

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is etiologically associated with the angioproliferative Kaposi's sarcoma (KS). KSHV infection and the expression of latency-associated nuclear antigen (LANA-1) upregulates the angiogenic multifunctional 123-amino-acid, 14-kDa protein angiogenin (ANG), which is detected in KS lesions and in KSHV-associated primary effusion lymphoma (PEL) cells. ANG knockdown or the inhibition of ANG's nuclear translocation resulted in decreased LANA-1 gene expression and reduced KSHV-infected endothelial and PEL cell survival (Sadagopan et al., J. Virol. 83:3342–3364, 2009). Further studies here demonstrate that LANA-1 and ANG colocalize and coimmunoprecipitate in de novo infected endothelial cells and in latently infected PEL (BCBL-1 and BC-3) cells. LANA-1 and ANG interaction occurred in the absence of the KSHV genome and other viral proteins. In gel filtration chromatography analyses of BC-3 cell lysates, ANG coeluted with LANA-1, p53, and Mdm2 in high-molecular-weight fractions, and LANA-1, p53, and Mdm2 also coimmunoprecipitated with ANG. LANA-1, ANG, and p53 colocalized in KSHV-infected cells, and colocalization between ANG and p53 was also observed in LANA-1-negative cells. The deletion constructs of ANG suggested that the C-terminal region of amino acids 104 to 123 is involved in LANA-1 and p53 interactions. Silencing ANG or inhibiting its nuclear translocation resulted in decreased nuclear LANA-1 and ANG levels, decreased interactions between ANG-LANA-1, ANG-p53, and LANA-1-p53, the induction of p53, p21, and Bax proteins, the increased cytoplasmic localization of p53, the downregulation of Bcl-2, the increased cleavage of caspase-3, and the apoptosis of cells. No such effects were observed in KSHV-negative BJAB cells. The phosphorylation of p53 at serine 15, which is essential for p53 stabilization and for p53's apoptotic and cell cycle regulation functions, was increased in BCBL-1 cells transduced with short hairpin RNA targeting ANG. Together, these studies suggest that the antiapoptosis observed in KSHV-infected cells and the suppression of p53 functions are mediated in part by ANG, and KSHV has probably evolved to utilize angiogenin's multiple functions for the maintenance of its latency and cell survival. Thus, targeting ANG to induce the apoptosis of cells latently infected with KSHV is an attractive therapeutic strategy against KSHV infection and associated malignancies.
INTRODUCTION
Kaposi's sarcoma (KS) is an angioproliferative and highly vascularized tumor with a microenvironment abundant with inflammatory cytokines, angiogenic molecules, and growth factors. Kaposi's sarcoma associated herpesvirus (KSHV) is etiologically associated with KS, where it is in a latent state in the proliferating spindle-shaped endothelial cells, and it expresses a limited set of genes, including ORF73 (latency-associated nuclear antigen [LANA-1]), ORF72 (v-CyclinD), ORF71 (vFLIP; K13), K12 (Kaposins), and >12 microRNAs (miRNAs) (3, 10, 14, 34). KSHV is also etiologically associated with B-cell proliferative neoplasms, such as primary effusion lymphoma (PEL) or body cavity B-cell lymphoma (BCBL) and multicentric Castleman's disease (MCD), each of which also shows robust angiogenesis (4, 5, 14). B-cell lines such as BCBL-1 and BC-3, established from PEL, grow indefinitely in vitro as suspension cells and carry >80 episomal copies of the KSHV latent genome. In addition to ORF73, ORF72, ORF71, K12, and miRNAs, these cells also express K10.5 (LANA-2), K1, and K2 (v-IL-6) genes (3, 10, 14, 34).
KSHV infection reprograms the host cell's transcriptional machinery to create an environment that is conducive for its latent intracellular parasitism (14, 30). KSHV latent genes efficiently modify and/or hijack several host molecules to evade host defense mechanisms, such as transcriptional blocks, DNA damage responses, interferons, apoptosis, autophagy, NK cells, and adaptive immune responses, and they inhibit the KSHV lytic cycle and ensure uninterrupted cell division and viral episome propagation (14). KSHV latent genes drive PEL cell proliferation, antiapoptosis, and pathogenesis, while both the latent and lytic cycles, along with infection-induced neoangiogenic inflammatory networks, are suggested to be involved in KS pathogenesis. Available treatment strategies to control KSHV infection-associated malignancies are limited and of low efficacy. Hence, there is a vital need for designing therapies that target viral infection and tumor formation.
Angiogenesis, the formation of new blood vessels from preexisting vessels, is a highly regulated event occurring during embryogenesis, development, inflammation, wound healing, and the female reproductive cycle. Viruses have been shown to regulate angiogenesis either by expressing their own proangiogenic factors or by modulating cellular proteins and signaling pathways (46). KSHV infection upregulates the transcription of many host genes involved in angiogenesis, such as vascular endothelial growth factor A (VEGF-A), VEGF-C, COX-2, etc., which promote KSHV latency and disease pathogenesis (30, 40, 42). KSHV LANA-1, one of the major players during latency, is involved in tethering the viral genome to the host chromosome, binding to and downregulating the functions of crucial host tumor suppressors, such as p53, Rb, and KSHV lytic switch protein RTA (ORF50) (2, 7, 12, 24, 33). LANA-1 also modifies transcriptional activity by altering the subcellular distribution of glycogen synthase kinase 3β (GSK-3β), a negative regulator of β-catenin (13). Proteomic approaches have identified several additional LANA-1-interacting host proteins (6, 20, 31).
Our previous studies have demonstrated that during the infection of primary human dermal microvascular endothelial (HMVEC-d) cells, KSHV upregulated the expression and secretion of angiogenin (ANG), a multifunctional angiogenic molecule, in a time- and dose-dependent manner beginning as early as 8 h postinfection, and this lasted until the fifth day of the observation period (35). Telomerase-immortalized human umbilical vein endothelial (TIVE) cells latently infected with KSHV (TIVE-LTC) secreted higher levels of angiogenin than TIVE cells (35). Significant ANG gene expression and secretion (250 to 400 pg/ml) were observed in KSHV+ PEL (BCBL-1 and BC-3) cells but not in Epstein-Barr virus-negative (EBV−) KSHV− or EBV+ KSHV− lymphoma cells (less than 30 pg/ml) (36). In addition, when expressed alone in HMVEC-d cells, latent ORF73 (LANA-1) and lytic ORF74 (vGPCR) genes induced significant ANG gene expression and secretion (35). These studies clearly suggested a specific association of ANG in KSHV biology and pathogenesis, which is highlighted by the detection of ANG in KS tissue sections (35).
ANG, a 14-kDa proangiogenic protein, is upregulated in many cancers. Unlike other angiogenic molecules, such as VEGF and angiopoietins that mediate their angiogenic effects via specific cell surface molecules, ANG is unique in that it mediates its effects at multiple sites in the cell, such as the plasma membrane, cytoplasm, nucleus, and nucleolus, and directly binds to DNA. ANG translocates into the nucleus in sub- and semiconfluent cells, moves into the nucleolus in subconfluent cells only, and helps to transcribe rRNA by binding to CT repeats in the rRNA gene promoter region and induces 45S rRNA transcription and cell proliferation (28, 47). It is an RNase and has ribonucleolytic activity toward tRNA and CpA and generates 18S and 28S rRNA (43).
ANG's nuclear translocation is critical not only for its angiogenic and proliferative effects but also for the angiogenic potential of VEGF and basic fibroblast growth factor (bFGF) (21). ANG's nuclear translocation, and hence its nuclear functions, requires the activation of phospholipase Cγ (PLCγ). Neomycin (neo), an FDA-approved aminoglycoside antibiotic, inhibits PLCγ and thus inhibits the nuclear translocation of ANG as well as ANG-induced cell proliferation and angiogenesis (19). Neomycin specifically interferes with bacterial protein synthesis and should not be confused with neomycin (G418), which inhibits protein synthesis in both prokaryotic and eukaryotic cells.
The inhibition of the nuclear translocation of ANG by neomycin resulted in reduced BCBL-1, BC-3, and TIVE-LTC cell survival and proliferation, while uninfected endothelial cells as well as EBV+ KSHV− or EBV− KSHV− cells were unaffected (36). ANG-induced ORF73 expression and PLC-γ and AKT phosphorylation were inhibited by neomycin treatment and ANG silencing. Neomycin also inhibited latent ORF73 gene expression and increased lytic ORF50 gene expression both during de novo infection and in latently infected cells (36). A greater quantity of infectious KSHV was detected in the supernatants of neomycin-treated BCBL-1 cells than in 12-O-tetradecanoylphorbol-13-acetate (TPA)-treated cells. Treatment with the conventional PLC-γ inhibitor U73122 showed similar results.
Silencing of ANG also resulted in reduced cell survival, ORF73 gene expression, and lytic gene activation in BCBL-1 and TIVE-LTC cells and during de novo infection. ANG silencing did not affect normal uninfected cells or cells infected with EBV, which clearly indicated a role for a PLCγ pathway in the maintenance of KSHV latency and survival (36). The detection of higher levels of ANG in KSHV-infected PEL cells, decreased survival of KSHV+ B cells, but not that of KSHV− or EBV+ B cells, upon the inhibition of nuclear angiogenin, together with the ability of LANA-1 to induce angiogenin expression and secretion clearly indicated a specific association of angiogenin with KSHV biology and suggested that KSHV has evolved to exploit ANG to its advantage via a PLCγ pathway for maintaining its latency. It is currently unknown whether ANG binds to the latency and ORF50 promoters and modulates their transcription. Promoter studies are hindered by the fact that all primary cells that express low levels of ANG are poorly transfectable, while the transformed cell lines that are readily transfectable express high levels of endogenous ANG.
The present study was designed to explain the potential mechanism behind the neomycin treatment and the disruption of KSHV latency and death of KSHV-infected cells induced by short hairpin RNA targeting ANG (sh-ANG). We analyzed the relationship between LANA-1 and ANG, and these studies demonstrate that ANG plays crucial roles in the antiapoptotic state of KSHV-infected cells by suppressing p53 functions, and the abolition of this property by sh-ANG and neomycin could be responsible for the disruption of KSHV latency and the death of KSHV-infected cells. These findings implicate ANG in herpesvirus (KSHV) latency and uncover a novel paradigm that demonstrates the evolution of KSHV's genome plasticity to utilize angiogenin for antiapoptosis, p53 modulation, latency, and survival advantages.
MATERIALS AND METHODS
Antibodies, reagents, and growth factors.
Rabbit antibodies against baculovirus expressing full-length LANA-1 were raised by us (1). Goat and rabbit polyclonal antibodies against human angiogenin; rabbit polyclonal antibody against Bax; and mouse monoclonal antibody against promyelocytic leukemia protein (PML), p53, Mdm2, p21, Bcl-2, myc, and green fluorescent protein (GFP) were from Santa Cruz Biotechnology Inc., Santa Cruz, CA. Cleaved caspase 3 (Asp175), cleaved caspase 3 (Alexa Fluor 488 conjugate), and p-p53 (Ser15) antibodies were from Cell Signaling, Danvers, MA. Anti-mouse fibrillarin antibody was from Abcam, Cambridge, MA. Antibodies against lamin B, tubulin, actin, and recombinant human angiogenin were from Sigma, St. Louis, MO.
Cells.
HMVEC-d cells (CC-2543; Clonetics, Walkersville, MD) were grown in endothelial basal medium 2 (EBM-2) with growth factors (Clonetics). HEK 293T, p53 null SaOS-2 (ATCC HTB-85), and wild-type p53 U2OS (ATCC HTB-96) human osteosarcoma cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and antibiotics. KSHV carrying PEL cells (BCBL-1 and BC-3) were cultured in RPMI 1640 (Gibco BRL) medium with 10% heat-inactivated FBS (HyClone, Logan, UT), 2 mM l-glutamine, and antibiotics.
Virus.
The induction of the KSHV lytic cycle in BCBL-1 cells, supernatant collection, and virus purification procedures were described previously (22), and purity was assessed according to general guidelines established in our laboratory (1, 22, 29). KSHV DNA was extracted from the purified virus, and copy numbers were quantified by real-time DNA PCR using primers amplifying the KSHV ORF73 gene (22). KSHV DNA copy numbers from untreated and neomycin-treated BCBL-1 cells were also quantitated by this method.
Quantitative real-time RT-PCR.
The expression of KSHV ORF73 and ORF50, p53, p21, and ANG was measured by real-time reverse transcription-PCR (RT-PCR) using SYBR green. ORF73 DNA copy numbers were detected by real-time RT-PCR using previously described TaqMan methods (22). cDNA was used as a template with the following primers specific for angiogenin: forward (Fw), CCGTTTCTGCGGACTTGTTC; reverse (Rev), GCCCATCACCATCTCTTCCA; p21 Fw, GCAGACCAGCATGACAGATTT; Rev, GGATTAGGGCTTCCTCTTGGA; p53 Fw, TCAACAAGATGTTTTGCCAACTG; Rev, GCATCTATCCCCCCTAAAGTGG; ORF 50 Fw, CGCAATGCGTTACGTTGTTG; Rev, GCCCGGACTGTTGAATCG; ORF73 Fw, CGCGAATACCGCTATGTACTCA; Rev, GGAACGCGCCTCATACGA; and 18S rRNA Fw, CGGCTACCACATCCAAGGAA; Rev, GCTGGAATTACCGCGGCT. 18S rRNA was used as an internal control. PCR was performed using an Applied Biosystems 7500 real-time PCR system. The standard amplification program included 40 cycles of two steps, each comprised of heating to 95 and 60°C. Fluorescent product was detected at the last step of each cycle. For SYBR green reactions, the final mRNA levels of the genes studied were normalized using the comparative threshold cycle (CT) method.
293T transfection studies.
HEK 293T cells were transfected with 5 μg of pCI-neo full-length ORF73 gene construct, full-length C-terminal GFP-tagged angiogenin construct, pcDNA-GFP empty vector gene construct, and ANG deletion constructs using the calcium phosphate precipitation method. The cells were incubated for 48 h in 3% CO2, harvested in radioimmunoprecipitation assay (RIPA) buffer, and subjected to immunoprecipitation (IP) and Western blotting.
Immunofluorescence assay (IFA).
Semiconfluent HMVEC-d cells in 8-well chamber slides were left uninfected or were infected with 20 KSHV DNA copes/cell at 37°C for 48 h. The cells were fixed with 4% paraformaldehyde for 10 min at room temperature, permeabilized with 0.4% Triton X-100 for 10 min at room temperature, and stained with primary antibody for 1 h at 37°C. Cells were washed and incubated with secondary antibodies for 1 h at room temperature. Nuclei were visualized using 4′,6-diamidino-2-phenylindole (DAPI) and viewed under a fluorescence microscope using a Metamorph digital imaging system. BCBL-1 cells were acetone fixed to glass slides and stained as described above.
Cleaved caspase 3 assay.
HMVEC-d cells grown to 50% confluence in 8-well chamber slides were serum starved for 6 to 8 h, followed by infection with live KSHV (20 KSHV DNA copes/cell) or angiogenin treatment (1 μg/ml) for 24 h or pretreatment with 200 μM neomycin for 1 h before KSHV infection or angiogenin treatment. Cells were washed with Hanks balanced salt solution (HBSS), incubated with cleaved caspase 3 antibody (Alexa Fluor 488 conjugated) in EBM-2 for 2 h at 37°C, stained with DAPI, and visualized. HMVEC-d cells were treated in a similar manner, harvested in RIPA buffer, and probed by Western blotting.
Nuclear extract preparation.
Nuclear extracts from BCBL-1 and TIVE-LTC cells were prepared using a nuclear extract kit (Active Motif Corp., Carlsbad, CA) per the manufacturer's instructions.
IP.
Cells were harvested in RIPA buffer (1% NP-40, 1.5% sodium deoxycholate, 0.1% SDS, 125 mM NaCl, 0.01 M sodium phosphate, 1 mM EDTA, 50 mM NaF, 1% protease inhibitor cocktail) and homogenized using a 22-gauge needle and syringe. Equal protein concentrations of cell lysates were subjected to IP with equal concentrations of rabbit anti-angiogenin, rabbit anti-LANA-1, or control rabbit IgG antibody using 20 μl of protein G Sepharose beads (GE Healthcare, Bio-Sciences).
Western blot reaction.
IP lysates, protein lysates, or nuclear extracts were resolved by SDS-PAGE, transferred to nitrocellulose membranes, blocked with 5% skim milk, and immunoblotted with the indicated antibodies. Species-specific horseradish peroxidase or alkaline phosphatase-conjugated secondary antibodies were used for detection.
Lentivirus production and infection of BCBL-1 cells.
Lentiviral infection was done as described before (45). Vesicular stomatitis virus G envelope-pseudotyped lentivirus was produced with a four-plasmid transfection system as previously described (8, 27). The sequences of sh-ANG used were CCGGTGCTGTCCTTGCCTTCCATTTCTCGAGA and AATGGAAGGCAAGGACAGCATTTTTG, and the sequence of sh-GFP used was TACAACAGCCACAACGTCTAT. BCBL-1 cells were infected in culture for 8 h, and the medium was replaced with complete growth medium and incubated for 72 h without any selection.
Cell survival analysis by fluorescence-activated cell sorting (FACS).
BCBL-1 cells transduced with sh-GFP or sh-ANG were collected after 3 days, washed, and stained with YO-PRO-1/propidium iodide (V13243; Invitrogen) for 30 min. Data were collected and analyzed at the RFUMS flow cytometry core facility.
Gel filtration chromatography.
BC-3 cell lysates were separated by gel filtration chromatography on a Superdex 200 HR column with a fast protein liquid chromatography (FPLC) system (Pharmacia Biotech, Uppsala, Sweden) as described previously (38).
Statistical analysis.
For Fig. 6 and 7, the statistical significance (t test) was conducted with respect to untreated cells. For Fig. 11, the statistical significance (t test) was conducted with respect to sh-GFP treated cells (*, P < 0.05; ***, P < 0.005).
Fig 6.

Effects of neomycin on LANA-1 and ANG in BCBL-1 cells. (A) BCBL-1 cells were left untreated or were treated with 200 or 500 μM neomycin for 3 days and stained for ANG (green), LANA (red), and DAPI for nuclei (blue), and the images were merged. White boxes are shown as enlarged images. (B) The percentage of LANA-1-positive cells was determined by manually counting the number of LANA-1 dot-positive cells in an average of five different fields with 20 cells per view. Values shown represent the means ± standard deviations from three independent experiments. (C) Levels of LANA-1 protein per cell were measured by examining the number of LANA-1 dots/BCBL-1 cell in an average of five different fields by manually counting the number of punctate nuclear LANA-1 dots in cells from three independent experiments. (D) DNA was extracted from BCBL-1 cells treated with neomycin as described above, and KSHV DNA copy numbers were quantitated by estimating the ORF73 copies by real-time DNA PCR.
Fig 7.

Effects of 3 days of neomycin treatment on LANA-1, ANG, and p53 (A to C) nuclear extracts were prepared from BCBL-1 cells that were either left untreated or treated with 200 μM neomycin for 3 days. Treated and untreated nuclear lysates were subjected to IP with rabbit anti-ANG IgG antibodies, separated with a 7.5% gel, and subjected to Western blotting for LANA-1 (A); subjected to IP with rabbit anti-LANA-1 IgG antibodies, separated in a 10% gel, and subjected to Western blotting for p53 (B); or subjected to IP with rabbit anti-ANG IgG antibodies and Western blotting for p53 (C). Input controls for ANG, LANA-1, and p53 are shown for all IP reactions. (D and E) BCBL-1 cells were treated with neomycin as described above, and the expression of ORF73 and ORF50 genes was determined by real-time RT-PCR. Values shown represent the means ± standard deviations from three independent experiments. *, P < 0.05; ***, P < 0.005. BJAB (F and G) and BCBL-1 (H and I) cells were either left untreated or were treated with 200 or 500 μM neomycin for 3 days, and the expression of p53 and p21 genes was measured by real-time RT-PCR.
Fig 11.

Reduction of cell survival upon angiogenin knockdown. (A) 293T cells were transfected with control sh-GFP and sh-Renilla (RL) plasmids or two sh-ANG (sh-ANG1 and sh-ANG2) plasmids, and ANG knockdown was checked by separating the lysates with a 12.5% gel and performing Western blotting for ANG. (B) ANG knockdown was also checked by real-time RT-PCR in sh-GFP- and sh-ANG1-transduced BCBL-1 cells. (C to E) BCBL-1 cells transduced with either sh-GFP or sh-ANG1 were subjected to Western blotting for p53, p-p53, p21, Bcl-2, Bax, caspase 3, and β-actin. Fold changes are indicated. (F) BCBL cells transduced with either sh-GFP or sh-ANG1 were stained with YO-PRO dye and analyzed by FACS for the percentage of live cells. Values shown represent the means ± standard deviations from three independent experiments. ***, P < 0.005.
RESULTS
LANA-1 and ANG interact in de novo infected HMVEC-d cells and in latently infected BCBL-1 cells.
We have recently shown that LANA-1 interacts with ANG in KSHV-infected endothelial TIVE-LTC cells (31). To extend this observation, uninfected HMVEC-d cells and HMVEC-d cells infected with purified KSHV (20 DNA copies/cell) for 48 h were immunoprecipitated with anti-ANG or control IgG antibodies and probed with anti-LANA-1 antibodies in Western blot reactions. We also performed immunoprecipitation with anti-LANA-1 or IgG antibodies and probed with anti-ANG antibodies. LANA-1 was detected in ANG IP and ANG in LANA-1 IP reactions only in KSHV-infected HMVEC-d cells (Fig. 1A and B, lanes 2). IP reactions with nuclear fractions from BCBL-1 cells also detected ANG in LANA-1 IP and LANA-1 in ANG IP reactions (Fig. 1C and D, lanes 2). An earlier study had shown that LANA-1 does not associate with the PML of BCBL cells (44). When we carried out IP reactions with anti-LANA-1 or IgG antibodies and subjected them to Western blotting for PML protein, we did not observe any specific association of LANA-1 with PML protein (Fig. 1E). Instead, we detected only a nonspecific band that was present in both the IgG control and LANA-1 IP reactions (Fig. 1E). These results demonstrated the specificity of LANA-1-ANG interactions occurring during the primary KSHV infection of endothelial cells and in KSHV latently infected PEL cells.
Fig 1.
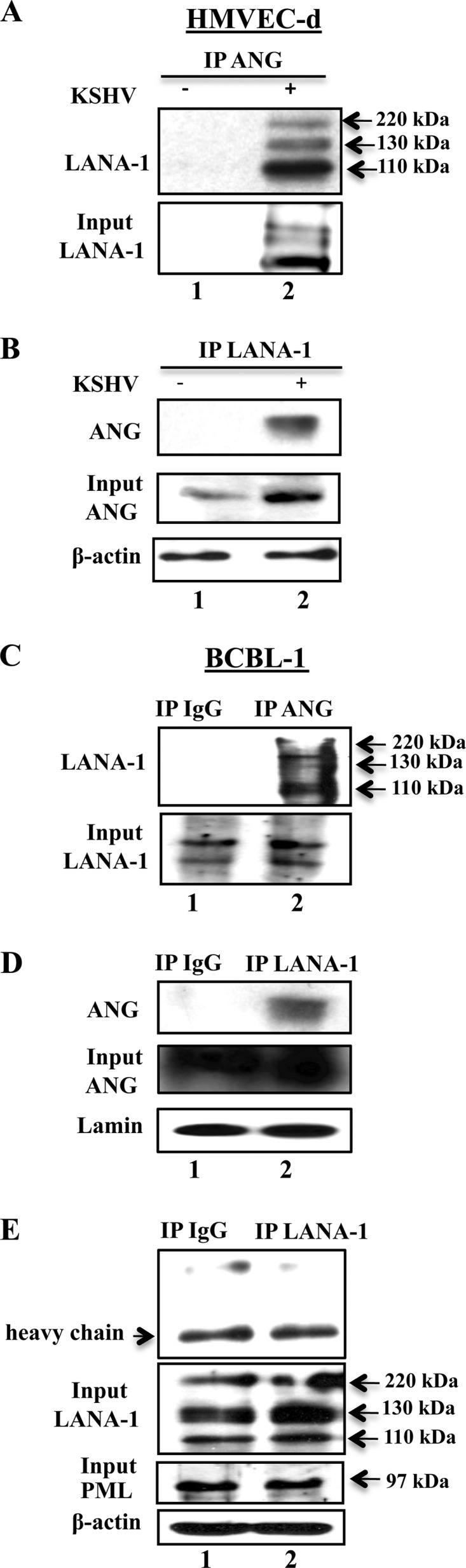
KSHV LANA-1 interaction with angiogenin in various KSHV-infected cells. (A and B) HMVEC-d cells grown to ∼60 to 70% confluence were serum starved for 8 h and either infected with 20 KSHV DNA copies/cell for 48 h or left uninfected. An equal quantity of protein from each lysate was immunoprecipitated with equal concentrations of either rabbit anti-ANG or anti-LANA-1 antibodies, separated with a 7.5% SDS-PAGE gel, and subjected to Western blotting with rat anti-LANA-1 monoclonal antibodies (A) or separated with a 12.5% gel and subjected to Western blotting with goat anti-ANG antibodies (B). (C and D) Nuclear extracts were prepared from BCBL-1 cells and immunoprecipitated with rabbit-IgG, anti-ANG, or anti-LANA-1 antibodies, separated with a 7.5% gel, and subjected to Western blotting for LANA-1 (C) or separated with a 12.5% gel and subjected to Western blotting for angiogenin (D). Five percent input and lamin loading controls are shown. (E) BCBL-1 cell lysates were immunoprecipitated with rabbit-IgG or anti-LANA-1 antibodies, separated with a 10% gel, and subjected to Western blotting for PML. Five percent input control for LANA-1, PML, and β-actin loading controls are shown.
LANA-1 and ANG colocalize in the nucleus of de novo infected HMVEC-d cells and in latently infected BCBL-1 and BC-3 cells.
When LANA-1 and ANG were visualized by IFA, no LANA-1 was detected in uninfected HMVEC-d cells (Fig. 2A, top, red), and a diffused cytoplasmic and nuclear staining of ANG was observed (Fig. 2A, top, green). In KSHV-infected HMVEC-d cells, LANA-1 appeared with its characteristic punctate nuclear fluorescence (Fig. 2A, second row, red). ANG also appeared as punctate nuclear staining in LANA-1-positive cells (Fig. 2A, second row, green). Interestingly, LANA-1 and ANG showed >90% colocalization in the nucleus (enlarged inset), and some noncolocalized spots were also observed. Similarly, in KSHV latently infected BCBL-1 and BC-3 cells, punctate nuclear LANA-1 was detected (Fig. 2A, third and fourth rows, red). ANG also appeared as punctate nuclear staining (Fig. 2A, third and fourth rows, green), and >90% colocalization of ANG and LANA-1 was seen. As reported previously (44), LANA-1 did not colocalize with PML bodies in the nucleus of KSHV-infected HMVEC-d cells (Fig. 2B), which demonstrated the specificity of the observed ANG-LANA-1 colocalization in the nucleus. LANA-1 and ANG appeared to colocalize predominately in the nonnucleolar compartment of the nucleus of infected HMVEC-d cells, since the majority of LANA-1 and ANG spots did not colocalize with the nucleolus marker fibrillarin (Fig. 2C, white spots).
Fig 2.

LANA-1 colocalization with ANG in the nucleus of KSHV-infected cells. (A) Serum-starved ∼60 to 70% confluent HMVEC-d cells were either infected with 20 KSHV DNA copies/cell for 48 h or left uninfected. After fixation and permeabilization, HMVEC-d, BCBL-1, and BC-3 cells were stained for LANA-1 (red, rabbit anti-LANA-1 antibodies) and angiogenin (green, goat anti-ANG antibodies). DAPI (blue) was used as a nuclear stain and merged with LANA-1 and ANG images. The white inserts are enlarged on the right. (B) HMVEC-d cells infected with KSHV as described for panel A were stained for LANA-1, PML bodies, and DAPI, and the images were merged. White inserts are shown as enlarged images. (C) HMVEC-d cells infected with KSHV were stained for LANA-1, ANG, DAPI, and the nucleolar marker fibrillarin, and the images were merged.
LANA-1 and ANG interaction occurs in the absence of KSHV genome and other KSHV proteins.
The punctate pattern of LANA-1 seen in cells latently infected with KSHV is believed to be due to its concentration at the site of the tethering of KSHV episomes to host chromatin (2, 7). Since ANG also appeared as punctate spots upon KSHV infection and colocalized with LANA-1 in the nucleus, to determine whether ANG-LANA-1 interaction required the presence of KSHV genome and/or other viral proteins, we overexpressed full-length LANA-1 along with GFP-tagged full-length ANG (designated FL ANG-GFP) in 293T cells. Since ANG's nuclear delivery is critical for it to be functional (19), we first examined whether the GFP tag hampered the nuclear entry of ANG. When nuclear and cytoplasmic extracts from 293T cells transfected with FL ANG-GFP were examined for GFP, a substantial amount of overexpressed ANG protein was detected both in the cytoplasm and in the nucleus (Fig. 3A, lanes 1 and 2). When the level of secreted ANG in the supernatants of 293T cells was measured by enzyme-linked immunosorbent assay (ELISA), a significant amount of endogenous ANG was detected (16,000 pg/ml; data not shown), which was comparable to the other transformed cells for which ANG levels have been shown to be highly upregulated (9, 49).
Fig 3.

KSHV LANA-1 and angiogenin interaction in overexpressed 293T cells. (A) Nuclear (nuc) and cytoplasmic (cyto) extracts were prepared from 293T cells transfected with full-length GFP tagged angiogenin (FL-ANG GFP), separated with a 10% gel, and subjected to Western blotting for GFP. The purity of the extracts was checked by Western blotting for β-actin and lamin. (B) U2OS cells were transfected with FL-ANG GFP plasmid and stained for GFP and DAPI after fixing and permeabilization. The white box is shown as an enlarged image. (C) U2OS cells transfected with ANG-GFP and LANA-1 plasmids were stained for ANG (green), LANA-1 (red), or DAPI (blue), and the images were merged. The white box is shown as an enlarged image. (D and E) 293T cells were transfected with pcDNA-GFP empty vector, FL-ANG GFP, or pcINeo full-length LANA-1 plasmids and subjected to IP with rabbit anti-LANA-1 IgG antibodies, separated with a 10% gel, and subjected to Western blotting with mouse anti-GFP monoclonal antibodies (D) or separated with a 12.5% gel and subjected to Western blotting with goat anti-ANG antibodies (E). (F) 293T cells transfected as described above were subjected to IP with rabbit anti-ANG antibodies, separated with a 7.5% gel, and subjected to Western blotting with rabbit anti-LANA-1 antibodies. Fiver percent input and β-actin loading controls are shown.
Although 293T cells are adherent cells, due to the presence of adenovirus E1A and E1B and simian virus 40 (SV40) T antigen, they lift up very easily from the slides and do not withstand the repeated washing that is required for immunofluorescence assays. Hence, we used p53+/+ adherent U2OS cells. ANG-GFP was also visualized in the nuclei of U2OS cells transfected with FL-ANG GFP (Fig. 3B). These results demonstrated that the GFP tag did not hinder the nuclear entry of overexpressed ANG. When LANA-1 and ANG were overexpressed in U2OS cells, both proteins were detected in the nucleus as well as in the cytoplasm, with several LANA-1 and ANG dots colocalizing mostly in the nucleus (Fig. 3C, yellow spots in the enlarged image).
To further confirm this interaction, 293T cells transfected with ANG and LANA-1 were subjected to IP with anti-LANA-1 antibodies and subjected to Western blotting for GFP. ANG-GFP immunoprecipitated with LANA-1 only in those cells where both proteins were overexpressed (Fig. 3D, lane 4). When LANA-1 immunoprecipitates were examined with anti-ANG antibodies detecting endogenous ANG, in addition to the cells overexpressing ANG and LANA-1 proteins (Fig. 3E, lane 4), ANG was also detected in cells where LANA-1 alone was overexpressed (Fig. 3E, lane 3). This demonstrated the interaction of LANA-1 with endogenous ANG. Similar results were observed when these lysates were subjected to IP with anti-ANG antibodies and Western blotted for LANA-1 (Fig. 3F, lanes 2 and 4), which further verified the interactions between ANG (endogenous and expressed) and LANA-1. These results demonstrated that the presence of the KSHV genome and/or other viral gene products are not required for LANA-1 and ANG interaction.
LANA-1, ANG, and p53 coelute in gel chromatography of BC-3 lysates.
To further characterize the LANA-1 and ANG interactions, latent uninduced BC-3 cell lysates were analyzed by gel filtration chromatography. As reported previously (39), LANA-1 eluted in fractions 2, 3, 4, and 5 with a molecular mass range of 300 to 669 kDa (Fig. 4A, top). ANG coeluted with LANA-1 and was detected in fractions 2, 3, and 4 (Fig. 4A, second gel). ANG was also detected in the first fraction, which is around 700 kDa. Since neomycin treatment and sh-ANG lentivirus transduction resulted in the death of KSHV-positive cells (36), we examined the distribution of p53 and Mdm2 in these fractions. Interestingly, p53, Mdm2, LANA-1, and ANG all coeluted in the 440- to 669-kDa fractions 2, 3, and 4 (Fig. 4A). LANA-1 and p53 also coeluted in fraction 5. ANG, Mdm2, and p53 were detected in the first fraction (700 kDa) without LANA-1. We also observed the presence of p53 in fractions 6 to 9 (Fig. 4A, third gel), suggesting the presence of p53 in complexes with other proteins, such as Mdm2 in fraction 9. The coelution of LANA-1, ANG, p53, and Mdm2 in the same fraction (fractions 2, 3, and 4) suggests that LANA-1-angiogenin could be in the same complex with p53 and Mdm2 to facilitate antiapoptosis in infected cells by blocking p53-mediated pathways.
Fig 4.

Gel chromatography for LANA-1, ANG, and p53 in KSHV-infected BC-3 cells. (A) Cell lysates from BC-3 cells were separated by Superdex 200 HR column gel filtration chromatography. Ten gel filtration fractions, including the whole-cell lysate (WCL), were immunoblotted for LANA-1 (7.5% gel), ANG (12.5% gel), and p53 (10%). (B and C) Nuclear extracts from BC-3 cells were subjected to IP with rabbit IgG and rabbit anti-angiogenin IgG antibodies, separated with a 7.5% gel, and subjected to Western blotting for LANA-1 (B) and for p53 (C). (D) Whole-cell lysates from 293T cells were subjected to IP as described above and subjected to Western blotting for p53. Input controls for p53 and ANG are shown. (E and F) Whole-cell lysates from SaOS-2 (p53 null) and U2OS (wild-type p53) cells were subjected to IP with ANG, separated with a 10% gel, and subjected to Western blotting for p53 (E) and Mdm2 (F). Input controls for p53, Mdm2, and β-actin loading controls are shown. (G) SaOS-2 and U2OS cells were transfected with pcDNA empty vector or full-length LANA-1 plasmids, subjected to IP with ANG, separated with a 7.5% gel, and subjected to Western blotting for LANA-1. Input control for p53 as well as LANA-1 and β-actin loading controls are shown.
To confirm the BC-3 cell gel filtration data, we performed IP reactions in BC-3 cell lysates with anti-ANG antibody or IgG control and performed Western blotting for LANA-1 and p53. Both LANA-1 (Fig. 4B, lane 2) and p53 (Fig. 4C, lane 2) were detected in ANG-IP reactions. To determine whether the ANG-p53 interaction occurs in uninfected cells, we performed IP reactions in 293T cells with anti-ANG antibody or IgG control and performed Western blotting for p53. We detected p53 in ANG immunoprecipitates (Fig. 4D, lane 2).
To further confirm this interaction, we used two additional cell lines, SaOS-2 (p53 null) and U2OS (wild-type p53), and performed IP reactions with anti-ANG antibodies. Both of these cell lines have high levels (1,300 pg/ml) of endogenous ANG (37). p53 was detected in ANG-IP reactions only in U2OS cells (Fig. 4E, lane 2). Mdm2 was detected in IP-Western blotting reactions of SaOS-2 and U2OS cell lysates with anti-ANG antibodies (Fig. 4F). However, a larger quantity was detected in p53+/+ U2OS cells than in p53−/− SaOS-2 cells. Since Mdm2 is also a target gene of p53, less of it was detected in SaOS-2 cells (Fig. 4F, lane 1). Hence, the lower level of interaction seen with ANG is most likely due to less input Mdm2. Taken together, these results suggested that ANG-p53 and ANG-Mdm2 interactions occur in uninfected cells independent of LANA-1 or KSHV infection.
To determine whether p53 is required for LANA-1 and ANG interaction, SaOS-2 and U2OS cells were transfected with empty vector or full-length LANA-1 plasmid. When cell lysates were subjected to IP with anti-ANG antibodies and then subjected to Western blotting, LANA-1 was detected in both SaOS-2 and U2OS cells transfected with LANA-1 (Fig. 4G, lanes 2 and 4). These results suggested that p53 is not required for LANA-1 and ANG interaction. A moderately higher level of LANA-1 detected in U2OS cells (Fig. 4G, input LANA-1, lanes 2 and 4) could be due to differences in the transfection efficiency of LANA-1 plasmid, as seen in the LANA-1 input blot.
Overall, these results demonstrated ANG-LANA-1 as well as ANG-p53 and ANG-Mdm2 interactions in the absence of LANA-1 and KSHV infection. Most transformed cells used for transfection contain high levels of ANG, and our studies demonstrate that LANA-1 not only induces ANG but also interacts with ANG. In light of these facts, the reported LANA-1 interaction with p53 in a LANA-1 and p53 overexpression system (12) could be via ANG-LANA-1 interaction, since the 14-kDa ANG protein was never looked for and was probably missed by the higher-percentage SDS-PAGE system used in the earlier studies.
LANA-1, ANG, and p53 colocalize in KSHV-infected cells.
To further evaluate these results, we carried out triple-colocalization IFA studies for ANG (green), LANA-1 (red), and p53 (blue) in TIVE-LTC cells. Since only about 20% of these cells are latently infected, they were used to observe both uninfected as well as latently infected cells. LANA-1 appeared as punctate nuclear staining, and ANG appeared as punctate nuclear dots only in LANA-1-positive cells (Fig. 5A and B). p53 was detected in LANA-1-positive cells as well as in negative uninfected cells (Fig. 5A). In LANA-1-positive cells, we observed the colocalization of LANA-1, ANG, and p53, as represented by white spots (Fig. 5B, white arrowheads). We also readily observed LANA-1 and ANG colocalization (yellow spots) without p53 (Fig. 5B, long white arrows). LANA-1 and p53 colocalization was also observed (Fig. 5B, image 5). We also observed colocalization between ANG and p53 in LANA-1-negative cells, which were more apprent only in the enlarged images, perhaps due to lower levels of ANG in the uninfected cells (Fig. 5A, enlarged red box). These results further suggested that ANG, p53, and LANA-1 could be present in the same complex.
Fig 5.

Immunofluorescence analyses of LANA-1, ANG, and p53 interactions and mapping of ANG domain interacting with LANA-1 and p53. (A) Permeabilized TIVE-LTC cells were stained for ANG (green), LANA-1 (red), and p53 (blue). The red-bordered box image from a LANA-1-negative cell is merged, and the enlarged image of p53 and ANG is shown on the left. Red arrows point to p53 and ANG colocalization. (B) The white-bordered box image from a LANA-1-positive cell was merged, and enlarged images of double and triple colocalization of LANA-1, ANG, and p53 are shown. White arrowheads indicate the triple colocalization of LANA-1, ANG, and p53 (white spots), while the long white arrows point to LANA-1 and ANG colocalization (yellow spots). A Venn diagram indicating the effects of different color combinations is provided to interpret double and triple colocalization results. 1, LANA-1 and ANG; 2, LANA-1 and p53; 3, LANA-1, ANG and p53; 4, Ang and p53. (C) Schematic diagram showing full-length (FL) angiogenin indicating the amino acid (aa) residues with reported important functions, nuclear localization signal area, and N- and C-terminal deletion constructs. (D) 293T cells were transfected with FL ANG-GFP or four C-terminal GFP-tagged ANG deletion constructs, and their expression was checked by Western blotting for GFP. (E) 293T cells were transfected with full-length LANA-1 along with the empty vector, full-length ANG, or four ANG deletion constructs. The lysates were subjected to IP with rabbit anti-LANA-1 IgG antibodies, separated with a 10% gel, and subjected to Western blotting for GFP. LANA-1 expression levels were checked by Western blotting for LANA-1 in the samples. (F) 293T cells were transfected with the empty vector, FL ANG-GFP, or four of its deletion constructs, and the lysates were subjected to IP with rabbit anti-p53 IgG antibodies, separated with a 10% gel, and subjected to Western blotting for GFP. Input control for p53 and β-actin loading controls are shown.
C-terminal region of ANG is involved in its interaction with LANA-1 and p53.
To reinforce the ANG-LANA-1 and ANG-p53 interaction results, we created four ANG deletion constructs with a C-terminal GFP tag (Fig. 5C). Full-length ANG protein consists of a nuclear localization signal (NLS; 31 to 35 amino acids [aa]), regions important for cell binding (60 to 68 aa and Asn109), and regions important for its ribonucleolytic activity (His13, Lys40, and His114) (Fig. 5C) (15). The expression of these deletion constructs in 293T cells was confirmed by Western blotting with anti-GFP antibodies (Fig. 5D). 293T cells were transfected with FL-LANA-1 plasmid along with the empty vector, FL ANG-GFP, or four ANG deletion constructs. These cell lysates were subjected to IP with anti-LANA-1 antibodies and Western blotted for GFP. The LANA-1-ANG interaction was observed even when the N-terminal 20 and 40 residues of ANG were deleted. In contrast, the LANA-1-ANG interaction was lost when 20 and 40 residues of ANG were deleted from the C terminus (Fig. 5E). Similarly, we transfected either the empty vector plasmid, FL ANG-GFP, or four ANG deletion constructs in 293T cells, immunoprecipitated the lysates with anti-p53 antibodies (endogenous p53), and subjected them to Western blotting for GFP. Similarly to the LANA-1 IP, we observed the loss of interaction between p53 and ANG when the C-terminal 20 and 40 residues of ANG were deleted (Fig. 5F). These data also suggested that the N-terminal 20 to 40 aa of ANG are moderately involved in ANG-p53 interaction. Overall, these results suggested that the C-terminal 104- to 123-amino-acid region of ANG plays a critical role in its interaction with both LANA-1 and p53.
Blocking angiogenin's nuclear transport by neomycin decreases nuclear LANA-1 levels, reduces ORF73, and upregulates ORF50.
We have shown previously that the aminoglycoside antibiotic neomycin, a known PLC-γ inhibitor, disrupts the nuclear translocation of ANG and ANG-induced cell proliferation, angiogenesis, and KSHV LANA-1 expression (19, 36). To determine the potential functional consequences of LANA-1-ANG interaction, BCBL-1 cells were either left untreated or were treated with 200 or 500 μM neomycin for 3 days, stained with ANG (green), LANA-1 (red), and DAPI (blue), and visualized under the fluorescence microscope. Compared to the untreated cells, where many LANA-1 and ANG dots colocalized, colocalization was reduced in neomycin-treated cells (Fig. 6A). The punctate nuclear staining of ANG was greatly reduced and the cytoplasmic staining was more prominent. We observed a neomycin dose-dependent reduction in LANA-1 dots, in which both the number of LANA-1-positive cells and the number of LANA-1 dots per cell were significantly decreased (Fig. 6B and C).
To determine whether there is a reduction in the number of KSHV genomes by neomycin treatment, ORF73 DNA copy numbers in BCBL-1 cells treated with neomycin were determined by real-time DNA PCR. We did observe a decrease in KSHV DNA copy numbers with 200 or 500 μM neomycin (Fig. 6D). However, since neomycin also induced the KSHV lytic cycle (22), a concomitant increase in KSHV genome copy numbers could be a reason for the absence of increased reduction in KSHV genome copy numbers. Nevertheless, these studies clearly indicated a critical role of nuclear ANG in regulating KSHV latency, probably by maintaining LANA-1 levels.
Blocking angiogenin nuclear transport affects ANG-LANA-1, LANA-1-p53, and ANG-p53 interactions, reduces ORF73, and upregulates ORF50.
Since nuclear ANG and LANA-1 were both affected by neomycin, we next determined whether their interactions with each other and with p53 are affected by neomycin treatment. Nuclear extracts from BCBL-1 cells treated with 200 μM neomycin for 3 days were subjected to IP with anti-ANG antibodies and subjected to Western blotting for LANA-1 (Fig. 7A). Compared to untreated cells, neomycin-treated cells had reduced LANA-1 levels (the input control was 88% less than that for untreated samples) and showed reduced interactions with ANG.
When these samples were subjected to IP with anti-LANA-1 antibodies and Western blotting for p53 (Fig. 7B), a reduction in LANA-1 and p53 interaction was observed in neomycin-treated samples along with a reduction in nuclear ANG levels (the input control was 60% less than that for untreated samples), while p53 levels were comparable in both samples. When subjected to IP with anti-ANG antibodies and Western blotting for p53, a reduction in ANG and p53 interaction was observed in neomycin-treated samples (Fig. 7C). These results suggested that ANG, LANA-1, and p53 are in the same complex, and the decrease in viability seen upon neomycin treatment in BCBL-1 cells could have been preceded by the disruption of ANG-p53, LANA-1-p53, and LANA-1-ANG interactions and the activation of p53. The detection of decreased interactions is probably due to a reduction in nuclear ANG and LANA-1 as opposed to neomycin's interference with the interactions among these proteins. ORF73 (LANA-1) gene expression was reduced by about 40 and 60% with 200 and 500 μM neomycin treatment, respectively (Fig. 7D). As reported earlier (36), there was a concomitant increase in lytic ORF50 gene expression (Fig. 7E). These results suggested that nuclear ANG is necessary to maintain LANA-1 levels in BCBL-1 cells and that the interaction between LANA-1 and ANG could be critical to maintaining latency in KSHV-infected cells.
Since neomycin treatment resulted not only in the observed decreased interactions between ANG-p53 and LANA-1-p53 but also in the death of KSHV-positive cells (36), we hypothesized that neomycin treatment releases p53 from negative regulation by LANA-1 and ANG, resulting in the functional activation of p53. To test this, we treated BJAB and BCBL-1 cells with 200 and 500 μM neomycin and determined the transcription levels of p53 and its target proapoptotic gene p21 by real-time RT-PCR. There were no differences between untreated and treated samples in BJAB cells (Fig. 7F and G). In contrast, after 3 days of neomycin treatment, we observed significant increases in both p53 (about 3- and 4-fold) and p21 (about 2.5- and 4-fold) transcript levels in BCBL-1 cells (Fig. 7H and I).
Neomycin treatment upregulates p53 and p21 protein levels in BCBL-1 and BC-3 cells, but not in BJAB cells, at earlier time points.
Neomycin treatment decreased p53-LANA-1 and p53-ANG interactions (Fig. 7). Since LANA-1 has been shown to bind and inactivate p53 (12), the observed increase in p53 and p21 levels could be due to decreased LANA-1 levels. Therefore, we determined the p53 and p21 protein levels at earlier time points. LANA-1 is a stable protein with a half-life of 24 h (23); hence, its levels were not affected by neomycin at earlier time points, as demonstrated in Fig. 8B (BCBL-1) and C (BC-3). We treated BJAB, BCBL-1, and BC-3 cells with 200 μM neomycin and subjected them to Western blotting for p53 and p21 proteins at 4, 6, 8, and 24 h posttreatment. Although a slight increase in p53 and p21 levels was observed in BJAB cells at 4 h after neomycin treatment, their levels were comparable to that of the untreated sample by 24 h (Fig. 8A). However, compared to that of the untreated BCBL-1 cells, p53 protein levels in neomycin-treated cells increased by 2.8-, 3.6-, 2.5-, and 2.9-fold, and p21 protein levels increased by 1.8-, 2.2-, 2.6-, and 3.2-fold, respectively (Fig. 8B). Since BCBL-1 cells have one allele of p53 mutated, we repeated this experiment in BC-3 cells that contain wild-type p53 (32). Neomycin treatment induced p53 by 2.6-, 1.3-, 2.3-, and 3.3-fold and p21 by 1.5-, 1.3-, 2.5-, and 3.2-fold (Fig. 8C) after 4, 6, 8, and 24 h posttreatment, respectively, compared to the untreated samples. These results suggested that p53 pathways are activated at earlier time points due to the inhibition of nuclear translocation of ANG, and perhaps the loss of LANA-1 could further activate p53 at later time points. This also explains the lack of neomycin effects on KSHV-negative cell lines.
Fig 8.

Early time point effects of neomycin treatment on p53, p21, and LANA-1 protein levels. BJAB cells (A), BCBL-1 cells (B), and BC-3 cells (C) were treated with 200 μM neomycin for 4, 6, 8, or 24 h or were left untreated, and cell lysates were subjected to Western blotting for p53 (10% gel), p21 (12.5% gel), LANA-1 (7.5% gel), and β-actin proteins.
Neomycin treatment causes induction and redistribution of p53 even at earlier time points.
Since functions of p53 are also dictated by its localization within the cell, we analyzed how neomycin treatment affected p53 distribution. Compared to the untreated BCBL-1 cell population, we observed an increased level of p53 spots in neomycin-treated cells (Fig. 9). More interestingly, in addition to the nucleus, p53 dots were detected in increasing quantities in the perinuclear and cytoplasmic regions as well (Fig. 9). These results demonstrated that blocking the nuclear transport of ANG results in the release of p53 from negative regulation by LANA-1 and ANG in the nucleus, resulting in the induction and redistribution of p53.
Fig 9.

Distribution of p53 upon neomycin treatment. BCBL-1 cells were either left untreated or were treated with 200 μM neomycin for 4, 8, and 24 h, fixed with acetone, and stained for p53 (red) and for nuclei (DAPI; blue). White boxes are shown as enlarged images.
Angiogenin provides antiapoptotic advantages to KSHV-infected cells.
Antiapoptosis is a prerequisite for cell growth and proliferation. Since inhibiting nuclear ANG with neomycin induced p53 and p21 levels, we next determined whether the cells that have increased ANG are able to survive better and suppress p53 functions. Apoptotic signals induce the cleavage of procaspase 3 into the active form (caspase 3), which acts downstream on the execution phase to trigger apoptosis. For the cleaved caspase 3 assay, we serum starved (SS) HMVEC-d cells for 8 h to induce apoptosis and either left them untreated and serum starved for another 24 h, treated them with 1 μg/ml of purified ANG protein, infected them with KSHV (20 DNA copies/cell), or pretreated the cells with neomycin for 1 h before carrying out the treatments described above. Both ANG-treated and KSHV-infected cells showed negligible cleaved caspase 3 staining, while the cells that were serum starved or pretreated with neomycin before ANG and KSHV infection showed distinct staining for cleaved caspase 3 in >50% of the cells (Fig. 10A). To quantify these results, we used Western blot assays for caspase 3 with an antibody that detects both the full-length and cleaved form of caspase 3. The cleavage of caspase 3 was observed in serum-starved cells and in cells pretreated with neomycin before ANG and KSHV infection (Fig. 10B, lanes 1, 3, and 5), while no cleaved caspase 3 was detected in ANG-treated, KSHV-infected cells and cells grown in full medium (Fig. 10B, lanes 2, 4, and 6). These observations verified our hypothesis that the survival advantage bestowed upon infected cells by KSHV is in part mediated by ANG.
Fig 10.

Apoptotic effects of neomycin and antiapoptotic effects of angiogenin. (A) Serum-starved (8 h) HMVEC-d cells in 8-well chamber slides were either left untreated, treated with 1 μg/ml ANG, infected with 20 KSHV DNA/cell, or pretreated with 200 μM neomycin (N) for 1 h and then infected with the virus or treated with 1 μg/ml ANG. At 24 h, the slides were stained with anti-cleaved caspase-3 antibody and visualized under the fluorescence microscope. (B) HMVEC-d cells were treated similarly, and the samples were separated with a 10% gel and subjected to Western blotting for cleaved caspase-3.
Silencing angiogenin has effects similar to neomycin treatment in KSHV-positive BCBL-1 cells.
Neomycin has been shown to block PLCγ and AKT phosphorylation in PEL cells, which are essential signaling pathways for survival (36). It can therefore be argued that the results seen with the neomycin treatment are due to its effects on the signaling pathways. To directly implicate ANG in the survival processes, we used lentivirus-based shRNA targeting ANG and shRNA targeting GFP as a control. We have previously shown the ANG mRNA and protein level knockdown efficacies of these lentiviruses (36). When we tested the efficiency of knockdown in 293T cells by Western blotting for ANG, sh-ANG1 showed appreciable knockdown (Fig. 11A), and shANG1 was used in all subsequent studies. We tested ANG knockdown using sh-ANG1 in BCBL-1 cells by real-time RT-PCR. ANG transcripts were reduced by about 50% (Fig. 11B).
Compared to the control cells, p53 was induced by about 3-fold with sh-ANG1 (Fig. 11C, top, lane 2). The phosphorylation of p53 at serine 15 impairs the ability of Mdm2 to bind p53 in response to DNA damage, and p53 is stabilized to control apoptotic and cell cycle regulation functions (41). Compared to the control cells, p53 phosphorylation at serine 15 was induced by 3.6-fold (Fig. 11C, lane 2) in sh-ANG1-transduced cells. This also suggested that ANG blocks p53 phosphorylation at serine 15. In addition, we also observed the upregulation of proapoptotic protein targets of p53, such as p21 and Bax, by 2- and 2.7-fold, respectively (Fig. 11C and D, lanes 2). In contrast, the antiapoptotic protein Bcl-2 was downregulated (Fig. 11D, top). Similar to neomycin-treated cells, we also observed an increased level of cleaved caspase-3 in ANG knockdown cells (Fig. 11E).
The induction and activation of p53, its proapoptotic gene targets, and the downregulation of its antiapoptotic target protein are an indication that the cells are undergoing apoptosis. To quantitatively determine the percentage of dead cells with sh-ANG, we stained BCBL-1 cells transduced with sh-GFP or sh-ANG with YO-PRO-1 and propidium iodide nucleic acid stains. We then analyzed them by FACS. YO-PRO-1 stain selectively passes through the plasma membranes of apoptotic cells and labels them with moderate green fluorescence. Necrotic cells exhibit red fluorescence with propidium iodide. We gated for viable cells and found that compared to sh-GFP control cells (represented as 100% viability), sh-ANG cell viability was decreased by about 60%. The averages from three experiments at 3 days after sh-ANG transduction are shown in Fig. 11F. These results clearly suggested that ANG provides survival advantages to KSHV-infected cells.
DISCUSSION
Our comprehensive studies presented here demonstrate that KSHV LANA-1 interacts with ANG in de novo infected endothelial cells and in latently infected B cells, and the presence of the KSHV genome and other viral proteins is not required for the interaction. The ability of herpesviruses to establish latency and exist for life in the host is due to the successful manipulation of host cell machinery to their advantage, hence any host protein that these viruses induce or interact with is not random or a coincidence. Viruses have evolved to target specific molecules and modulate their functions to facilitate their survival, and KSHV is no exception (14, 30). LANA-1 is one of KSHV's primary latency-associated proteins that is employed to interact with many host proteins to manipulate their functions (24). Our earlier studies showed that LANA-1 expression induces ANG, which plays roles in angiogenesis, 45S rRNA synthesis, and cell proliferation (35). Blocking ANG's nuclear translocation with neomycin or knocking down ANG resulted in the selective death of KSHV-infected cells, while uninfected or EBV+ cells were not affected (35, 36). However, the mechanisms behind the reduced viability and the relationship between LANA-1 and ANG were largely unexplored.
Our very recent study (31) involving the mass spectrometric analysis of LANA-1- and ANG-immunoprecipitated samples from TIVE-LTC cells identified 28 cellular proteins that were immunoprecipitated by both proteins, which implied that LANA-1 and ANG are present in a complex with other proteins. This is supported by the present study, which demonstrates the presence of ANG and LANA-1 in the same high-molecular-weight complex along with p53 and Mdm2 in the gel filtration analysis of BC-3 cells. It is interesting that LANA-1, p53, and Mdm2 have been shown to be in the same high-molecular-weight fractions of the BC-3 lysates (39). The detection of ANG in these fractions suggests that ANG is in that complex and helps to mediate crucial functions. This is also supported by the detection of LANA-1-p53-ANG complexes by IFA in TIVE-LTC cells. Although IFA images do not definitively indicate interaction, the observed colocalization suggested that LANA-1, ANG, and p53 are in close proximity to each other. Almost all of the ANG dots appeared to be tied up with LANA-1, some of which also colocalized with p53. ANG and LANA-1 interacted in LANA-1-transfected SaOS-2 cells that are devoid of p53, indicating that p53 is not required for LANA-1-ANG interaction and confirming the IFA results. ANG-p53 colocalization was readily seen in LANA-1-positive cells due to the induction of the ANG gene by LANA-1. Although the detection of ANG was weaker in LANA-1-negative cells, we could still observe some level of colocalization between ANG and p53. This was also supported by the detection of ANG and p53 without LANA-1 in the first fraction of the BC-3 cell lysate gel filtrates. Furthermore, p53 was pulled down by ANG in U2OS cells that contain wild-type p53 but not in SaOS-2 cells that do not have p53. Since Mdm2 is a target gene of p53, less Mdm2 protein was detected in SaOS-2 cells, and it was pulled down more by ANG in U2OS cells. These results confirm the BC-3 gel filtration data and also suggest that these complexes are present in uninfected cells.
The detection of LANA-1 and p53 together in the fifth fraction without ANG suggests the presence of different combinations of complexes. It is possible that the difference in antibody sensitivity also affected the detection of these proteins in different fractions. Nevertheless, the presence of LANA-1, ANG, p53, and Mdm2 in the same complex in KSHV-infected endothelial cells and PEL cells suggests that ANG is mediating the LANA-1-p53 interaction and/or directly interacting with p53 to suppress its functions. A previous study showing LANA-1-p53 interaction was done in SaOS-2 cells by overexpressing LANA-1 and p53 (12). Since these cells have high levels of endogenous ANG, the LANA-1-p53 interaction detected could have been mediated by ANG. The observation that the deletion of aa 104 to 123 in the C-terminal region of ANG resulted in the loss of interaction with both LANA-1 and p53 further strengthens this hypothesis.
The inhibition of ANG nuclear transport by neomycin in BCBL-1 cells resulted in reduced ANG and LANA-1, ANG and p53, as well as LANA-1 and p53 interactions, which is most likely due to a decrease in nuclear ANG and LANA-1 as opposed to interference with the interactions among these proteins. The inhibition of the ORF73 gene and the induction of the ORF50 gene in neomycin-treated BCBL-1 cells and sh-ANG-treated cells after 3 days (22) suggest that this is a consequence of reduced ANG and LANA-1, ANG and p53, and LANA-1 and p53 interactions. The observed upregulation of p53 and its target gene p21 at the same time point in BABL-1 cells but not in BJAB cells suggests that the decrease in ANG and LANA-1, ANG and p53, and LANA-1 and p53 interactions, the reduction of LANA-1 and ANG, and the induction of ORF50 result in the functional activation of p53. Since LANA-1 is a stable protein (36), neomycin did not affect its levels at earlier time points. Therefore, the induction of p53 and p21 proteins seen at earlier time points of neomycin treatment in BCBL-1 and BC-3 cells is probably due to the inhibition of the PLCγ pathway and nuclear translocation of ANG. BC-3 cells contain wild-type p53, while BCBL-1 cells have one mutated p53 allele and are more resistant to treatment with drugs such as nutlin and doxorubicin (32, 39). The ability of neomycin to effectively induce p53 and p21 in both cell lines suggests that neomycin can be used to treat nutlin- and doxorubicin-resistant PEL cases.
Interestingly, p53 and p21 levels in the KSHV− lymphoma cell line BJAB were not induced by neomycin. These results support our earlier observations that 200 μM neomycin treatment results in a reduction in BCBL-1 cell survival, while various KSHV− B-cell lines (Akata, Ramos, Loukes, BJAB, LCL, and Raji) showed no reduction in cell survival even at higher concentrations (36). Most of these cell lines contain mutations in one or both alleles of p53 as well as very low levels of ANG (11). Hence, the activation of p53 pathways by neomycin only in KSHV+ cells regardless of p53 mutation status (BCBL-1 with mutated p53 and BC-3 with wild-type p53) could be one of the explanations for the observed reduction in viability. This also suggests that the level of angiogenin correlates better with neomycin sensitivity than the p53 mutation status. The increased detection of p53 spots in the perinuclear and cytoplasmic regions after neomycin treatment is corroborated by the detection of comparable amounts of p53 in neomycin-treated and untreated nuclear fractions of BCBL-1 cells. The main function of p53 is in the nucleus, where it acts as a nuclear transcription factor regulating genes involved in apoptosis, cell cycle regulation, and numerous other processes. However, there is an accumulating body of research that has unraveled important functions of p53 in the cytoplasm, where it triggers apoptosis and inhibits autophagy (16). By their interactions, LANA-1 and ANG most likely sequester p53 in the nucleus in an inactive form. Knocking down ANG also induced p53 and p21 levels, which suggests that the induction seen with neomycin is not due to any nonspecific effects.
The ability of KSHV to confer survival advantages to the starved, infected HMVEC-d cells is clearly demonstrated by Western blotting and staining reactions for cleaved caspase 3. This could be mediated in part by ANG, as demonstrated by the near absence of caspase 3 activation in ANG-treated, uninfected, serum-starved cells and its abolition and cell death by pretreatment with neomycin. The antiapoptotic role of ANG in BCBL-1 cells was also apparent by the increased phosphorylation of p53 at serine 15, indicating p53 activation, and also by the upregulation of proapoptotic proteins p21 and Bax, the downregulation of antiapoptotic protein Bcl-2, and the induction of cell death upon ANG knockdown. These results show that ANG provides prosurvival signals possibly through p53-mediated pathways, and this function is exploited by KSHV to maintain the survival of infected cells and its latency. Studies in neuronal cells have demonstrated that ANG inhibits the nuclear translocation of apoptosis-inducing factor (AIF), attenuates a series of Bcl-2-dependent events such as caspase 3 activation and poly(ADP-ribose) polymerase-1 (PARP-1) cleavage, and mediates neuroprotective functions (25). ANG treatment upregulated the expression of both the mRNA and protein of NF-κB in embryonal carcinoma P19 cells (26). The role of NF-κB in cell survival and the latency maintenance of KSHV is well recognized (17, 48). Thus, ANG could also be mediating its prosurvival effects via NF-κB pathways, which need to be studied further.
It is apparent that the diverse roles of angiogenin in signal transduction, angiogenesis, cell proliferation, and survival that are of high significance to proliferating cells have been hijacked by KSHV. It is possible that in KSHV-infected cells, LANA-1 inserts itself into ANG-p53 complexes. ANG and p53 interacted not only in BC-3 cells but also in KSHV− 293T cells and U2OS cells. ANG could be complexing with p53 in uninfected subconfluent and semiconfluent endothelial cells to suppress p53 functions and thereby enhance 45S rRNA synthesis, cell survival, proliferation, and endothelial cell tube formation. Our recent studies show that ANG forms complexes with p53 in uninfected HMVEC-d cells, 293T cells, and several adherent cancer cell lines (37). In uninfected cells expressing ANG and p53, ANG-p53 interaction was detected mainly in the nucleus. The increased expression of ANG in p53-negative SaOS-2 cells transfected with p53 expression plasmid resulted in the inhibition of p53 serine phosphorylation, a higher level of interaction between Mdm2 and p53, and the increased ubiquitination of p53. ANG expression also inhibited p53-dependent p53 promoter activity. These findings in uninfected cells together with our results in KSHV-positive cells suggest that KSHV, by inducing ANG expression and secretion and by interacting with it via LANA-1, has developed another avenue to suppress p53 functions while also taking advantage of ANG's functions in maintaining its latency and other activities. Since neomycin treatment resulted in decreased nuclear LANA-1, the observed LANA-1-ANG interaction could function to stabilize LANA-1 levels in the nucleus. The reduction in cell survival seen upon neomycin or sh-ANG treatment is most likely due to a combination of various events, such as (i) the inhibition of PLCγ-AKT pathways, (ii) reduction of LANA-1 and activation of RTA, and (iii) decreases in LANA-1-ANG, LANA-1-p53, and ANG-p53 interactions and subsequent activation of p53. Since p53 is a central checkpoint protein, it is not surprising that KSHV utilizes multiple, overlapping avenues, such as those via LANA-1, v-FLIP, NF-κB, etc., to modulate its functions, and angiogenin could be yet another host protein that is induced to increase interaction with LANA-1 and p53 to help repress p53's functions. A summary of our findings have been presented as a schematic model that shows the events in B cells latently infected with KSHV in the context of ANG and the consequences of silencing angiogenin or inhibiting PLCγ activation by neomycin (Fig. 12).
Fig 12.

Schematic model showing the events occurring in B cells latently infected with KSHV in the context of angiogenin and the consequences of silencing angiogenin or inhibiting PLCγ activation by neomycin. In BCBL-1 cells, LANA-1 expression induces ANG, which activates the PLCγ, ERK, and AKT pathways. PLCγ activation is required for the nuclear translocation of ANG. The present study shows that in the nucleus, LANA-1 interacts with ANG and p53 and forms a complex. There are also other complexes consisting of ANG-p53, LANA-1-p53, and LANA-1-ANG. These interactions suppress p53 functions, leading to enhanced cell survival and latency maintenance. When ANG is silenced or its nuclear transport via PLCγ activation is inhibited by neomycin treatment, AKT signaling is decreased while the ERK pathway is unaffected. ANG's nuclear translocation is inhibited, which results in decreased LANA-1 expression; decreased interactions between LANA-1-ANG-p53, LANA-1-p53, ANG-p53, and LANA-1-ANG, leading to the activation of p53; and increased detection of p53 free of ANG and LANA-1 in the cytoplasm, resulting in the induction of apoptosis, KSHV lytic cycle, and cell death.
Since angiogenin is selectively upregulated in cancer cells and B cells with KSHV infection, neomycin treatment provides target specificity that spares normal cells, which could be used to target p53 functions as well. Although neomycin is oto- and nephrotoxic, its derivative, neamine (18), does not possess these side effects and is safe. Studies are under way to test the efficacy of neomycin and neamine in animal models. In recent years, targeting angiogenesis for cancer treatment has been a prevailing concept, and our data showing the interaction and regulation of p53 functions by an angiogenic molecule that is also a target for viral manipulation sheds new light on angiogenic proteins and cancer treatment. Hence, ANG appears to be an exciting molecular target, and using FDA-approved neomycin/neamine or other alternatives could lead to novel therapeutic solutions for KSHV-associated malignancies (Fig. 12).
ACKNOWLEDGMENTS
This study was supported in part by Public Health Service grant R56 AI091767 and the RFUMS-H. M. Bligh Cancer Research Fund to B.C.
We thank Keith Philibert for critically reading the manuscript and Bob Dickinson for FACS analysis at the RFUMS core facility. We also thank Rolf Rene for his generous gifts of TIVE and TIVE-LTC cells.
Footnotes
Published ahead of print 21 March 2012
REFERENCES
- 1. Akula SM, Pramod NP, Wang FZ, Chandran B. 2002. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407–419 [DOI] [PubMed] [Google Scholar]
- 2. Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644 [DOI] [PubMed] [Google Scholar]
- 3. Cai X, et al. 2005. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:5570–5575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 5. Chang Y, et al. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 6. Chen W, Dittmer DP. 2011. Ribosomal protein S6 interacts with the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 85:9495–9505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cotter MA, II, Robertson ES. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254–264 [DOI] [PubMed] [Google Scholar]
- 8. Dull T, et al. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eppenberger U, et al. 1998. Markers of tumor angiogenesis and proteolysis independently define high- and low-risk subsets of node-negative breast cancer patients. J. Clin. Oncol. 16:3129–3136 [DOI] [PubMed] [Google Scholar]
- 10. Fakhari FD, Dittmer DP. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213–6223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farrell PJ, Allan GJ, Shanahan F, Vousden KH, Crook T. 1991. p53 is frequently mutated in Burkitt's lymphoma cell lines. EMBO J. 10:2879–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friborg J, Jr, Kong W, Hottiger MO, Nabel GJ. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889–894 [DOI] [PubMed] [Google Scholar]
- 13. Fujimuro M, et al. 2003. A novel viral mechanism for dysregulation of beta-catenin in Kaposi's sarcoma-associated herpesvirus latency. Nat. Med. 9:300–306 [DOI] [PubMed] [Google Scholar]
- 14. Ganem D. 2007. Kaposi's sarcoma-associated herpesvirus, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 15. Gao X, Xu Z. 2008. Mechanisms of action of angiogenin. Acta Biochim. Biophys. Sin. 40:619–624 [DOI] [PubMed] [Google Scholar]
- 16. Green DR, Kroemer G. 2009. Cytoplasmic functions of the tumour suppressor p53. Nature 458:1127–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guasparri I, Keller SA, Cesarman E. 2004. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 199:993–1003 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18. Hirukawa S, Olson KA, Tsuji T, Hu GF. 2005. Neamine inhibits xenografic human tumor growth and angiogenesis in athymic mice. Clin. Cancer Res. 11:8745–8752 [DOI] [PubMed] [Google Scholar]
- 19. Hu GF. 1998. Neomycin inhibits angiogenin-induced angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 95:9791–9795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaul R, Verma SC, Robertson ES. 2007. Protein complexes associated with the Kaposi's sarcoma-associated herpesvirus-encoded LANA. Virology 364:317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kishimoto K, Liu S, Tsuji T, Olson KA, Hu GF. 2005. Endogenous angiogenin in endothelial cells is a general requirement for cell proliferation and angiogenesis. Oncogene 24:445–456 [DOI] [PubMed] [Google Scholar]
- 22. Krishnan HH, et al. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwun HJ, et al. 2007. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J. Virol. 81:8225–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lan K, Kuppers DA, Verma SC, Robertson ES. 2004. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J. Virol. 78:6585–6594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li S, Yu W, Hu GF. 2011. Angiogenin inhibits nuclear translocation of apoptosis inducing factor in a Bcl-2-dependent manner. J. Cell. Physiol. 227:1639–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li S, Yu W, Kishikawa H, Hu GF. 2010. Angiogenin prevents serum withdrawal-induced apoptosis of P19 embryonal carcinoma cells. FEBS J. 277:3575–3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. 1998. Development of a self-inactivating lentivirus vector. J. Virol. 72:8150–8157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moroianu J, Riordan JF. 1994. Nuclear translocation of angiogenic proteins in endothelial cells: an essential step in angiogenesis. Biochemistry 33:12535–12539 [DOI] [PubMed] [Google Scholar]
- 29. Naranatt PP, Akula SM, Zien CA, Krishnan HH, Chandran B. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J. Virol. 77:1524–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Naranatt PP, et al. 2004. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 64:72–84 [DOI] [PubMed] [Google Scholar]
- 31. Paudel N, Sadagopan S, Balasubramanian S, Chandran B. 2011. Kaposi's sarcoma associated herpesvirus latency associated nuclear antigen and angiogenin interact with common host proteins including annexin A2, which is essential for survival of latently infected cells. J. Virol. 86:1589–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Petre CE, Sin SH, Dittmer DP. 2007. Functional p53 signaling in Kaposi's sarcoma-associated herpesvirus lymphomas: implications for therapy. J. Virol. 81:1912–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Radkov SA, Kellam P, Boshoff C. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6:1121–1127 [DOI] [PubMed] [Google Scholar]
- 34. Renne R, et al. 2001. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:458–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sadagopan S, et al. 2009. Kaposi's sarcoma-associated herpesvirus upregulates angiogenin during infection of human dermal microvascular endothelial cells, which induces 45S rRNA synthesis, antiapoptosis, cell proliferation, migration, and angiogenesis. J. Virol. 83:3342–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sadagopan S, Valiya Veettil M, Paudel N, Bottero V, Chandran B. 2011. Kaposi's sarcoma-associated herpesvirus-induced angiogenin plays roles in latency via the phospholipase C gamma pathway: blocking angiogenin inhibits latent gene expression and induces the lytic cycle. J. Virol. 85:2666–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sadagopan S, et al. 23 January2012. Angiogenin functionally interacts with p53 and regulates p53-mediated apoptosis and cell survival. Oncogene [Epub ahead of print.] doi:10.1038/onc.2011.648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sarek G, Jarviluoma A, Ojala PM. 2006. KSHV viral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood 107:725–732 [DOI] [PubMed] [Google Scholar]
- 39. Sarek G, et al. 2007. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J. Clin. Investig. 117:1019–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharma-Walia N, et al. 2010. Kaposi's sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog. 6:e1000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shieh SY, Ikeda M, Taya Y, Prives C. 1997. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91:325–334 [DOI] [PubMed] [Google Scholar]
- 42. Sivakumar R, et al. 2008. Kaposi's sarcoma-associated herpesvirus induces sustained levels of vascular endothelial growth factors A and C early during in vitro infection of human microvascular dermal endothelial cells: biological implications. J. Virol. 82:1759–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. St. Clair DK, Rybak SM, Riordan JF, Vallee BL. 1988. Angiogenin abolishes cell-free protein synthesis by specific ribonucleolytic inactivation of 40S ribosomes. Biochemistry 27:7263–7268 [DOI] [PubMed] [Google Scholar]
- 44. Szekely L, et al. 1999. Human herpesvirus-8-encoded LNA-1 accumulates in heterochromatin-associated nuclear bodies. J. Gen. Virol. 80:2889–2900 [DOI] [PubMed] [Google Scholar]
- 45. Vart RJ, et al. 2007. Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoietin-2 expression in lymphatic endothelial cells. Cancer Res. 67:4042–4051 [DOI] [PubMed] [Google Scholar]
- 46. Vrancken K, Vervaeke P, Balzarini J, Liekens S. 2011. Viruses as key regulators of angiogenesis. Rev. Med. Virol. 21:181–200 [DOI] [PubMed] [Google Scholar]
- 47. Xu ZP, Tsuji T, Riordan JF, Hu GF. 2003. Identification and characterization of an angiogenin-binding DNA sequence that stimulates luciferase reporter gene expression. Biochemistry 42:121–128 [DOI] [PubMed] [Google Scholar]
- 48. Ye FC, et al. 2008. Kaposi's sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-kappaB-mediated suppression of the AP-1 pathway: a novel mechanism of virus control of latency. J. Virol. 82:4235–4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yoshioka N, Wang L, Kishimoto K, Tsuji T, Hu GF. 2006. A therapeutic target for prostate cancer based on angiogenin-stimulated angiogenesis and cancer cell proliferation. Proc. Natl. Acad. Sci. U. S. A. 103:14519–14524 [DOI] [PMC free article] [PubMed] [Google Scholar]