge.net/apps/mediawiki/amos) was performed with the 16S rRNA sequence from S. pneumoniae TIGR4 as the template.
DNA sequencing.
The bacteria for which the relevant sequences were not already available in the respective databases were cultivated on Todd-Hewitt agar (Difco Laboratories, Detroit, MI) incubated for 2 days at 37°C in an anaerobic chamber. For preparation of crude genomic DNA, a 1-μl loopful of bacteria harvested from an agar plate culture was suspended in 100 μl H2O and 400 μl 0.05 M NaOH was added; after incubation at 60°C for 45 min, the sample was neutralized by adding 46 μl 1 M Tris-HCl (pH 7.0), and 5 μl of a 1:100 dilution of this was used for the PCR. Primers for amplification of the 16S rRNA fragment corresponding to positions 10 to 30 and 501 to 522 were 5′-TGGCTCAGGACGAACGCTGGC-3′ and 5′-CGGCTGCTGGCACGTAGTTAGC-3′. Sequences of the housekeeping genes map, pfl, ppaC, pyk, rpoB, sodA, and tuf were determined as described by Bishop et al. (5) (http://viridans.emlsa.net). All primers were purchased from DNA Technology (Aarhus, Denmark). For the PCRs we used approximately 1 ng whole-cell DNA as the template and Ready-To-Go PCR beads (Amersham Pharmacia Biotech, Uppsala, Sweden). The temperature profile for the PCR was an initial denaturation at 94°C for 5 min, followed by 30 cycles at 94°C for 1 min, 60°C for 1 min, and 72°C for 2 min, followed by a final extension at 72°C for 8 min. Amplicons were purified using Wizard minicolumns (Promega, Madison, WI). Sequencing of both strands of the amplified fragments was achieved with the same primers at GATC Biotech.
Sequencing of each of the four individual copies of the 16S rRNA gene in the genomes of selected strains was performed as described previously (23). Briefly, genome fragments each containing one copy of the 16S rRNA gene were separated by pulsed-field gel electrophoresis (PFGE) of genomic DNA prepared from bacteria harvested from a Todd-Hewitt broth culture and cleaved in the agar plugs with 1 U of the intronic endonuclease I-CeuI (New England BioLabs, Hertfordshire, United Kingdom), which recognizes a sequence unique to 23S rRNA genes (39). The individual fragments were purified from the gel using a Qiagen gel extraction kit (Qiagen) and subjected to amplification and sequencing of the 16S rRNA gene as described above.
Sequence comparisons and MLSA.
All sequences were aligned in MEGA version 5.05. Housekeeping gene sequences were then trimmed to match the templates from http://viridans.emlsa.net, concatenated, and subjected to phylogenetic analysis using the Minimum evolution algorithm (Jones-Taylor-Thornton model) with the pairwise deletion option. Identification of strains was done according to their clustering relative to designated type strains of relevant Streptococcus species.
Restriction endonuclease analysis.
A 286-bp fragment of the 16S rRNA gene (positions 81 to 366 in the S. pneumoniae strain R6 16S rRNA gene) was amplified using the primers Pn-16S-up (ATGAGTTGCGAACGGGTGAGTAA) and Pn-16S-do (ATTGCCGAAGATTCCCTACTGCT). The PCR was performed on approximately 0.5 ng genomic DNA in a 25-μl reaction volume containing 10 pmol of each primer, using PuReTaq Ready-To-Go PCR beads (GE Healthcare). PCR parameters were 94°C for 5 min, 30 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 2 min, and then an extension at 72°C for 8 min. The PCR products were analyzed by agarose gel electrophoresis. Digestion with the restriction endonuclease BsiHKAI (New England BioLabs) was performed in a 50-μl volume containing 20 μl PCR product, 3 μl 10× NEBuffer 4, 5 μl bovine serum albumin (BSA) (10 mg/ml), and 20 U enzyme. After incubation at 65°C for 1 h, the DNA fragments were analyzed by agarose gel electrophoresis.
RESULTS
Identity of strains.
Based on the phylogenetic analysis of concatemers of housekeeping genes, 440 strains were identified as S. pneumoniae, 60 as S. mitis, 41 as S. oralis, 17 as S. oligofermentans, 25 as S. infantis, 10 as belonging to the so-called “S. mitis biovar 2” (which constitutes a distinct, yet-to-be described species closely related to S. oralis), and five as S. pseudopneumoniae. Type strains and single additional representatives of the remaining species of the mitis group formed distinct lineages in the tree. A total of 28 strains formed separate clusters unrelated to recognized species. A tree based on a selection of strains (to facilitate visibility of individual lineages) is shown in Fig. 1.
Fig 1.
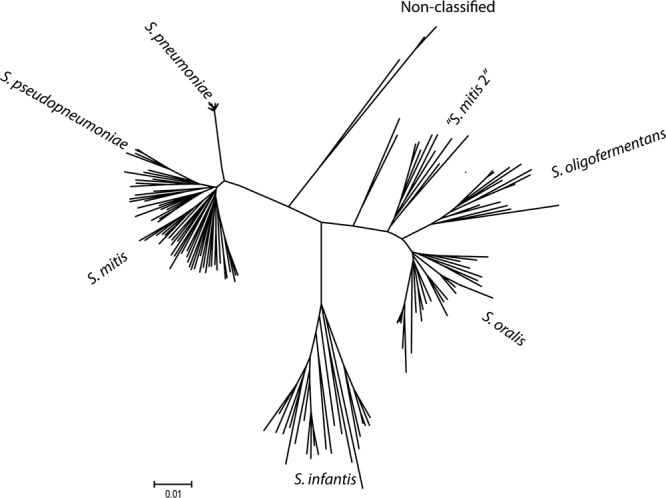
Phylogenetic tree based on concatenated sequences of seven housekeeping gene sequences, constructed with the Minimum evolution algorithm and used as the “gold standard” for identification of the 634 mitis group streptococcal strains included in the study. The clusters were named according to the clustering of designated type strains of the respective species.
Comparison of the 16S rRNA gene sequences showed that all 440 strains verified as S. pneumoniae based on the MLSA analysis had a cytosine (C) at position 203 (relative to the annotated 16S rRNA gene sequence in the genome of strain R6 [accession no. AE007317.1]; the positions in the four 16S rRNA genes are 15366, 1696983, 1794533, and 1856458). All other strains had an adenine (A) in this position. We have previously demonstrated intragenomic polymorphisms among the four 16S rRNA genes in mitis group streptococci, but in no case was polymorphism involving the mentioned signatures at position 203 seen. With three exceptions, all 440 S. pneumoniae strains showed a fully conserved stretch of 15 nucleotides flanking the signature C at position 203. The sequence and exceptions are shown in Table 1.
Table 1.
Sequences (positions 193 to 209) flanking the S. pneumoniae-specific cytosine at position 203 in the 16S rRNA genes of mitis group Streptococcus species
| Species | Allele or strain(s) | Sequencea | Comment |
|---|---|---|---|
| S. pneumoniae | Major allele | AAAAGGTGCACTTGCAT | Includes type strain |
| EU NPO1 | AAAAAGTGCACTTGCAT | Accession no. AGQH01000017 | |
| VN O27 | AAAAGGTGAACTTGCAT | Accession no. AY525794.1 | |
| 06-01-003MEF | AAAAAAGGTGCATGCAT | Accession no. AEGF00000000.1 | |
| S. mitis | Major allele | AAAAGGTGCAATTGCAT | Includes type strain |
| A1435,SK626,SK636, SK674 | AAAAGGTGCAAATGCAT | ||
| SK624 | GAAAGGTGCAACTGCAC | ||
| SK629 | GAAAGATGCAATTGCAT | ||
| S. pseudopneumoniae | AAAAGGTGCAAATGCAT | ||
| S. oralis | Major allele | AAAAGGTGCAATTGCAT | Includes type strain |
| SK664 | GAAAGGTGCAATTGCAT | ||
| S. mitis 2 | Major allele | GAAAGGTGCAAATGCAT | |
| SK79, SK429 | AAAAGGTGCAAATGCAT | ||
| S. oligofermentans | GAAAGGTGCAAATGCAT | Includes type strain | |
| S. infantis | Major allele | GAAAGGTGCAATTGCAC | Includes type strain |
| SK660 | GAAAGGTGCAAATGCAC | ||
| SK385,48291 | AAAAGGTGCAATTGCAT | ||
| SK655 | GAAAGGTGCAACTGCAC | ||
| S. peroris | AAAAGGTGCAATTGCAC | Includes type strain | |
| S. sanguinis | GAAAGGGGCAATTGCTC | Type strain | |
| S. gordonii | GAAAGGTGCAATTGCAC | Type strain | |
| S. australis | GAAAGGGGCAATTGCTC | Type strain | |
| S. parasanguinis | GAAAGGTGCAAATGCAT | Type strain | |
| S. cristatus | GAAAGGTGCAAATGCAC | Type strain | |
| S. sinensis | AAAAGATGCAATTGCAT | Type strain | |
| S. lactis | GAAAGATGCAATTGCAT | Type strain |
The nucleotide at position 203 is in bold.
A BLASTN search of the NCBI database identifies 17 16S rRNA gene sequences of strains designated S. pneumoniae that deviated from the mentioned patterns: strains NCTC 7466 (accession no. AJ001248), HK P116 (accession no. AY525791.1), SI P25 (accession no. AY525793.1), HK P55 (accession no. AY525790.1), Kor 145 (accession no. AY525788.1), SI P7 (accession no. AY525792.1), HER 1055 (accession no. AJ617796.1), 578 (accession no. GU326243.1), 101/87 (accession no. GU326241.1), 1504 (accession no. GU326242.1), Ejeff (accession no. GU561401.1), C30 (accession no. GU907525.1), PTO6 (accession no. GU561400.1), CCARM 4033 (accession no. GU045397.1), ToTT (accession no. FJ823147.1), CCRI-15796 (accession no. EU156782.1), and CCRI-14740 (accession no. EU156780.1). Strain NCTC 7466 is the same as strain D39 and its rough variant R6, both of which have been genome sequenced (accession numbers NC008533 and NC003098). All copies of the 16S rRNA sequences retrieved from these two genomes show the S. pneumoniae-characteristic cytosine at position 203 and do not confirm the 16S rRNA sequence under accession number AJ001248. Five sequences (HK P116, SI P25, HK P55, Kor 145, and SI P7) originated in a Korean study of “phenotypically and genotypically aberrant Streptococcus pneumoniae strains” isolated from Asian countries (24). Analysis of the deposited partial sequences of the glucose-6-phosphate dehydrogenase (gdh) genes of these strains supports that the identification was incorrect and that they are S. mitis strains. Strain HER 1055 is referred to and validated as S. mitis in the publication from which sequence accession no. AJ617796.1 originates (35) but is incorrectly listed as S. pneumoniae in the NCBI database. Accordingly, 9 out of 10 other sequences of strain HER 1055 in the NCBI database are labeled S. mitis. The strain corresponding to the sequence CCARM 4033 (accession no. GU045397.1) was identified based on single-locus sequencing (rpoA) (31), which according to other reports is insufficient (17). Among the remaining sequences, the identity of strain 101/87 is discussed in several studies, one of which concludes that the strain “may represent either an atypical or incorrectly classified organism” (40). Another, more recent study concludes that strain 101/87 “is as distant (or close) from pneumococci as it is from S. mitis” (35). There are no publications corresponding to the remaining sequences that deviate from the pattern, and the validity of the identifications therefore cannot be further analyzed.
A search of human microbiome data at NCBI and the RDP-II database identified the following sequences generated from uncultured material: clone 4V4 (accession no. AM157442.1) from an analysis of bacteria in breast milk (26) and ATC H22 37 (GU425360.1) from a comprehensive mapping of the human oral microbiome (10). Both sequences were annotated as S. pneumoniae, which according to our findings is incorrect. Contrasting examples were also disclosed among sequences generated in the human oral microbiome study (10). The sequences CMC176, CMC177, CMC178, CMC179, CMC183, CMC186, CMC188, CMC189, CMC192, CMC193, CMC195, CMC196, CMC199, CMC784, CMC795, CMC794, CMC792, CMC791, CMC787, CMC786, CMC785, CMC781, CMC778, CMC796, CMC782, and CMC783 were annotated as S. mitis but, according to the characteristic cytosine at position 203, belong to S. pneumoniae. A search in the RDP-II database did not reveal any strains annotated as S. infantis, S. oralis, S. peroris, S. pseudopneumoniae, S. sanguinis, S. gordonii, S. parasanguinis, or S. cistatus that had the pneumococcal signature cytosine.
Detection of the signature C by restriction enzyme analysis.
The restriction endonuclease BsiHKAI (New England BioLabs) cleaves the sequence 5′…GWGCW↓C…3′ (W = A or T; C corresponds to the signature cytosine at position 203) and thus recognizes the sequence that includes the C at position 203 in all 440 confirmed strains of S. pneumoniae except strain 06-01-003MEF (Table 1). To examine whether restriction fragment length pattern analysis would be useful for validation of identification of S. pneumoniae, we digested PCR amplicons of the relevant 16S rRNA gene sequence in 114 validated strains of S. pneumoniae and 57 strains of S. mitis, S. oralis, S. pseudopneumoniae, and S. infantis. With one exception (S. pneumoniae clinical isolate 183/95), all strains of S. pneumoniae showed the expected banding pattern, while the PCR amplicons of all the strains representing other species remained uncleaved (Fig. 2). To analyze why the 16S rRNA gene amplicon of this strain was only partially cleaved by the endonuclease BsiHKAI, the amplicon was sequenced. In both directions, the sequence showed clear evidence of a mixture of T and C at position 201, suggesting different alleles in the four rRNA operons in the genome. Only the sequence with C at this position is recognized by the restriction endonuclease.
Fig 2.
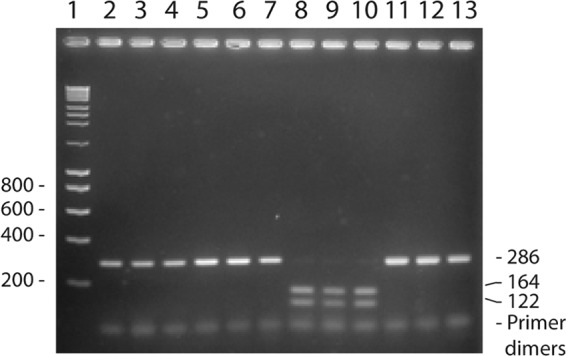
Detection of the signature C at position 203 in the 16S rRNA gene in S. pneumoniae. A 286-bp fragment of the 16S rRNA gene was amplified by PCR and subsequently cleaved with the restriction endonuclease BsiHKAI, which recognizes a sequence including the C at position 203. Lane 1, molecular weight marker; lanes 2 to 7, uncleaved PCR products; lanes 8 to 13, PCR products cleaved with BsiHKAI. Lanes 2 and 8, S. pneumoniae TIGR4; lanes 3 and 9, S. pneumoniae R6; lanes 4 and 10, S. pneumoniae SK856; lanes 5 and 11, S. oralis SK100; lanes 6 and 12, S. oralis SK304; lanes 7 and 13, S. mitis SK142 (NCTC 12261T).
DISCUSSION
Frequent homologous recombination within and between Streptococcus species renders identification based on sequences of single loci problematic (5, 14, 15, 17, 18, 23). With this background, it is remarkable that the cytosine at position 203 in the 16S rRNA gene of S. pneumoniae appears to be fully conserved in the population of S. pneumoniae while strains of all other mitis group streptococci have an adenosine at this position. The evidence supporting this conclusion is based on our analysis of 440 strains of S. pneumoniae and 194 strains representing other species, all identified by multilocus sequence analysis and with the inclusion of relevant designated type strains. This distinguishing sequence signature that is directly detectable by sequence analysis or, alternatively, by restriction fragment length polymorphism (RFLP) analysis of a PCR amplicon of the 16S rRNA gene provides a simple and reliable means of identification or confirmation of S. pneumoniae isolates.
Our results demonstrate several examples of published studies in which strains of S. mitis were misidentified as S. pneumoniae, resulting in incorrect conclusions (24), and of published and unpublished sequences in databases incorrectly labeled as S. pneumoniae. This is a source of confusion and may result in further misidentifications when such sequences are used as reference in BLASTN analyses of sequences generated from unidentified bacterial isolates. One notable example is the 16S rRNA gene sequence of S. pneumoniae strain NCTC 7466 (accession no. AJ001248.1), which shows an adenosine at position 203. Analysis of all four 16S rRNA gene sequences in the genomes of both strain D39, which is the original designation for NCTC 7466, and strain R6, which is an acapsular derivative of the same strain, fail to confirm this. All sequences in these genomes show the S. pneumoniae-specific cytosine at position 203 and are in full agreement with our conclusions.
Automated BLAST analysis of 16S rRNA gene sequences has become a widely used method for molecular mapping of complex microbiotas, e.g., in the Human Microbiome Project, which often involves thousands or millions of sequences. In such studies, the species assignment of sequences is usually done automatically according to their closest fit in the databases. However, due to the relative conservation of 16S rRNA sequences among streptococci, particularly in S. pneumoniae, S. mitis, S. oralis, and S. infantis, separation to species level is often not achievable or may result in misidentifications. This study revealed several examples of misidentified sequences generated and annotated as part of studies of the human oral microbiome and of bacteria present in human breast milk (10, 26). As distinction between S. pneumoniae and commensal streptococci is important for understanding their ecology and relationships with the host, we recommend that the S. pneumoniae-specific signature be used as an integrated part of the annotation procedure employed in studies where these species are relevant.
The conserved C corresponding to position 203 in the 16S rRNA sequence of S. pneumoniae strain R6 is located in helix 11 in the equivalent Escherichia coli 16S rRNA sequence (accession no. J01695) (37). Conservation of this nucleotide, which is unique to S. pneumoniae among mitis group streptococci, is intriguing and unexpected in a species that is known to exchange DNA with its close relatives. The bacterial ribosome is a very complex structure and consists of 23S, 16S, and 5S rRNAs and about 50 proteins. The start codon of mRNA molecules becomes positioned in the ribosome by base pairing between the 3′ end of 16S rRNA and the Shine-Dalgarno sequence just upstream of the mRNA start codon, although the precise mechanism of translation initiation is still unclear (36). We speculate that an interaction between a pneumococcus-specific ribosomal protein and helix 11 in the 16S rRNA molecule is crucial and thereby precludes mutations in this region without simultaneous changes in the protein.
In conclusion, this study identified a remarkable conservation of a cytosine residue at position 203 in the 16S rRNA gene of S. pneumoniae, which in all other species of mitis group streptococci is replaced by an adenosine. Detection of this S. pneumoniae-specific sequence signature by sequence analysis or, indirectly, by RFLP analysis using the restriction endonuclease BsiHKAI is an easy and reliable means of identification of S. pneumoniae in clinical microbiology and as an integrated part the annotation procedure in high-throughput molecular analyses of complex microbiotas based on 16S rRNA sequencing.
ACKNOWLEDGMENT
This study was supported by grant 10-083748 from the Danish Research Council for Health and Disease.
Footnotes
Published ahead of print 21 March 2012
REFERENCES
- 1. Abdeldaim G, et al. 2009. Is quantitative PCR for the pneumolysin (ply) gene useful for detection of pneumococcal lower respiratory tract infection? Clin. Microbiol. Infect. 15:565–570 [DOI] [PubMed] [Google Scholar]
- 2. Arbique JC, et al. 2004. Accuracy of phenotypic and genotypic testing for identification of Streptococcus pneumoniae and description of Streptococcus pseudopneumoniae sp. nov. J. Clin. Microbiol. 42:4686–4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balsalobre L, et al. 2006. Molecular characterization of disease-associated streptococci of the mitis group that are optochin susceptible. J. Clin. Microbiol. 44:4163–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berrón S, Fenoll A, Ortega M, Arellano N, Casal J. 2005. Analysis of the genetic structure of nontypeable pneumococcal strains isolated from conjunctiva. J. Clin. Microbiol. 43:1694–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bishop CJ, et al. 2009. Assigning strains to bacterial species via the internet. BMC Biol. 7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bosshard PP, Abels S, Altwegg M, Böttger EC, Zbinden R. 2004. Comparison of conventional and molecular methods for identification of aerobic catalase-negative gram-positive cocci in the clinical laboratory. J. Clin. Microbiol. 42:2065–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carvalho MGS, Steigerwalt AG, Thompson T, Jackson D, Facklam RR. 2003. Confirmation of nontypeable Streptococcus pneumoniae-like organisms isolated from outbreaks of epidemic conjunctivitis as Streptococcus pneumoniae. J. Clin. Microbiol. 41:4415–4417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chi F, Nolte O, Bergmann C, Ip M, Hakenbeck R. 2007. Crossing the barrier: evolution and spread of a major class of mosaic pbp2x in Streptococcus pneumoniae, S. mitis and S. oralis. Int. J. Med. Microbiol. 297:503–512 [DOI] [PubMed] [Google Scholar]
- 9. Croucher NJ, et al. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dewhirst FE, et al. 2010. The human oral microbiome. J. Bacteriol. 192:5002–5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Drancourt M, Roux V, Fournier PE, Raoult D. 2004. rpoB gene sequence-based identification of aerobic Gram-positive cocci of the genera Streptococcus, Enterococcus, Gemella, Abiotrophia, and Granulicatella. J. Clin. Microbiol. 42:497–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Glazunova OO, Raoult D, Roux V. 2010. Partial recN gene sequencing: a new tool for identification and phylogeny within the genus Streptococcus. Int. J. Syst. Evol. Microbiol. 60:2140–2148 [DOI] [PubMed] [Google Scholar]
- 13. Godoy D, et al. 2003. Multilocus sequence typing and evolutionary relationships among the causative agents of melioidosis and glanders, Burkholderia pseudomallei and Burkholderia mallei. J. Clin. Microbiol. 41:2068–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hakenbeck R, et al. 2001. Mosaic genes and mosaic chromosomes: intra- and interspecies genomic variation of Streptococcus pneumoniae. Infect. Immun. 69:2477–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanage WP, Fraser C, Spratt BG. 2005. Fuzzy species among recombinogenic bacteria. BMC Biol. 7;. 3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hanage WP, et al. 2005. Using multilocus sequence data to define the pneumococcus. J. Bacteriol. 187:6223–6230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoshino T, Fujiwara T, Kilian M. 2005. Use of phylogenetic and phenotypic analyses to identify nonhemolytic streptococci from bacteremic patients. J. Clin. Microbiol. 43:6073–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jensen A, Kilian M. 2012. Delineation of Streptococcus dysgalactiae, its subspecies, and its clinical and phylogenetic relationship to Streptococcus pyogenes. J. Clin. Microbiol. 50:113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnston C, et al. 2010. Detection of large numbers of pneumococcal virulence genes in streptococci of the mitis group. J. Clin. Microbiol. 48:2762–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaijalainen T, Rintamäki S, Herva E, Leinonen M. 2002. Evaluation of gene-technological and conventional methods in the identification of Streptococcus pneumoniae. J. Microbiol. Methods 51:111–118 [DOI] [PubMed] [Google Scholar]
- 21. Kawamura Y, Hou Sultana X-GF, Miura H, Ezaki T. 1995. Determination of 16S rRNA Sequences of Streptococcus mitis and Streptococcus gordonii and phylogenetic relationships among members of the genus Streptococcus. Int. J. Syst. Bacteriol. 45:406–408 [DOI] [PubMed] [Google Scholar]
- 22. Kawamura Y, Whiley RA, Shu SE, Ezaki T, Hardie JM. 1999. Genetic approaches to the identification of the mitis group within the genus Streptococcus. Microbiology 145:2605–2613 [DOI] [PubMed] [Google Scholar]
- 23. Kilian M, et al. 2008. Evolution of Streptococcus pneumoniae and its close commensal relatives. PLoS One 3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ko KS, et al. 2005. Phenotypic and genotypic discrepancy of Streptococcus pneumoniae strains isolated from Asian countries. FEMS Immunol. Med. Microbiol. 45:63–70 [DOI] [PubMed] [Google Scholar]
- 25. Martens M, et al. 2008. Advantages of multilocus sequence analysis for taxonomic studies: a case study using 10 housekeeping genes in the genus Ensifer (including former Sinorhizobium). Int. J. Syst. Evol. Microbiol. 58:200–214 [DOI] [PubMed] [Google Scholar]
- 26. Martin R, et al. 2007. Cultivation-independent assessment of the bacterial diversity of breast milk among healthy women. Res. Microbiol. 158:31–37 [DOI] [PubMed] [Google Scholar]
- 27. Neeleman C, Klaassen CH, Klomberg DM, de Valk HA, Mouton JW. 2004. Pneumolysin is a key factor in misidentification of macrolide-resistant Streptococcus pneumoniae and is a putative virulence factor of S. mitis and other streptococci. J. Clin. Microbiol. 42:4355–4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nielsen XC, Justesen US, Dargis R, Kemp M, Christensen JJ. 2009. Identification of clinically relevant nonhemolytic streptococci on the basis of sequence analysis of 16S-23S intergenic spacer region and partial gdh gene. J. Clin. Microbiol. 47:932–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nørskov-Lauritsen N, Bruun B, Kilian M. 2005. Multilocus sequence phylogenetic study of the genus Haemophilus with description of Haemophilus pittmaniae sp. nov. Int. J. Syst. Evol. Microbiol. 55:449–456 [DOI] [PubMed] [Google Scholar]
- 30. Park HK, et al. 2010. Identification of the cpsA gene as a specific marker for the discrimination of Streptococcus pneumoniae from viridans group streptococci. J. Med. Microbiol. 59:1146–1152 [DOI] [PubMed] [Google Scholar]
- 31. Park HK, Yoon JW, Shin JW, Kim JY, Kim W. 2010. rpoA is a useful gene for identification and classification of Streptococcus pneumoniae from the closely related viridans group streptococci. FEMS Microbiol. Lett. 305:58–64 [DOI] [PubMed] [Google Scholar]
- 32. Pop M, Phillippy A, Delcher AL, Salzberg SL. 2004. Comparative genome assembly. Brief Bioinform. 5:237–248 [DOI] [PubMed] [Google Scholar]
- 33. Postic D, Garnier M, Baranton G. 2007. Multilocus sequence analysis of atypical Borrelia burgdorferi sensu lato isolates—description of Borrelia californiensis sp. nov., and genomospecies 1 and 2. Int. J. Med. Microbiol. 297:263–271 [DOI] [PubMed] [Google Scholar]
- 34. Poyart C, Quesne G, Coulon S, Berche P, Trieu-Cuot P. 1998. Identification of streptococci to species level by sequencing the gene encoding the manganese-dependent superoxide dismutase. J. Clin. Microbiol. 36:41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Romero P, López R, García E. 2004. Characterization of LytA-like N-acetylmuramoyl-l-alanine amidases from two new Streptococcus mitis bacteriophages provides insights into the properties of the major pneumococcal autolysin. J. Bacteriol. 186:8229–8239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmeing TM, Ramakrishnan V. 2009. What recent ribosome structures have revealed about the mechanism of translation. Nature 461:1234–1242 [DOI] [PubMed] [Google Scholar]
- 37. Van de Peer Y, Nicolaï S, De Rijk P, De Wachter R. 1996. Database on the structure of small ribosomal subunit RNA. Nucleic Acids Res. 24:86–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Verhelst R, et al. 2003. Comparison of five genotypic techniques for identification of optochin-resistant pneumococcus-like isolates. J. Clin. Microbiol. 41:3521–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vincze T, Posfai J, Roberts RJ. 2003. NEBcutter: a program to cleave DNA with restriction enzymes. Nucleic Acids Res. 31:3688–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Whatmore AM, et al. 2000. Genetic relationships between clinical isolates of Streptococcus pneumoniae, Streptococcus oralis, and Streptococcus mitis: characterization of “atypical” pneumococci and organisms allied to S. mitis harboring S. pneumoniae virulence factor-encoding genes. Infect. Immun. 68:1374–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zbinden A, Köhler N, Bloemberg GV. 2011. recA-based PCR assay for accurate differentiation of Streptococcus pneumoniae from other viridans streptococci. J. Clin. Microbiol. 49:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]