

Abstract
The Tf1 retrotransposon represents a group of long terminal repeat retroelements that use an RNA self-primer for initiating reverse transcription while synthesizing the minus-sense DNA strand. Tf1 reverse transcriptase (RT) was found earlier to generate the self-primer in vitro. Here, we show that this RT can remove from the synthesized cDNA the entire self-primer as well as the complete polypurine tract (PPT) sequence (serving as a second primer for cDNA synthesis). However, these primer removals, mediated by the RNase H activity of Tf1 RT, are quite inefficient. Interestingly, the integrase of Tf1 stimulated the specific Tf1 RT-directed cleavage of both the self-primer and PPT, although there was no general enhancement of the RT's RNase H activity (and the integrase by itself is devoid of any primer cleavage). The RTs of two prototype retroviruses, murine leukemia virus and human immunodeficiency virus, showed only a partial and nonspecific cleavage of both Tf1-associated primers with no stimulation by Tf1 integrase. Mutagenesis of Tf1 integrase revealed that the complete Tf1 integrase protein (excluding its chromodomain) is required for stimulating the Tf1 RT primer removal activity. Nonetheless, a double mutant integrase that has lost its integration functions can still stimulate the RT's activity, though heat-inactivated integrase cannot enhance primer removals. These findings suggest that the enzymatic activity of Tf1 integrase is not essential for stimulating the RT-mediated primer removal, while the proper folding of this protein is obligatory for this function. These results highlight possible new functions of Tf1 integrase in the retrotransposon's reverse transcription process.
INTRODUCTION
Long terminal repeat (LTR) retrotransposons are retrovirus-like elements found in a broad variety of eukaryotic cells (8, 9, 13). The structure and mechanisms of propagation of these elements are related to retroviruses, and similarly to them, they encode the Gag, protease, reverse transcriptase (RT), and integrase (IN) proteins. Like all retroviruses, the RTs of LTR retrotransposons are essential for synthesizing the double-stranded and integration-competent viral cDNA from their plus-sense, single-stranded RNA genome. This reverse transcription process is catalyzed by the two RT activities, the DNA polymerase (which copies both RNA and DNA) and the RNase H that, concurrently with DNA synthesis, cleaves the RNA in the formed DNA-RNA heteroduplex. In all retroviruses and LTR retrotransposons, the synthesis of plus-strand DNA is initiated from an RNA primer that is complementary to a specific genomic RNA sequence, designated the primer binding site (PBS), which is located near the 5′ end of the RNA genome. In almost all retroelements, this RNA primer is a specific cellular tRNA recruited by the virus (8, 9, 26, 38).
An alternative tRNA-independent priming mechanism was suggested for the LTR retrotransposon Tf1 of the fission yeast Schizosaccharomyces pombe (6, 9, 27–30). Here, a self-complementarity within the RNA genome can allow an intramolecular base-pairing near the 5′ end of the genomic RNA, thus causing this end to fold back. Accordingly, there is an intramolecular 11-bp RNA duplex that after cleavage between the 11th and 12th nucleotides of the RNA transcript releases an 11-nucleotide (nt) self-primer (SP) for initiating the minus-strand cDNA synthesis. Indeed, genetic studies of Tf1 have suggested that this segment provides the primer for DNA synthesis. Mutagenesis has also suggested that the RNase H domain of Tf1 RT is required for this specific cleavage (28). Consequently, the ability of Tf1 RT to perform in vitro this unique activity of nicking the RNA segment that mimics the 5′ end of genomic Tf1 RNA was confirmed recently by one of us (16). Product generation was specific to the recombinantly expressed Tf1 RT, since other RTs were unable to generate this specific product. As could be expected, an RNase H-deficient Tf1 RT mutant did not exhibit the SP-nicking activity. Moreover, the SP produced served as a functional primer, since in the presence of deoxynucleoside triphosphates (dNTPs) it was extended by the RT-associated DNA polymerase activity.
After self-priming was suggested for Tf1, a similar priming mechanism was also proposed for several other LTR retroelements, such as the highly homologous Tf2 and Maggy, Skippy, Cft-1, Boty, copia of maize, and Tf1/sushi of vertebrates, all of which belong to a single lineage of the Ty3/gypsy group of LTR retrotransposons (9, 31). These elements are believed to have diverged early in the evolution of LTR retrotransposons, well before retroviruses. Therefore, it is likely that the mechanism of self-priming represents an early form of initiating reverse transcription in LTR retroelements (26), as it was probably advantageous for these retroelements to use a tRNA-independent mechanism of priming cDNA synthesis.
The genomic retroviral RNA that participates in the complex process of reverse transcription is removed after being copied throughout reverse transcription by RT-associated RNase H activity (8, 14). This is true also for the RNA segments that serve as primers for DNA synthesis. These primers are the tRNA and polypurine tract (PPT) that initiate minus- and plus-strand DNA syntheses, respectively. Since in Tf1 the cellular tRNA is replaced by the SP, one could expect by analogy that the SP would also be removed by the RT-associated RNase H activity. Surprisingly, there is evidence that only a small fraction of the in vivo-generated Tf1 cDNA has the SP removed (2). This finding highlights another potentially exclusive property of the reverse transcription process of Tf1. One explanation for this finding is that Tf1 RT is inefficient in its primer removal activity. In the study presented here, we tested this assumption. A specific assay was developed to directly examine the full in vitro removal by Tf1 RT of the SP and PPT from the appropriate chimeric RNA-DNA strands (annealed to their complementary DNAs). Tf1 RT-directed primer removals were tested in comparison with two other prototype retroviral RTs (those of human immunodeficiency virus type 1 [HIV-1] and of murine leukemia virus [MLV]).
The results presented show that the removal of both the SP and PPT by RT is quite inefficient. The other two tested RTs did not properly remove either of the two Tf1 primers. Due to the low primer removal by Tf1 RT, we have tested whether other viral proteins can upregulate this RT activity. Consequently, the integrase (IN) enzyme of Tf1 was found to have a unique and previously unrecognized role in promoting the precise removal of both RNA primers by Tf1 RT activity. There are many previously documented cases of the structural and functional interplays between retroviral RT and IN (for example, see references 15, 23, and 33 to 35). However, as far as we know, the removal of RNA primers from the produced cDNA was never shown to be regulated by IN. This result may shed light on new roles for IN in the complex reverse transcription process in Tf1 and related LTR retrotransposons and possibly in retroviruses.
(The results presented are in partial fulfillment of a Ph.D. thesis by E. Herzig at Tel Aviv University.)
MATERIALS AND METHODS
Substrates for assaying the primer RT-directed removals.
The substrates are described schematically in Fig. 1. The RNA oligonucleotides that were used include the following: a synthetic 11-nt-long Tf1 SP (with the sequence 5′-AGUUCAGUUAU-3′) (16). Based on the Tf1 genome sequence and the known PPT sequences of other retroviruses, the specific Tf1 PPT sequence was also predicted to be 11 nt long (5′-GGGGAGGGCAA-3′). Also used was the 15-nt MLV PPT (5′-AGAAAAAGGGGGGAA-3′) (36). All RNA primers were prelabeled at their 5′ end with polynucleotide kinase and [γ-32P]ATP and then annealed to the appropriate minus-strand DNA segments of Tf1 or MLV. These DNA oligonucleotides were as follows: for the SP substrate, the 40-nt template with the sequence 5′-CAAATACCAAACTGCGTAGCTAACAATAACTGAACTCTTG-3′; for the PPT substrate, the 61-nt-long oligonucleotide with the sequence 5′-ACGTAGTGTATCATAGCGTAGTGTAGTATTGCTGACATTGCCCTCCCCAAGATTTTCTAT-3′; and for the MLV PPT substrate, the 62-nt oligonucleotide with the sequence 5′-GCTTGCCAAACCTACAGGTGGGGTCTTTCATTCCCCCCTTTTTCTGGAGACTAAATAAAATC-3′. Following the annealing, the formed heteroduplexes were elongated by the Klenow fragment of Escherichia coli DNA polymerase I that lacks the 3′→5′ exonuclease (purchased from NEB) in the presence of all four dNTPs. This generated the full-length chimeric RNA-DNA duplexes. The double-stranded substrates produced (36, 48, or 45 bp long for the Tf1 SP, Tf1 PPT, or MLV PPT removal, respectively) were then purified using a DNA extraction kit (purchased from RBC) and served as the substrates for SP or PPT removal by the tested RTs, as described below.
Fig 1.
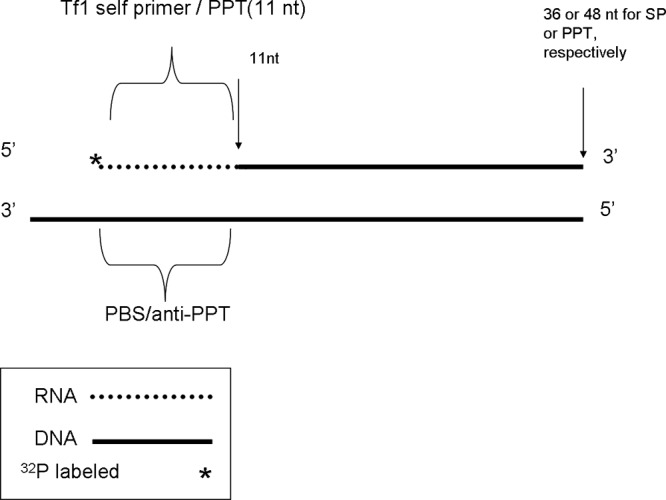
Schematic description of the substrates used for assaying the removal of Tf1 SP or PPT substrates. The substrates were prepared as described in detail in Materials and Methods. The 11-nt-long RNA segments with sequences of either the Tf1 SP or PPT were 5′ end labeled with 32P. The labeled RNA was then annealed to Tf1-derived DNA segments with the appropriate 3′ ends that match either the SP sequences (the PBS-derived 40-nt-long DNA) or the PPT (61-nt-long DNA) These heteroduplexes were then elongated with the Klenow fragment of E. coli polymerase I (lacking the 3′→5′ exonuclease), thus producing full-length substrates.
Enzymes. (i) Reverse transcriptases.
All RTs used here were prepared by us as recombinant six-histidine-tagged proteins. These RTs were expressed in bacteria and then purified to homogeneity. These are the wild-type RTs of Tf1 (25), HIV-1 (21, 37), and MLV (4, 18). In addition, we have used a mutant Tf1 RT that lacks its RNase H activity (designated RH-RT). It was prepared by introducing a single residue exchange (D362N), already shown to lack the RNase H activity and to be incapable of generating the SP (16).
(ii) Integrases.
The full-length Tf1 IN (477 residues long) and the Tf1 IN that lacks its 71-residue-long C-terminal chromodomain (designated CD-IN) were expressed and purified as described previously (20). In addition, we have engineered new expression vectors that induce the bacterial expression of various Tf1 IN mutants (all described schematically below in Fig. 7). These mutants are as indicated. The C-terminally truncated IN (designated CT-IN) was prepared by removing 178 residues from the IN′s C terminus; the N-terminally truncated IN (NT-IN) was expressed after removing 117 residues from the N terminus; the catalytic core domain of Tf1 IN (CCD IN) was composed of residues 118 to 320 of the full-length IN; the double mutant D194N/E230Q was modified at the active site of Tf1 IN (DM IN) and lost its IN activities (data not shown). The genes encoding all truncated IN versions were amplified by PCR, cloned, and sequenced for verification. The gene encoding the DM IN was synthesized by overlapping PCR from two separate plasmids, each containing the full-length Tf1 retrotransposon with a single IN mutation (these two plasmids were a generous gift from H. Levin of NIH). HIV-1 IN was prepared as described previously (34).
Fig 7.

A schematic description of the Tf1 IN variants generated and used in this study. Wild-type Tf1 IN was genetically modified either by introducing mutations in the IN active site (the DM IN) or by truncating parts of the protein in the expression vectors for generating the different variants of Tf1 IN (see Materials and Methods). Except for the first residue, all other residues that specify the boundaries between the different domains are the last residue in each domain.
Enzymatic assays. (i) RNase H activity of Tf1 RT.
The release of [3H]AMP from the [3H]poly(rA)-poly(dT) substrate into the trichloroacetic acid (TCA)-soluble fraction was measured as described by us previously (22). The reactions were performed under linear conditions, where only a fraction of the substrate was hydrolyzed by the RT-associated RNase H activity (data not shown). Each reaction was performed at a final volume of 40 μl with 0.25 μg Tf1 RT in 50 mM Tris-HCl (pH 8.5), 50 mM KCl, 0.5 mM MnCl2, 2.5 mM dithiothreitol (DTT), and 20 μg/ml acetylated bovine serum albumin (BSA) at 37°C for 60 min. Then, the reactions were stopped by transferring the reaction mixtures onto ice and adding 50 μg of carrier herring sperm DNA. Ice-cold TCA (to a final concentration of 10% [wt/vol]) was added to remove the unhydrolyzed macromolecules, followed by centrifugation for 5 min at 15,000 × g. The supernatants, containing the released [3H]AMP, were transferred into new tubes with 16 μl 0.1 M NaOH to neutralize the TCA. The samples were then analyzed in a β-scintillation counter after adding scintillation liquid.
(ii) The RT-associated SP/PPT removal activity.
The produced chimeric RNA-DNA heteroduplexes served as the substrates for SP and PPT removals. Each reaction mixture contained 200 ng of the purified RT, i.e., wild-type Tf1 RT, Tf1 RH-RT, HIV-1 RT, or MLV RT, and ∼0.1 pmol of each of the labeled substrates. This means that, in order to achieve maximal primer removals under the assay conditions employed, we had to use an excess of the various RTs over the substrates, with molar ratios of 15- to 30-fold. This ratio is within the same general range of RT over cDNA or viral RNA found in retrovirus-infected cells (8). All reactions were performed in final volumes of 12.5 μl in 50 mM Tris-HCl (pH 8.5), 2 mM DTT, 50 mM NaCl, 10 mM CHAPS {3-[(3-cholamidylpropyl)-dimethylammonio]-1-propanesulfonate}, 100 μg/ml acetylated BSA, and either 5 mM MgCl2 (for HIV-1 RT) or 0.5 mM MnCl2 (for the other tested RTs). After 30 min at 37°C, the reactions were stopped by adding an equal amount of formamide dye mix, and the reaction mixtures were denatured at 100°C for 5 min, cooled on ice, and then analyzed by high-voltage electrophoresis through 12% polyacrylamide-urea in Tris-borate and EDTA sequencing gels, as described previously (34). For calculating the molecular ratios between Tf1 IN and the various RTs used, we assumed that Tf1 IN is homodimeric and that Tf1 and MLV RTs are monomeric, while HIV-1 RT is heterodimeric.
(iii) Quantification of the cleavage products.
After the gels were visualized by autoradiography, the cleavage products were quantified using ImageJ software (National Institutes of Health). Individual gel lanes were quantified as rectangular objects. The obtained values are the means with the indicated standard deviations (SDs) of three independent experiments.
RESULTS AND DISCUSSION
The removal of the self-primer by Tf1 RT.
Our previous study on Tf1 RT has shown that recombinant Tf1 RT can introduce nicks at the end of a duplexed region at the 5′ end of Tf1 genomic RNA, substantiating the prediction that this enzyme is responsible for generating this RNA SP (16). In the present study, we generated a synthetic substrate that mimics the appropriate substrate for testing the complete removal of the SP. This substrate was constructed as described in Materials and Methods and schematically in Fig. 1. It is apparent from the data presented in Fig. 2A that, after incubating the substrate with Tf1 RT, there is a clear and specific, albeit very weak, removal of the whole 11-nt-long SP (without further cleavage to shorter RNA segments) (lane 3). This precise cleavage in the RNA-DNA boundary depends on the presence of an active RT, since heat-inactivated RT does not generate the 11-nt product (lane 2). Moreover, as already shown previously by us with the folded RNA substrate (16), the primer cleavage activity depends on the Tf1 RT-associated RNase H activity, since the RT mutant (D362N) that lacks this activity (RH-RT) cannot release the SP (lane 4).
Fig 2.

SP removal by Tf1 RT without and with IN. (A) Primer removal was assayed for 30 min at 37°C with 200 ng of either native wild-type Tf1 RT (lane 3, marked RT), heat-inactivated RT (lane 2, HI), or RNase H-deficient mutant (lane 4, RH-RT). S, substrate only (lane 1). (B) Reactions with constant amounts of wild-type Tf1 RT in the absence (lane 2) or the presence of Tf1 IN, with increasing molar ratios of IN to RT ranging from 0.25:1 to 2:1 (lanes 3 to 6). 2(HI), a 2:1 IN/RT ratio of Tf1 RT with heat-inactivated Tf1 IN (lane 7); 2(NR), with the same concentration of Tf1 IN with no RT present (lane 8); 2(HIV), a 2:1 IN/RT molecular ratio of HIV-1 IN with Tf1 RT (lane 9).
It should be noted, based on the interactions between RT and the RNA-DNA substrate, that the RNase H activity associated with RTs has three distinct modes (7, 14). The RNase H-related primer removal activity found here is DNA polymerization independent, since all SP and PPT removal assays were performed in the absence of dNTPs, and when dNTPs were added, no differences in primer removals were observed (data not shown). Therefore, this cleavage possibly belongs to the RNA 5′-directed RNase H cleavage mode. In this mode of activity, the RT's polymerase domain binds the DNA strand internally in a site that is opposite to, or near, the 5′ end of the recessed RNA strand in the DNA-RNA heteroduplex. Accordingly, the RNase H domain in most RTs is usually positioned 13 to 19 nt from the RNA's 5′ end (7, 14), while in this specific case of Tf1 RT-mediated primer cleavage, the distance is shorter (11 nt). The same considerations and conclusions apply also to the Tf1-RT-directed PPT cleavage of the 11-nt-long PPT, as shown below in Fig. 3A.
Fig 3.

PPT removal by Tf1 RT in the absence or in the presence of IN. (A) Primer removal was assayed for 30 min at 37°C with 200 ng of wild-type Tf1 RT (lane 3, marked RT), heat-inactivated RT (lane 2, HI), or RNase H-deficient mutant (lane 4, RH-RT). S, substrate only (lane 1). (B) Reactions with constant amounts of wild-type Tf1 RT in the absence (lane 2) or the presence of Tf1 IN, with increasing molar ratios of IN to RT from 0.25:1 to 2:1 (lanes 3 to 6). 2(HI), a 2:1 IN/RT ratio of Tf1 RT with heat-inactivated IN (lane 7); 2(NR), with the same concentration of Tf1 IN with no RT present (lane 8); 2(HIV), a 2:1 IN/RT molecular ratio of HIV-1 IN with Tf1 RT (lane 9).
The effects of Tf1 IN on the SP removal by Tf1 RT.
The cleavage of the Tf1 SP by the homologous RT, as shown in Fig. 2A, is relatively weak. Therefore, we checked whether this activity could be affected by Tf1 IN. A constant amount of Tf1 RT with increasing Tf1 IN concentrations (with molar ratios relative to Tf1 RT ranging from 0.25:1 to 2:1) were added to the cleavage reaction mixtures. The results presented in Fig. 2B show that there is a gradual rise in SP cleavage as IN increases (lanes 3 to 6). A quantitative analysis of the extent of enhancement of the RT-associated SP removal by Tf1 IN indicated that at a 2:1 IN/RT molar ratio a 13.0- ± 0.1-fold increase of activity was achieved. This increase in SP cleavage results from the presence of a native IN, since the heat-inactivated IN, even at the highest tested IN/RT molar ratio of 2:1, was devoid of any significant cleavage induction (lane 7). Moreover, the IN by itself (at the highest tested IN concentration) lacks any detectable cleavage activity (lane 8). The IN effect is not due to protein-dependent nonspecific RT stabilization, because all reaction mixtures contained a large excess of BSA (see Materials and Methods). It should be noted that in retroviruses the molar ratios between the RT and IN subunits are expected to be similar, since the two originate from the same polyprotein precursor. Assuming that Tf1 RT is monomeric (25) and IN is homodimeric (39) and that their intracellular stabilities are similar, the biologically relevant IN/RT ratio is roughly 0.5:1. Therefore, the in vitro conditions applied here are quite close to those that probably exist in vivo, and the maximal IN/RT ratio of 2:1, employed in this experiment, is sufficient.
When another retroviral IN (i.e., that of HIV-1) was tested in a setting similar to the previously described experiment, there was no apparent effect on SP removal by Tf1 RT (Fig. 2B, lane 9). This result suggests that the demonstrated enhancement of SP removal by Tf1 RT is specific to the homologous IN.
The removal of the PPT by Tf1 RT and the effect of Tf1 IN.
So far, we have tested both here and in our previous study (16) the capacity of Tf1 RT to cleave the SP. Therefore, it was of interest to test also whether Tf1 RT can remove the PPT. To this end, we generated a substrate that mimics Tf1 PPT in a chimeric RNA-DNA molecule annealed to the appropriate viral DNA segment (similar to the one described above for the SP removal [Fig. 1]). The results of the initial experiment testing PPT cleavage by Tf1 RT are shown in Fig. 3A. Similarly to SP removal (Fig. 2A), there is an RT-dependent removal of the whole 11-nt-long PPT, with no further cleavage to shorter RNA segments (lane 3). As in the case of the SP cleavage (Fig. 2A), this apparently very weak cleavage is at the exact boundary between the RNA and DNA in the chimeric RNA-DNA strand. Apparently, it depends on the enzymatically active RT, as heat-inactivated RT lost the activity (lane 2). This activity is also mediated by the RT's RNase H activity, as the D362N mutant Tf1 RT does not show any PPT removal (lane 4). As shown for the SP removal (Fig. 2A), we have also tested the effects of Tf1 IN on PPT removal (Fig. 3B). The data for PPT removal in the presence of increasing ratios of Tf1 IN to RT (from 0.25:1 to 2:1) imply that, as in the case of SP removal by Tf1 RT, Tf1 IN increasingly stimulates the PPT removal by Tf1 RT (lanes 3 to 6). A quantitative analysis of enhancement of the RT-associated PPT removal by Tf1 IN shows that at the highest IN/RT molar ratio tested (2:1), a 5.4- ± 0.8-fold increase of activity was achieved. Here also, the IN by itself does not show any significant PPT removal activity (lane 8) while the heat-inactivated IN lost its capacity to stimulate the RT-associated PPT removal (lane 7). Moreover, the enhancement of the Tf1 RT-mediated primer removal by Tf1 IN was shown to be specific to Tf1 IN, since, as in the previous case of SP removal (Fig. 2B), no augmentation of PPT cleavage was apparent when HIV-1 IN was used instead of Tf1 IN (Fig. 3B, lane 9). In all, the data presented here and in Fig. 2B support the idea that only the cognate IN can stimulate the primer removal capacity of Tf1 RT. It should be also emphasized that all Tf1 RT-associated primer removal activities, shown in this study, as well as the enhancements of this activity by Tf1 IN could be achieved with only Mn2+ and not with Mg2+ (data not shown). This is not surprising, since both Tf1 enzymes have a profound preference for the former divalent cation (16, 20, 25).
The effect of Tf1 IN on the general RNase H activity of Tf1 RT.
We showed above that the removal of both primers by Tf1 RT is mediated by the RT's RNase H activity. Therefore, a possible explanation for the observed Tf1 IN-driven enhancement of primer removal is that IN has a general activity of stabilizing or augmenting nonspecifically the RT-associated RNase H activity. To check this supposition, we have assessed the Tf1 RT-associated general RNase H activity in the presence of Tf1 IN. The quantitative results, shown in Fig. 4, indicate that there is no stimulation of the RT's RNase H activity at any of the IN/RT ratios shown above to increase primer removals (Fig. 2 and 3). In addition, we have tested whether Tf1 IN has any effects on the RT's DNA polymerase activity. Here again, no significant change in the Tf1 RT DNA polymerase activity was detected (data not shown). Apparently, this information implies that the effect of Tf1 IN on the primer removals by Tf1 RT is specific to only this function.
Fig 4.
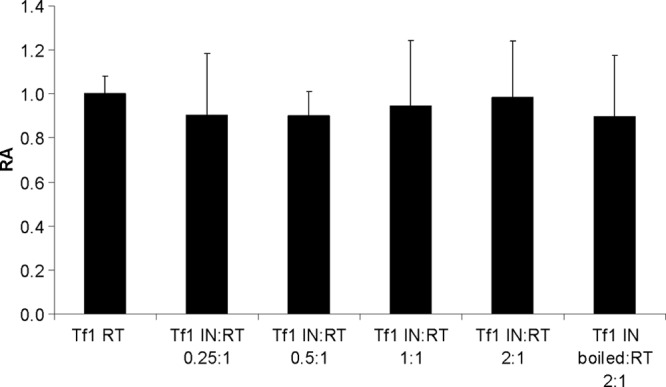
The RNase H activity of Tf1 RT in the absence or presence of increasing Tf1 IN/RT molar ratios. The soluble RNase H assay that measures the release of [3H]AMP from [3H]poly(rA)-poly(dT) substrate into the TCA-soluble fraction was carried out as described in Materials and Methods. The activity of Tf1 RT by itself with no IN present has a value of 1, and all other IN/RT ratios exhibit activities relative to this value (relative activity [RA]). The numbers represent the results of three independent experiments after subtracting the nonspecific backgrounds (with the indicated SD).
The ability of the RTs of HIV-1 and MLV to properly remove the Tf1 SP and PPT and the effects of Tf1 IN on these two RTs.
In order to assess how specific the full primer removal is to Tf1 RT, we tested whether RTs from other retroviruses can also exhibit this activity. The RTs of two prototype retroviruses were tested. These viruses are MLV, representing the gammaretroviruses, and the lentivirus HIV-1. The results obtained with these RTs show that both enzymes cleave the SP and PPT RNA primers nonspecifically in the RNA-DNA heteroduplex substrates used (Fig. 5), and unlike the Tf1 RT-mediated cleavage, MLV and HIV-1 RTs do not cut precisely at the boundaries between the RNA and DNA in the chimeric RNA-DNA strand. Thus, with both RTs (and with both the SP- and PPT-containing substrates), the sizes of produced RNA segments are appreciably shorter than the expected 11-nt complete primer generated by Tf1 RT. Both MLV and HIV-1 RTs generate 7- and 8-nt-long end-labeled products with the SP substrate (Fig. 5A, lanes 6 and 8, respectively) and 4- to 10-nt-long labeled products with the PPT substrate (Fig. 5B, lanes 6 and 8). Clearly, MLV RT tends to produce fragments that are significantly shorter (and less homogeneous) than those generated by HIV-1 RT. These cleavage products are probably the final ones, since they were observed regardless of the incubation time. Even after very short incubations of 1 min longer, products were not observed (data not shown). Therefore, we believe that no initial cuts between the RNA and DNA were performed prior to the generation of the shorter RNA products. Probably the most interesting result is that there is definitely no significant enhancement of the relatively nonspecific cleavage of the primers by MLV and HIV-1 RTs by Tf1 IN. Therefore, the observed enhancement of RNA primer removals by Tf1 RT is restricted to only the homologous Tf1 IN. Moreover, HIV-1 IN had no effect on HIV-1 and MLV RTs regarding the pattern and amount of primer cleavage (data not shown). Importantly, Fig. 5 shows also that the stimulation of primer removal of Tf1 RT by Tf1 IN is restricted to only wild-type Tf1 RT, as the RH-RT mutant cannot be activated by the IN (lanes 4 and 5 in Fig. 5A and B).
Fig 5.

The removal of the SP (A) or the PPT (B) by various RTs in the absence or presence of Tf1 IN. Primer removal activities of wild-type Tf1, HIV-1, and MLV RTs as well as of the Tf1 RT that lacks RNase H activity (RH-RT) were tested in the absence or presence of Tf1 IN (at IN/RT molar ratios of 2:1).
The ability of Tf1 RT to remove other nonspecific primers.
To further study whether the primer removal activity of Tf1 RT is limited to only the Tf1-related primers, we have performed the experiment described in Fig. 6. Here, we used a substrate that mimics the 15-nt-long MLV PPT in a chimeric RNA-DNA molecule annealed to the appropriate viral DNA segment. The results show that Tf1 RT was not capable of removing the MLV PPT to any significant extent. Moreover, the presence of Tf1 IN did not lead to any enhancement of PPT removal. In contrast, both MLV and HIV-1 RTs could cleave this primer (Fig. 6, lanes 9 and 10). However, similar to the case of removing the Tf1-related SP and PPT (Fig. 5), the labeled RNA segments produced were shorter than the expected 15-nt full-length primer. In another experiment, we have tested in a similar setting whether Tf1 RT can remove an artificial RNA primer that was derived from the sequence of HIV-1 U5. Here again, neither significant primer cleavage nor enhancement by Tf1 IN was observed (data not shown).
Fig 6.
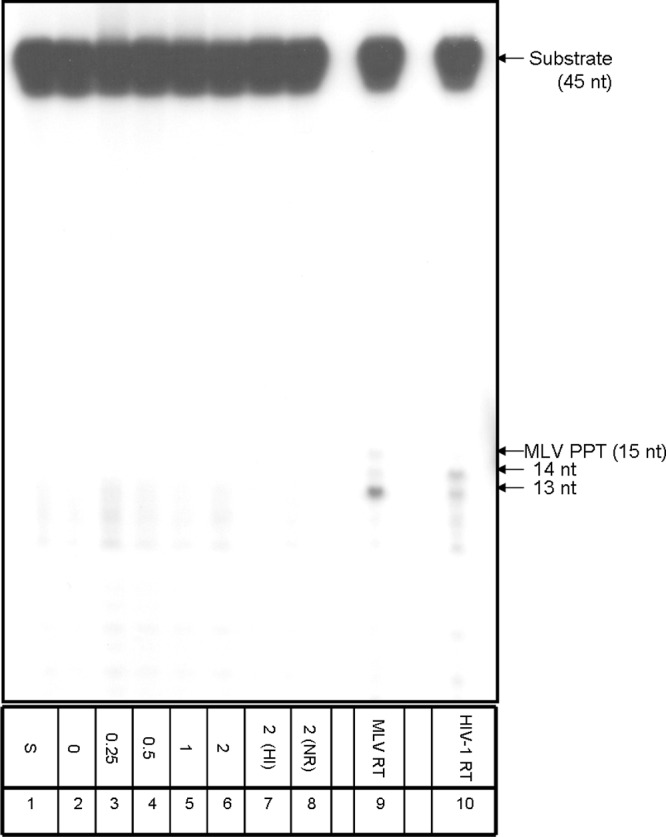
The removal of the nonhomologous MLV primer by Tf1 RT. All reactions were performed with a constant amount of wild-type Tf1 RT in the absence (lane 2) or the presence of Tf1 IN, with increasing molar ratios of IN to RT ranging from 0.25:1 to 2:1 (lanes 3 to 6). 2(HI), a 2:1 IN/RT ratio of Tf1 RT with heat-inactivated Tf1 IN (lane 7); 2(NR), with the same concentration of Tf1 IN with no RT present (lane 8); MLV RT, a 2:1 Tf1 IN/MLV RT molar ratio (lane 9); HIV-1 RT, a 2:1 Tf1 IN/HIV-1 RT ratio (lane 10). S, substrate only (lane 1).
The effects of Tf1 IN mutants on the RNA primer removals by Tf1 RT.
Tf1 IN is unique among INs of retroelements in having a chromodomain at its C terminus. We have shown previously that this domain regulates the catalytic activities of Tf1 IN, as the IN that lacks the chromodomain had significantly higher activities that are accompanied by substantially reduced substrate specificity (20). Another feature common to all retroviral INs is the clear designations of different parts of the IN molecule. All INs have three protease-resistant domains, and the functions of these domains have been studied extensively (10, 24). The N-terminal domain contains a conserved motif similar to zinc fingers. Although this domain does bind zinc, little is known about its function. The central (core) domain is the catalytic domain that contains highly conserved acidic residues, Asp, Asp, and Glu (the DD35E motif). This central domain of HIV-1 IN was shown in vitro to be sufficient for the reverse reaction of the strand transfer activity, known as disintegration (5). The C-terminal domain is less conserved but has a nonspecific DNA binding activity. Only recently, the crystal structure of the full-length IN from the prototype foamy virus in complex with its cognate DNA was resolved (12).
We have tested at this stage which segments of the Tf1 IN molecule participate in stimulating the Tf1 RT-associated primer removal. To this aim, we generated several IN-derived mutants or segments, as described schematically in Fig. 7 and in Materials and Methods. These IN variants are the enzymatically active chromodomain-minus IN (CD-IN), the truncated IN molecule that lacks its N terminus (NT-IN), the IN devoid of its C terminus (CT-IN), and the catalytic core domain (CCD IN). In addition, we created the Tf1 IN double mutant (D194N/E230Q, designated DM IN) that was mutated in two of the highly conserved IN residues and, therefore, is devoid of any IN catalytic activities (data not shown). All IN versions were recombinantly expressed and purified to homogeneity (see Materials and Methods). Similarly to wild-type Tf1 IN (Fig. 2B and 3B, lanes 8), all Tf1 IN variants had no intrinsic primer removal activity (data not shown). These IN variants were tested for their effects on the Tf1 RT-associated primer removal, all at the IN/RT molar ratio of 2:1, shown above to be effective in enhancing primer removal by Tf1 RT. The results, shown in Fig. 8, indicate that both the SP and PPT removals are affected by the IN versions quite similarly. Only the double mutant and the CD-IN versions enhanced considerably the RT-associated primer removals. All other IN versions tested (all devoid of integration activity) did not stimulate the RT-associated primer removals. Interestingly, the double mutant that also lost its IN activity was still fully active as an RT inducer. It should be also emphasized that, as shown above (Fig. 2B and 3B, lanes 7), heat-inactivated Tf1 IN completely lost its primer removal enhancement activity. Therefore, the most likely conclusion is that the properly folded IN molecule (even if it is enzymatically inactive, as in the case of the DM IN) is required for stimulating the RT-associated primer removal activity, while the enzymatic activity of the IN per se is not involved in stimulating this activity. As for the IN molecule, it is apparent that all the major domains of the IN protein (except for its chromodomain) are required for enhancing the Tf1 RT primer removal activity. It should also be noted, from the results presented in Fig. 8, that the CD-IN and the DM IN seem to function as efficiently as the wild-type IN in stimulating the RT-associated SP removal, while they were less efficient than the wild-type IN in enhancing the PPT removal by the RT. This may suggest a possible mechanistic subtle difference between the IN-dependent enhancement of the SP and PPT removals by the RT.
Fig 8.

SP and PPT removal by Tf1 RT and Tf1 IN mutants. (A) Removal of the SP. (B) Removal of the PPT. In both panels, lane 1 shows substrate only. In all other lanes, reactions were performed with a constant amount of Tf1 RT in the absence (−, lanes 2) or the presence of the different Tf1 IN variants, assayed with a 2:1 molar ratio of the IN variant over Tf1 RT (lanes 3 to 8). WT, wild-type IN (lanes 3); CD-IN, chromodomain-minus IN (lanes 4); DM IN, double mutant IN (lanes 5); NT-IN, N-terminally truncated IN (lanes 6); CT-IN, C-terminally truncated IN (lanes 7); CCD IN, catalytic core (lanes 8).
In summary, the data presented here suggest an interesting new role for Tf1 IN in affecting reverse transcription in Tf1. It is likely that in this case, IN is part of the machinery that participates in removing the two RNA primers used for the synthesis of both minus and plus DNA strands. The catalytic activity of Tf1 IN is not a prerequisite for its unique effect on the primer removal by its related RT, although the properly folded IN is required for this activity. As far as we know, the information presented here is the first direct evidence for the involvement of IN in promoting the RNase H RT-mediated primer removal.
The potential functional interactions between RT and IN in LTR retroelements are supported by many studies that show cooperation between RT and IN proteins. Direct interactions between these proteins in MLV were already shown 25 years ago (23). Other studies have also found these mutual IN-RT contacts in HIV-1 (15, 33–35). Moreover, in many alpharetroviruses, the IN sequence is part of two different proteins, the free IN molecule and the C terminus of the larger RT subunit (14, 17, 19). Also, we and others found that HIV-1 RT or HIV-1 RT-derived peptides could bind HIV-1 IN and affect its activities (33–35, 41). It was also shown that HIV-1 IN stimulates early steps of reverse transcription by increasing the processivity of HIV-1 RT and suppressing the formation of pause products (11). An in vivo study with the LTR retrotransposon Ty3 implied that IN is required for initiating reverse transcription, as mutations in both N-terminal and C-terminal domains of IN led to a drop in the accumulation of full-length cDNA in the virus-like particles. This reduction in cDNA was accompanied by smaller amounts of early intermediates, such as minus-strand, strong-stop DNA (32). Another in vivo study with Ty1, which belongs to another retrotransposon lineage, suggested a domain between IN residues 233 and 520 that inhibits RT activities, whereas another domain located between residues 521 and 607 enhances RT activity (40). This C-terminal IN domain probably affects the proper folding of RT and stabilizes its activity. As for Tf1, two-hybrid system experiments suggested the existence of interactions between IN and RT (39). Nonetheless, it may be that the effects reported here of Tf1 IN on Tf1 RT also result from indirect protein interactions due to substrate binding. Therefore, we are now testing the protein-protein binding of Tf1 IN with Tf1 RT in the absence and in the presence of nucleic acid substrates containing the SP and PPT. We hope that these ongoing studies will shed light on new roles of IN in the complex reverse transcription process in Tf1 and related LTR retrotransposons and possibly in other retroelements.
Future studies may also include in vivo studies with IN-deleted Tf1. As could be expected from the mechanism of retroviral reverse transcription, this lack of IN should not have a substantial theoretical effect on total cDNA production. Indeed, this result was also found experimentally (1, 3). A potential complexity to this direction of research may stem from the finding that deleting IN resulted in the in vivo accumulation of RT molecules fused to the upstream Tf1 protease, at the expense of the free RT (1). Since the enzymatic activities of this 72-kDa protease-RT molecule may potentially differ from those of the 60-kDa free RT, any result obtained may indirectly reflect a catalytic difference between the two RTs, rather than the absence of Tf1 IN.
ACKNOWLEDGMENTS
We are thankful to Henry Levin from NIH for providing the plasmids with the complete Tf1 retrotransposon with the two core domain IN mutations, from which the DM integrase was generated. We also thank Iris Oz-Gleenberg from our laboratory for carefully reading the manuscript.
This research was supported in part by a grant from the Israel Science Foundation (grant no. 411/07). A. Hizi is an incumbent of the Gregorio and Dora Shapira Chair for the Research of Malignancies.
Footnotes
Published ahead of print 4 April 2012
REFERENCES
- 1. Atwood-Moore A, Ejebe K, Levin HL. 2005. Specific recognition and cleavage of the plus-strand primer by reverse transcriptase. J. Virol. 79:14863–14875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Atwood-Moore A, Yan K, Judson RL, Levin HL. 2006. The self primer of the long terminal repeat retrotransposon Tf1 is not removed during reverse transcription. J. Virol. 80:8267–8270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atwood A, Choi J, Levin HL. 1998. The application of a homologous recombination assay revealed amino acid residues in an LTR-retrotransposon that were critical for integration. J. Virol. 72:1324–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Avidan O, Loya S, Tonjes RR, Sevilya Z, Hizi A. 2003. Expression and characterization of a recombinant novel reverse transcriptase of a porcine endogenous retrovirus. Virology 307:341–357 [DOI] [PubMed] [Google Scholar]
- 5. Bushman FD, Engelman A, Palmer I, Wingfield P, Craigie R. 1993. Domains of the integrase protein of human immunodeficiency virus type 1 responsible for polynucleotidyl transfer and zinc binding. Proc. Natl. Acad. Sci. U. S. A. 90:3428–3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Butler M, Goodwin T, Simpson M, Singh M, Poulter R. 2001. Vertebrate LTR retrotransposons of the Tf1/sushi group. J. Mol. Evol. 52:260–274 [DOI] [PubMed] [Google Scholar]
- 7. Champoux JJ, Schultz SJ. 2009. Ribonuclease H: properties, substrate specificity and roles in retroviral reverse transcription. FEBS J. 276:1506–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coffin JM, Hughes SH, Varmus HE. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 9. Craig NL, Craigie R, Gellert M, Lambowitz AM. 2002. Mobile DNA II. ASM Press, Washington, DC [Google Scholar]
- 10. Craigie R. 2001. HIV integrase, a brief overview from chemistry to therapeutics. J. Biol. Chem. 276:23213–23216 [DOI] [PubMed] [Google Scholar]
- 11. Dobard CW, Briones MS, Chow SA. 2007. Molecular mechanisms by which human immunodeficiency virus type 1 integrase stimulates the early steps of reverse transcription. J. Virol. 81:10037–10046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. 2010. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464:232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Havecker ER, Gao X, Voytas DF. 2004. The diversity of LTR retrotransposons. Genome Biol. 5:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herschhorn A, Hizi A. 2010. Retroviral reverse transcriptases. Cell. Mol. Life Sci. 67:2717–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herschhorn A, Oz-Gleenberg I, Hizi A. 2008. Quantitative analysis of the interactions between HIV-1 integrase and retroviral reverse transcriptases. Biochem. J. 412:163–170 [DOI] [PubMed] [Google Scholar]
- 16. Hizi A. 2008. The reverse transcriptase of the Tf1 retrotransposon has a specific novel activity for generating the RNA self-primer that is functional in cDNA synthesis. J. Virol. 82:10906–10910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hizi A, Herschhorn A. 2008. Retroviral reverse transcriptases (other than those of HIV-1 and murine leukemia virus): a comparison of their molecular and biochemical properties. Virus Res. 134:203–220 [DOI] [PubMed] [Google Scholar]
- 18. Hizi A, Hughes SH. 1988. Expression in Escherichia coli of a Moloney murine leukemia virus reverse transcriptase whose structure closely resembles the viral enzyme. Gene 66:319–323 [DOI] [PubMed] [Google Scholar]
- 19. Hizi A, Leis JP, Joklik WK. 1977. RNA-dependent DNA polymerase of avian sarcoma virus B77. II. Comparison of the catalytic properties of the alpha, beta2, and alphabeta enzyme forms. J. Biol. Chem. 252:2290–2295 [PubMed] [Google Scholar]
- 20. Hizi A, Levin HL. 2005. The integrase of the long terminal repeat-retrotransposon tf1 has a chromodomain that modulates integrase activities. J. Biol. Chem. 280:39086–39094 [DOI] [PubMed] [Google Scholar]
- 21. Hizi A, McGill C, Hughes SH. 1988. Expression of soluble, enzymatically active, human immunodeficiency virus reverse transcriptase in Escherichia coli and analysis of mutants. Proc. Natl. Acad. Sci. U. S. A. 85:1218–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hizi A, Tal R, Shaharabany M, Loya S. 1991. Catalytic properties of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2. J. Biol. Chem. 266:6230–6239 [PubMed] [Google Scholar]
- 23. Hu SC, Court DL, Zweig M, Levin JG. 1986. Murine leukemia virus pol gene products: analysis with antisera generated against reverse transcriptase and endonuclease fusion proteins expressed in Escherichia coli. J. Virol. 60:267–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jaskolski M, Alexandratos JN, Bujacz G, Wlodawer A. 2009. Piecing together the structure of retroviral integrase, an important target in AIDS therapy. FEBS J. 276:2926–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kirshenboim N, Hayouka Z, Friedler A, Hizi A. 2007. Expression and characterization of a novel reverse transcriptase of the LTR retrotransposon Tf1. Virology 366:263–276 [DOI] [PubMed] [Google Scholar]
- 26. Le Grice SF. 2003. “In the beginning”: initiation of minus strand DNA synthesis in retroviruses and LTR-containing retrotransposons. Biochemistry 42:14349–14355 [DOI] [PubMed] [Google Scholar]
- 27. Levin HL. 1995. A novel mechanism of self-primed reverse transcription defines a new family of retroelements. Mol. Cell. Biol. 15:3310–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levin HL. 1996. An unusual mechanism of self-primed reverse transcription requires the RNase H domain of reverse transcriptase to cleave an RNA duplex. Mol. Cell. Biol. 16:5645–5654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin JH, Levin HL. 1997. A complex structure in the mRNA of Tf1 is recognized and cleaved to generate the primer of reverse transcription. Genes Dev. 11:270–285 [DOI] [PubMed] [Google Scholar]
- 30. Lin JH, Levin HL. 1997. Self-primed reverse transcription is a mechanism shared by several LTR-containing retrotransposons. RNA 3:952–953 [PMC free article] [PubMed] [Google Scholar]
- 31. Malik HS, Eickbush TH. 1999. Modular evolution of the integrase domain in the Ty3/Gypsy class of LTR retrotransposons. J. Virol. 73:5186–5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nymark-McMahon MH, Beliakova-Bethell NS, Darlix JL, Le Grice SF, Sandmeyer SB. 2002. Ty3 integrase is required for initiation of reverse transcription. J. Virol. 76:2804–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oz Gleenberg I, Herschhorn A, Goldgur Y, Hizi A. 2007. Inhibition of human immunodeficiency virus type-1 reverse transcriptase by a novel peptide derived from the viral integrase. Arch. Biochem. Biophys. 458:202–212 [DOI] [PubMed] [Google Scholar]
- 34. Oz Gleenberg I, Avidan O, Goldgur Y, Herschhorn A, Hizi A. 2005. Peptides derived from the reverse transcriptase of human immunodeficiency virus type 1 as novel inhibitors of the viral integrase. J. Biol. Chem. 280:21987–21996 [DOI] [PubMed] [Google Scholar]
- 35. Oz I, Avidan O, Hizi A. 2002. Inhibition of the integrases of human immunodeficiency viruses type 1 and type 2 by reverse transcriptases. Biochem. J. 361:557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schultz SJ, Zhang M, Champoux JJ. 2003. Specific cleavages by RNase H facilitate initiation of plus-strand RNA synthesis by Moloney murine leukemia virus. J. Virol. 77:5275–5285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sevilya Z, Loya S, Hughes SH, Hizi A. 2001. The ribonuclease H activity of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2 is affected by the thumb subdomain of the small protein subunits. J. Mol. Biol. 311:957–971 [DOI] [PubMed] [Google Scholar]
- 38. Skalka AM, Goff SP. 1993. Reverse transcriptase. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 39. Steele SJ, Levin HL. 1998. A map of interactions between the proteins of a retrotransposon. J. Virol. 72:9318–9322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilhelm M, Wilhelm FX. 2005. Role of integrase in reverse transcription of the Saccharomyces cerevisiae retrotransposon Ty1. Eukaryot. Cell 4:1057–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wilkinson TA, et al. 2009. Identifying and characterizing a functional HIV-1 reverse transcriptase-binding site on integrase. J. Biol. Chem. 284:7931–7939 [DOI] [PMC free article] [PubMed] [Google Scholar]