

Abstract
Foamy viruses (FV) synthesize Pol from a spliced pol mRNA independently of Gag, unlike orthoretroviruses, which synthesize Pol as a Gag-Pol protein that coassembles with Gag. We found that prototype FV (PFV) mutants expressing Gag and Pol only as a Gag-Pol protein without the spliced Pol contain protease activity equivalent to that of wild-type (WT) Pol. Regardless of the presence or absence of the spliced Pol, the PFV Gag-Pol proteins can assemble into virus-like particles (VLPs), in contrast to the orthoretroviral Gag-Pol proteins, which cannot form VLPs. However, the PFV Gag-Pol VLPs have aberrant morphologies and are not infectious. In the absence of the spliced Pol, coexpression of a PFV Gag-Pol protein with Gag can produce infectious virions. Our results suggest that enzymes encoded by PFV pol (protease, reverse transcriptase, and integrase) are enzymatically active if they are synthesized as part of a Gag-Pol protein.
INTRODUCTION
The foamy virus (FV) genome encodes the three major viral proteins Gag, Pol, and Env that are common to all other retroviruses, in addition to two regulatory proteins, Bet and Tas. However, FVs are classified as one of two subfamilies of the Retroviridae, the Spumaretrovirinae, whose viral replication pathway is distinct from that of Orthoretrovirinae. Major differences include the mode of Pol expression and its encapsidation into virions. Orthoretroviruses, such as human immunodeficiency virus type 1 (HIV-1) and murine leukemia virus (MLV), synthesize Pol as a Gag-Pol fusion protein. Gag-Pol is produced by translational readthrough, which occurs by either ribosomal frameshifting or suppression of a stop codon at the C terminus of Gag. Readthrough occurs at a frequency of approximately 5 to 10% relative to gag translation (reviewed in reference 9). The Gag-Pol protein coassembles with Gag through Gag assembly domains (23, 30). In HIV-1, when the Gag/Gag-Pol ratio (normally about 20:1) was altered to increase the amount of Gag-Pol relative to Gag, viral assembly was disrupted, possibly due to steric hindrance caused by an excess of Gag-Pol during the assembly process (10, 29). Expression of orthoretroviral Gag-Pol protein alone does not lead to the production of viral particles (7, 14, 22, 35). In contrast, FV Pol is expressed from a singly spliced mRNA (38) and Pol expression is regulated at the transcriptional level, using a suboptimal 3′ splice site (3′ss) for the pol gene (15). Because FV Pol is synthesized independently of Gag, the mechanism of FV Pol incorporation into virions is different from that of orthoretroviruses. cis-acting sequences in the genomic RNA are required for FV Pol packaging (12, 24). Previously, we proposed that protein-protein interactions between Gag and Pol are involved in Pol packaging and a Gag/Pol complex binds to genomic RNA for encapsidation into virions (17).
Compared to orthoretroviruses, in FV, there are limited proteolytic cleavages of both Gag and Pol. FV Gag is not cleaved into separate matrix (MA), capsid (CA), and nucleocapsid (NC) proteins that are found in mature orthoretroviruses. FV Gag is partially cleaved once near the C terminus to release a p3 peptide (Fig. 1A). This cleavage is required for efficient viral infectivity (6, 41). Another distinct feature of FV Gag is that it does not contain the conserved cysteine and histidine motifs that are present in orthoretroviral NC, but instead contains two or three glycine/arginine-rich (GR) boxes upstream of the p3 cleavage site near the C terminus of Gag. The first two GR boxes are involved in essential steps of viral replication, including genomic RNA packaging, Pol packaging, reverse transcription, and particle morphology (17, 20, 31). GR box 3 (GR3) is absent in some FV isolates and, as yet, has no known function (34, 36), which suggests that GR3 might be dispensable. FV Pol is synthesized as a precursor polyprotein containing protease (PR), reverse transcriptase (RT), and integrase (IN), and Pol is cleaved only once by PR between RT and IN, producing an IN and a PR-RT fusion protein (26) (Fig. 1A). Examination of mutants lacking the cleavage site between RT and IN showed that virus replication is dependent on this cleavage (27).
Fig 1.

Construction of PFV Gag-Pol fusion proteins. (A) Diagrams of WT Gag and Pol proteins and the Gag-Pol fusion protein. Both the 5′ and 3′ splice sites (5′ss and 3′ss) for pol mRNA are indicated. The three glycine/arginine-rich (GR) boxes are indicated by small black boxes near the C terminus of Gag. The WT Pol protein is translated in the +1 reading frame relative to Gag. ATG indicates the start codon for each open reading frame. A deletion of 1 nt brings Gag and Pol into the same reading frame to generate a Gag-Pol fusion protein. The proteolytic cleavage sites for viral protease are indicated by arrows and dotted lines. PR, protease; RT, reverse transcriptase; IN, integrase. (B) The amino acid sequences at the fusion junctions of the Gag-Pol fusion proteins. Amino acids are shown in the single-letter code. The recognition sequences for the p3 cleavage site at the C terminus of Gag are shown in boxes: WT sequences are in gray boxes, and the mutated sequences are in open boxes. The coding sequences for Pol proteins are underlined.
In the present study, we created several prototype FV (PFV) mutants encoding Gag-Pol fusion proteins. These fusion proteins were expressed alone or together with the Gag protein, to mimic the mode of orthoretroviral assembly. We examined the expression and packaging of viral proteins into virions and the enzymatic activities of Pol during viral assembly and replication. While we were writing this article, another group published an article describing similar PFV Gag-Pol fusion proteins (33).
MATERIALS AND METHODS
Construction of recombinant DNAs.
The prototype foamy virus (PFV) used in this study is a chimpanzee FV isolated from a human-derived cell culture, which was previously designated human FV (HFV). PFV Gag-Pol fusions were generated in the context of a full-length proviral clone containing a cytomegalovirus (CMV) immediate-early promoter, pcPFV (31). A deletion of 1 nucleotide (nt) just downstream of the p3 cleavage site was used to make an in-frame Gag-Pol fusion protein [Fig. 1A and B; Gag-Pol (G-P)]. Besides this 1-nt deletion, we also made additional modifications that led to changes in the amino acid sequences of the viruses we created. In all of the in-frame Gag-Pol fusion constructs, 17 amino acids (aa) were removed from the C terminus of Gag, because the coding region for this portion of Gag overlaps with the coding region for the N terminus of Pol (Fig. 1B). The region encoding 7 aa downstream of the p3 cleavage site was also altered from Gln-Ser-Ala-Thr-Ser-Ser-Thr to Arg-Val-Pro-Arg-Pro-Pro-Gln in all of the Gag-Pol fusion constructs. The deletion of GR box 3 in the ΔGR3 G-P mutant starts at nucleotide position 2811 of pcPFV and ends at position 2905. The amino acid sequences around the p3 cleavage site were mutated from Val-Asn-Thr-Val-Thr to Val-Gln-Tyr-Arg-Asp in both mp3 G-P and ΔGR3/mp3 G-P mutants (Fig. 1B). An adenine nucleotide at position 2459 was changed to cytosine in order to eliminate the 3′ splice site of the pol gene in a Δ3′ss mp3 G-P mutant. All of the site-directed mutations in the PFV genome were generated by two rounds of PCR using four oligonucleotides. Briefly, the two outer oligonucleotides were designed to anneal to the gag or pol gene and contain unique restriction sites at each end. The two inner mutagenic oligonucleotides (in either forward or reverse orientation) were designed to be complementary to the gag sequences near the p3 cleavage site, except for the desired mutations. Each mutant construct was sequenced to confirm the presence of the correct mutations. Primer sequences will be supplied upon request.
Cell cultures and transfections.
293T, HT1080 (human fibrosarcoma cells), and FAB cells (39) were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% bovine growth serum and 1% penicillin and streptomycin. Transient transfection was done using 1 mg/ml polyethyleneimine (Polysciences, Warrington, PA) as previously described (4). Cells and viral supernatants were harvested between 45 and 48 h posttransfection and prepared as previously described (17). Briefly, culture supernatants were collected, and cell debris was cleared by low-speed centrifugation and filtration through a 0.45-μm-pore-diameter syringe filter. Viral particles were pelleted through 20% sucrose cushions at 25,000 rpm for 2 h using an L7 ultracentrifuge (Beckman). After removing the supernatants, cells were scraped off plates in 1× SDS sample buffer (12.5% 50 mM Tris-HCl [pH 6.8], 10% glycerol, 2% SDS, 5% 2-mercaptoethanol, 0.01% bromophenol blue), and lysates were prepared by using Qiashredder (Qiagen) in accordance with the manufacturer's protocol. To measure infectivity, a foamy virus-activated β-galactosidase (β-Gal) expression assay was performed as previously described (39). Briefly, the FAB indicator cell line has a single integrated copy of a PFV long terminal repeat (LTR)-driven β-Gal gene. Infection of these cells leads to the transactivation of the PFV LTR promoter by the viral Tas protein.
Western blot analysis.
Cell lysates and viral pellets were resuspended in 1× SDS sample buffer before being loaded onto 9% SDS–polyacrylamide gels. In some experiments, viral pellets were resuspended in 1× phosphate-buffered saline (PBS) solution and proteolytically digested with subtilisin (Sigma-Aldrich), according to previously described protocols (32). Western blot analysis was performed as previously described (1), using a 1:5,000 dilution of rabbit polyclonal anti-Gag antibody (1), 1:800 dilution of mouse monoclonal anti-Pol antibody (31), 1:5,000 dilution of rabbit polyclonal anti-IN antibody (27), 1:1,000 dilution of mouse monoclonal anti-SU antibody (40), 1:1,000 dilution of mouse monoclonal anti-leader peptide (anti-LP) antibody, or 1:2,000 dilution of mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Mouse monoclonal antibody (IgG2a subtype) against PFV LP was generated using the GST-LP fusion protein. Proteins were visualized using the Odyssey detection system (Li-Cor, Lincoln, NE), according to the manufacturer's protocol. Intracellular protein levels were normalized to the level of GAPDH protein as an internal control.
RT-PCR.
Forty-eight hours after transfection, both cells and virus-containing supernatants were collected and used to extract RNA. The cells were scraped off plates with PBS and centrifuged for 10 min at 500 × g. Cell pellets were lysed, and total cellular RNA was extracted using the RNeasy minikit (Qiagen). The amounts of viral supernatants to be pelleted were determined according to each sample's intracellular Gag level that was quantified by Western blots. The Gag level in the cell was normalized by the GAPDH level. Equal numbers of virus particles, as determined by extracellular Gag levels, were treated with RNase-free DNase I, and viral RNA was isolated using a QIAamp viral RNA minikit (Qiagen). Extracted cellular or viral RNA (0.7 μg) was reverse transcribed using a poly(A) · poly(dT)12 primer and ThermoScript RT (Invitrogen), according to the manufacturer's protocol. The cellular levels of viral RNA were measured by PCR amplification using the same reverse primer for both the unspliced genomic RNA and the spliced pol mRNA (5′-TCCACGTCCTCCTCCGTACC-3′) and a forward primer specific for each RNA (5′-AGATAATCAAACAAGAGC-3′ for the unspliced genomic RNA and 5′-ACTACTCGCTGCGTCGAGAG-3′ for the spliced pol mRNA), each giving a PCR amplification product of ∼300 nt. gapdh mRNA was used as an internal control for cellular RNA. The level of viral genomic RNA packaged into virions was measured using primers around the 5′ splice site (5′-GGAGCTCTTCACTACTCGCTGCGTCGAG-3′ and 5′-CAACCAGAGCTTCAACATCAAG-3′), yielding PCR products of 501 nt. Before thermal cycling, samples were denatured for 5 min at 95°C. Temperatures for denaturing, annealing, and extension were 95°C, 48°C, and 72°C, respectively, for 45 s for each cycle and run for 25 cycles. The final extension reaction was done at 72°C for 10 min.
Immunofluorescence microscopy.
Transfected cells were fixed with freshly made 3% paraformaldehyde at room temperature for 15 min. Cells were quenched and blocked as previously described in reference 40. Samples were incubated with the primary antibodies at room temperature for 1 h. After washing, cells were incubated with Alexa Fluor-conjugated secondary antibodies at 1:5,000 at room temperature for 45 min. Samples were then washed in PBS and mounted in Vectashield (Vector Laboratories, Burlingame, CA). Fluorescence was visualized using a fixed-stage Nikon Eclipse E800 microscope.
EM analysis.
293T cells were transfected with the wild type (WT) or the mp3 G-P fusion construct in the context of the full-length proviral genome. Forty-five to 48 h after transfection, the cells were washed with PBS and fixed with half-strength Karnovosky's fixative (2% paraformaldehyde, 2.5% glutaraldehyde) at room temperature for 1 h. Cells were scraped off the plates and collected by centrifugation at 1200 rpm for 5 min at 4°C. The cell pellets were rinsed in half-strength Karnovosky's fixative and dehydrated in graded ethanol (50 to 100%). The pellet was processed for electron microscopy (EM) analysis as described in reference 16. Digital images were obtained with an electron microscope (JOEL 1230) equipped with a digital camera (Gatan).
Particle density analysis.
293T cells were seeded in 10-cm plates a day prior to transfection. The supernatants of cells transfected with the WT or the mp3 G-P fusion construct were harvested between 45 and 48 h posttransfection and pelleted through 20% sucrose cushions by ultracentrifugation at 25,000 rpm for 2 h. The viral pellets were resuspended in PBS and loaded onto a discontinuous density gradient consisting of 20, 30, 40, and 50% (wt/vol) iodixanol. The samples were subjected to ultracentrifugation at 32,000 rpm for 19 h. Fourteen fractions were collected from the top, and the density of each fraction was measured. Proteins from each fraction were precipitated with 10% trichloroacetic acid (TCA) and resuspended in 1× SDS sample buffer for Western blot analysis. The Western blot membranes were probed with a polyclonal anti-Gag antibody for detection of the WT Gag protein and a monoclonal anti-Pol antibody for detection of the mp3 G-P Gag-Pol fusion protein.
RESULTS
Construction of the PFV Gag-Pol fusion proteins.
Processing by FV protease (PR) is limited compared to processing by orthoretroviral PRs, which completely cleave Gag to matrix (MA), capsid (CA), and nucleocapsid (NC). Figure 1A shows proteolytic processing of the prototype FV (PFV) Gag and Pol proteins. The partial cleavage near the C terminus of Gag releases a p3 peptide, converting a 71-kDa Gag protein to a 68-kDa protein. The other cleavage is in the Pol protein between reverse transcriptase (RT) and integrase (IN). Cleavage produces a 40-kDa IN and an 85-kDa PR-RT fusion from a 125-kDa precursor Pol protein.
Normally, the FV pol gene is in the +1 reading frame with respect to the gag gene. We created a deletion of GR box 3 (ΔGR3) near the C terminus of PFV Gag. In this mutant, by deletion of one extra nucleotide, the Gag and Pol coding regions were brought into the same reading frame, producing a ΔGR3 Gag-Pol fusion protein (Fig. 1B; ΔGR3 G-P). The 1-nt deletion downstream of the p3 cleavage site led to a change in 7 aa downstream of the p3 cleavage site in Gag. Gln-Ser-Ala-Thr-Ser-Ser-Thr was changed to Arg-Val-Pro-Arg-Pro-Pro-Gln in this mutant (Fig. 1B). The in-frame Gag-Pol fusion near the C terminus of Gag resulted in a truncation of the 17 aa at the C terminus of Gag because normally the segment encoding this portion of Gag overlaps with the coding region for the N terminus of Pol (Fig. 1B). We also made several other Gag-Pol fusion constructs. The Gag-Pol (G-P) mutant was constructed by the insertion of a segment encoding GR box 3 into the ΔGR3 G-P construct (Fig. 1B). In the mp3 G-P construct, the p3 cleavage site was mutated to prevent Pol from being released from a Gag-Pol fusion protein (Fig. 1B). Mutations in both the p3 cleavage site and GR box 3 were introduced to create the ΔGR3/mp3 G-P construct (Fig. 1B).
Expression of the PFV Gag-Pol fusion proteins.
To determine the ability of the Gag-Pol fusion proteins to participate in virus assembly and support replication, these Gag-Pol fusion constructs were inserted into a full-length PFV plasmid in place of the regions encoding Gag and Pol. Expression of the viral sequences is under the control of the cytomegalovirus immediate-early promoter (pcPFV) (31). 293T cells were transfected with constructs expressing the Gag-Pol fusion proteins, and Western blot analyses were performed on both cell lysates and pelleted supernatants. Unlike in orthoretroviruses, FV Env is required for virus budding (1, 8). A PFV mutant with a deletion of the env gene (ΔEnv) was used as a negative control to ascertain the level of nonspecific release of viral proteins that could be caused by cell lysis. Cells transfected with the ΔEnv construct expressed Gag in cells at levels equivalent to those in the WT, but no Gag was released into the supernatant (Fig. 2A, lanes 3 and 10). This indicates that any Gag or Pol detected in the supernatants was not the result of nonspecific release. The two Gag-Pol fusion mutants containing cleavage sites in both Gag and Pol showed a cleaved 68-kDa Gag when probed with anti-Gag antibody (Fig. 2A, lanes 4 and 7; ΔGR3 G-P and G-P). The mp3 G-P and ΔGR3/mp3 G-P mutants that disrupted the p3 cleavage site expressed Gag-Pol fusion proteins but no Gag. These Gag-Pol proteins were more readily detected with anti-Pol antibody than anti-Gag antibody for unknown reasons (Fig. 2A and B, lanes 5 and 6). The amounts of Gag-Pol fusion proteins present in cells were significantly larger than the amounts of Pol present in cells transfected with WT (Fig. 2B, compare lanes 4 to 7 with lane 2).
Fig 2.

Analyses of intracellular and extracellular viral proteins by Western blot analyses. Forty-five to 48 h after the transfection of 293T cells with expression constructs for either WT or mutant viruses, cell lysates and viral supernatants were collected. Viral pellets were digested with subtilisin before being fractionated on 9% SDS–polyacrylamide gels. The proteins were transferred to a membrane, and the membrane was probed with anti-Gag antibody (A), anti-Pol antibody (B), or anti-IN antibody (C). The cellular protein GAPDH was used as a loading control (data not shown). PrPol, precursor Pol (PR-RT-IN); Pol, cleaved Pol (PR-RT). Molecular mass markers (MM) are shown in kilodaltons.
The PFV protease is active when expressed as a Gag-Pol fusion protein in the absence of the spliced Pol protein.
The sizes of the Gag-Pol proteins expressed in the p3 cleavage site mutants, judged by the position to which they migrate on gels, are consistent with a partial processing by PFV PR at the cleavage site between RT and IN of Gag-Pol proteins (Fig. 1A). This cleavage would result in a 193-kDa full-length Gag-PR-RT-IN fusion protein and a 150-kDa Gag-PR-RT fusion lacking IN (Fig. 2B, lanes 5 and 6). This interpretation was confirmed by detection of cleaved IN with anti-IN antibody in the pelleted supernatants (Fig. 2C). PR activity was also detected in the Gag-Pol fusion mutants that had cleavage sites in both Gag and Pol. The ΔGR3 G-P and G-P mutants produced Pol products that migrated on SDS-PAGE, in a manner similar to that of the corresponding WT products, yielding an approximately 80-kDa Pol (PR-RT) and a 125-kDa Pol precursor (PR-RT-IN; PrPol) (Fig. 2B, lanes 4 and 7). Similar Pol products were also detected in the p3 cleavage site mutants (Fig. 2B, lanes 5 and 6). These results suggest that in these mutants, Pol was also expressed from a spliced pol mRNA independently of the Gag-Pol fusion protein. Thus, to determine whether protease is active in the context of Gag-Pol, it was necessary to eliminate the spliced Pol product.
To eliminate the expression of WT Pol, we introduced into the mp3 G-P mutant a mutation at the 3′ splice site used to generate pol mRNA. This new Gag-Pol construct (Δ3′ss mp3 G-P) lacks both the 3′ss and the p3 cleavage site near the C terminus of Gag, resulting in the expression of only a Gag-Pol fusion protein. 293T cells transfected with this new Gag-Pol construct were harvested, and the levels of unspliced genomic RNA and spliced pol mRNA in cells were measured by RT-PCR. Both the mp3 G-P and the Δ3′ss mp3 G-P mutants expressed unspliced genomic RNA at levels equivalent to those in the WT (Fig. 3A, lanes 2 to 4). However, the 3′ss mutant did not express the spliced pol mRNA, while the mp3 G-P synthesized approximately the same amount of the spliced pol mRNA as the WT (Fig. 3A, lanes 6 to 8). Western blot analyses showed that in the cells expressing the Δ3′ss mp3 G-P protein, both 80-kDa Pol and 125-kDa precursor Pol bands were absent (Fig. 3C, compare lane 5 with lane 4). Proteolytic cleavage at the site between RT and IN was seen by detection of cleaved IN with anti-IN antibody in the pelletable supernatants (Fig. 3D, lane 9). The results indicate that the protease expressed in the context of a Gag-Pol fusion protein is active in the absence of WT Pol.
Fig 3.

Analyses of intracellular RNA and expression and incorporation of viral proteins into virions of the Δ3′ss mp3 G-P mutant. (A) Forty-five to 48 h after the transfection of 293T cells with expression constructs for either WT or mutant viruses, total cellular RNA was extracted, reverse transcribed, and PCR amplified using a pair of specific primers for an unspliced genomic RNA and a spliced pol mRNA. gapdh mRNA was used as an internal standard. Cells and viral supernatants were collected from transfected cells and prepared for Western blot analyses. Viral pellets were resuspended in PBS and divided into two aliquots. One aliquot was digested with subtilisin before being loaded onto 9% SDS–polyacrylamide gels, and the other aliquot without subtilisin digestion ran on the gel. The blot membranes were probed with anti-Gag antibody (B), anti-Pol antibody (C), anti-IN antibody (D), anti-SU antibody (E), or anti-LP antibody (F). The cellular protein GAPDH was used as a loading control (data not shown). For Western blot analyses (B to D), half the amounts of WT viral supernatants compared to those of the mutants were used to prepare samples with or without subtilisin digestion. PrPol, precursor Pol (PR-RT-IN); Pol, cleaved Pol (PR-RT). DNA ladders (in base pairs) and molecular mass markers (MM) (in kilodaltons) are shown.
The PFV Gag-Pol fusion protein can assemble into VLPs.
To examine whether PFV mutants expressing only a Gag-Pol protein can assemble virus particles, the supernatants of transfected cells were concentrated by sedimentation through a 20% sucrose cushion. Virus pellets were proteolytically digested with a non-membrane-permeable endoprotease, subtilisin, to eliminate nonspecific aggregates and cellular debris (32). Expression of the ΔGR3 G-P and G-P fusion proteins led to the release of a cleaved 68-kDa Gag into the medium (Fig. 2A, lanes 11 and 14). This was expected because the Gag p3 cleavage sites are intact in these forms of Gag-Pol proteins. Despite the fact that there are high levels of the Gag-Pol fusion proteins from the p3 cleavage site mutants in the cells, regardless of presence or absence of the WT Pol, only small amounts of 150-kDa Gag-Pol proteins were detected in the supernatants (Fig. 2B, lanes 12 and13; Fig. 3C, lanes 12 and 14). These results contrast with the efficient assembly and release of Pol in the WT and the two Gag-Pol fusion mutants that contain cleavage sites in both Gag and Pol (Fig. 2B, compare lanes 12 and 13 with lanes 9, 11, and 14; Fig. 3C, lane 8 versus lanes 12 and 14). However, the amounts of subtilisin-resistant Gag-Pol proteins present in the pellets were significant compared to the amounts seen in the absence of Env (Fig. 2B and 3C, lanes 10), suggesting that the Gag-Pol fusion proteins can assemble, albeit inefficiently, into virus-like particles (VLPs) protected by membranes.
In order to examine whether the VLPs released from the Gag-Pol fusions are particulate, release of the Env protein was examined with anti-LP antibody. PFV Env is synthesized as a precursor protein and is subsequently processed by cellular protease to yield the particle-associated leader peptide (LP), surface (SU), and transmembrane (TM) subunits (3). After digestion of virus pellets with subtilisin, the SU bands that were shown in the absence of subtilisin disappeared (Fig. 3E, compare lanes 3, 7, and 9 with lanes 2, 6, and 8), indicating the efficacy of subtilisin digestion. LP proteins were trimmed outside the membrane after digestion with subtilisin and the smaller p14LP bands were detected in the supernatants of both the mp3 G-P and Δ3′ss mp3 G-P as well as the WT (Fig. 3F, lanes 3, 7, and 9), supporting release of VLPs from the Gag-Pol fusion mutants.
The PFV Gag-Pol fusion particles are not infectious.
The infectivity of Gag-Pol fusion viruses was measured using the FAB assay (39) and summarized in Table 1. The G-P mutant that retains the PR cleavage sites in both Gag and Pol produced approximately the same amount of infectious viruses as the WT. The titer of the ΔGR3 G-P mutant was about 10-fold lower than that of the WT, implying that GR box 3 might contribute to infectivity. All of the p3 cleavage site mutants, including the ΔGR3/mp3 G-P, mp3 G-P, and Δ3′ss mp3 G-P constructs, in which the p3 cleavage site was mutated, did not produce infectious viruses (Table 1). These results indicate that the Gag-Pol fusion viruses were able to replicate as long as PR-mediated proteolytic cleavages took place in both Gag and Pol of the fusion proteins. However, the Gag-Pol mutants, in which Gag is fused with most of Pol except IN, are not replication competent, regardless of the presence or absence of the spliced Pol.
Table 1.
Infectivity of extracellular viruses in supernatants from cells transfected with Gag-Pol proviruses
| WT or mutant construct | Change(s) from WT | Presence of spliced Pol | Infectivity (U/ml)a | Relative infectivity (%) |
|---|---|---|---|---|
| WT | Yes | (2.5 ± 0.5) × 105 | 100 | |
| ΔEnv | No Env | Yes | <10 | <0.004 |
| G-P | Gag-Pol fusion | Yes | (2.2 ± 0.9) × 105 | 91 |
| ΔGR3 G-P | Gag-Pol fusion lacking GR box 3 | Yes | (2.3 ± 0.5) × 104 | 9 |
| mp3 G-P | Gag-Pol fusion lacking p3 cleavage site | Yes | <10 | <0.004 |
| ΔGR3/mp3 G-P | Gag-Pol fusion lacking both GR box 3 and p3 cleavage site | Yes | <10 | <0.004 |
| Δ3′ss mp3 G-P | Gag-Pol fusion lacking p3 cleavage site and 3′ splice site for pol mRNA | No | <10 | <0.004 |
Infectivity was measured by the FAB assay as described in reference 39. The values shown are the average ± standard deviation from four independent assays.
VLPs produced from expression of the PFV Gag-Pol fusion protein have aberrant morphologies in cells.
We examined intracellular localization of the Gag-Pol fusion proteins, using both immunofluorescence microscopy and electron microscopy (EM). Previously, our laboratory reported that PFV Gag assembly occurs at a perinuclear region (40). Gag proteins accumulate in a perinuclear region and colocalize with γ-tubulin, a subcellular marker for the microtubule organizing center (MTOC). When HT1080 cells were transfected with a WT proviral vector and were stained with anti-Gag and anti-Pol antibodies, Gag localized in a perinuclear region and accumulated at a site called “the site for Gag assembly” (40) (Fig. 4A). Although less Pol was expressed than Gag, Pol colocalized with Gag at the assembly site (Fig. 4A, “Merge” panel). In cells transfected with either the mp3 G-P or the Δ3′ss mp3 G-P constructs, the Gag-Pol fusion proteins localized in a perinuclear region (Fig. 4B and C). EM analysis showed large amounts of electron-dense cores in 293T cells transfected with the WT expression construct (Fig. 5A and B). The intracellular virions appear as capsids lacking Env spikes in the viral membrane. WT virions that budded out through the cell membranes of transfected cells contained Env and were uniform in size and shape (Fig. 5C and D). Cells transfected with the mp3 G-P mutant showed electron-dense material near the nucleus, but there were no regularly shaped capsids visible in the EM (Fig. 5E to H). In cells transfected with the mp3 G-P mutant, it was difficult to detect VLPs budding through the cell membrane.
Fig 4.
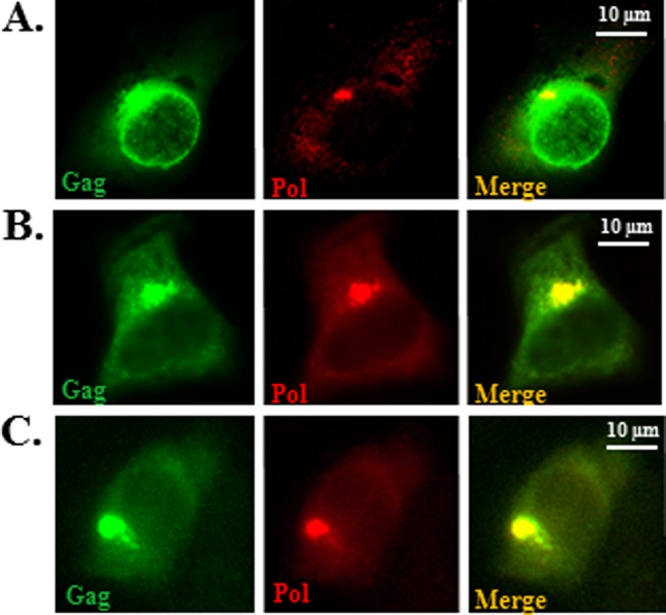
Immunofluorescence microscopy of the cells transfected with the PFV Gag-Pol constructs. 293T cells were transfected with the WT (A), mp3 G-P (B), or Δ3′ss mp3 G-P (C) constructs and costained with rabbit polyclonal anti-Gag antibody (green) and mouse monoclonal anti-Pol antibody (red). Overlapped images for each set of panels are shown in the “Merge” panels. The scale bars are shown in the right corner of each merged image. All images were captured using a Nikon E800 microscope.
Fig 5.
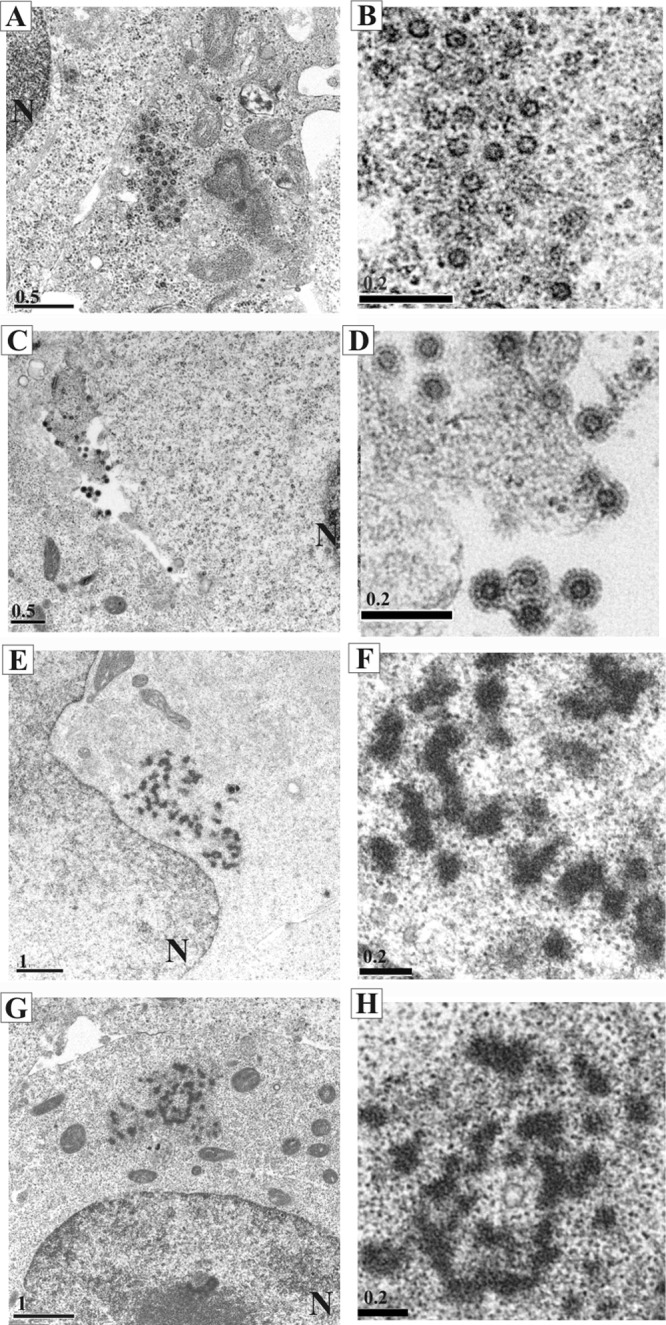
Electron microscopic analyses of cells transfected with the PFV Gag-Pol construct. 293T cells were transfected with constructs expressing WT or the mp3 G-P Gag-Pol protein. The images are presented in pairs. Each image in the right column shows a close up of the electron-dense materials seen in the images shown in the left column. The WT images are in the top 4 panels (A to D), and the mp3 G-P images are in the lower 4 panels (E to H). The nuclei are indicated as “N.” The scale bars in μm are shown in the left corner of each image. Digital images were obtained with a JOEL 1230 electron microscope.
The PFV Gag-Pol VLPs have a density slightly less than that of the WT and are deficient in genomic RNA.
To determine whether the Gag-Pol proteins found in supernatants (detected by Western blot analysis) were in VLPs, we performed isopycnic gradient centrifugation analyses. The supernatants of 293T cells transfected with the WT or the mp3 G-P construct were pelleted 2 days after transfection and prepared for analysis on iodixanol density gradients. The densities of each number fraction prepared for the two samples were comparable (Fig. 6A). The WT Gag proteins were recovered in fractions 5 to 9 with densities of 1.12 to 1.2 g/ml, peaking in fractions 5 and 6 with densities of 1.12 and 1.14 g/ml (Fig. 6B), as predicted for PFV particles (17, 38). About a 4-times-larger volume of viral supernatant was collected from cells transfected with the mp3 G-P construct than the WT and used in the density analyses after pelleting. The VLPs of the mp3 G-P were slightly lighter than the WT VLPs, with densities ranging from 1.1 to 1.14 g/ml (Fig. 6C). The results imply that VLPs that contain the Gag-Pol fusions have a density slightly lower than that of the WT VLPs.
Fig 6.
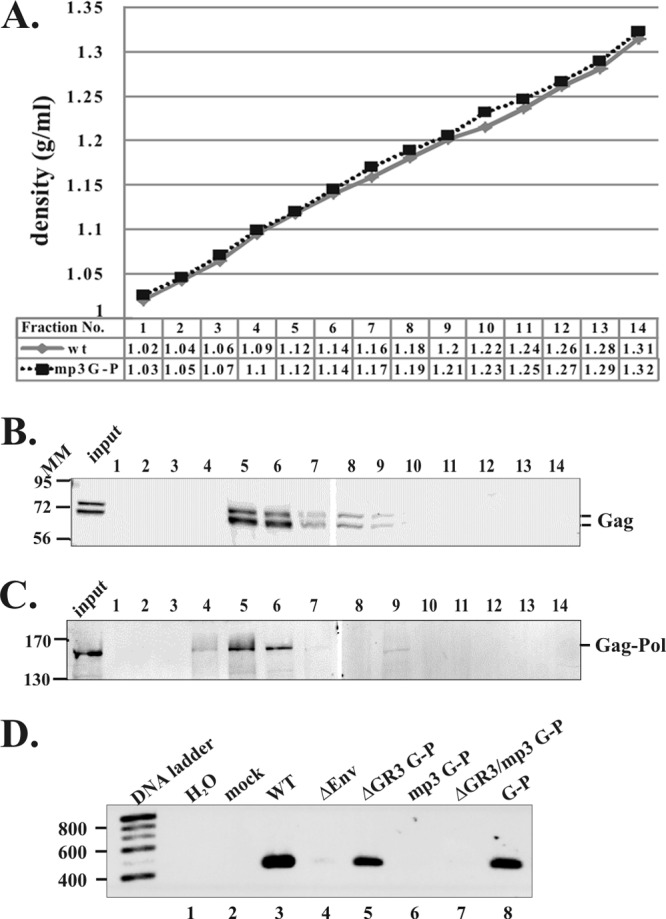
Particle density analyses of virions released from transfected cells expressing the PFV Gag-Pol. 293T cells were transfected with constructs expressing WT or the mp3 G-P. Viral samples were applied to an isopycnic gradient and subjected to ultracentrifugation. A total of 14 fractions were collected from the top of the each sample gradient. The numbers in the table below the x axis of the graph (A) show densities of each fraction in both gradients in g/ml. The viral proteins from each fraction were precipitated with 10% TCA and fractionated on a 9% SDS–polyacrylamide gel. The proteins were transferred to a membrane, and the Western blot was done with anti-Gag antibody to detect the WT protein (B) or anti-Pol antibody to detect the mp3 G-P protein (C). (D) RT-PCR was performed to measure the levels of genomic RNA packaged into virions. Molecular mass markers (MM) are shown in kilodaltons. DNA ladders in base pairs are indicated.
RT-PCR after DNase treatment was performed to measure the ability of the Gag-Pol mutants to encapsidate genomic RNA into virions. No viral RNA was detected in supernatants of the ΔEnv control (Fig. 6D). The levels of unspliced genomic RNA in the virions released from the ΔGR3 G-P and G-P fusions (which retained the p3 cleavage sites) were comparable to that of the WT (Fig. 6D, lanes 3, 5, and 8). However, neither of the p3 cleavage site mutants, mp3 G-P and ΔGR3/mp3 G-P, was able to encapsidate detectable levels of genomic RNA into virions (Fig. 6D, lanes 6 and 7). Thus, the VLPs that are slightly lighter than WT VLPs are deficient in genomic RNA.
PFV Gag-Pol can coassemble with Gag into virions to produce infectious particles.
Since orthoretroviral Gag can assemble into VLPs and Gag-Pol coassembles with Gag (reviewed in reference 9), we asked whether, in the presence of PFV Gag, the PFV Gag-Pol protein could coassemble with Gag into virus particles. Equimolar amounts of a plasmid expressing PFV Gag (pGag) and Gag-Pol proviral plasmids (mp3 G-P or Δ3′ss mp3 G-P) were cotransfected into 293T cells, and we measured the levels of the viral proteins in cells and viral assembly and release by Western blot analyses. Both the PFV Gag and Gag-Pol proteins were detected in the cotransfected cells and in the supernatants (Fig. 7A and B, lanes 5, 7, 12, and 14). Thus, VLPs were released from cells that were cotransfected with the pGag plasmid and the Gag-Pol mutant constructs. In Δ3′ss mp3 G-P cells that coexpressed Gag, although WT Pol protein was absent, the protease expressed in the Gag-Pol fusion cleaved Gag (Fig. 7A, lane 7). Virus titers from cotransfected cells with the Gag and Gag-Pol proteins were measured. We have previously shown that a PFV Gag mutant that has an R50A mutation in the cytoplasmic targeting/retention signal (CTRS) prevents virus assembly at the MTOC, resulting in no virus production (5). When the R50A full-length proviral construct was cotransfected with pGag, capsid assembly and virus budding were restored and the titer was about 11% of that of the WT (Table 2). In order to compare virus titers, we used this R50A PFV that was released from cells cotransfected with pGag as a control since the Gag-Pol fusions are deficient in assembling normal VLPs (Table 2). The virus titers from cells cotransfected with an equimolar ratio of plasmids expressing pGag and either the mp3 G-P or Δ3′ss mp3 G-P were 9.7 × 103 and 4.6 × 103 U/ml; these are 36 and 17% of the R50A PFV cotransfected with pGag, respectively (Table 2). When molar ratios of the expression plasmids for pGag and the Δ3′ss mp3 G-P fusion were changed to increase the amounts of Gag in the transfected cells, the titers were higher (Table 2). Cells cotransfected at a molar ratio of 20:1 (pGag/Δ3′ss mp3 G-P fusion), which should give a ratio of Gag and the Gag-Pol proteins that is similar to the ratio found in orthoretroviruses produced infectious viruses comparable to the levels seen when the Gag expression plasmid was cotransfected with R50A PFV. These results show that expression of a free Pol was not needed for PFV infectivity and the Gag-Pol protein can provide the enzymatic activities required for PFV replication.
Fig 7.
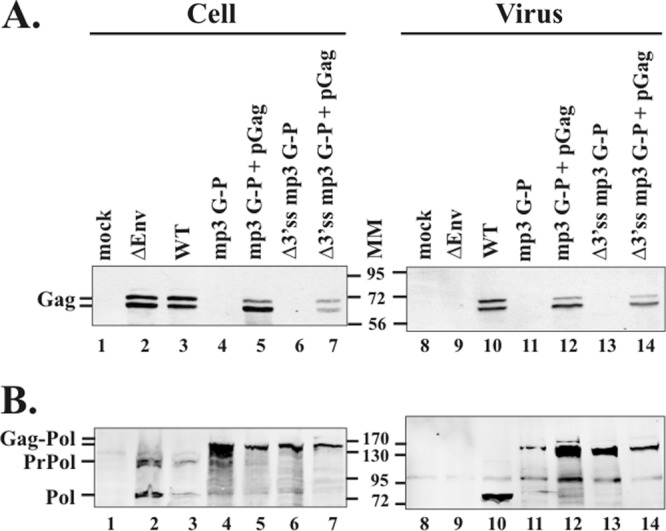
Expression and packaging of Gag and Pol proteins from cells cotransfected with the Gag-Pol construct and the pGag plasmid. Virus pellets were digested with subtilisin before being loaded onto 9% SDS–polyacrylamide gels. Viral proteins were visualized by Western blot analyses using anti-Gag antibody (A) or anti-Pol antibody (B). PrPol, precursor Pol (PR-RT-IN); Pol, cleaved Pol (PR-RT); Gag-Pol, Gag-PR-RT-IN and Gag-PR-RT. Molecular mass markers (MM) are shown in kilodaltons.
Table 2.
Infectivity of viruses produced from cells cotransfected with Gag-Pol proviruses and the pGag Gag expression vector
| WT or mutant construct | Infectivity (U/ml)a | Relative infectivity (%) |
|---|---|---|
| WT | (2.4 ± 0.5) × 105 | 885 |
| pGag | <10 | <0.04 |
| R50A PFV | <10 | <0.04 |
| mp3 G-P | <10 | <0.04 |
| Δ3′ss mp3 G-P | <10 | <0.04 |
| pGag/R50A PFV (1:1) | (2.7 ± 0.7) × 104 | 100 |
| pGag/mp3 G-P (1:1) | (9.7 ± 2.2) × 103 | 36 |
| pGag/Δ3′ss mp3 G-P (1:1) | (4.6 ± 0.5) × 103 | 17 |
| pGag/Δ3′ss mp3 G-P (5:1) | (1.8 ± 1.1) × 104 | 65 |
| pGag/Δ3′ss mp3 G-P (10:1) | (2.6 ± 0.9) × 104 | 97 |
| pGag/Δ3′ss mp3 G-P (20:1) | (3.4 ± 1.2) × 104 | 126 |
Infectivity was measured by the FAB assay as described in reference 39. The numbers are the average ± standard deviation from three independent assays.
DISCUSSION
FV Pol is synthesized from a spliced pol mRNA and does not contain Gag sequences. This is unlike orthoretroviruses, in which Pol is synthesized as part of a Gag-Pol protein. In orthoretroviruses, if the Gag-Pol protein is expressed by itself, it cannot assemble into virus particles. In the present study, we examined the behavior of PFV Gag and Pol in the context of a Gag-Pol protein. We found that regardless of coexpression of the spliced Pol protein, a PFV Gag-Pol protein can assemble into VLPs, although these particles have aberrant morphology and are not infectious. In the absence of normal spliced pol, the viral protease encoded in PFV Gag-Pol is active and PFV Gag-Pol coassembled with Gag leads to the production of infectious viruses. We do not know whether PFV Gag-Pol is incorporated into virions by binding to Gag as in orthoretroviruses or by binding to RNA as does FV WT Pol.
When we cotransfected cells with an equimolar ratio of PFV Gag and a Gag-Pol plasmid (in the absence of WT Pol), the resultant viruses had 17% infectivity compared to a control virus that coexpressed R50A PFV and Gag protein (Table 2). Another laboratory also reported that coexpression of equal amounts of PFV Gag-Pol and Gag, using a PFV 4-plasmid expression system, yielded a virus with a titer 10% of that of the WT (33). Because all of the viral enzymatic activities are required for the production of infectious particles, all three individual enzymes of Pol (PR, RT, and IN) must be active when expressed as a Gag-Pol fusion in the absence of spliced pol. In the WT virus, PR cleaves Gag once near the C terminus of Gag at the p3 cleavage site and Pol once at the site between RT and IN (Fig. 1A). Previously, our laboratory showed that IN needs to be cleaved from precursor Pol protein for viral replication (27). Both cleavages can occur when PR is expressed as part of a Gag-Pol protein, consistent with results published by Löchelt et al. (19). Like orthoretroviral PRs, PFV PR is active only as a homodimer (11, 18, 37). This implies that PFV Pol-Pol interactions can occur within the context of the Gag-Pol protein and that these interactions give rise to protease activity.
When the Gag-Pol proteins were expressed in the absence of Gag from HIV-1 and spleen necrosis virus, the Gag-Pol proteins were efficiently processed by the viral protease (14, 22, 25, 35). On the contrary, the Gag-Pol protein of MLV was not processed by PR if it was expressed in the absence of Gag (7). In all cases, there was no virion assembly or VLP release when orthoretroviral Gag-Pol proteins were expressed in the absence of Gag. In contrast, PFV Gag-Pol protein can assemble into VLPs with aberrant morphologies (Fig. 5). Recently Swiersy et al. (33) also reported that expression of similar PFV Gag-Pol proteins (with an inactivated protease) gave rise to aberrant intracellular structures, but the Gag-Pol proteins were unable to assemble into normal capsid structures. In reference 33, Gag-Pol proteins pelleted from the supernatants were mostly degraded by subtilisin digestion, suggesting Gag-Pol proteins did not lead to the production of fully formed membrane-enclosed VLPs in the absence of Gag. In contrast, the Gag-Pol VLPs that we detected are subtilisin resistant. It is not clear why the results obtained in the experiments done in the two laboratories using similar PFV Gag-Pol proteins were different. One possible explanation might be the differential expression of Gag-Pol proteins detected in the two studies. In our experiments, when Pol was expressed as a Gag-Pol fusion, the level of Gag-Pol in the cell was significantly higher than the level of Pol seen with a WT virus. This was expected because expression of Pol from the spliced message is regulated at a transcriptional level. However, the level of Gag-Pol using a PFV 4-plasmid expression system reported by Swiersy et al. (33) was similar to the level of WT Pol, and this level may have been too low for efficient assembly.
Although PFV Gag-Pol can assemble aberrant VLPs that are released into the supernatants, the Gag-Pol mutants do not efficiently encapsidate genomic RNA into VLPs. Domains in the C terminus of Gag are involved in genomic RNA packaging (17, 31). The encapsidation of Pol is not a prerequisite for RNA packaging (2, 17). There are three GR boxes near the C terminus of PFV Gag. These motifs are characteristic of FV and are thought to be functionally equivalent to the Cys-His motifs found in the NC protein of orthoretroviruses that are involved in several essential steps in the viral life cycle, including RNA packaging (reviewed in reference 13). The FV GR boxes have been shown to be involved in multiple steps of viral replication, including the encapsidation of genomic RNA, Pol packaging, reverse transcription, and viral assembly (17, 20, 31). Because the fusion of Gag and Pol could prevent the GR boxes from functioning in RNA encapsidation, we were surprised to find that VLPs produced from a Gag-Pol protein had peak fractions of similar densities as WT virus, although perhaps a bit lighter than the WT, despite the deficiency of genomic RNA. Although RNA is a structural element in orthoretroviral particles (21), viral genomic RNA is not required. Viral particles that do not package viral RNA package cellular mRNAs (28). We have not yet examined the presence of cellular mRNAs in PFV Gag-Pol viruses.
We found that the expression of Gag-Pol proteins that contain cleavage sites in both Gag and Pol led to the production of virions with a titer similar to the WT (G-P in Table 1). This implies that PFV Gag-Pol expression alone is compatible with viral assembly and replication as long as there is appropriate processing of the fusion protein in cells. This is consistent with recent results obtained by others using similar PFV Gag-Pol constructs (33). We coexpressed Gag-Pol with Gag in the absence of Pol, varying the ratios of Gag to Gag-Pol from 20:1 (similar to that found in orthoretroviruses) to 1:1 (Table 2). In HIV-1, when the normal ratio of Gag to Gag-Pol (around 20:1) was altered by expressing increased amounts of Gag-Pol, viral assembly was disrupted and infectivity was greatly reduced (10, 14, 29). In contrast to what is seen with orthoretroviruses, increasing the amounts of PFV Gag-Pol relative to Gag did not abrogate infectivity. Taken together, the data show that FV can use an orthoretroviral-like Gag-Pol expression mechanism to make infectious viruses, even though, under normal circumstances, spumaretroviruses have evolved to use a different mechanism for Pol synthesis and assembly. Wild-type FV reverse transcribes RNA during or after viral assembly and budding, leading to infectious DNA virions. It will be of interest to determine whether FV RT expressed in a Gag-Pol context is also active at this stage.
ACKNOWLEDGMENTS
We thank Steve Hughes and Shannon Murray for critical reading of the manuscript.
This research was funded by NIH grant R01 CA 18282 to M.L.L., and A. Sinicrope was supported by the NCI grant Continuing Umbrella of Research Experience (CURE) 3 P30 CA015704-36S2.
Footnotes
Published ahead of print 4 April 2012
REFERENCES
- 1. Baldwin DN, Linial ML. 1998. The roles of Pol and Env in the assembly pathway of human foamy virus. J. Virol. 72:3658–3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baldwin DN, Linial ML. 1999. Proteolytic activity, the carboxy terminus of Gag, and the primer binding site are not required for Pol incorporation into foamy virus particles. J. Virol. 73:6387–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Duda A, et al. 2004. Prototype foamy virus envelope glycoprotein leader peptide processing is mediated by a furin-like cellular protease, but cleavage is not essential for viral infectivity. J. Virol. 78:13865–13870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Durocher Y, Perret S, Kamen A. 2002. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eastman SW, Linial ML. 2001. Identification of a conserved residue of foamy virus Gag required for intracellular capsid assembly. J. Virol. 75:6857–6864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Enssle J, et al. 1997. Carboxy-terminal cleavage of the human foamy virus Gag precursor molecule is an essential step in the viral life cycle. J. Virol. 71:7312–7317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Felsenstein KM, Goff SP. 1988. Expression of the gag-pol fusion protein of Moloney murine leukemia virus without gag protein does not induce virion formation or proteolytic processing. J. Virol. 62:2179–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fischer N, et al. 1998. Foamy virus particle formation. J. Virol. 72:1610–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goff SP. 2007. Retroviridae: the retroviruses and their replication, p 1999–2069 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 10. Haraguchi H, et al. 2010. Intracellular localization of human immunodeficiency virus type 1 Gag and GagPol products and virus particle release: relationship with the Gag-to-GagPol ratio. Microbiol. Immunol. 54:734–746 [DOI] [PubMed] [Google Scholar]
- 11. Hartl MJ, et al. 2010. Formation of transient dimers by a retroviral protease. Biochem. J. 427:197–203 [DOI] [PubMed] [Google Scholar]
- 12. Heinkelein M, et al. 2002. Pregenomic RNA is required for efficient incorporation of Pol polyprotein into foamy virus capsids. J. Virol. 76:10069–10073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jewell NA, Mansky LM. 2000. In the beginning: genome recognition, RNA encapsidation and the initiation of complex retrovirus assembly. J. Gen. Virol. 81:1889–1899 [DOI] [PubMed] [Google Scholar]
- 14. Karacostas V, Wolffe EJ, Nagashima K, Gonda MA, Moss B. 1993. Overexpression of the HIV-1 gag-pol polyprotein results in intracellular activation of HIV-1 protease and inhibition of assembly and budding of virus-like particles. Virology 193:661–671 [DOI] [PubMed] [Google Scholar]
- 15. Lee E-G, et al. 2008. A premature termination codon mutation at the C terminus of foamy virus Gag downregulates the levels of spliced pol mRNA. J. Virol. 82:1656–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee E-G, Linial ML. 2006. Deletion of a Cys-His motif from the Alpharetrovirus nucleocapsid domain reveals late domain mutant-like budding defects. Virology 347:226–233 [DOI] [PubMed] [Google Scholar]
- 17. Lee E-G, Linial ML. 2008. The C terminus of foamy retrovirus Gag contains determinants for encapsidation of Pol protein into virions. J. Virol. 82:10803–10810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee E-G, et al. 2011. Foamy retrovirus integrase contains a Pol dimerization domain required for protease activation. J. Virol. 85:1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Löchelt M, Flügel RM. 1996. The human foamy virus pol gene is expressed as a Pro-Pol polyprotein and not as a Gag-Pol fusion protein. J. Virol. 70:1033–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mullers E, et al. 2011. Novel functions of prototype foamy virus Gag glycine-arginine-rich boxes in reverse transcription and particle morphogenesis. J. Virol. 85:1452–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Muriaux D, Mirro J, Harvin D, Rein A. 2001. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. U. S. A. 98:5246–5251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park J, Morrow CD. 1991. Overexpression of the gag-pol precursor from human immunodeficiency virus type 1 proviral genomes results in efficient proteolytic processing in the absence of virion production. J. Virol. 65:5111–5117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Park J, Morrow CD. 1992. The nonmyristylated Pr160gag-pol polyprotein of human immunodeficiency virus type 1 interacts with Pr55gag and is incorporated into viruslike particles. J. Virol. 66:6304–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peters K, Wiktorowicz T, Heinkelein M, Rethwilm A. 2005. RNA and protein requirements for incorporation of the Pol protein into foamy virus particles. J. Virol. 79:7005–7013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pettit SC, Everitt LE, Choudhury S, Dunn BM, Kaplan AH. 2004. Initial cleavage of the human immunodeficiency virus type 1 GagPol precursor by its activated protease occurs by an intramolecular mechanism. J. Virol. 78:8477–8485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pfrepper KI, et al. 1998. Molecular characterization of proteolytic processing of the Pol proteins of human foamy virus reveals novel features of the viral protease. J. Virol. 72:7648–7652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roy J, Linial ML. 2007. Role of the foamy virus Pol cleavage site in viral replication. J. Virol. 81:4956–4962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rulli SJ, Jr, et al. 2007. Selective and nonselective packaging of cellular RNAs in retrovirus particles. J. Virol. 81:6623–6631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shehu-Xhilaga M, Crowe SM, Mak J. 2001. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 75:1834–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith AJ, Srinivasakumar N, Hammarskjold M-L, Rekosh D. 1993. Requirements for incorporation of Pr160gag-pol from human immunodeficiency virus type 1 into particles. J. Virol. 67:2266–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stenbak CR, Linial ML. 2004. Role of the C terminus of foamy virus Gag in RNA packaging and Pol expression. J. Virol. 78:9423–9430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stirnnagel K, et al. 2010. Analysis of prototype foamy virus particle-host cell interaction with autofluorescent retroviral particles. Retrovirology 7:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Swiersy A, Wiek C, Reh J, Zentgraf H, Lindemann D. 2011. Orthoretroviral-like prototype foamy virus Gag-Pol expression is compatible with viral replication. Retrovirology 8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tobaly-Tapiero J, et al. 2000. Isolation and characterization of an equine foamy virus. J. Virol. 74:4064–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weaver TA, Talbot KJ, Panganiban AT. 1990. Spleen necrosis virus gag polyprotein is necessary for particle assembly and release but not for proteolytic processing. J. Virol. 64:2642–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Winkler I, et al. 1997. Characterization of the genome of feline foamy virus and its proteins shows distinct features different from those of primate spumaviruses. J. Virol. 71:6727–6741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wlodawer A, et al. 1989. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science 245:616–621 [DOI] [PubMed] [Google Scholar]
- 38. Yu SF, Baldwin DN, Gwynn SR, Yendapalli S, Linial ML. 1996. Human foamy virus replication—a pathway distinct from that of retroviruses and hepadnaviruses. Science 271:1579–1582 [DOI] [PubMed] [Google Scholar]
- 39. Yu SF, Linial ML. 1993. Analysis of the role of the bel and bet open reading frames of human foamy virus by using a new quantitative assay. J. Virol. 67:6618–6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu SF, Eastman SW, Linial ML. 2006. Foamy virus capsid assembly occurs at a pericentriolar region through a cytoplasmic targeting/retention signal in Gag. Traffic 7:966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zemba M, et al. 1998. The carboxy-terminal p3(Gag) domain of the human foamy virus gag precursor is required for efficient virus infectivity. Virology 247:7–13 [DOI] [PubMed] [Google Scholar]