

Abstract
EBNA1 is the only nuclear Epstein-Barr virus (EBV) protein expressed in both latent and lytic modes of infection. While EBNA1 is known to play several important roles in latent infection, the reason for its continued expression in lytic infection is unknown. Here we identified two roles for EBNA1 in the reactivation of latent EBV to the lytic cycle in epithelial cells. First, EBNA1 depletion in latently infected cells was shown to positively contribute to spontaneous EBV reactivation, showing that EBNA1 has a role in suppressing reactivation. Second, when the lytic cycle was induced, EBNA1 depletion decreased lytic gene expression and DNA amplification, showing that it positively contributed to lytic infection. Since we have previously shown that EBNA1 disrupts promyelocytic leukemia (PML) nuclear bodies, we investigated whether this function could account for the effects of EBNA1 on lytic infection by repeating the experiments with cells lacking PML proteins. In the absence of PML, EBNA1 did not promote lytic infection, indicating that the EBNA1-mediated PML disruption is responsible for promoting lytic infection. In keeping with this conclusion, PML silencing was found to be sufficient to induce the EBV lytic cycle. Finally, by generating cells with single PML isoforms, we showed that individual PML isoforms were sufficient to suppress EBV lytic reactivation, although PML isoform IV (PML IV) was ineffective because it was most efficiently degraded by EBNA1. Our results provide the first function for EBNA1 in lytic infection and show that EBNA1 interactions with PML IV lead to a loss of PML nuclear bodies (NBs) that promotes lytic infection.
INTRODUCTION
Epstein-Barr virus (EBV) is a gammaherpesvirus that infects most people worldwide and is maintained for life through a combination of latent and lytic modes of infection in B lymphocytes and epithelial cells. While EBV is most often found in a latent mode of infection in B cells, lifelong persistence of EBV in infected individuals involves occasional reactivation of the virus to the lytic state, and a partial (or abortive) lytic infection also appears to be important in EBV-induced B-cell and epithelial tumors (8, 31, 32, 66). In addition, in epithelial cells of the orthopharynx, reactivation of EBV to lytic replication is necessary to produce the viral particles responsible for host-to-host spread (50). However, it is currently unclear how viral reactivation is triggered in vivo.
Latent infection in proliferating cells involves expression of a small subset of EBV proteins that always includes Epstein-Barr nuclear antigen 1 (EBNA1) and sometimes is limited to EBNA1. EBV genomes are maintained in these cells as double-stranded circular DNA molecules that replicate once per cell cycle and segregate stably to the daughter cells such that a constant copy number is maintained (1, 72). EBNA1 contributes to these processes through its binding to the family of repeats (FR) and dyad symmetry (DS) elements of the latent origin oriP, which govern segregation and DNA replication, respectively (47, 73, 74). EBNA1 binding to the FR is also important for the activation of expression of some of the other latency genes (25, 49). In addition, EBNA1 can affect cellular processes in ways that promote cell proliferation and survival (35, 53, 63), and the combination of expression of EBNA1, other latency proteins, and RNA molecules leads to cell immortalization typical of EBV latent infection. As a result of these effects on cell growth, EBV latent infection is known to contribute to the development of several types of B-cell lymphomas and at least two types of carcinomas, nasopharyngeal carcinoma (NPC) and gastric carcinoma of the upper stomach (23, 46).
The switch from EBV latent to lytic infection involves turning on the expression of the EBV protein BZLF1 (also known as ZEBRA, ZTA, Z, and EB1), since its induction or exogenous expression is sufficient to initiate the cascade of lytic protein expression and DNA replication (36, 42). BZLF1 is a DNA-binding protein that transactivates its own promoter (Zp) and the other immediate-early promoter, Rp, inducing the production of BRLF1 transcription factor (24, 37, 42). BZLF1 and BRLF1 then act synergistically to induce the expression of subsequent EBV lytic genes (20). In addition, BZLF1 binding to specific elements in the origin of lytic DNA replication, oriLyt, recruits the virus-encoded DNA polymerase/primase and polymerase processivity factors that are essential for lytic replication (21, 26, 39). Replication then results in the amplification of the viral genomes and production of linear DNA concatemers that are subsequently cleaved into unit length and packaged into capsids.
While EBNA1 is generally thought of as a latency protein due to its multiple important roles in latency, it is the only nuclear latency protein that continues to be expressed in lytic infection. This is similar to the EBV latent membrane protein 1 (LMP1), which is important for cell transformation in latent infection but is also expressed in lytic infection, where it plays a critical role in virus release (4). The expression of EBNA1 in lytic infection involves a switch from the latency promoters that control EBNA1 expression in different types of latency to the Fp lytic promoter (7, 38, 58, 77). In addition, EBNA1 has been shown to localize to viral replication compartments during the lytic cycle (11). The fact that there is a mechanism to ensure EBNA1 expression in lytic infection suggests that EBNA1 has a function in this stage of infection, although the role of EBNA1 in lytic infection has not been elucidated.
In previous studies, we have shown that EBNA1 disrupts promyelocytic leukemia (PML) nuclear bodies (NBs) in cells latently infected with EBV by inducing the degradation of PML proteins (62, 63). This mechanism involves EBNA1 binding to and rerouting of two cellular proteins, ubiquitin-specific protease 7 (USP7) (also called HAUSP) and casein kinase 2 (CK2) (30, 61–63). The EBNA1-CK2 interaction results in increased phosphorylation of PML proteins, which triggers polyubiquitylation and degradation (56, 57, 61). PML NBs are comprised of six different isoforms of PML (9), and coimmunoprecipitation experiments indicate that the association of EBNA1 with PML NBs is predominantly through PML isoform IV (63). PML NBs govern multiple cellular processes, including apoptosis, p53 activation, and DNA repair (6, 45, 51), and accordingly, EBNA1 was found to disrupt these processes in the context of latent infection.
PML proteins are also part of the interferon-mediated antiviral response and have long been known to be associated with lytically replicating viruses, including EBV (5, 15, 27). Considerable evidence indicates that PML NBs suppress lytic infection of herpesviruses and that these suppressive effects are counteracted by specific viral proteins that either induce the degradation of PML proteins or interfere with the PML protein interactions necessary to form NBs (13, 14). For example, the ICP0 protein of herpes simplex virus type 1 (HSV-1) induces the degradation of PML proteins and is required for efficient lytic infection in the presence of PML (16, 18). Moreover, in keeping with an active role in viral suppression, in the absence of ICP0, PML NBs have been shown to move to incoming HSV-1 genomes (17). Similarly, PML NBs suppress lytic protein expression and replication by human cytomegalovirus (CMV), and these effects are counteracted by the viral IE1 protein, which interferes with PML NB formation (3, 67, 68).
It is not yet clear whether PML similarly inhibits the EBV lytic cycle. EBV lytic infection differs from that of HSV and CMV in that primary EBV infection first results in latent infection and the virus then must reactivate to enter the lytic cycle. Our finding that EBNA1 induces the loss of PML NBs, combined with the reports of others that EBV lytic genomes are associated with PML NBs and that EBNA1 continues to be expressed in lytic infection, raises the possibility that PML NBs may inhibit EBV reactivation and that EBNA1 may contribute to lytic infection by depleting them. In this study, we have investigated the contribution of EBNA1 to EBV reactivation in EBV-positive gastric carcinoma and NPC cell lines and identified two distinct roles for EBNA1: a role in suppressing spontaneous reactivation and a role in facilitating lytic infection once the lytic switch is initiated. We have also shown that PML is suppressive to lytic reactivation, and we present data indicating that the lytic mode-activating role of EBNA1 is due to its ability to induce the loss of PML.
MATERIALS AND METHODS
Cell lines.
AGS-EBV cells, generated by coculturing AGS cells with Akata Burkitt's lymphoma cells carrying recombinant neomycin-resistant EBV as described previously (64), were a generous gift from Christopher Dawson and Lawrence Young and were cultured in RPMI 1640 (Sigma) supplemented with 10% fetal calf serum and 1% l-glutamine. HONE-Akata cells were derived from HONE-1 cells by infection with the Akata strain of EBV (28) and maintained in alpha minimal essential medium (alpha-MEM) (Gibco) supplemented with 10% fetal calf serum and 1% l-glutamine. Both cell lines were also maintained in G418 (Invitrogen; 400 μg/ml) to select for cells containing recombinant EBV. CNE2 cells (also called CNE2Z) are NPC cells that have lost the EBV (65), and these were maintained in alpha-MEM (Gibco) supplemented with 10% fetal calf serum and 1% l-glutamine. CNE2 cell lines expressing single PML isoforms are described by Sarkari et al. (55).
siRNA transfections and induction of EBV reactivation.
Approximately 5 × 105 AGS-EBV or HONE-Akata cells were plated in 5 mL of RPMI medium in 10-cm dishes. They were immediately transfected with 100 pmol of small interfering RNA (siRNA) targeted against either EBNA1 (GGAGGUUCCAACCCGAAAUTT) or green fluorescent protein (GFP) (GAACUUCAGGGUCAGCUUGCCG) as a negative control using 2 μl of Lipofectamine 2000 (Invitrogen). In the experiments shown in Fig. 1, an alternative EBNA1 siRNA (EBNA1a; GGACTACCGACGAAGGAACTT) (75) and AllStars negative-control siRNA (Qiagen) were also included. The cells were subjected to second and third rounds of the same transfection after 24 and 48 h. Twenty-four hours after the third round of transfections (48 h for HONE-Akata), the cells were treated with 3 mM sodium butyrate (NaB) (dissolved in distilled water [dH2O]) and 20 ng/ml 12-O-tetradecanoylphorbol-13-acetate (TPA) (dissolved in dimethyl sulfoxide [DMSO]) to induce EBV reactivation or with an equivalent amount of DMSO as a negative control. Floating and adherent cells were harvested 20 to 24 h post-drug treatment and either analyzed by Western blotting, processed for immunofluorescence microscopy, or used to quantify EBV lytic DNA replication as described below.
Fig 1.

EBNA1 expression inhibits spontaneous EBV reactivation. AGS-EBV cells were treated with siRNA against GFP (siGFP) or EBNA1 (siEBNA1) or left untreated (AGS-EBV). (A) Cells were fixed and stained with antibodies against EBNA1 (middle panels) and BZLF1 (right panels), and the percentage of BZLF1-positive cells was determined, where more than 100 cells were counted under each condition. Images with the same antibody treatment were captured using the same exposure times. The bar graph shows average values from three separate experiments with standard deviations. The bar graph on the right shows results using an alternative siRNA against EBNA1 (siEBNA1a) and AllStars negative-control siRNA (siAS). **, P value < 0.01. (B) Equal amounts of cell lysates from untreated AGS-EBV (lane 3) or AGS-EBV treated with siGFP, siEBNA1, siEBNA1a, or AllStars siRNA (siAS) were compared by Western blotting using antibodies for EBNA1, BMRF1, BZLF1, and actin (loading control). (C) Steady-state transcript levels of BZLF1 were monitored by isolating total RNA, synthesizing cDNA, and quantifying transcript levels using primers specific to BZLF1. Values were normalized to the GAPDH housekeeping gene. Average levels of the BZLF1 transcript from three separate experiments are shown for siEBNA1 treatment (with standard deviations) relative to those for the siGFP treatment (set to 1). (D) EBV genomic DNA was quantified by isolating total DNA, amplifying the EBV DS sequence, and normalizing to GAPDH. Average levels (with standard deviations) for siEBNA1 treatment were compared to those for siGFP treatment (set to 1; left graph), and results for siEBNA1a and siAllStars treatment was compared to results for untreated AGS-EBV (set to 1; left panel). ***, P < 0.001.
Immunofluorescence microscopy.
Cells grown on coverslips were fixed using 3% formaldehyde for 15 to 20 min at room temperature and then rinsed briefly using phosphate-buffered saline (PBS). Cells were permeabilized using 1% Triton in PBS for 5 min, followed by two 5-min rinses in PBS. Coverslips were blocked for 20 min in 4% bovine serum albumin (BSA) in PBS. Samples were incubated with primary antibodies against EBNA1 (R4 rabbit serum at 1:300 [30]), PML (PG-M3 at 1:50; Santa Cruz), or BZLF1 (1:100 of monoclonal antibody [Jaap Middeldorp] or 1:50 monoclonal antibody sc-53904 [Santa Cruz]) for 30 min in the humidifying chamber and rinsed twice for 5 min (each) in PBS. Samples were incubated with the secondary antibodies goat anti-rabbit Alexa Fluor 555 (1:800; Molecular Probes) and goat anti-mouse Alexa Fluor 488 (1:800; Molecular Probes) in 4% BSA for 30 min and washed twice for 5 min (each). Coverslips were mounted on slides using ProLong Gold antifade medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Images were obtained using the 40× oil objective on a Leica inverted fluorescence microscope and processed using the OpenLAB (ver.X.0) software program. BZLF1-positive cells were quantified by counting more than 100 cells per sample, and experiments were performed in triplicate.
Western blotting.
Cells were lysed in either 9 M urea–10 mM Tris (pH 6.8) followed by sonication or in RIPA (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM MgCl2, 10% glycerol, 1% Triton X-100, and protease inhibitors) for 20 min on ice. Thirty to fifty micrograms of clarified lysates were loaded onto a 10% SDS-PAGE gel and transferred onto nitrocellulose. Membranes were blocked in 5% nonfat dry milk in PBT-T (PBS with 0.1% Tween) for 1 h, followed by incubation with primary antibody in blocking buffer overnight at room temperature. The primary antibodies used were EBNA1 K67-3 (1:1000; a gift from Jaap Middeldorp), BMRF1 (1:10,000, MAB8186; Chemicon), BZLF1 (1:500 monoclonal antibody [from Jaap Middeldorp] or 1:50 sc-53904 [Santa Cruz]), PML (1:2000, A301-167A; Bethyl) and actin (1:10,000, Ab-1; Oncogene Research Products). Membranes were washed three times with PBS-T and then incubated with goat-anti-mouse (1:3,000 dilution) or goat-anti-rabbit (1:5,000 dilution) secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology) for 1 h. Membranes were washed three times with PBS-T, and signals were detected by enhanced chemiluminescence (ECL) assay (Perkin Elmer Life and Analytical Sciences).
EBV genome quantification.
Approximately 1 × 106 AGS-EBV or HONE-Akata cells, treated as described above, were processed for real-time PCR (RT-PCR) quantification of the EBV genomes as a measure of lytic DNA replication as described previously (78). Briefly, cells were lysed in RIPA buffer on ice for 20 min, and clarified lysates were diluted in RIPA to a final volume of 400 μl and incubated with 8 μl of proteinase K (20 mg/ml) for 2 h at 50°C. The DNA was phenol-chloroform extracted, ethanol precipitated, resuspended in 50 μl of 50 mM Tris, and diluted to 1 ml with water. EBV DNA from each sample was quantified in a Rotorgene qPCR system (Corbett Research) using 2 μl of the template (sample), 5 μl of Lightcycler 480 SYBR green I master mix (Roche), and 2 μl of primers for the DS region of EBV (12) or the GAPDH cellular locus (76) in a 10-μl final reaction volume. Values from the DS region were normalized to values from GAPDH.
Isolation of total RNA and reverse-transcription PCR.
AGS-EBV cells were snap-frozen, and then total RNA was isolated using either TRIzol (Invitrogen) or the Qiagen RNeasy minikit (catalog no. 74 104) according to the manufacturer's instructions. The quantity and quality of the extracted RNA were determined by reading the optical densities at 260 and 280 nm (OD260/280) in a NanoDrop spectrophotometer (Thermo Scientific). One microgram total RNA was reverse transcribed in a 20-μl reaction mixture using the Transcriptor First Strand cDNA synthesis kit catalog no. 04379012001; (Roche) with random hexamer primers according to the manufacturer's instructions. Quantitative real-time PCR was performed using 1 μl of the cDNA to analyze the levels of BZLF1 and GAPDH (endogenous control) along with 5 μl of the LightCycle SYBR green I master mix (Roche) in a Rotorgene quantitative PCR (qPCR) system (Corbett Research). Primers used for quantification of BZLF1 (70) and GAPDH (76) mRNAs were as described previously.
Generation of and experiments with AGS-EBVshPML cells.
pLKO.shPML1, expressing anti-PML short hairpin RNA (shRNA) (shPML) (18), was kindly provided by Roger Everett and is described in detail elsewhere (10). This plasmid was used to generate lentiviruses as previously described (19). The negative-control (puromycin-resistant) lentivirus expressing shRNA against GFP was a gift from Jason Moffat. For infections, 1 ml of filtered culture medium containing the lentivirus was added to 1 × 105 AGS-EBV cells with Polybrene (Sigma) at a final concentration of 8 μg/μl and, after 24 h, it was replaced with medium containing 2 μg/ml puromycin. Seventy-two hours later, puromycin was removed and cells were processed for immunofluorescence microscopy and Western blotting as described above to verify the silencing of PML. Light microscopy images of the cells with and without lentivirus infection were obtained using the 40× objective on a Leica inverted fluorescence microscope and processed using the OpenLAB (ver.X.0) software program. When cells were compared for their ability to reactivate EBV, approximately 5 × 106 cells in 10-cm dishes were treated for 24 h with 3 mM NaB or 20 ng/ml TPA (in DMSO) or a combination of both and compared to cells treated with the amount of DMSO present in the TPA sample. Floating and adherent cells were harvested and processed for Western blotting and EBV genome quantification as described above.
Generation of AGS-EBV cells expressing single PML isoforms.
AGS-EBVshPML cells (1 × 105) were infected with lentiviruses expressing individual PML isoforms I to VI (from a truncated CMV promoter; described in in reference 10) in six-well plates with coverslips as described previously (10, 55). Seventy-four hours later, cells were fixed and processed for immunofluorescence microscopy as described above. One hundred PML-positive cells for each PML isoform were scored for whether or not BZLF1 expression was detected, and experiments were carried out in triplicate. For experiments involving EBNA1 silencing with single PML isoforms, AGS-EBVshPML cells were transfected with siRNA against EBNA1 as described above and 24 h later were infected with lentivirus expressing single PML isoforms; 24 h later, they were transfected with a second round of EBNA1 siRNA.
EBNA1-mediated disruption of NBs formed by single PML isoforms.
CNE2 cells expressing single PML isoforms are described by Sarkari et al. (54). These cells (1.5 × 105) were transfected with 2 μg of the EBNA1 expression plasmid pc3OriPE (71) using Lipofectamine 2000 (Invitrogen), and 48 h later, cells were fixed and processed for immunofluorescence imaging as described above. Samples were scored by counting PML NBs in 100 EBNA1-positive and EBNA1-negative cells for each PML isoform. Experiments were performed in triplicate.
RESULTS
EBNA1 expression inhibits spontaneous EBV reactivation in gastric carcinoma cells.
To investigate the possible role of EBNA1 in the EBV lytic cycle, we used a gastric carcinoma cell line containing recombinant EBV, AGS-EBV, which is permissive for EBV reactivation. Immunofluorescence microscopy (IF) for the BZLF1 protein showed that 2 to 3% of the cells spontaneously expressed this lytic switch protein before and after treatment with negative-control siRNA against GFP (Fig. 1A, top two panels and histogram). Expression of BZLF1 and the EBV DNA replication protein, BMRF1 (DNA polymerase processivity factor), was also detected at low levels by Western blotting (Fig. 1B, lane 1), consistent with a small percentage of the cells entering the lytic cycle. When EBNA1 was downregulated with siRNA treatment, the percentage of cells expressing BZLF1 and therefore entering the lytic cycle increased to 8 to 9% (Fig. 1A, bottom panel and histogram). EBNA1 depletion was confirmed both by IF (Fig. 1A) and by Western blotting (Fig. 1B; compare lanes 1 and 2 in the top panel). Consistent with the IF results, Western blotting showed that EBNA1 depletion increased BZLF1 levels (Fig. 1B, compare lanes 1 and 2 in the third panel) and also increased expression of BMRF1 (Fig. 1B, compare lanes 1 and 2 in second panel). To verify that these results were not due to off-target effects of the siRNA, the experiments were repeated with a second siRNA against EBNA1 (siEBNA1a) and a second negative-control siRNA (AllStars siRNA), yielding the same results as the first set of siRNAs (Fig. 1A and B). These observations suggest that EBNA1 suppresses spontaneous viral reactivation.
To determine whether EBNA1 increased BZLF1 levels by affecting the transcription of the BZLF1 gene as opposed to affecting the stability of the spontaneously produced BZLF1 protein, we examined how EBNA1 depletion affected the level of BZLF1 transcripts. To this end, total mRNA was isolated from cells treated with siRNA against GFP (siGFP) or against EBNA1 (siEBNA1), and BZLF1 transcripts were quantified by quantitative real-time PCR (RT-PCR). EBNA1 depletion was found to cause a 2- to 3-fold increase in BZLF1 transcript levels compared to results for the siGFP control (Fig. 1C). This suggests that EBNA1 decreases EBV reactivation at least in part by repressing the expression of BZLF1.
We then examined whether the effect of EBNA1 on immediate-early (BZLF1) and early (BMRF1) gene expression affected lytic DNA replication, which would be expected if the complete lytic cycle was activated. Since EBV lytic replication (unlike latent replication) results in amplification of the viral genomes, lytic replication was assessed by quantification of the level of the EBV genomes per cell. To this end, DNA was isolated from cells treated with siGFP or siEBNA1, and EBV genomes were quantified by quantitative RT-PCR using primers specific for the DS element present in oriP; these results were normalized to the levels of GAPDH. This revealed a consistent 2-fold increase in lytic EBV DNA replication upon EBNA1 depletion (with either siRNA) compared to results with siGFP or siAllStars treatment (Fig. 1D), indicating the productive cycle was activated by EBNA1 downregulation. Therefore, EBNA1 appears to be a factor in promoting EBV latency.
EBNA1 positively contributes to the lytic cycle after initial reactivation in gastric carcinoma cells.
The percentage of cells spontaneously entering the lytic cycle in the above studies is very low, and EBV reactivation is typically studied by treating the cells with drugs that increase histone acetylation (e.g., sodium butyrate or trichostatin A [TSA]) and activate protein kinase C (TPA), thereby overcoming chromatin suppression and enabling the efficient expression and phosphorylation of BZLF1 (24, 42, 43). Therefore, we also examined the role of EBNA1 in the lytic cycle when EBV was reactivated by sodium butyrate (NaB)–TPA treatment. To this end, AGS-EBV cells were transfected with siRNA against EBNA1 or GFP or left untransfected, and then the lytic cycle was induced by the addition of NaB and TPA. The number of cells that entered the lytic cycle after drug treatment was first examined, by staining the cells for BZLF1 (Fig. 2A). On average, 45% of the cells that were untreated or treated with siGFP expressed BZLF1, whereas on average only 22% of the cells treated with siEBNA1 expressed BZLF1. This suggests that EBNA1 positively contributes to EBV reactivation after NaB-TPA treatment.
Fig 2.

EBNA1 positively contributes to the EBV lytic cycle after NaB-TPA induction. AGS-EBV cells were treated with siRNA against GFP (siGFP) or EBNA1 (siEBNA1) or left untreated, followed by induction of the EBV lytic cycle by NaB-TPA treatment. (A) Cells were fixed and stained with antibodies against EBNA1 (middle panels) and BZLF1 (right panels), and the percentage of BZLF1-positive cells was determined, where more than 100 cells were counted for each condition. The bar graph shows average values from three separate experiments with standard deviations. ***, P < 0.001. Images with the same antibody treatment were captured using the same exposure times. (B) Equal amounts of cell lysates from AGS-EBV cells treated with siGFP or siEBNA1 with and without NaB-TPA induction were compared by Western blotting using antibodies against EBNA1, PML, BMRF1, BZLF1, and actin. (C) EBV genomic DNA was quantified after NaB-TPA treatment as described for 1D.
This effect was also investigated by Western blotting for EBV lytic proteins. Consistent with the previous observation, in the absence of drug treatment, EBNA1 depletion increased the expression of both the BZLF1 and BMRF1 proteins (Fig. 2B, third panel, compare lanes 1 and 2). As expected, NaB-TPA treatment resulted in a robust induction of the lytic cycle proteins BZLF1 and BMRF1 (Fig. 2B, third panel, compare lanes 1 and 3). However, when EBNA1-depleted cells received the same NaB-TPA treatment, the expression of both the BZLF1 and BMRF1 proteins was reduced compared to that for the cells expressing EBNA1 (Fig. 2B, third panel, compare lanes 3 and 4). This confirms the IF data that EBNA1 positively contributes to the expression of EBV lytic proteins after induced reactivation. The effect of EBNA1 depletion on EBV lytic DNA replication after NaB-TPA treatment was also examined by comparing the levels of the EBV genomes after siGFP and siEBNA1 treatments. Consistent with the effect on the lytic protein levels, downregulation of EBNA1 resulted in decreased lytic EBV DNA replication compared to results with the siGFP treatment (Fig. 2C), further suggesting that EBNA1 positively contributes to the lytic cycle under these conditions.
Since PML proteins are known to be suppressive for the replication of some herpesviruses, and EBNA1 can induce degradation of PML proteins in latent infection, we also examined the effect of EBNA1 depletion on PML proteins after lytic induction. We found that EBNA1 depletion resulted in increased levels of the PML proteins (seen as the ladder of bands in the 60- to 120-kDa range) both before (Fig. 2B, second panel, compare lanes 1 and 2) and after (Fig. 2B, second panel, compare lanes 3 and 4) NaB-TPA treatment. This shows that EBNA1 lowers PML levels in both the latent and lytic modes of infection, raising the possibility that this may be how EBNA1 promotes lytic infection after NaB-TPA treatment. We also noticed that a single PML isoform or modified form migrating at approximately 60 kDa was consistently upregulated in EBNA1-silenced cells that were induced for EBV reactivation. The nature of this PML protein is unknown, but its size is consistent with PML V or VI (9).
EBNA1 inhibits spontaneous reactivation but is required for NaB-TPA-induced EBV reactivation in NPC cells.
We next investigated whether the contributions of EBNA1 that we identified in lytic reactivation in AGS-EBV are also observed in EBV-positive NPC cells. To this end, we examined the effects of siEBNA1 and siGFP treatments in HONE-Akata cells. In the absence of NaB-TPA treatment, EBNA1 depletion increased the basal levels of the BZLF1 and BMRF1 lytic proteins (even though the levels of the PML proteins were increased)(Fig. 3A). This suggests that, as in AGS-EBV cells, EBNA1 acts to suppress spontaneous reactivation of EBV. Consistent with the increase in lytic proteins, we consistently detected a small increase in the level of spontaneous lytic DNA replication when EBNA1 was downregulated (Fig. 3C, −NaB/TPA). However, as in AGS-EBV cells, EBNA1 depletion had the opposite effect after NaB-TPA treatment. After this induction of the lytic cycle, EBNA1 depletion decreased BZLF1 and BMRF1 expression (Fig. 3B) and similarly decreased EBV lytic DNA replication compared to results with the siGFP negative-control treatment (Fig. 3C, NaB-TPA samples). Under these conditions, EBNA1 depletion upregulated PML, consistent with the possibility that the increase in PML could potentiate the observed decrease in lytic protein expression and DNA replication. Therefore, the results in HONE-Akata cells support those in AGS-EBV cells, that EBNA1 has different effects before and after lytic induction.
Fig 3.
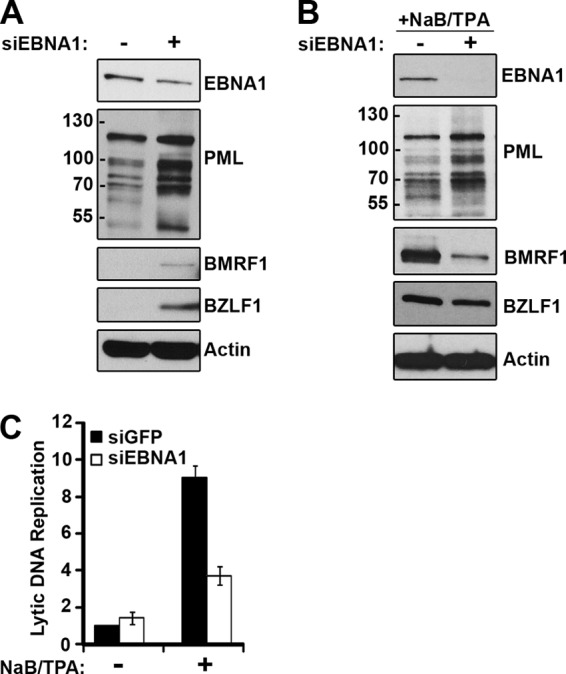
EBNA1 effects on EBV reactivation in NPC cells. HONE-Akata cells were treated with siRNA against GFP or EBNA1. (A and B) Equal amounts of cell lysates before (A) or after (B) NaB-TPA treatment were compared by Western blotting using antibodies against EBNA1, PML, BMRF1, BZLF1, and actin. (C) EBV genomic DNA was quantified before and after NaB-TPA treatment as described for Fig. 1D. Average levels are shown with standard deviations; the value for siGFP samples without NaB-TPA treatment was set to 1.
PML represses EBV reactivation.
The observation that EBNA1 silencing after NaB-TPA treatment increased PML levels and decrease lytic infection suggests that higher levels of PML may be inhibitory to the EBV lytic cycle. To test this possibility, AGS-EBV cells were infected with a lentivirus carrying shRNA designed to silence all PML isoforms (18), and cells without detectable PML, AGS-EBVshPML, were generated. The absence of PML was verified by both IF (Fig. 4A) and Western blotting (Fig. 4B) using antibodies that recognize all PML isoforms. During routine light microscopy inspection of the cell lines, we noticed that the PML-silenced cells consistently had a higher percentage of cells that were enlarged and rounded, as is typical of cells undergoing lytic viral replication, than the parental AGS-EBV cells (Fig. 4C). To verify that these cells were indeed spontaneously reactivating EBV, BZLF1 and BMRF1 levels were examined by Western blotting. Both of these lytic protein markers were upregulated in AGS-EBVshPML cells compared to levels for the parental cells with PML (Fig. 4D, compare lanes 1 and 3), although not to as high a level as that seen after NaB-TPA treatment of AGS-EBV cells (Fig. 4D, lane 4). This was not a reaction to lentivirus infection or shRNA in general, since BZLF1 and BMRF1 were not induced when AGS-EBV cells were infected with a lentivirus expressing shRNA against GFP (Fig. 4D, lane 2), nor did infection with this lentivirus result in the enlarged, rounded cells seen with the shPML lentivirus (data not shown). Therefore, the data show that PML negatively affects EBV reactivation.
Fig 4.

PML represses EBV reactivation. AGS-EBV (A-EBV) cells were infected with a lentivirus carrying shRNA against all PML isoforms and were selected for cells containing the lentivirus to generate AGS-EBVshPML cells (A-EBVshPML). (A) Cells were fixed and stained with antibodies against EBNA1 (right panels) and PML (middle panels). Images with the same antibody treatment were captured using the same exposure times. (B) Equal amounts of cell lysates from AGS-EBV (wt) and AGS-EBVshPML (shPML) were compared by Western blotting using an antibody that recognized all isoforms of PML or actin (loading control). (C) Light microscopy images of AGS-EBV and AGS-EBVshPML cells from two experiments (I and II). Examples of enlarged, rounded cells in the AGS-shPML cells are seen in the bottom panels. (D) Equal amounts of cell lysates from AGS-EBV (before and after NaB-TPA treatment) and AGS-EBVshPML (no NaB-TPA treatment) were compared by Western blotting using antibodies that recognize BMRF1, BZLF1, and actin. Lysates from AGS-EBV cells containing a negative-control lentivirus expressing shGFP are also shown (AGS-EBVshGFP, lane 2, no NaB-TPA treatment). (E) Equal amounts of cell lysates from AGS-EBV (PML+) and AGS-EBVshPML (PML−) were compared post-treatment with NaB, TPA, and NaB-TPA by Western blotting using antibodies against PML (all isoforms), EBNA1, BMRF1, BZLF1, and actin. Light (LE) and dark (DE) exposures of the same blot are shown for BMRF1. (F) EBV genomic DNA was quantified post-treatment with DMSO, NaB, TPA, or NaB-TPA as described for Fig. 1D. Average levels with standard deviations after each treatment are shown; values were normalized to AGS-EBV cells treated with DMSO (set to 1).
We also examined whether PML silencing acted synergistically with NaB and TPA drug treatments to reactivate EBV. To this end, AGS-EBV and AGS-EBVshPML cells were treated with NaB or TPA or both NaB and TPA, and the effects of these treatment on BZLF1 and BMRF1 expression were examined by Western blotting (Fig. 4E). While treatments with NaB or TPA alone had little effect on lytic protein expression in AGS-EBV each single treatment resulted in some induction of BZLF1 and BMRF1 in AGS-EBVshPML cells, including generation of a slower-migrating form of BMRF1 (Fig. 4E, fifth and sixth panels, compare lanes 1 and 2 and lanes 3 and 4). In addition, the AGS-EBVshPML cells produced more of the lytic proteins when treated with both NaB and TPA than AGS-EBV cells (Fig. 4E, fourth and sixth panels, compare lanes 5 and 6). Western blots for PML and EBNA1 are also shown for all of the treatments, confirming that PML remained silenced after the drug treatments and that EBNA1 expression levels were similar before and after PML silencing (Fig. 4E, panels 1 and 2). We also examined the effect of the NaB and TPA treatments on amplification of the EBV genomes (Fig. 4F). In keeping with the lytic protein levels, the AGS-EBVshPML cells showed increased EBV amplification compared to that for AGS-EBV under all conditions tested, including the DMSO negative-control treatment. These data show that PML is a negative regulator of EBV reactivation that is not overcome by NaB-TPA treatments.
Effects of EBNA1 on EBV reactivation in cells lacking PML.
We next used the AGS-EBVshPML cells to examine whether the ability of EBNA1 to disrupt PML NBs could account for the effect of EBNA1 in increasing virus reactivation after NaB-TPA treatment. We rationalized that if this was the case, then EBNA1 should not increase EBV reactivation after drug treatment in cells that lacked PML. Therefore, EBNA1 was silenced in AGS-EBVshPML cells, and the effect on BZLF1 and BMRF1 protein induction was examined by Western blotting before and after NaB-TPA treatment. Consistent with the results in the AGS-EBV cells, in the absence of NaB-TPA treatment, EBNA1 silencing increased the spontaneous reactivation of EBV as detected by increased levels of BZLF1 and BMRF1 (Fig. 5A, second panel, compare lanes 1 and 2). Note that a much shorter exposure was used for the BMRF1/BZLF1 blot than that in Fig. 4D, which is why BMRF1 and BZLF1 expression is not seen in lane 1. In addition, EBNA1 depletion in AGS-EBVshPML cells, under these conditions, led to increased levels of EBV genomes (Fig. 5B). These results show that the role of EBNA1 in maintaining latency in the absence of drug treatments is independent of PML.
Fig 5.
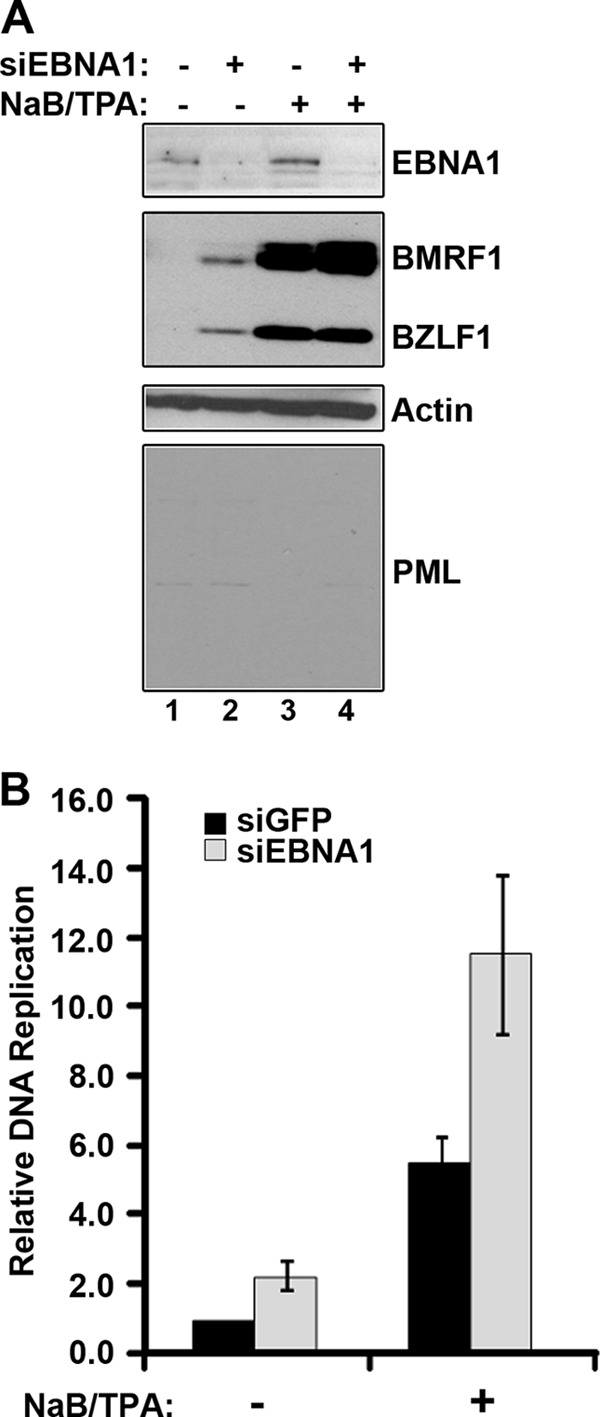
The positive role of EBNA1 in EBV lytic infection is not observed in the absence of PML. (A) AGS-EBVshPML cells were treated with siRNA against GFP (−siEBNA1) or EBNA (+siEBNA1) and then treated with NaB-TPA (+) or the DMSO control (−NaB-TPA). Equal amounts of cell lysates were compared by Western blotting using the indicated antibodies. (B) EBV genomic DNA was quantified in each sample as described for Fig. 1D. The AGS-EBVshPML sample treated with siGFP and DMSO was set to 1 and used to normalize all other samples.
However, the effect of EBNA1 silencing after NaB-TPA treatment in AGS-EBVshPML cells was considerably different from that observed in AGS-EBV cells. Specifically, in the AGS-EBVshPML cells, downregulation of EBNA1 did not decrease the expression of BZLF1 and BMRF1 (Fig. 5A, second panel, compare lanes 3 and 4). In fact, EBNA1-depleted samples had slightly higher levels of lytic protein expression after NaB-TPA treatment, particularly for BMRF1 (Fig. 5A, second panel, compare lanes 3 and 4). Similar results were obtained when the level of the EBV genomes was quantified, in that EBNA1 silencing in AGS-EBVshPML cells promoted genome amplification after NaB-TPA treatment (Fig. 5B), an effect that is similar to that observed in the absence of NaB-TPA treatment. Taken together, the data support the hypothesis that the role of EBNA1 in activating lytic infection, which is revealed after NaB-TPA treatment, is due to the ability of EBNA1 to overcome the suppressive effects of PML NBs.
Effects of individual PML isoforms on EBV reactivation and sensitivity to EBNA1.
PML consists of six different nuclear isoforms (I to VI) that can differ in their abilities to mediate PML-associated functions, including the ability to inhibit HSV-1 lytic infection (10). Therefore, we examined the abilities of individual PML isoforms to inhibit EBV reactivation. To this end, we individually infected the AGS-EBVshPML cells with lentiviruses expressing individual PML isoforms containing silent mutations making them resistant to silencing by the initial PML shRNA. These PML lentiviruses have been previously described and were used to determine effects of PML isoforms on HSV-1 infection (10). It was also previously shown that the individual PML isoforms all form nuclear bodies, albeit of various sizes and numbers (10, 55), and this is also shown in the context of the AGS-EBV cells (Fig. 6A). We then examined the degree to which cells lacking PML or containing individual PML isoforms spontaneously reactivated EBV as measured by staining for BZLF1 and determining the percentage of cells expressing BZLF1 (Fig. 6A, right panels, and 6B). Note that for the single-PML-isoform cells, only cells that were observed to express the PML protein were counted, since all of the cell populations contained some nonexpressing cells. PML silencing was found to induce a 3- to 4-fold increase in the number of cells expressing BZLF1 compared to that for AGS-EBV cells with wild-type PML (Fig. 6B, first two bars), and this increased reactivation was rescued by expression of any of the individual PML isoforms, with the exception of PML IV (Fig. 6B). However, IF imaging of the PML NBs formed by the individual PML isoforms showed that PML IV formed the fewest and smallest PML NBs in the context of the AGS-EBV cells, although this is not typical of PML IV when expressed in EBV-negative cells (10, 55). This suggests that PML IV may be less effective at inhibiting EBV reactivation because it is more sensitive to degradation by EBNA1.
Fig 6.
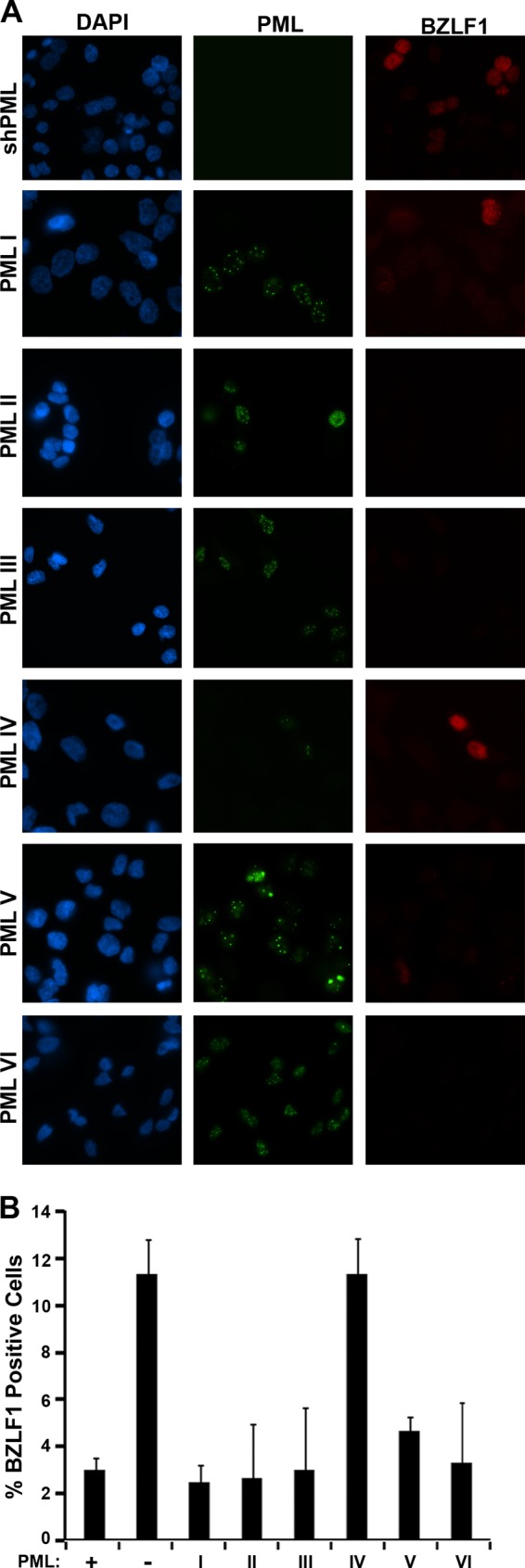
Effect of single PML isoforms on EBV reactivation and lytic infection. AGS-EBVshPML (shPML) cells were infected with lentivirus expressing individual PML isoforms I to VI. (A) The cells were fixed and stained 72 h postinfection for PML (green) and BZLF1 (red). (B) AGS-EBVshPML cells (−), parental AGS-EBV cells (+) and the single PML isoform cells (I to VI) were scored for whether or not they expressed BZLF1, and the percentages of BZLF1-positive cells were determined. For the single PML isoform samples, only the PML-positive cells were scored. Average values with standard deviations from three independent experiments are shown.
We then investigated the sensitivities of PML NBs formed by the individual PML isoforms to EBNA1. To this end, we engineered an EBV-negative NPC cell line, CNE2, to express single PML isoforms as previously described by Sarkari et al. (55). We then transfected these cells with an EBNA1 expression plasmid and examined EBNA1 expression and PML NBs by IF (Fig. 7A). The number of PML NBs was counted in cells expressing or not expressing EBNA1, and average values from multiple experiments are shown in Fig. 7B. PML NB counts and intensity (reflected by ease of IF detection) in the absence of EBNA1 confirmed that PML IV was better able to form NBs in the absence of EBV infection. Comparison of the number of PML NBs in the presence and absence of EBNA1 showed that while EBNA1 modestly decreased the NBs formed by any PML isoform, it had the greatest effect on PML IV NBs, which were reduced in number 3-fold in the presence of EBNA1 (Fig. 7B). These results confirm that EBNA1 preferentially disrupts NBs formed by PML IV and suggest that the reason that PML IV did not appear to repress EBV reactivation is due to its efficient disruption by EBNA1.
Fig 7.

EBNA1 preferentially disrupts PML NBs formed by PML IV. CNE2 cells expressing single PML isoforms I to VI were generated as in the work of Sarkari et al. (55) and were transfected with an EBNA1 expression plasmid. (A) Cells were stained with antibodies against PML (green) and EBNA1 (red) and visualized by IF microscopy. These images were also merged with a DAPI counterstain (right panels). (B) The number of PML NBs per cell was counted for EBNA1-positive cells 48 h after EBNA1 transfection (white bars) and EBNA1-negative cells (control; black bars), and average values from three independent experiments (with standard deviations) are shown.
To further confirm that the ineffectiveness of PML IV in suppressing EBV reactivation is due to the presence of EBNA1 as opposed to some other property of PML IV, we compared the BZLF1 expression frequencies in AGS-EBV cells lacking PML or expressing only PML IV, both before and after depleting EBNA1 with siRNA treatment (Fig. 8). Consistent with the results shown in Fig. 6B, PML IV and PML-null cells had similar frequencies of EBV spontaneous reactivation in the presence of EBNA1, but after EBNA1 depletion, PML IV cells had a lower frequency of spontaneous reactivation than PML-null cells. In contrast, cells expressing only PML I were repressive for EBV reactivation (relative to PML-null cells) both in the presence and absence of EBNA1. The results confirm that PML IV can suppress EBV reactivation in the absence of EBNA1.
Fig 8.
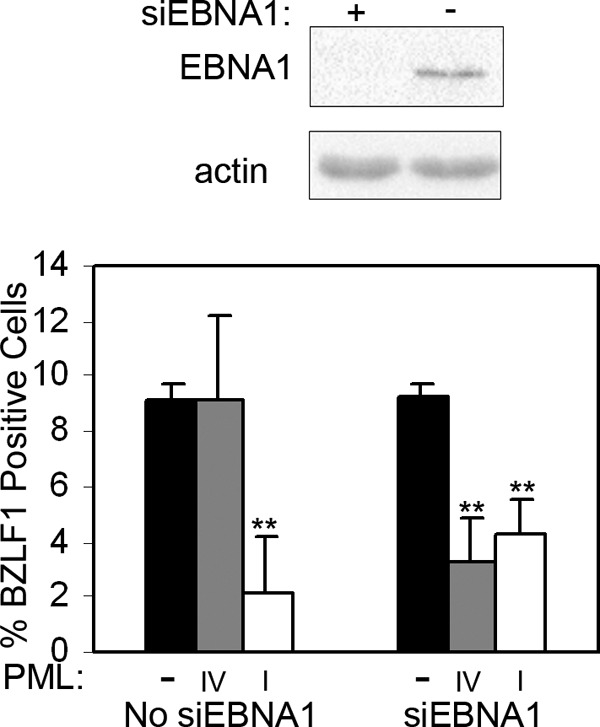
PML IV can suppress EBV reactivation in EBNA1-depleted cells. AGS-EBVshPML cells were treated with siRNA against EBNA1 (siEBNA1) or not treated (no siEBNA1), and then cells were reconstituted with PML IV or PML I or left with no PML (−). Cells were then imaged for BZLF1 and PML as described for Fig. 6A, and the percentage of cells expressing BZLF1 was plotted in the bar graph. The Western blot on the top was performed with AGS-EBVshPML cell extracts prior to introducing PML IV or PML I to confirm the EBNA1 depletion. **, P < 0.01.
DISCUSSION
While EBNA1 is known to have multiple roles in EBV latency, its role in lytic infection has not been investigated. Here we have examined the contributions of EBNA1 in reactivating EBV to the lytic cycle in epithelial cells and identified two distinct roles for EBNA1. First, EBNA1 suppresses spontaneous reactivation (seen in the absence of NaB-TPA treatment) by decreasing BZLF1 transcripts and protein (and possibly the expression of other EBV genes). This mechanism is independent of PML, since the same EBNA1 effect was observed in cells with and without PML expression. Since expression of the BZLF1 gene is known to be controlled by the chromatin state of its promoter (34, 36) and EBNA1 is known to interact with proteins that affect chromatin modifications (e.g., USP7, TAF-I, and NAP1) (54, 69), we speculate that this effect of EBNA1 is due to chromatin effects. After reactivation is induced by NaB-TPA treatment (which overcomes the chromatin suppression of the BZLF1 gene), a second role for EBNA1 was revealed in promoting lytic infection, affecting both the expression level of viral lytic proteins and the degree of amplification of the EBV genomes. These effects of EBNA1 on EBV lytic infection are due largely to the ability of EBNA1 to induce the degradation of PML proteins, and hence the loss of PML NBs, because the positive effects of EBNA1 on lytic infection were not observed in the same cells lacking PML proteins.
Our studies also provide evidence that PML proteins suppress EBV reactivation. This is in keeping with findings of studies of other herpesviruses showing that PML actively suppresses primary lytic infections (18, 27, 48, 67, 68) and with the observation that PML NBs are disrupted in EBV lytic infection (5). Our findings also fit well with those of Sides et al. (60), who recently reported that treatment of an NPC cell line containing EBV with arsenic trioxide, a chemical known to disrupt PML NBs, resulted in increased expression of BZLF1 and conferred susceptibility to ganciclovir, an inhibitor of EBV lytic DNA replication. Our studies also show that EBNA1 is a key player in the disruption of PML NBs. The ability of EBNA1 to disrupt PML NBs may be the basis for the observations of Shannon-Lowe et al. (59), who found that infection of epithelial cells with an EBV lacking EBNA1 resulted in decreased BZLF1 protein expression compared to infection with the wild-type virus. This study followed the effects of primary EBV infection, conditions under which BZLF1 is expressed without the need for NaB-TPA to overcome chromatin effects. However, EBNA1 may not act alone in facilitating lytic infection by disrupting PML NBs, since high levels of BZLF1 have been reported to disrupt PML NBs, in addition to its other important roles in lytic activation (2, 52). Unlike EBNA1, BZLF1 disrupts the PML NBs, by inhibiting interactions between PML proteins, but does not induce the degradation of PML proteins. Therefore, the two proteins might cooperate in overcoming the suppressive effects of PML.
PML NBs are normally comprised of six different PML isoforms, and understanding how these multiple isoforms contribute to various PML-associated processes is an active area of study. For example, PML IV has been shown to be responsible for p53 interactions leading to p53 activation (22, 29). In addition, PML isoform IV was recently shown to be responsible for the inhibition of varicella-zoster virion production by entrapping the viral capsid protein in PML cages (48) and for inhibition of encephalomyocarditis virus replication through sequestration of the viral 3D polymerase (40). In contrast, examination of the effect of PML isoforms on HSV-1 infection showed that no single isoform was as effective as wild-type PML NBs in suppressing infection by ICP0-null HSV-1 (10); however, PML I or II each partially suppressed infection. PML II also appears to mediate the rearrangement of PML NBs into tracks, as induced by the adenovirus type 5 E4orf3 protein (33). Here we have shown that any individual PML isoform, with the possible exception of PML IV, can suppress the first step in EBV reactivation (BZLF1 expression) in epithelial cells as effectively as wild-type PML NBs, as well as additional steps after lytic induction. The failure of PML IV to inhibit BZLF1 expression is likely not a property intrinsic to this PML protein but rather appears to be due to its more efficient degradation by EBNA1. This fits with our previous observation that EBNA1 preferentially interacts with PML IV (63) and suggests that this interaction with PML IV is important for inducing the degradation of the multiple PML isoforms in the NB.
Activation of the lytic cycle in AGS-EBV cells after EBNA1 depletion resulted in the induction of a PML protein migrating at approximately 60 kDa (Fig. 2B). This observation suggests that reactivation results in the induction of this PML form but that this induction is normally counteracted by EBNA1-mediated PML degradation. The induced PML form is smaller than PML I to IV but is consistent with the size of PML isoform V or VI. Alternatively, it could be a form of PML I to IV that lacks SUMO modifications, resulting in a faster migration in the gel, or a highly modified form of the cytoplasmic PML VII, which is the smallest of the PML isoforms. On that note, it is interesting that a cytoplasmic form of PML has been shown to inhibit HSV-1 replication (41). However, we found that in HONE-Akata cells, a different PML band (migrating at about 80 kDa) was preferentially induced upon lytic activation after EBNA1 depletion, suggesting that the PML forms induced by lytic infection may vary in different cells. Nonetheless, the results suggest that the profile of PML proteins changes in response to EBV lytic infection. This is reminiscent of a report by Nojima et al. (44) that showed that HSV-2 infection, through the action of the ICP27 protein, alters the splicing of PML mRNA, causing a switch from production of PML II to PML V.
In summary, we have shown that the presence of EBNA1 in latent epithelial cell infection suppresses EBV reactivation, but once the lytic cycle is initiated, EBNA1 promotes lytic infection by inducing the loss of PML NBs. This suggests that the reason that EBNA1 expression is maintained in the EBV lytic cycle (by switching to the Fp promoter) is to disrupt PML NBs, thereby preventing their antiviral effects and allowing efficient EBV replication and virion production. Interestingly, while EBNA1 has been clearly shown to disrupt PML NBs in epithelial cells (62, 63), it is not clear if it does so in B lymphocytes (5). Epithelial cells are the principal site of lytic EBV infection in the body, whereas EBV infection of B lymphocytes is most often latent. Thus, EBNA1-induced PML protein degradation in epithelial cells may be a major factor in determining permissivity for lytic infection.
ACKNOWLEDGMENTS
We thank Lawrence Young and Chris Dawson for the AGS-EBV cells, Fei-Fei Liu for the HONE-Akata cells, Jason Moffat for the anti-GFP shRNA (shGFP)-expressing negative-control lentivirus, and Jaap Middeldorp for antibodies against EBNA1 and BZLF1. We are also grateful to Roger Everett for generously providing the lentivirus constructs for PML silencing and expression of silencing-resistant PML isoforms.
This work was funded by an operating grant (12477) from the Canadian Institutes of Health Research to L.F. N.S. was supported by a Canadian Institutes for Health Research doctoral award. L.F. is a tier 1 Canada Research Chair in Molecular Virology.
Footnotes
Published ahead of print 4 April 2012
REFERENCES
- 1. Adams A. 1987. Replication of latent Epstein-Barr virus genomes. J. Virol. 61:1743–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adamson AL, Kenney S. 2001. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 75:2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahn JH, Hayward GS. 2000. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 274:39–55 [DOI] [PubMed] [Google Scholar]
- 4. Ahsan N, Kanda T, Nagashima K, Takada K. 2005. Epstein-Barr virus transforming protein LMP1 plays a critical role in virus production. J. Virol. 79:4415–4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bell P, Lieberman PM, Maul GG. 2000. Lytic but not latent replication of Epstein-Barr virus is associated with PML and induces sequential release of nuclear domain 10 proteins. J. Virol. 74:11800–11810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bernardi R, Pandolfi PP. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8:1006–1016 [DOI] [PubMed] [Google Scholar]
- 7. Brink AA, Meijer CJ, Nicholls JM, Middeldorp JM, van den Brule AJ. 2001. Activity of the EBNA1 promoter associated with lytic replication (Fp) in Epstein-Barr virus associated disorders. Mol. Pathol. 54:98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cochet C, et al. 1993. Expression of the Epstein-Barr virus immediate early gene, BZLF1, in nasopharyngeal carcinoma tumor cells. Virology 197:358–365 [DOI] [PubMed] [Google Scholar]
- 9. Condemine W, et al. 2006. Characterization of endogenous human promyelocytic leukemia isoforms. Cancer Res. 66:6192–6198 [DOI] [PubMed] [Google Scholar]
- 10. Cuchet D, et al. 2011. PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J. Cell Sci. 124:280–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Daikoku T, et al. 2004. In vivo dynamics of EBNA1-oriP interaction during latent and lytic replication of Epstein-Barr virus. J. Biol. Chem. 279:54817–54825 [DOI] [PubMed] [Google Scholar]
- 12. Deng Z, et al. 2005. Inhibition of Epstein-Barr virus OriP function by tankyrase, a telomere-associated poly-ADP ribose polymerase that binds and modifies EBNA1. J. Virol. 79:4640–4650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Everett RD. 2001. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 20:7266–7273 [DOI] [PubMed] [Google Scholar]
- 14. Everett RD. 2006. Interactions between DNA viruses, ND10 and the DNA damage response. Cell Microbiol. 8:365–374 [DOI] [PubMed] [Google Scholar]
- 15. Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830 [DOI] [PubMed] [Google Scholar]
- 16. Everett RD, et al. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Everett RD, Murray J. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 79:5078–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Everett RD, et al. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80:7995–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Everett RD, Young DF, Randall RE, Orr A. 2008. STAT-1- and IRF-3-dependent pathways are not essential for repression of ICP0-null mutant herpes simplex virus type 1 in human fibroblasts. J. Virol. 82:8871–8881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feederle R, et al. 2000. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 19:3080–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fixman ED, Hayward GS, Hayward SD. 1992. trans-acting requirements for replication of Epstein-Barr virus ori-Lyt. J. Virol. 66:5030–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fogal V, et al. 2000. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 19:6185–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fukayama M. 2010. Epstein-Barr virus and gastric carcinoma. Pathol. Int. 60:337–350 [DOI] [PubMed] [Google Scholar]
- 24. Gabelova A, et al. 2007. Sensitivity of different endpoints for in vitro measurement of genotoxicity of extractable organic matter associated with ambient airborne particles (PM10). Mutat. Res. 620:103–113 [DOI] [PubMed] [Google Scholar]
- 25. Gahn T, Sugden B. 1995. An EBNA1 dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Epstein-Barr virus LMP gene. J. Virol. 69:2633–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao Z, et al. 1998. The Epstein-Barr virus lytic transactivator Zta interacts with the helicase-primase replication proteins. J. Virol. 72:8559–8567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J. Interferon Cytokine Res. 31:145–158 [DOI] [PubMed] [Google Scholar]
- 28. Glaser R, et al. 1989. Two epithelial tumor cell lines (HNE-1 and HONE-1) latently infected with Epstein-Barr virus that were derived from nasopharyngeal carcinomas. Proc. Natl. Acad. Sci. U. S. A. 86:9524–9528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guo A, et al. 2000. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2:730–736 [DOI] [PubMed] [Google Scholar]
- 30. Holowaty MN, et al. 2003. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J. Biol. Chem. 278:29987–29994 [DOI] [PubMed] [Google Scholar]
- 31. Hong GK, et al. 2005. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 79:13993–14003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hong GK, et al. 2005. Epstein-Barr virus lytic infection is required for efficient production of the angiogenesis factor vascular endothelial growth factor in lymphoblastoid cell lines. J. Virol. 79:13984–13992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hoppe A, Beech SJ, Dimmock J, Leppard KN. 2006. Interaction of the adenovirus type 5 E4 Orf3 protein with promyelocytic leukemia protein isoform II is required for ND10 disruption. J. Virol. 80:3042–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jenkins PJ, Binne UK, Farrell PJ. 2000. Histone acetylation and reactivation of Epstein-Barr virus from latency. J. Virol. 74:710–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kennedy G, Komano J, Sugden B. 2003. Epstein-Barr virus provides a survival factor to Burkitt's lymphomas. Proc. Natl. Acad. Sci. U. S. A. 100:14269–14274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kenney SC. 2007. Reactivation and lytic replication of EBV. In Arvin A, et al. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 37. Kolman JL, Taylor N, Gradoville L, Countryman J, Miller G. 1996. Comparing transcriptional activation and autostimulation by ZEBRA and ZEBRA/c-Fos chimeras. J. Virol. 70:1493–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lear AL, et al. 1992. The Epstein-Barr virus (EBV) nuclear antigen 1 BamHI F promoter is activated on entry of EBV-transformed B cells into the lytic cycle. J. Virol. 66:7461–7468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lieberman PM, Hardwick JM, Sample J, Hayward GS, Hayward SD. 1990. The zta transactivator involved in induction of lytic cycle gene expression in Epstein-Barr virus-infected lymphocytes binds to both AP-1 and ZRE sites in target promoter and enhancer regions. J. Virol. 64:1143–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maroui MA, Pampin M, Chelbi-Alix MK. 2011. Promyelocytic leukemia isoform IV confers resistance to encephalomyocarditis virus via the sequestration of 3D polymerase in nuclear bodies. J. Virol. 85:13164–13173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McNally BA, Trgovcich J, Maul GG, Liu Y, Zheng P. 2008. A role for cytoplasmic PML in cellular resistance to viral infection. PLoS One 3:e2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. 2007. Lytic cycle switches of oncogenic human gammaherpesviruses(1). Adv. Cancer Res. 97:81–109 [DOI] [PubMed] [Google Scholar]
- 43. Moser MS, Abu-Laban RB, van Beek CA. 2004. Attitude of emergency department patients with minor problems to being treated by a nurse practitioner. CJEM 6:246–252 [DOI] [PubMed] [Google Scholar]
- 44. Nojima T, et al. 2009. Herpesvirus protein ICP27 switches PML isoform by altering mRNA splicing. Nucleic Acids Res. 37:6515–6527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pearson M, et al. 2000. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406:207–210 [DOI] [PubMed] [Google Scholar]
- 46. Raab-Traub N. 2002. Epstein-Barr virus in the pathogenesis of NPC. Semin. Cancer Biol. 12:431–441 [DOI] [PubMed] [Google Scholar]
- 47. Rawlins DR, Milman G, Hayward SD, Hayward GS. 1985. Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA1) to clustered sites in the plasmid maintenance region. Cell 42:859–868 [DOI] [PubMed] [Google Scholar]
- 48. Reichelt M, et al. 2011. Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus. PLoS Pathog. 7:e1001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reisman D, Sugden B. 1986. trans activation of an Epstein-Barr viral transcriptional enhancer by the Epstein-Barr viral nuclear antigen 1. Mol. Cell. Biol. 6:3838–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rickinson AB, Kieff E. 1996. Epstein-Barr virus, p 2397–2446 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology, 3rd ed Lippincott-Raven Publishers, Philadelphia, PA [Google Scholar]
- 51. Salomoni P, Ferguson BJ, Wyllie AH, Rich T. 2008. New insights into the role of PML in tumour suppression. Cell Res. 18:622–640 [DOI] [PubMed] [Google Scholar]
- 52. Salsman J, Zimmerman N, Chen T, Domagala M, Frappier L. 2008. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog. 4:e1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saridakis V, et al. 2005. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1: implications for EBV-mediated immortalization. Mol. Cell 18:25–36 [DOI] [PubMed] [Google Scholar]
- 54. Sarkari F, et al. 2009. EBNA1-mediated recruitment of a histone H2B deubiquitylating complex to the Epstein-Barr virus latent origin of DNA replication. PLoS Pathog. 5:e1000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sarkari F, Wang X, Nguyen T, Frappier L. 2011. The herpesvirus associated ubiquitin specific protease, USP7, is a negative regulator of PML proteins and PML nuclear bodies. PLoS One 6:e16598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Scaglioni PP, et al. 2006. A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell 126:269–283 [DOI] [PubMed] [Google Scholar]
- 57. Scaglioni PP, et al. 2008. CK2 mediates phosphorylation and ubiquitin-mediated degradation of the PML tumor suppressor. Mol. Cell. Biochem. 316:149–154 [DOI] [PubMed] [Google Scholar]
- 58. Schaefer BC, Strominger JL, Speck SH. 1995. The Epstein-Barr virus BamHI F promoter is an early lytic promoter: lack of correlation with EBNA1 gene transcription in group 1 Burkitt's lymphoma cell lines. J. Virol. 69:5039–5047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shannon-Lowe C, et al. 2009. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: viral genome expression, genome maintenance, and genome amplification. J. Virol. 83:7749–7760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sides MD, et al. 2011. Arsenic mediated disruption of promyelocytic leukemia protein nuclear bodies induces ganciclovir susceptibility in Epstein-Barr positive epithelial cells. Virology 416:86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sivachandran N, Cao JY, Frappier L. 2010. Epstein-Barr virus nuclear antigen 1 hijacks the host kinase CK2 to disrupt PML nuclear bodies. J. Virol. 84:11113–11123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sivachandran N, et al. 2012. Contributions of the Epstein-Barr virus EBNA1 protein to gastric carcinoma. J. Virol. 86:60–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sivachandran N, Sarkari F, Frappier L. 2008. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog. 4:e1000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stewart S, et al. 2004. Epstein-Barr virus-encoded LMP2A regulates viral and cellular gene expression by modulation of the NF-kappaB transcription factor pathway. Proc. Natl. Acad. Sci. U. S. A. 101:15730–15735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sun Y, et al. 1992. An infrequent point mutation of the p53 gene in human nasopharyngeal carcinoma. Proc. Natl. Acad. Sci. U. S. A. 89:6516–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tao Q, Chan AT. 2007. Nasopharyngeal carcinoma: molecular pathogenesis and therapeutic developments. Expert Rev. Mol. Med. 9:1–24 [DOI] [PubMed] [Google Scholar]
- 67. Tavalai N, Papior P, Rechter S, Leis M, Stamminger T. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 80:8006–8018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tavalai N, Stamminger T. 2011. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res. 157:128–133 [DOI] [PubMed] [Google Scholar]
- 69. Wang S, Frappier L. 2009. Nucleosome assembly proteins bind to Epstein-Barr virus nuclear antigen 1 and affect its functions in DNA replication and transcriptional activation. J. Virol. 83:11704–11714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wen W, et al. 2007. Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J. Virol. 81:1037–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu H, Kapoor P, Frappier L. 2002. Separation of the DNA replication, segregation, and transcriptional activation functions of Epstein-Barr nuclear antigen 1. J. Virol. 76:2480–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yates JL, Guan N. 1991. Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J. Virol. 65:483–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yates JL, Warren N, Reisman D, Sugden B. 1984. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 81:3806–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yates JL, Warren N, Sugden B. 1985. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 313:812–815 [DOI] [PubMed] [Google Scholar]
- 75. Yin Q, Flemington EK. 2006. siRNAs against the Epstein Barr virus latency replication factor, EBNA1, inhibit its function and growth of EBV-dependent tumor cells. Virology 346:385–393 [DOI] [PubMed] [Google Scholar]
- 76. Yuan J, Cahir-McFarland E, Zhao B, Kieff E. 2006. Virus and cell RNAs expressed during Epstein-Barr virus replication. J. Virol. 80:2548–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zetterberg H, Stenglein M, Jansson A, Ricksten A, Rymo L. 1999. Relative levels of EBNA1 gene transcripts from the C/W, F and Q promoters in Epstein-Barr virus-transformed lymphoid cells in latent and lytic stages of infection. J. Gen. Virol. 80(Pt 2):457–466 [DOI] [PubMed] [Google Scholar]
- 78. Zhou J, Snyder AR, Lieberman PM. 2009. Epstein-Barr virus episome stability is coupled to a delay in replication timing. J. Virol. 83:2154–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]