

Abstract
Genome packaging is functionally coupled to replication in RNA viruses pathogenic to humans (Poliovirus), insects (Flock house virus [FHV]), and plants (Brome mosaic virus [BMV]). However, the underlying mechanism is not fully understood. We have observed previously that in FHV and BMV, unlike ectopically expressed capsid protein (CP), packaging specificity results from RNA encapsidation by CP that has been translated from mRNA produced from replicating genomic RNA. Consequently, we hypothesize that a physical interaction with replicase increases the CP specificity for packaging viral RNAs. We tested this hypothesis by evaluating the molecular interaction between replicase protein and CP using a FHV-Nicotiana benthamiana system. Bimolecular fluorescence complementation in conjunction with fluorescent cellular protein markers and coimmunoprecipitation assays demonstrated that FHV replicase (protein A) and CP physically interact at the mitochondrial site of replication and that this interaction requires the N-proximal region from either amino acids 1 to 31 or amino acids 32 to 50 of the CP. In contrast to the mitochondrial localization of CP derived from FHV replication, ectopic expression displayed a characteristic punctate pattern on the endoplasmic reticulum (ER). This pattern was altered to relocalize the CP throughout the cytoplasm when the C-proximal hydrophobic domain was deleted. Analysis of the packaging phenotypes of the CP mutants defective either in protein A-CP interactions or ER localization suggested that synchronization between protein A-CP interaction and its subcellular localization is imperative to confer packaging specificity.
INTRODUCTION
Many icosahedral viruses with single-stranded, positive-sense RNA genomes cause serious diseases in humans, animals, insects, and plants. Assembly of infectious mature virions, a prerequisite for successful dissemination of a given viral infection to a healthy host, is a carefully orchestrated process maintaining a high degree of precision and requires protein subunits interacting with each other and with viral nucleic acids. Since mature virions predominantly contain viral RNAs, it is reasonable to assume that viruses have evolved to use mechanisms to exclude packaging of RNAs other than those associated with the virus itself. Information gleaned from recent studies with eukaryotic RNA viruses pathogenic to humans and animals (e.g., Poliovirus and Venezuelan equine encephalitis virus) (20, 30, 44), insects (e.g., Flock house virus [FHV]) (42), and plants (e.g., Brome mosaic virus [BMV]) (1) revealed that the mechanism of genome packaging in these taxonomically distinct viral systems is commonly shared and is functionally coupled to replication.
FHV, a positive-strand RNA virus of a coleopteran insect origin, is a member of the insect virus family Nodaviridae (37). The genome organization in FHV is simple, consisting of two RNAs packaged in a single, nonenveloped icosahedral virion with T=3 symmetry (34). RNA1 (F1; 3.1 kb) encodes the viral RNA-dependent RNA polymerase (protein A; 112 kDa), and RNA2 (F2; 1.4 kb) encodes the viral coat protein precursor α (CP-α; 43 kDa) (36). F1 is capable of independent replication, whereas the replication of F2 is F1 dependent (5). During replication, the 3′ end of F1 synthesizes a subgenomic RNA3 (sgF3; 0.4 kb) coding for two overlapping proteins B1 and B2. The function of B1 is unknown, while B2 is the designated suppressor of RNA silencing to overcome host defense response and maintain sustained replication (22). Studies of FHV infection in Drosophila cells identified mitochondria as the key cellular organelle involved in RNA replication by inducing the formation of membrane bound spherules in the mitochondrial intermembrane space (27, 28).
FHV CP-α must be translated from a replicating F2 in order for the FHV genome to be specifically packaged (3, 42). CP-α and the two genomic RNAs assemble into a noninfectious precursor particle called provirion (35). Provirions mature and acquire infectivity by spontaneous cleavage of the CP-α into protein β (38 kDa) and γ (5 kDa) subunits. Deletion analysis of FHV CP identified critical regions required for packaging specificity. For example, deletion of C-terminal residues 382 to 407 resulted in packaging large amounts of random cellular RNA and various amounts of viral RNA (35), while deletion of N-terminal residues 2 to 31 affected the packaging efficiency of F2 but not F1 (26). Analysis that is more recent showed that the packaging of F1 is determined by an N-terminal arginine-rich motif (ARM) region located between positions 32 and 50 (43). Based on these observations in conjunction with confocal microscopic analyses, it was proposed that an ARM is required for the trafficking of CP to mitochondria where FHV RNA replication occurs actively (43). Despite these advances, the underlying molecular mechanism regulating genome-packaging specificity is not well understood.
Although FHV is inherently an insect virus (37), it represents a unique example of a eukaryotic RNA virus that crosses the kingdom barrier and multiplies in a variety of cells, including plants (3, 38), yeast (27, 28), Drosophila (37), and Caenorhabditis elegans (23). In the present study, we opted to use Nicotiana benthamiana plants as a host for the macromolecular interactions involved FHV packaging for the following reasons: (i) analogous to insect cells, FHV replication occurs on the outer mitochondrial membrane of N. benthamiana cells (3); (ii) it has been shown that FHV is not only competent to replicate in several monocotyledonous and dicotyledonous plant species without producing symptoms but also to assemble virions that can infect insect cells (38); (iii) a simple, robust, and synchronized delivery of multiple plasmids to the same plant cell via agroinfiltration (3, 17, 25) and development of a wide range of vectors suitable for evaluating protein-protein interactions using bimolecular fluorescent complementation (BiFC) (8, 19) make the plant system an ideal platform for research questions we sought to examine here.
The simple genome organization of FHV is attractive for elucidating the mechanism of genome packaging in RNA viruses. Despite significant advancement in understanding the various factors regulating genome packaging in FHV (3, 42, 43), the following key issues still remain unanswered. First, the packaging phenotypes exhibited by the FHV CP translated from a replicating versus nonreplicating F2 are distinct (3, 42). However, it is not known whether this differential packaging is due to variation in the subcellular localization sites of the respective CP forms. Second, although protein A and CP has been shown to colocalize to replication sites (i.e., mitochondria), it is not known whether these two macromolecules physically interact to regulate packaging specificity. Therefore, to find answers to these questions, we systematically analyzed and identified various viral and subcellular components involved in the regulation of packaging specificity in FHV. We exploited the inherent salient features of agroinfiltration (2) for efficient transient expression of proteins and BiFC assay (8) and fluorescent cellular marker proteins for evaluating interactions between a pair of desired FHV proteins and their subcellular localization in living cells. We found that FHV CP directly interacts with protein A at the mitochondrial site of replication and the N-proximal arginine residues located in the region from amino acids 1 to 31 or the region from amino acids 32 to 50 of the CP promote this interaction. In addition, we also found that the C terminus of CP, which contains a hydrophobic domain, is critical for endoplasmic reticulum (ER) localization of the CP. Finally, in vivo evaluation of selected CP mutants affecting the interaction with protein A and ER localization revealed their significance in packaging specificity of the FHV genome.
MATERIALS AND METHODS
Construction of YFP fusion proteins for ectopic expression.
Agrobacterium-based binary plasmids PZPc-nYFP, PZPc-cYFP, PZPn-nYFP, or PZPn-cYFP, amenable for BiFC assays in planta, were used to assemble N and C-terminal yellow fluorescent protein (YFP) fusion proteins (see Fig. 1). Either the N-terminal region of YFP (encompassing amino acids 1 to 156 [nYFP]) or C-terminal region of YFP (encompassing amino acids 157 to 239 [cYFP]) was amplified by PCR using appropriate primer sets and the resulting product was inserted into the StuI-digested PZP vector. Adding nucleotides for six histidines and an AGG sequence at the 3′ ends of the primers used to amplify nYFP and cYFP regenerated a StuI site. The resulting constructs were referred to as PZPn-nYFP and PZPn-cYFP, respectively (see Fig. 1). FHV protein A and CP open reading frames (ORFs) were amplified by PCR and inserted into PZPc-nYFP, PZPc-cYFP, PZPn-nYFP, or PZPn-cYFP utilizing StuI and SpeI sites. Deletions and amino acid substitutions were engineered into either N- or C-terminal domains of CP by PCR using appropriate primer sets. Each CP mutant was inserted downstream of nYFP or GFP to express the desired fusion protein. To construct fluorescent cellular markers specific for the ER, nucleus, or mitochondria, respective subcellular targeting signals were engineered into a desired fluorescent protein by PCR using the appropriate primer sets. The resulting PCR products were inserted into StuI- and SpeI-digested PZP vector. These constructs were referred to as pMito-RFP (red fluorescent protein), pNLS-CFP (cyan fluorescent protein), and pER-GFP (green fluorescent protein) that will be used, respectively, to visualize the mitochondria, nucleus and ER (see Fig. 3A). Variants of full-length wild-type (wt) F2 clones harboring CP mutations were assembled using PCR. To construct pF2-ARM1&2 R→A, a 5′ 520-bp fragment from wt F2 was amplified by using the primers F2-NRA-5′-Fw and F2-519-BamHI-Rv and the cDNA of CP ARM1&2 R→A as a template and inserted into pCassRz vector digested with StuI and BamHI (2, 3). The resulting construct was referred to as pF2-5′500RA. Then, the wt F2 sequence was amplified with the primers F2-5′-Fw and F2-3′-BamHI-Rv, and the resulting PCR product was digested with SpeI and BamHI and inserted into SpeI- and BamHI-digested pF2-5′500RA. To construct pF2-C6Thr and pF2-C3Thr, the 3′ end region of the CP gene harboring nucleotides (nt) 1065 to 1250 was first amplified with the primers F2-1065-Fw and F2-CThr-1250-Rv and CP C-6Thr or CP C-3Thr as templates, respectively. The resulting PCR products were gel purified and used as megaprimers for the following steps. The combination of a primer F2-3′-BamHI-Rv and the megaprimers as prepared above was used in a PCR to amplify a region spanning the 3′ sequence of F2 from nt 1065 to 1400. The resulting PCR products were digested with BglII and BamHI and inserted into BglII- and BamHI-digested pF2. The nature of all recombinant clones was verified by DNA sequencing. The composition of the oligonucleotides used in the construction of the above-described recombinant clones is available upon request.
Fig 1.

Schematic representation of fluorescent protein fusion constructs used in the present study. Two pairs of basal BiFC binary vectors amenable for agroinfiltration—one pair for generating N-terminal fusions (PZPn-nYFP and PZPn-cYFP [A]) and the other pair for generating C-terminal fusions (PZPc-nYFP and PZPc-cYFP [B])—were constructed. ORFs of protein A and CP were fused in-frame to each pair of binary vectors using StuI and SpeI sites. Each binary vector contained in sequential order, a left border of T-DNA (LB); a double 35S promoter (35Sx2); a tobacco etch virus (TEV) translation enhancer leader sequence (TL), six-histidine tag (His×6), a fragment of N-terminal 157 residues of yellow fluorescent protein (nYFP), a fragment of C-terminal 83 residues of YFP (nYFP), a 35S terminator (T35S), and a right border of T-DNA (RB).
Fig 3.
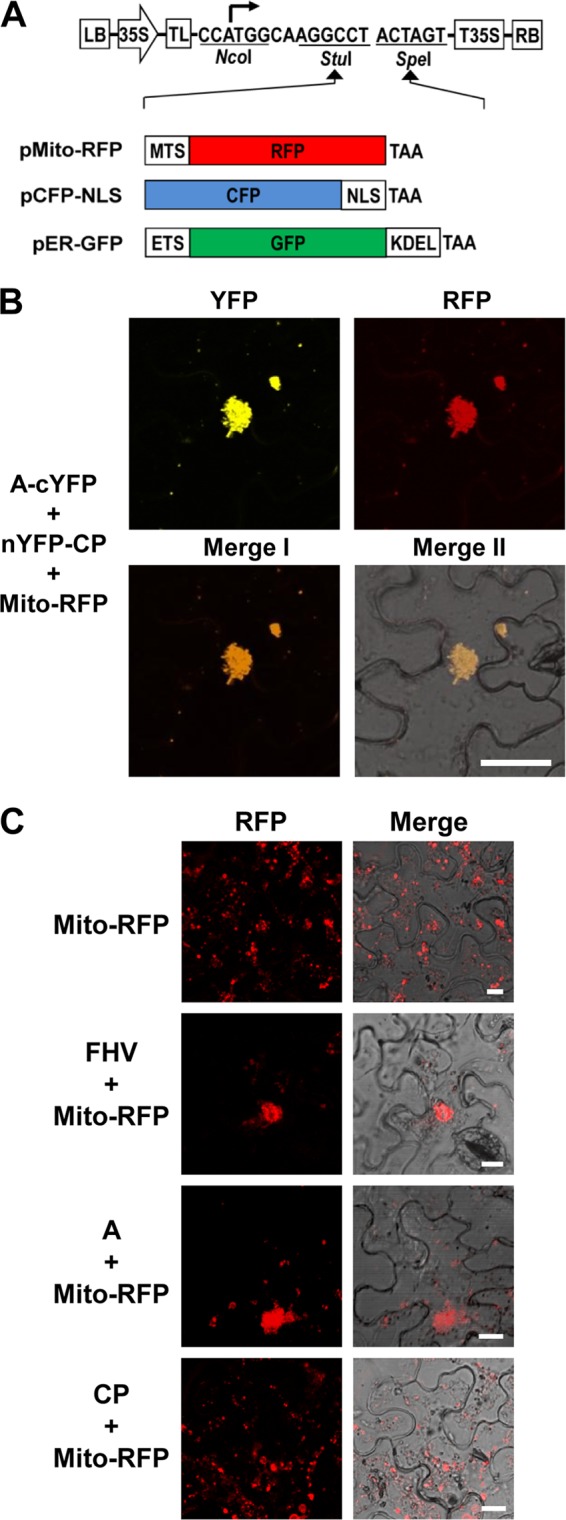
Protein A-CP interaction occurs on the mitochondria. (A) Schematic representation of binary vectors designed to express fluorescent subcellular markers. Three different subcellular marker genes were fused in-frame into the vector using StuI and SpeI sites. Plasmid pMito-RFP was designed specifically for mitochondria to emit red fluorescence by fusing ORF of red fluorescent protein (RFP) to mitochondrial targeting signal (MTS), plasmid pCFP-NLS was designed specifically for the nucleus to emit cyan fluorescence by fusing the ORF of cyan fluorescent protein (CFP) to the nuclear localization signal of simian virus 40 T antigen (NLS), and plasmid pER-GFP was specifically designed for the ER to emit green fluorescence by fusing the ORF of green fluorescent protein (GFP) to the ER targeting signal (ETS) and ER retention sequence (KDEL). (B) Visualization of subcellular localization of the protein A-CP interaction. A mixture of agro-constructs as indicated on the left side of the image was delivered into N. benthamiana leaves by agroinfiltration. The fluorescence signals were observed in the epidermal cells using confocal microscopy at 3 dpi. Bar, 30 μm. (C) Protein A mediates the clustering of mitochondria in plant cells. N. benthamiana leaves were infiltrated with the indicated agrotransformants. FHV denotes a mixture of F1 and F2 agrotransformants (3). The fluorescence signals were observed in the epidermal cells using confocal microscopy at 3 dpi. Mitochondria emit red fluorescence. Bar, 15 μm. Agrotransformants of protein A fused to YFP (A-YFP) and CP fused to GFP or RFP (GFP-CP or RFP-CP) were mixed with agrotransformants expressing the desired fluorescent subcellular markers and infiltrated into N. benthamiana leaves. The fluorescence signals were observed in the epidermal cells using confocal microscopy at 3 dpi. Mitochondria, nuclei, and ER emit red, cyan, and green fluorescence, respectively. Bar, 15 μm.
Agroinfiltration and confocal microscopy.
After transformation of the binary vectors into Agrobacterium strain GV3101, infiltration into N. benthamiana leaves was performed as described previously (2, 39). At 3 days postinfiltration (dpi), epidermal cells of agroinfiltrated leaves were observed for emission of fluorescence using a Leica SP2 laser scanning confocal microscope (Leica, Germany) equipped with a specific laser/filter combination to detect CFP (excitation at 458 nm), GFP (excitation at 488 nm), YFP (excitation at 514 nm), and RFP (excitation at 594 nm).
Coimmunoprecipitation assays.
Total protein extracts were prepared from healthy and agroinfiltrated N. benthamiana leaves as described previously (7, 14), with minor modifications. Briefly, at 3 dpi, infiltrated leaves were ground in liquid nitrogen by using a mortar and pestle and homogenized in 3 volumes of protein extraction buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5% Triton X-100, 0.2% 2-mercaptoethanol, 5% glycerol, proteinase inhibitor cocktail [Sigma, USA]). Cell debris was removed by centrifugation at 18,000 × g for 20 min at 4°C. The resulting supernatants were incubated with anti-protein A antibody at 1:100 dilution (kindly provided by Paul Ahlquist) for 8 h at 4°C. Then, a 30-μl aliquot of protein G-agarose beads (Santa Cruz Biotechnology, USA) was added to each tube, followed by incubation for 2 h at room temperature. The immunocomplexes were then precipitated by centrifugation for 1 min at 10,000 × g and washed three times in 1 ml of phosphate-buffered saline (0.1 M NaCl, 90 mM sodium phosphate [pH 7.0]). For RNase A treatment, the precipitated proteins were treated with RNase A (50 μg/ml) for 2 h at 25°C. The resulting precipitated proteins were eluted from the beads by boiling in SDS-PAGE sample buffer for 3 min. Equal volumes of protein samples were analyzed by SDS-PAGE, followed by immunoblot analysis with anti-protein A and anti-CP antibodies.
Bioinformatic analyses.
Predictions of transmembrane domain (TMD) and hydrophobicity in the sequence of FHV CP was performed using the programs TMHMM v.2 (21) and ProtScale (according to the Kyte and Doolittle method) (16), respectively.
Purification of FHV virions.
At 5 dpi, infiltrated leaves were ground vigorously in buffer A (50 mM HEPES [pH 7.0], 5 mM CaCl2, 0.5% [wt/vol] Nonidet P-40, 0.2% [wt/vol] 2-mercaptoethanol). After removing the debris by centrifugation at 18,000 × g for 20 min at 4°C, the resulting supernatant was incubated with 10 μg of RNase A/ml for 1 h at 27°C. The incubated mixture was subjected to centrifugation at 18,000 × g for 20 min at 4°C, and the virus was pelleted by centrifuging the supernatant at 280,000 × g for 3 h at 4°C. The pellet was resuspended in buffer A by shaking for 16 h at 4°C. Debris was removed by centrifugation at 18,000 × g for 20 min at 4°C, and the supernatant was overlaid on a 30% (wt/wt) sucrose cushion prepared in buffer B (50 mM HEPES [pH 7.0], 5 mM CaCl2, 0.1% bovine serum albumin, 0.2% 2-mercaptoethanol) and centrifuged at 280,000 × g for 3 h at 4°C. The final pellet was resuspended in buffer A.
FHV progeny analysis.
Total RNA and protein preparations were extracted from infiltrated leaves using TRIzol reagent (Invitrogen, USA) according to the manufacturer's instructions. FHV RNA in either total cell extracts or purified virions was analyzed as described previously (3). FHV RNA in total and purified virions was detected by Northern blot hybridization using riboprobes complementary to either full-length F1 or F2 as described previously (3) using appropriate primers containing the T7 promoter sequence.
RESULTS
Protein A and CP physically interact at the mitochondrial site of replication.
As schematically shown in Fig. 1, two split fragments of YFP, the N-terminal residues 1 to 156 (nYFP) and C-terminal residues 157 to 239 (cYFP) were fused to target proteins, protein A and CP. First, to examine the self-interaction of either protein A or CP, four pairs of nYFP and cYFP terminal fusions of protein A (A-nYFP+A-cYFP, A-nYFP+cYFP-A, nYFP-A+A-cYFP, and nYFP-A+cYFP-A) or CP (CP-nYFP+CP-cYFP, CP-nYFP+cYFP-CP, nYFP-CP+CP-cYFP, and nYFP-CP+cYFP-CP) (Fig. 2) were infiltrated into N. benthamiana, and the reconstituted YFP signal was monitored. For protein A self-interaction, among the four pairs, only one pair coexpressing A-nYFP and A-cYFP resulted in YFP fragment complementation confirming protein A self-interaction (Fig. 2D). Furthermore, unlike free YFP that was distributed throughout the cytoplasm of epidermal cells (Fig. 2A), we found that yellow fluorescence representing protein A self-interaction was restricted to a specific location in the cytoplasm that was later identified to be the mitochondria (see below).
Fig 2.

Visualization in vivo of protein A and CP interactions. Each binary construct was transformed into Agrobacterium strain GV3103 and infiltrated into the abaxial side of N. benthamiana leaves in either single or pairwise combinations, as indicated on the left side of each panel. The reconstructed YFP signals were observed in the epidermal cells using confocal microscopy at 3 days postinfiltration (dpi). Representative confocal fluorescence images shown. (A to C) Three sets of control samples. (D and E) Self-interaction of protein A and CP. (F and G) Protein A-CP interactions. Fluorescent signals from expression of YFP (in panel A) represent a positive control. Subcellular images for fluorescence emitted by YFP and the merged images under the transmitted-light mode are shown. Images a′, b′, c′, d′, e′, f′, g′, and h′, in either single or pairwise combinations, are enlargements that correspond to the areas indicated in panels a, b, c, d, e, f, g, and h, respectively. The fluorescence signals were observed in the epidermal cells using confocal microscopy at 3 dpi. Bar, 30 μm. (H) Summary of self- and cross-interactions. The presence or absence of yellow fluorescence was indicated by “+” and “−” symbols, respectively.
For self-interaction of CP, among the four pairs tested, only one pair coexpressing nYFP-CP and cYFP-CP resulted in YFP fragment complementation confirming CP self-interaction (Fig. 2E). The subcellular distribution site of CP self-interaction was distinct from that of free YFP, and later it was identified to be the ER (see below). Collectively, the data provided a convincing proof-of-concept for the efficacy of BiFC approach in detecting self-interaction of protein A and CP.
A previous confocal analysis has shown that protein A and CP colocalizes on mitochondria (43), but it is not known whether a physical interaction between these two macromolecules exists. Thus, using BiFC we sought to examine the interaction between protein A and CP in living cells. To this end, eight possible pairs of N- and C-terminal YFP fusions of protein A and CP were assembled and infiltrated into N. benthamiana and monitored for fluorescence as described above. Among the eight pairs infiltrated, we were able to detect BiFC signals from two pairs, A-nYFP+cYFP-CP (Fig. 2F) and A-cYFP+nYFP-CP (Fig. 2G), indicating that protein A and CP interact.
It has been previously demonstrated that protein A localizes on the outer mitochondrial membranes in both insect and plant cells (3, 28). Furthermore, mitochondria, which are normally distributed in the cytoplasm, are clustered into the perinuclear region following nodavirus infection (15, 43). Therefore, it is reasonable to surmise that the reconstituted YFP signals resulting from interaction of protein A and CP might be localized on the clustered mitochondria. To substantiate this assumption, we assembled fluorescent subcellular markers that are specifically targeted to mitochondria (pMito-RFP, characterized by the fusion of red fluorescent protein to mitochondrial targeting signal) (Fig. 3A). When a pair of bona fide interacting partners of protein A and CP (i.e., A-cYFP and nYFP-CP) was coexpressed with pMito-RFP, the reconstituted YFP signals were colocalized with RFP signals (Fig. 3B), demonstrating that the clustered mitochondria is the subcellular localization site of protein A and CP interaction. These results were consistently reproduced in at least three independent experiments.
Ectopic expression and subcellular localization of FHV protein A and CP.
One of the hallmark features of nodavirus infection is the perinuclear clustering of mitochondria (15, 43), the recognized site of FHV replication (28). Similar mitochondrial clustering was observed when protein A and CP were coexpressed transiently in N. benthamiana (Fig. 3B). It was previously shown that the mitochondrial clustering is mediated by protein A in yeast cells (27). To verify this in plant cells, protein A or CP was transiently expressed with the mitochondrial marker. In contrast to the autonomous expression of Mito-RPF resulting in cytoplasmic distribution of mitochondria, a clustering of mitochondria was observed in leaves coexpressing Mito-RPF and either wt FHV or protein A but not with CP (Fig. 3C). Taken together, these results confirm previous observations in yeast cells (27) that protein A is the sole determining factor for mitochondrial clustering in N. benthamiana.
Next, we sought to examine and visualize the precise subcellular localization sites of protein A and CP in plant cells. Consequently, these proteins were tagged to a fluorescent protein (protein A was fused to YFP, while CP was fused with either GFP or RFP) and coexpressed with fluorescent subcellular markers in N. benthamiana leaves. The results are summarized in Fig. 4A and B. When A-YFP was coexpressed with Mito-RFP, as expected YFP signals were colocalized with RFP signals, indicating that protein A localized on the clustered mitochondria (Fig. 4A). Subcellular localization of FHV CP was examined in Drosophila cells, where its expression was contingent on replication (42), and was shown to closely associate with the ER network (43). However, during FHV infection, not only did the mitochondria cluster around perinuclear region, but the ER was also found to retract significantly toward perinuclear region (43). Therefore, to verify whether the subcellular localization of CP would be different when it is expressed independent of replication, CP tagged with either GFP or RFP (GFP-CP and RFP-CP, respectively) was expressed ectopically in plant cells. As shown in Fig. 4B, GFP-CP showed a distinct subcellular localization pattern reminiscent of the ER distribution. Coexpression with fluorescent subcellular markers revealed that CP was predominantly localized on the ER as punctate bodies (Fig. 4B, middle panel) but not on mitochondria (Fig. 4B, bottom panel). However, when GFP-CP was coexpressed with CFP-NLS, some overlapping fluorescence signals for GFP and CFP were observed (Fig. 4B, top panel). Since in plant cells the perinuclear region is highly structured with the ER network (18, 40), the observed colocalization of GFP and CFP signals could be due to the accumulation of GFP-CP on ER surrounding the nucleus.
Fig 4.
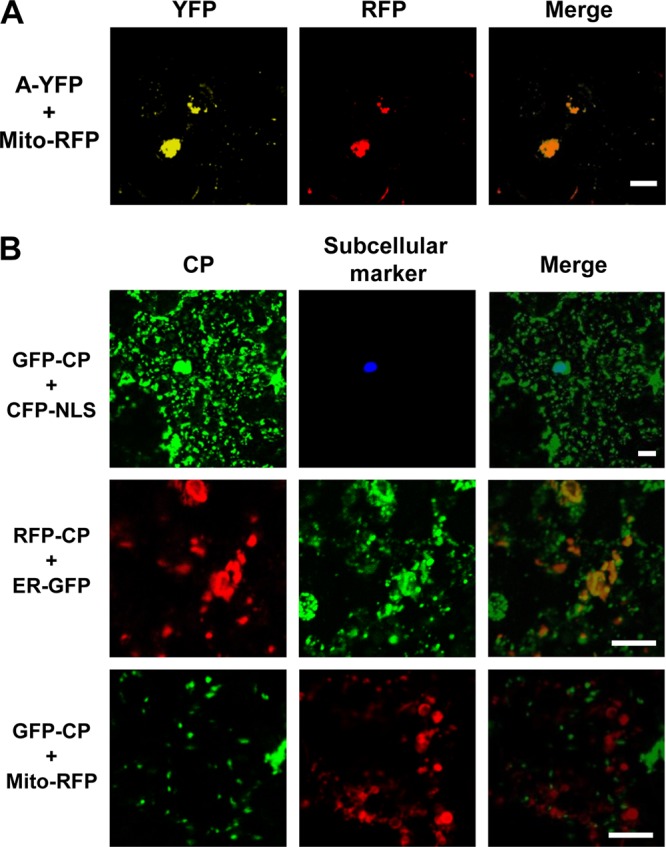
Subcellular distribution pattern of protein A and CP in plant cells. (A and B) Agrotransformants of protein A fused to YFP (A-YFP) and CP fused to GFP or RFP (GFP-CP or RFP-CP) were mixed with agrotransformants expressing the desired fluorescent subcellular markers (see Fig. 3A) and infiltrated into N. benthamiana leaves. The fluorescence signals were observed in the epidermal cells using confocal microscopy at 3 dpi. Mitochondria, nuclei, and ER emit red, cyan, and green fluorescence, respectively. Bar, 15 μm.
Arginine residues at the N terminus of CP are required to promote interaction with protein A.
To identify the domains of FHV CP required for its interaction with protein A, based on the previous data (26), we first focused on characterizing the N-proximal region spanning amino acids 1 to 50 and the C-proximal region spanning amino acids 381 to 407. The N-proximal 50-amino-acid region of CP is very basic and is commonly referred to as an arginine-rich motif (ARM). However, to precisely delineate and characterize the critical part of this region with respect to its interaction with protein A, we divided the N-proximal region from amino acids 1 to 50 into ARM1 (spanning residues 1 to 31) and ARM2 (spanning residues 32 to 50) (Fig. 5A). As shown in Fig. 5B, a series of deletions and amino acid substitutions were engineered into the N-proximal ARM1 and ARM2, as well as the C-proximal region (spanning amino acids 364 to 407). Engineered deletions are classified into three sets. First set consisted of three deletion mutants, each lacking either ARM1 (CPΔ1-31) or ARM2 (CPΔ32-50) or both (CPΔ1-50) (Fig. 5B). The second set consisted of two deletion mutants: mutant CPΔ137-407 lacked amino acids 137 to 407, whereas in mutant CPΔ382-407, the C-proximal amino acids located between amino acids 382 and 407 were deleted (Fig. 5B). One of the two deletion mutants of the third set is characterized by lacking both N-proximal ARMs (amino acids 1 to 50) and the C-proximal 26 amino acids (CPΔ1-50:382-407), while the other mutant lacked amino acids 1 to 363 (CPΔ1-363) (Fig. 5B). Since one of two bona fide pairs of interacting partners of protein A and CP being A-cYFP and nYFP-CP (Fig. 2G), each CP deletion mutant was subcloned into a genetic background of nYFP-CP and coinfiltrated with A-cYFP into N. benthamiana plants for evaluating the interaction between protein A and each mutant CP by BiFC. The results are summarized in Fig. 5D to K.
Fig 5.

Identification of CP domains required for the interaction with protein A. (A) Schematic representation of CP variant containing deletions and amino acid substitutions. Characteristic features of a binary vector, as described in the Fig. 1 legend, containing either nYFP or GFP is shown. A wt FHV CP (wt CP) of 407 amino acids is shown. The N-terminal region from amino acids 1 to 50 is divided into two regions designated ARM1 (amino acids 1 to 30 containing 5 arginine residues indicated in red font) and ARM2 (amino acids 32 to 50 containing 12 arginine residues indicated in red font) and are indicated by blue boxes. The region spanning mature β-subunit (amino acids 1 to 363) and the γ-subunit (amino acids 364 to 407) of the CP is indicated. The C terminus of the CP located between amino acids 382 and 407, indicated by orange shading, contains a high proportion of hydrophobic amino acid residues. (B) Dashed lines represent the extent of engineered deletions in the CP ORF. (C) Arginine residues located in ARM1, ARM2, or both were substituted with alanine residues (indicated in red font). nYFP fusions for each CP mutant (shown in Fig. 5A to C) were constructed by using the binary vector shown in Fig. 1. (D to O) Each nYFP-CP mutant was coexpressed with A-cYFP in N. benthamiana leaves by agroinfiltration. The reconstructed YFP signals were observed in the epidermal cells using confocal microscopy at 3 dpi. Images emitting yellow fluorescence by reconstituted YFP are shown to the left, and images merged with those taken under the transmitted-light mode are shown to the right. Bar, 15 μm.
As expected, control infiltrations involving coexpression of A-cYFP and nYFP-CP interacted, emitting a yellow fluorescence signal (Fig. 5D). Among the three deletion mutants encompassing the N-terminal ARMs, two mutants CPΔ1-31 and CPΔ32-50, but not CPΔ1-50, interacted with protein A (Fig. 5E to G). This result was unexpected since only ARM2 (i.e., encompassing amino acids 32 to 50) has been shown to be responsible for localizing CP to the RNA replication sites (43). The critical involvement of the N-proximal region encompassing the ARMs, but not the C-proximal region, in interaction with protein A was further accentuated by detection of YFP signals reconstructed by interactions between protein A and either CPΔ137-407 or CPΔ382-407, whereas no YFP signals were detectable in cells coexpressing protein A and either CPΔ1-50:382-407 or CPΔ1-363 (Fig. 5E to K). Taken together, these results firmly established that a CP domain required to interact with protein A is located in the N terminus extending from residues 1 to 136.
To further examine the significance of specific arginine residues of the N-proximal ARM1 and/or ARM2 in interaction with protein A, four additional amino acid substitution mutants were engineered into the genetic background of selected deletion mutants shown in Fig. 5A and B and are shown in Fig. 5C. First, all 12 arginine residues in ARM2 were substituted with alanine residues in the genetic background of either wt CP or CPΔ1-31, respectively, creating the variants CPARM2 R→A and CPΔ1-31 ARM2 R→A (Fig. 5C); similarly, the five arginine residues of ARM1 were substituted with alanine residues in the genetic background of CPΔ32–50 creating CPΔ32-50 ARM1 R→A (Fig. 5C); finally, all 17 arginine residues collectively located in the N-proximal ARM1 and ARM2 were substituted with alanine residues into the genetic background of wt CP, creating CP ARM1&2 R→A (Fig. 5C). Each of these mutants was fused with nYFP and coexpressed with A-cYFP in N. benthamiana and evaluated for the reconstituted yellow fluorescence by confocal microscopy. The results are shown in Fig. 5L to O. Among four mutants, yellow fluorescence was detected only with CP ARM2 R→A (Fig. 5L) but not with the other three mutants (Fig. 5M to O), suggesting either N-proximal ARM1 or ARM2, but not both, having positively charged arginine residues is necessary to promote interaction with protein A.
Coimmunoprecipitation assays confirm protein A and CP interaction.
N. benthamiana leaves were infiltrated to coexpress protein A and either wt CP or three selected ARM mutants (CP ARM1&2 R→A, CP ARM2 R→A, and CP ARM1 R→A). At 3 dpi, total proteins were extracted from infiltrated leaves and incubated with anti-protein A antibodies, followed by the precipitation of the complex with protein G-agarose. Western blotting with anti-protein A or CP antibodies detected the resulting coimmunoprecipitated products. Consistent with the BiFC assay (Fig. 5), wt CP, CP ARM2 R→A, and CP ARM1 R→A, but not CP ARM1&2 R→A, were coprecipitated with protein A (Fig. 6). These results further validated the results of our BiFC assays showing that protein A interacts with CP and that the arginine residues of either ARM1 or ARM2 are intimately involved in promoting this interaction. In addition, RNase A treatment did not disrupt the coprecipitation of protein A and CP (Fig. 6, lanes 7 to 9), suggesting that RNA is not involved in promoting the interaction between protein A and CP.
Fig 6.
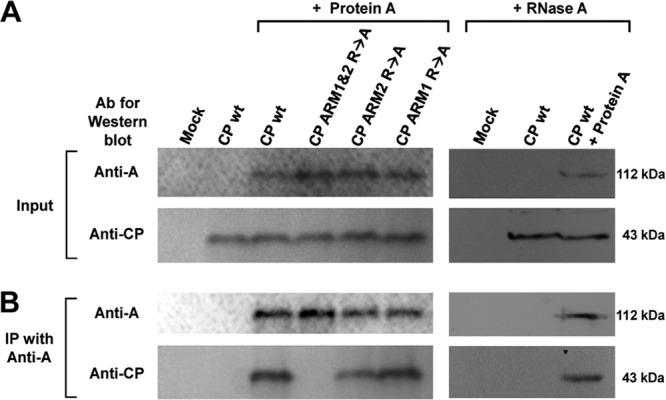
Coimmunoprecipitation assay. Protein A was coexpressed with either wt CP or its indicated mutants in N. benthamiana leaves by agroinfiltration. (A) Expression of protein A and CP in total protein extracts was confirmed by Western blotting with anti-protein A and anti-CP antibodies, respectively. Total protein extracts from leaves infiltrated with the empty vector or CP only were used as controls. (B) For coimmunoprecipitation, total protein extracts of each sample were incubated first with anti-protein A antibodies, followed by complex precipitation with protein G-agarose. The resulting coimmunoprecipitated products were subjected to Western blotting with anti-protein A and anti-CP antibodies. The effect of RNase A treatment on coimmunoprecipitated products was verified by Western blot analysis as described above.
Hydrophobic amino acids at the C terminus of FHV CP are required for the specific subcellular localization of FHV CP.
As shown above (Fig. 4B), CP tagged with fluorescent proteins specifically localized on the ER. We extended this approach to characterize the domain required for in the subcellular localization of CP by constructing GFP-fused CP mutants having either deletions or substitutions (Fig. 5A to C). The GFP-fused CP mutants were expressed in N. benthamiana via agroinfiltration. The results are summarized in Fig. 7. The pattern of GFP distribution for N-terminal deletion mutants (i.e., GFP-CPΔ1-31, GFP-CPΔ32-50, and GFP-CPΔ1-50) or substitutions (i.e., CP ARM1&2 R→A) was indistinguishable from that of control infiltrations performed with wt CP. In contrast, the pattern of GFP distribution for the CP variants, (CPΔ382-407) lacking the C-proximal region or CPΔ1-363 having amino acids encompassing the CP γ-subunit, was identical to that of free GFP (Fig. 7A). Taken together, our results show that, although the N-proximal region containing ARM1 and ARM2 is dispensable for ER localization, the C-terminal domain of FHV CP is necessary but not sufficient for specific ER subcellular localization.
Fig 7.

Identification of the CP domain required for specific subcellular localization. (A) CP mutants were fused to GFP as shown in Fig. 5A to C. The indicated GFP-tagged CP mutants were expressed in N. benthamiana leaves by agroinfiltration. The GFP signals were observed in the epidermal cells using confocal microscopy at 3 dpi. Bar, 15 μm. (B and C) Prediction of the TMD (B) and the hydrophobic domain (C) of FHV CP. The TMHMM v.2 and ProtScale (the Kyte and Doolittle method) programs were used to predict the TMD and the hydrophobicity of CP, respectively. (D) Binary plasmids designed to express CP containing mutations (indicated in green font) in the C-terminal hydrophobic domain as GFP fusions. (E) After agroinfiltration of C-terminal mutants of CP, GFP signals were observed in the epidermal cells using confocal microscopy at 3 dpi. Bar, 15 μm.
In eukaryotic cells, TMDs mediate the secretory pathway, including the sorting, targeting, and membrane retention of proteins (9, 10, 24, 33). Thus, we analyzed the FHV CP amino acid sequence using the programs to identify potential TMDs and hydrophobic regions. First, the TMHMM v.2 program predicted that FHV CP has a putative TMD at the C terminus (Fig. 7B). This region was also predicted to have the highest hydrophobicity in the FHV CP ORF by using the ProtScale program (with the Kyte and Doolittle method) (Fig. 7C). Based on this prediction, we constructed three CP mutants with the substitution of threonines (a hydrophilic but uncharged amino acid) for hydrophobic amino acids at the C terminus (Fig. 7D). These included the following: (i) variant CP C-6Thr contains six threonines at positions 398, 400, 401, 402, 405, and 407, and (ii) variant CP C-3Thr contains three threonines at positions 398, 400, and 401. (iii) Moreover, since it was shown that FHV CP with alanine substitutions at positions 402, 405, and 407 was deficient for specific encapsidation of FHV RNAs (35), we also constructed an additional CP variant, CP C-3FA, with three alanines at positions 402, 405, and 407. All of these CP mutants were predicted, using the above-described programs (data not shown), to lose the putative TMD at the C terminus. These CP mutants tagged with GFP were expressed in N. benthamiana, and we examined their subcellular localization. Interestingly, the GFP distribution pattern displayed by all CP substitution mutants losing the putative TMD was indistinguishable from that of free GFP (Fig. 7E). This result suggested that the hydrophobic domain at the C terminus of FHV CP, with a putative TMD, is important for the specific subcellular localization of CP.
Effects of mutations disrupting interaction of CP with protein A and its subcellular localization on packaging specificity.
To investigate the biological significance of the CP mutants defective in either interaction with protein A or ER localization, we subcloned three selected CP mutants into the genetic background of a biologically active F2 agro-construct (3). The selected CP mutants were CP ARM1&2 R→A (defective in interaction with protein A [Fig. 5C and O]) and CP C-6Thr and CP C-3Thr (exhibited altered subcellular localization compared to the wt [Fig. 7D and E]), and the resulting F2 mutants were designated pF2-ARM1&2 R→A, pF2-C6Thr, and pF2-C3Thr, respectively. N. benthamiana leaves were infiltrated with Agrobacterium cultures containing pF1 and either pF2 wt or its mutants. At 5 dpi, infiltrated leaves from each inoculum were divided into two lots. One lot was used to extract the total RNA, while the other was used to obtain RNA from purified virions. FHV RNA profiles of total RNA extracts were analyzed by Northern blot hybridization. The results are shown in Fig. 8A. As expected, total RNA preparations of wt and each mutant inocula contained all three FHV RNAs. However, quantitative analysis revealed that, compared to wt, progeny accumulation for mutant pF2-ARM1&2 R→A was reduced by 39% for F1+F2+sgF3 and by 47% for F1+F2. In contrast, progeny accumulation for mutants pF2-C6Thr and pF2-C3Thr was near wt levels (Fig. 8A). Sequence analysis of reverse transcription-PCR products of progeny F2 confirmed that engineered mutations were maintained during replication in plant cells (data not shown).
Fig 8.

Effect of the N- and C-terminal mutants of CP on replication and packaging. (A) Northern blot analysis of FHV RNA replication in plant cells. The agrotransformant of F1 was mixed with either wt F2 or the indicated mutants and infiltrated into N. benthamiana. At 5 dpi, the total RNAs were extracted and subjected to Northern blot hybridization with riboprobes specific for F1 and F2. The FHV RNA accumulation levels shown below the Northern blot were normalized against the wt as 100%. (B) Virion yield. FHV virions were purified from the infiltrated leaves, and the yield was quantitated and normalized against the wt as 100%. The data shown represent averages of three independent virion preparations. (C) Analyses of RNAs extracted from purified FHV virions. At 5 dpi, virions were purified from the infiltrated leaves, and encapsidated RNA was isolated. Virion RNA was subjected to 1% agarose electrophoresis and visualized by staining with ethidium bromide. An asterisk indicates a heterogeneous population RNA of cellular origin. (D) The RNA profile shown in panel C was subjected to Northern blot hybridization with a mixture of riboprobes complementary to full-length F1 and F2. The positions of F1, F2, and sgF3 are shown to the right. In panel A, rRNA indicates a loading control. Encapsidated FHV progeny was quantitated as described above.
Virions were purified from wt or mutant FHV infiltrated leaves, and the yield was quantitated. Compared to the wt, the virion yield recovered from leaves infiltrated with mutant pF2-ARM1&2 R→A was reduced by 60%, while that of mutants pF2-C6Thr and pF2-C3Thr was near wt levels (Fig. 8B). To verify the packaging efficiency and specificity, encapsidated RNA was extracted from purified virions and subjected to analytical gel electrophoresis for visualization by ethidium bromide staining, followed by Northern blot hybridization. The results are shown in Fig. 8C and D. Ethidium bromide staining revealed that wt FHV virions packaged F1 and F2 exclusively, a profile identical to that observed in Drosophila cells (26), and packaging of no other RNAs was evident (Fig. 8C). In contrast, virions of mutant CP ARM1&2 R→A packaged a significantly lower amount of F1 and F2 and a faster-migrating heterogeneous population of RNAs varying in size (Fig. 8C). On the other hand, unlike virions of CP ARM1&2 R→A, those of CP C-6Thr and CP C-3Thr packaged detectable levels of progeny F1 and F2 in addition to a faster-migrating heterogeneous RNA population (Fig. 8C). To identify the nature of faster-migrating RNA, virion RNA was subjected to Northern blot hybridization using a mixture of riboprobes complementary to full-length F1 and F2. Northern analysis revealed that FHV RNA specific riboprobes hybridized to full-length F1 and F2 (Fig. 8D), suggesting that faster-migrating heterogeneous RNA is not of viral but of cellular origin. A comparative quantitative analysis of FHV RNA accumulated in total RNA preparations versus those packaged further revealed that mutant virions of pF2-ARM1&2 R→A packaged the FHV RNA poorly due to the absence of the N-proximal ARM region required for viral RNA binding. As expected, the presence of the N-proximal ARM region in the mutants CP C-6Thr and CP C-3Thr promoted near wt level packaging of F1 and F2. However, despite variations in replication and packaging efficiency, the presence of cellular RNA suggested that both N-proximal ARM domains are required not only for RNA binding but also for its interaction with protein A and that the C-proximal domain required for precise subcellular localization of CP is important for FHV packaging specificity.
DISCUSSION
The major impetus for evaluating the interaction between viral replicase and CP leading to packaging specificity in RNA viruses was based on the following two observations. First, in FHV and BMV, the packaging specificity is dependent on the translation of CP from a replication-derived mRNA. Second, packaging exhibited by CP expressed in the presence of replication is clearly distinct from packaging exhibited in its absence. Therefore, we hypothesize that, before encapsidation, a physical interaction between viral replicase and CP is obligatory for dictating packaging specificity. Testing this hypothesis by the application of agroinfiltration and BiFC in combination with fluorescent cellular marker proteins has revealed that packaging specificity is dependent on a physical interaction between protein A and CP occurring at the mitochondrial sites of replication promoted by either the ARM1 or the ARM2 region of the CP (Fig. 5 and 8).
Viral CPs have evolved to selectively package their genomes in the presence of a large pool of cellular RNAs. RNA-binding motifs such as ARMs found in several viral CPs (32, 41) in cooperation with the origin-of-assembly sequences (OAS) in viral nucleic acids have been shown to contribute to assembly and genome packaging (31). However, other evidence argues that the presence of either ARM or OAS is not always sufficient to guarantee assembly or packaging. For example, a BMV CP variant with an unaltered N-proximal ARM that was defective in β-hexamer formation failed to assemble in vitro; however, the environment provided by in planta expression of the β-hexamer mutant CP via replication-dependent transcription and translation supported in the assembly of RNA containing virions indistinguishable from those of the wt (12). Furthermore, similar to BMV and FHV, packaging of viral progeny by a CP translated from a replication-derived mRNA was demonstrated in other RNA viruses such as poliovirus (30), Kunjin virus (20), and Venezuelan equine encephalitis virus (44). Collectively, these observations suggested that some macromolecular interactions occurring in vivo at or near a subcellular compartment that are integral to replication are likely to also play an important role in controlling packaging specificity. Clearly, in the present study, BiFC assays have not only revealed one such macromolecular interaction but also identified where the interaction occurs, i.e., the interaction of CP with protein A occurs on the outer membranes of mitochondrial sites of replication.
The question that now needs to be addressed is how FHV CP is trafficked to the mitochondria. Based on the subcellular localization of FHV CP examined using a replication-dependent expression system, Venter et al. (43) proposed that trafficking of CP to a mitochondrial site of replication is mediated through the ARM region. Since FHV CP has no recognizable mitochondrial localization signals and considering our observation that ectopic expression of FHV CP with an intact ARM was predominantly localized on the ER (Fig. 4B), it is difficult to envision how the ARM could direct trafficking of CP to mitochondria. Since protein A has a mitochondrial localization signal (29), it is more logical to propose that trafficking of CP to mitochondria is mediated through a protein A-CP interaction, as demonstrated in the present study (Fig. 2 and 5). Our results also show that apart from a protein A and CP interaction, another characteristic feature integral to packaging is the ER localization of CP mediated by its C-proximal hydrophobic domain (Fig. 7). Subcellular localization of CP performed in a replication-based expression system revealed that deletion of the C-proximal region of CP (referred as Δγ381) encompassing the hydrophobic domain did not result in significant reduction in mitochondrial colocalization of CP and protein A (43). Since FHV infection results in dramatic cytopathological changes that include mitochondrial clustering (28) and significant retraction of the ER toward the perinuclear region (43), the innate characteristic of subcellular localization regulated by the C-proximal domain was masked in a replication-dependent expression system. Indeed, the precise role of the C-proximal domain in regulating the subcellular localization of CP became apparent when either the wt FHV CP or a mutant lacking the C-proximal domain was compared in ectopically expressed N. benthamiana cells, i.e., wt CP displayed a punctate distribution on the ER, whereas mutant CP was distributed throughout the cytoplasm (Fig. 7).
In conclusion, in addition to the results presented here, BiFC has been used successfully for evaluating the interactions of HIV Gag proteins (6), herpes simplex virus glycoproteins (4), and paramyxovirus fusion and hemagglutinin-neuraminidase proteins (11). Therefore, the BiFC approach can be extended to test whether packaging specificity regulated by a replicase-CP interaction is widely conserved in other viruses with positive-sense RNA genomes.
ACKNOWLEDGMENTS
We thank Shou-Wei Ding for providing FHV clones and anti-CP antibody Paul Ahlquist for providing anti-protein A antibody. The confocal microscopy analysis was performed at IIGB core facilities located at University of California, Riverside. We also thank Shou-Wei Ding, Theo Dreher, and Deb Mathews for editorial comments.
Footnotes
Published ahead of print 21 March 2012
REFERENCES
- 1. Annamalai P, Rao AL. 2006. Packaging of brome mosaic virus subgenomic RNA is functionally coupled to replication-dependent transcription and translation of coat protein. J. Virol. 80:10096–10108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Annamalai P, Rao AL. 2005. Replication-independent expression of genome components and capsid protein of brome mosaic virus in planta: a functional role for viral replicase in RNA packaging. Virology 338:96–111 [DOI] [PubMed] [Google Scholar]
- 3. Annamalai P, Rofail F, Demason DA, Rao AL. 2008. Replication-coupled packaging mechanism in positive-strand RNA viruses: synchronized coexpression of functional multigenome RNA components of an animal and a plant virus in Nicotiana benthamiana cells by agroinfiltration. J. Virol. 82:1484–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Atanasiu D, et al. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ball LA. 1995. Requirements for the self-directed replication of flock house virus RNA 1. J. Virol. 69:2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boyko V, et al. 2006. Coassembly and complementation of Gag proteins from HIV-1 and HIV-2, two distinct human pathogens. Mol. Cell 23:281–287 [DOI] [PubMed] [Google Scholar]
- 7. Bracha-Drori K, et al. 2004. Detection of protein-protein interactions in plants using bimolecular fluorescence complementation. Plant J. 40:419–427 [DOI] [PubMed] [Google Scholar]
- 8. Citovsky V, et al. 2006. Subcellular localization of interacting proteins by bimolecular fluorescence complementation in planta. J. Mol. Biol. 362:1120–1131 [DOI] [PubMed] [Google Scholar]
- 9. Cocquerel L, et al. 1999. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J. Virol. 73:2641–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cocquerel L, Meunier JC, Pillez A, Wychowski C, Dubuisson J. 1998. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. 72:2183–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Connolly SA, Leser GP, Jardetzky TS, Lamb RA. 2009. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: implications for the mechanism of fusion triggering. J. Virol. 83:10857–10868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Wispelaere M, Chaturvedi S, Wilkens S, Rao AL. 2011. Packaging and structural phenotype of brome mosaic virus capsid protein with altered N-terminal beta-hexamer structure. Virology 419:17–23 [DOI] [PubMed] [Google Scholar]
- 13. Reference deleted.
- 14. Fujioka Y, Utsumi M, Ohba Y, Watanabe Y. 2007. Location of a possible miRNA processing site in SmD3/SmB nuclear bodies in Arabidopsis. Plant Cell Physiol. 48:1243–1253 [DOI] [PubMed] [Google Scholar]
- 15. Garzon S, Strykowski H, Charpentier G. 1990. Implication of mitochondria in the replication of Nodamura virus in larvae of the Lepidoptera, Galleria mellonella (L.) and in suckling mice. Arch. Virol. 113:165–176 [DOI] [PubMed] [Google Scholar]
- 16. Gasteiger E, et al. 2005. Protein identification and analysis tools on the ExPASy server, p 571–607 In Walker JM. (ed), The proteomics protocols handbook. Humana Press, Clifton, NJ: [DOI] [PubMed] [Google Scholar]
- 17. Gopinath K, Kao CC. 2007. Replication-independent long-distance trafficking by viral RNAs in Nicotiana benthamiana. Plant Cell 19:1179–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heath MC. 2000. Advances in imaging the cell biology of plant-microbe interactions. Annu. Rev. Phytopathol. 38:443–459 [DOI] [PubMed] [Google Scholar]
- 19. Kerppola TK. 2008. Bimolecular fluorescence complementation: visualization of molecular interactions in living cells. Methods Cell Biol. 85:431–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khromykh AA, Varnavski AN, Sedlak PL, Westaway EG. 2001. Coupling between replication and packaging of flavivirus RNA: evidence derived from the use of DNA-based full-length cDNA clones of Kunjin virus. J. Virol. 75:4633–4640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580 [DOI] [PubMed] [Google Scholar]
- 22. Li H, Li WX, Ding SW. 2002. Induction and suppression of RNA silencing by an animal virus. Science 296:1319–1321 [DOI] [PubMed] [Google Scholar]
- 23. Lu R, et al. 2005. Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature 436:1040–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Machamer CE. 1993. Targeting and retention of Golgi membrane proteins. Curr. Opin. Cell Biol. 5:606–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marillonnet S, et al. 2004. In planta engineering of viral RNA replicons: efficient assembly by recombination of DNA modules delivered by Agrobacterium. Proc. Natl. Acad. Sci. U. S. A. 101:6852–6857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marshall D, Schneemann A. 2001. Specific packaging of nodaviral RNA2 requires the N terminus of the capsid protein. Virology 285:165–175 [DOI] [PubMed] [Google Scholar]
- 27. Miller DJ, Ahlquist P. 2002. Flock house virus RNA polymerase is a transmembrane protein with amino-terminal sequences sufficient for mitochondrial localization and membrane insertion. J. Virol. 76:9856–9867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller DJ, Schwartz MD, Ahlquist P. 2001. Flock house virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J. Virol. 75:11664–11676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller DJ, Schwartz MD, Dye BT, Ahlquist P. 2003. Engineered retargeting of viral RNA replication complexes to an alternative intracellular membrane. J. Virol. 77:12193–12202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nugent CI, Johnson KL, Sarnow P, Kirkegaard K. 1999. Functional coupling between replication and packaging of poliovirus replicon RNA. J. Virol. 73:427–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rao AL. 2006. Genome packaging by spherical plant RNA viruses. Annu. Rev. Phytopathol. 44:61–87 [DOI] [PubMed] [Google Scholar]
- 32. Rao AL, Grantham GL. 1996. Molecular studies on bromovirus capsid protein. II. Functional analysis of the amino-terminal arginine-rich motif and its role in encapsidation, movement, and pathology. Virology 226:294–305 [DOI] [PubMed] [Google Scholar]
- 33. Rayner JC, Pelham HR. 1997. Transmembrane domain-dependent sorting of proteins to the ER and plasma membrane in yeast. EMBO J. 16:1832–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reddy VS, Johnson JE. 2005. Structure-derived insights into virus assembly. Adv. Virus Res. 64:45–68 [DOI] [PubMed] [Google Scholar]
- 35. Schneemann A, Marshall D. 1998. Specific encapsidation of nodavirus RNAs is mediated through the C terminus of capsid precursor protein alpha. J. Virol. 72:8738–8746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schneemann A, Reddy V, Johnson JE. 1998. The structure and function of nodavirus particles: a paradigm for understanding chemical biology. Adv. Virus Res. 50:381–446 [DOI] [PubMed] [Google Scholar]
- 37. Scotti PD, Dearing S, Mossop DW. 1983. Flock House virus: a nodavirus isolated from Costelytra zealandica (White) (Coleoptera: Scarabaeidae). Arch. Virol. 75:181–189 [DOI] [PubMed] [Google Scholar]
- 38. Selling BH, Allison RF, Kaesberg P. 1990. Genomic RNA of an insect virus directs synthesis of infectious virions in plants. Proc. Natl. Acad. Sci. U. S. A. 87:434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seo JK, Kwon SJ, Choi HS, Kim KH. 2009. Evidence for alternate states of Cucumber mosaic virus replicase assembly in positive- and negative-strand RNA synthesis. Virology 383:248–260 [DOI] [PubMed] [Google Scholar]
- 40. Staehelin LA. 1997. The plant ER: a dynamic organelle composed of a large number of discrete functional domains. Plant J. 11:1151–1165 [DOI] [PubMed] [Google Scholar]
- 41. Tan R, Frankel AD. 1995. Structural variety of arginine-rich RNA-binding peptides. Proc. Natl. Acad. Sci. U. S. A. 92:5282–5286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Venter PA, Krishna NK, Schneemann A. 2005. Capsid protein synthesis from replicating RNA directs specific packaging of the genome of a multipartite, positive-strand RNA virus. J. Virol. 79:6239–6248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Venter PA, Marshall D, Schneemann A. 2009. Dual roles for an arginine-rich motif in specific genome recognition and localization of viral coat protein to RNA replication sites in flock house virus-infected cells. J. Virol. 83:2872–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Volkova E, Gorchakov R, Frolov I. 2006. The efficient packaging of Venezuelan equine encephalitis virus-specific RNAs into viral particles is determined by nsP1-3 synthesis. Virology 344:315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]