

Abstract
The human APOBEC3 family consists of seven cytidine deaminases (A3A to A3H), some of which display potent antiretroviral activity against HIV-1 and other retroviruses. Studies that analyzed the effect of A3G on human T-lymphotropic virus type 1 (HTLV-1) infectivity resulted in conflicting findings, and our knowledge of HTLV-1 restriction by other A3 proteins remains limited. Since HTLV-1, much like HIV, targets CD4+ T cells, we hypothesized that A3 proteins other than A3G restrict HTLV-1. All seven human A3 proteins were tested in HTLV-1 reporter and HIV-1 infectivity assays. We show that A3A, A3B, and A3H haplotype 2 (A3H hapII) acted as potent inhibitors of HTLV-1. Wild-type HIV-1, in contrast, was restricted by A3B and A3H hapII, but not by A3A. Catalytic site mutants of A3A, A3B, and A3H hapII showed that A3A and A3B restriction of HTLV-1 required deaminase activity. However, A3H hapII acted in a deaminase-independent manner when restricting HTLV-1, while requiring deaminase activity for HIV-1 restriction. We also analyzed A3 editing of HTLV-1 in five T-cell lines obtained from HTLV-1-infected patients. These cell lines contained extensively edited HTLV-1 sequences with G-to-A mutations in dinucleotide contexts suggestive of APOBEC3 mutagenesis. Comparison of the A3-induced mutations from reporter cells and the patient-derived cell lines indicate that A3G but also other A3 members, possibly A3A and A3B, affect HTLV-1 in vivo. Taken together, our data indicate that HTLV-1 is a likely target for multiple A3 proteins.
INTRODUCTION
The deltaretrovirus human T-lymphotropic virus type 1 (HTLV-1) was the very first human retrovirus discovered (67). The results of phylogenetic analyses indicate that HTLV-1 was introduced into human populations thousands of years ago (39, 84). This long interaction between HTLV-1 and humans stands in contrast with HIV-1, which is estimated to have crossed the species barrier only a century ago (92).
Estimations suggest that approximately 15 to 20 million people are infected with HTLV-1 worldwide (15, 24). HTLV-1 infections are endemic in Southern Japan, sub-Saharan Africa, parts of South America, and the Caribbean islands (reviewed in reference 68). HTLV-1 transmission occurs through mother-to-child transmission (breast milk), sexual transmission, blood transfusion, and intravenous drug use (37, 38, 71). Adult T-cell leukemia/lymphoma (ATLL) develops in up to 5% of HTLV-1-positive individuals (47), and in an additional 1 to 2% of these individuals, HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) is observed (18, 29). HTLV-1 primarily targets CD4+ T lymphocytes, and the spread of infection takes place by cell-to-cell transmission through virological synapses, extracellular viral assemblies, or cellular conduits (27, 42, 53, 63, 85). Upon primary infection, only limited productive replication takes place, but HTLV-1 multiplies by enhancing the proliferation of infected cells (47, 83). As a result, HTLV-1 sequence diversity is extremely low compared to HIV-1.
There is currently no cure for HTLV-1 infection, and the lack of efficient antiviral drugs for HTLV-1 offers limited management options. Furthermore, the use of nucleoside analogues in HTLV-1 therapy is subjected to criticism because HTLV-1 infection in vivo is accompanied by limited viremia (8).
The human cytidine deaminase family of apolipoprotein B mRNA editing enzyme catalytic polypeptide 3 (APOBEC3, A3) consists of seven proteins (A3A, A3B, A3C, A3D, A3F, A3G, and A3H) that can inhibit retroviruses, endogenous retroelements, and DNA viruses to different degrees (10, 55, 76, 80, 86). The human A3 locus on chromosome 22 is polymorphic on a genome level with a large structural variation deleting the entire A3B coding region as well as several single-nucleotide polymorphisms in introns and exons of each cytidine deaminase (2, 32, 70). Haplotypes with distinct antiviral phenotypes have been reported only for A3H: A3H haplotype 2 (A3H hapII) confers strong anti-HIV-1 activity, while most other A3H variants are unstable proteins that lack inhibitory activity (12, 22, 58).
The enzymatic activity of virion-packaged A3 molecules results in deamination of cytosine bases in the single-stranded viral DNA during reverse transcription, which leads to guanosine (G)-to-adenosine (A) mutations in the provirus (4, 23, 36, 43, 45). Of note, A3 proteins have also been shown to restrict through deaminase-independent mechanisms by reducing reverse transcription products and integration (3, 25, 28, 49, 50, 57). The accessory HIV-1 protein Vif counteracts the restriction of several A3 proteins (e.g., A3F and A3G) by mediating its proteasomal degradation in the producer cell (11, 46, 51, 77, 93) or by preventing their packaging into virions (21, 31).
HTLV-1 encounters A3 proteins in vivo as it replicates in the same CD4+ T cells as HIV-1, but it lacks a functional Vif to counteract them (42). Yet, several groups reported that wild-type HTLV-1 is largely resistant to A3G restriction in cell culture assays (41, 56, 59). Only one group reported that HTLV-1 is mildly sensitive to A3G and that its catalytic deaminase activity was dispensable for restriction (74). Moreover, proviruses with multiple G-to-A mutations suggestive of A3 action are frequently detected in HIV-1-infected individuals (30, 33), but HTLV-1 proviruses carrying footprints of past deamination are rarely found in cell culture or HTLV-1-infected patients. In the few cases in which G-to-A mutations have been found, they have been attributed to A3G activity based on the dinucleotide context in which they occur (17, 41, 44).
Derse and colleagues reported that HTLV-1 evolved a strategy different from HIV to counteract APOBEC3 proteins (14). In contrast to HIV-1 Vif-mediated proteosomal degradation, a portion of the HTLV-1 nucleocapsid results in A3G exclusion from the virion. Wild-type HTLV-1 particles failed to encapsidate A3G, whereas a deletion of a HTLV-1-specific 20-amino-acid (20-aa) region near the C terminus of NC resulted in enhanced A3G virion encapsidation and restriction, suggesting that this domain excludes A3G from packaging (14). We hypothesized that one or more A3 cytidine deaminases other than A3G may escape this NC peptide-mediated exclusion from virions and reduce HTLV-1 infectivity. In this study, we tested all seven human A3 proteins for their ability to restrict HTLV-1. We found that A3A, A3B, and A3H hapII potently decrease HTLV-1 infectivity. A3A and A3B required catalytic deaminase activity for restriction, whereas A3H hapII restricted HTLV-1 in a deaminase-independent manner. Analysis of HTLV-1 sequences in cell lines derived from ATLL and HAM/TSP patients revealed multiple independently mutated proviruses, suggesting that HTLV-1 is targeted by several A3 proteins, such as A3A, A3B, and A3G.
MATERIALS AND METHODS
Cell culture.
TZM-bl cells were provided by J. C. Kappes and X. Wu through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health, NIH Reagent program (90). The human cell lines HEK-293T and TZM-bl were maintained at 37°C in a humidified atmosphere of 5% CO2 in Dulbecco's high-glucose modified Eagle's medium, supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin. Jurkat cells were cultured at 37°C in a humidified atmosphere of 5% CO2 in Roswell Park Memorial Institute medium 1640 (RPMI 1640), supplemented with 10% FBS, 2 mM glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin.
Patient-derived HTLV-1-infected cell lines.
Cell lines derived from HTLV-1-infected patients have been established from peripheral blood mononuclear cells (PBMCs) obtained from patients diagnosed with ATLL or HAM/TSP as described previously (65, 72, 88). Cell lines derived from ATLL patients (StEd, Champ, and PaBe cell lines) and from HAM/TSP patients (Eva and Xpos cell lines) were cultured in RPMI 1640 containing 40% Panserin 401 (PAN Biotech), 20% FBS, l-glutamine (0.35 g/liter), and streptomycin-penicillin (0.12 g/liter each). Media for the different cell lines were supplemented with interleukin 2 (IL-2) (Roche Diagnostics) as follows: 40 U of IL-2/ml for StEd cells and 20 U of IL-2/ml for Champ, PaBe, Eva, and Xpos cell lines. Patient-derived cell lines were in continuous culture for at least 4 months.
DNA and RNA isolation from PBMCs.
PBMCs were isolated from fresh human or Macaca mulatta blood (Animal Facility Department, Paul-Ehrlich-Institute, Germany) by Histopaque-1077 gradient centrifugation (Sigma-Aldrich). Mononuclear cells at the interface were collected and washed twice with phosphate-buffered saline (PBS), and DNA was isolated by using the DNeasy DNA isolation kit (Qiagen). Macaca mulatta PBMCs were activated with phytohemagglutinin (3 μg/ml) for 3 days in RPMI 1640 medium containing 10% FBS, 5 × 105 mM 2-mercaptoethanol, 2 mM l-glutamine, and 100 units of human recombinant IL-2 per ml at 37°C and 5% CO2. Total RNA was isolated using the RNeasy RNA kit (Qiagen).
APOBEC3 expression plasmids.
All the expression plasmids encoded human A3 proteins (A3s) as triple-carboxy-terminal hemagglutinin (HA)-tagged proteins. A3A, A3B, A3C, A3D, A3F, A3G, A3H hapI, and A3H hapII were PCR amplified using primers carrying 5′ HindIII and 3′ XbaI restriction sites and a 3× HA tag, and inserted in the mammalian expression vector PTR600, as previously described (22). Site-directed mutagenesis was performed to construct the A3A E72A, A3B E68A, A3B E225A, A3B E68A/E255A, and A3H hapII E56A cytidine deaminase mutants, as previously described (22). The PCR products were cloned into the PTR600 vector and confirmed by sequencing. Primer sequences are available upon request.
Cloning of A3A from Macaca mulatta.
cDNA was synthesized from 1 μg of total Macaca mulatta PBMC RNA using oligo(dT)18 primers and the RevertAid first strand cDNA synthesis kit (Fermentas). A3A cDNA was amplified with forward 5′-ATGGACGGCAGCCCAGCATC-3′ and reverse 5′-GTTTCCCTGATTCTGGAGAATGGC-3′ primers, using recombinant Pfu polymerase (Fermentas) with the following PCR conditions: (i) 95°C for 3 min; (ii) 32 cycles, with 1 cycle consisting of 95°C for 30 s, 58°C for 30 s; and (iii) 72°C for 4 min. The PCR product was used for a second-round PCR with primers 5′ HindIII forward (5′-CCAAGCTTATGGACGGCAGCCCAGC-3′) and the 3′ SmaI HA tag (5′-CCCCCGGGTTAAGCGTAATCTGGAACATCGTATGGGTAGTTTCCCTGATTCTGGAG-3′) primer (HA tag underlined) and cloned into the PTR600 vector. The SV and SVR amino acid insertions in human A3A were cloned in the PTR600 vector using site-directed mutagenesis using overlapping primers. Site-directed mutagenesis was used to delete SV at positions 27 and 28 and SVR at positions 27 to 29 from Macaca mulatta A3. The inserts were confirmed by sequencing. Primer sequences are available upon request.
Plasmids used for viral infectivity assays.
The HTLV-1 packaging plasmid pCMVHT1-ΔEnv (CMV stands for cytomegalovirus) (13), which encodes all HTLV-1 genes except env, and the HTLV-1 reporter vector HTLV1-inLuc (Luc stands for luciferase) (48) were kindly provided by Gisela Heidecker and David Derse, NCI-Frederick. HTLV-1 viruses were pseudotyped using the vesicular stomatitis virus G glycoprotein (VSV-G) expression pMD.G plasmid (54). The replication-competent molecular clones NL4-3 (1) and NL4-3 ΔVif (19) were provided by the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health.
HTLV-1 infectivity assay.
Transfections into HEK-293T cells (2 × 105 cells/well in a 12-well plate) were performed with Lipofectamine LTX (Invitrogen) or Fugene 6 (Roche Diagnostics) according to the manufacturer's instructions. Standard transfection experiments consist of 800 ng of the HTLV-1 packaging construct (pCMVHT1Δenv), 800 ng of the HTLV-1 reporter vector HTLV1-inLuc, 100 ng of the VSV-G expression plasmid for viral pseudotyping, and 100 ng of the A3 expression plasmids. Green fluorescent protein (GFP)-expressing PTR600 (GFP-PTR600) vectors and LacZ expression plasmids were used as controls. After 24 h, cells were washed with PBS and 105 HEK-293T cells were mixed and cocultured with 105 Jurkat cells. Cells were collected 72 h later, and luciferase activity was measured using SteadyliteHTS luciferase reagent (Perkin Elmer) in black 96-well plates. Infections were done in triplicate, and at least three independent experiments were performed for each A3 protein. HTLV-1 virion release was detected by an anti-HTLV-1 p19 antibody (clone TP-7; Zeptometrix).
HIV-1 infectivity assay.
HEK-293T cells (3 × 105) were cotransfected with 500 ng of wild-type pNL4-3 (pNL4-3 WT) or pNL4-3 ΔVif and 50 ng of the respective A3 expression plasmids using 4 μg/ml polyethylenimine (PEI) (Polysciences, Inc.). Viral supernatants were collected 48 h posttransfection, clarified by centrifugation, and used to infect 1 × 104 TZM-bl cells in black 96-well plates. Infections were done in triplicate with viral supernatants from three independent experiments for each A3 protein tested. Infectivity of the virus particles in the TZM-bl cells was assessed 48 h postinfection by detecting β-galactosidase activity using the Galacto-Star System (Applied Biosystems).
HTLV-1 virion isolation.
HEK-293T cells (6 × 105 cells/well in a 6-well plate) were transfected with 900 ng pCMVHT1Δenv vector or 900 ng empty pCRV1 control plasmid and 100 ng A3 expression plasmids using Fugene 6 transfection agent (Roche Diagnostics) according to the manufacturer's instructions. Forty-eight hours posttransfection, the supernatants were filtered (0.45-μm pore size) and concentrated by centrifugation through a sucrose cushion (20% sucrose in PBS) at 20,000 × g for 3 h at 4°C. The pellets were gently dissolved in PBS and digested with subtilisin A according to the protocol from David Ott (62). Virions were mixed with 1 volume of 2× digestion buffer (40 mM Tris-HCl [pH 8.0], 2 mM CaCl2, 2 mg/ml subtilisin A [Sigma-Aldrich]) and incubated at 37°C for 18 h. Subtilisin was inhibited by the addition of the protease inhibitor phenylmethylsulfonyl fluoride (PMSF) (Boehringer Mannheim Biochemicals) (final concentration of 5 μg/ml), and virions were reisolated through a 20% sucrose cushion as described above. Pellets were dissolved in LDS sample buffer (NuPAGE; Invitrogen) and analyzed by immunoblotting.
Immunoblot analysis.
Two days posttransfection, HEK-293T cells were washed with PBS and lysed in radioimmunoprecipitation assay buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate). Protein concentration was measured using Bradford reagent (Bio-Rad). The concentrated virions were also resuspended in radioimmunoprecipitation assay buffer. Lysates containing 20 μg of protein were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were probed with mouse anti-HA antibody (1:10,000 dilution) (MMS-101P; Covance), mouse anti-p19 (1:1,000 dilution) (clone TP-7; ZeptoMetrix), or goat antiserum to HTLV-I (1:1,000 dilution), and mouse antitubulin antibody (1:4,000 dilution; clone B5-1-2; Sigma-Aldrich) to ensure equal protein loading. The goat antiserum to HTLV-I was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, from P. Szecsi, H. Halgreen, and J. Tang.
Detection of overexpressed A3-mediated HTLV-1 editing.
HEK-293T cells (2 × 105 cells/well in a 12-well plate) were transfected with 800 ng of pCMVHT1Δenv (a construct which expresses all HTLV-1 proteins except Env), 800 ng of the HTLV1-inLuc plasmid, 200 ng of A3 expression plasmids or GFP-PTR600 vector as a control, and 100 ng of VSV-G expression vector. Twenty-four hours posttransfection, HEK-293T cells were washed with PBS and cocultured with an equal amount of Jurkat cells (1:1 ratio). After 15 h of coculture, DNA was isolated using the DNeasy DNA isolation kit. A 515-bp fragment within the spliced luciferase gene was amplified using the primers 5′-CCGGGAAAACGCTGGGC-3′ and 5′-GGCGATCTTTCCGCCCTTCTTGG-3′. For selective amplification of the hypermutated products, the PCR denaturation temperatures were lowered stepwise from 88°C to 84°C (84, 85.3, 86.3, 87, 87.5, and 88°C) using a gradient PCR thermocycler. The PCR parameters were as follows: (i) 95°C for 1 min; (ii) 40 cycles, with 1 cycle consisting of 84 to 88°C for 30 s, 58°C for 30 s, and 72°C for 1 min; (iii) 10 min at 72°C. PCRs were performed with recombinant Taq DNA polymerase (Fermentas). PCR products from the lowest denaturation temperatures were cloned into pJet1.2/blunt vector (Fermentas) and sequenced. The nucleotide sequences of at least eight independent clones were analyzed with the Hypermut software (http://www.hiv.lanl.gov/content/sequence/HYPERMUT/hypermut.html).
Detection of HIV-1 editing by overexpressed A3G.
Viral stocks were generated by transfecting NL4-3 ΔVif (500 ng) and pTR600-A3G variants (50 ng). Viral supernatants were collected 48 h posttransfection, clarified by centrifugation, and used to infect TZM-bl cells in 24-well tissue culture plates in the presence of DNase I (Invitrogen). After 12 h, cells were washed extensively with PBS, and genomic DNA was extracted using a DNeasy DNA isolation kit (Qiagen). A 1,905-nucleotide-long region of pol (HXB2, nucleotides 2928 to 4833 [22]) was amplified by PCR using Taq DNA polymerase (Qiagen) and gel extracted using the Qiaprep kit (Qiagen). The PCR products were used as a template in the differential DNA denaturation PCR (3DPCR) using different denaturing temperatures (80.5, 80.9, 81.2, 81.5, 81.2, and 83.2°C). PCR products were gel extracted and cloned using a StrataClone kit as previously described (22). Inserts form eight clones were sequenced, manually aligned using Bioedit, and compared using Hypermut software.
Detection of A3-mediated editing of HTLV-1 in patient-derived cell lines.
Genomic DNA from the ATLL-derived (StEd, Champ, and PaBe) and HAM/TSP-derived (Xpos and Eva) cell lines were isolated by standard methods. For selective amplification of the HTLV-1 tax gene, two rounds of PCR were performed. The first-round PCR parameters were 95°C for 3 min, followed by 35 cycles (1 cycle consists of 95°C for 1 min, 59.2°C for 30 s, and 72°C for 1 min), and 5 min at 72°C. Second-round nested 3DPCR was performed using 0.5 μl of the first-round PCR products as input and a gradient denaturation temperature between 88°C to 84°C (84, 85.3, 86.3, 87, 87.5, and 88°C). The reaction parameters were 95°C for 1 min, followed by 40 cycles (1 cycle consists of 88 to 84°C for 30 s, 54°C for 30 s, and 72°C for 1 min), and 5 min at 72°C. First-round PCR primers tax-fwd (fwd stands for forward) (5′-CAGCCCACTTCCCAGGGTTTGGAC-3′) and tax-rev (5′-GTGTGAGAGTAGAAATGAGGGGT-3′) (88) amplify an 881-bp fragment. Second-round PCR was performed with primers tax-nested-fwd (5′-TAGGCCTTGGTTTGAAATTTGTG-3′) and tax-nested-rev (5′-CCTCCAGGCCATGCGCAAA-3) using recombinant Taq DNA polymerase. The PCR products from the lowest denaturation (88°C) temperature were cloned into pJet1.2/blunt vector and sequenced. The inserts of eight independent clones were analyzed with the Hypermut software (http://www.hiv.lanl.gov/content/sequence/HYPERMUT/hypermut.html). All sequences were compared with the tax template sequence derived after PCR amplification using a recombinant Pfu polymerase (Fermentas) and tax-fwd and tax-rev primers. The PCR conditions were 95°C for 3 min, followed by 30 cycles (1 cycle consists of 95°C for 30 s, 59.2°C for 30 s, and 72°C for 2 min), and 10 min at 72°C.
3DPCR amplification of the human myc exon 2.
The myc gene exon 2 was amplified from genomic DNA (500 ng) obtained from HTLV-1-infected T-cell lines (StEd, Champ, PaBe, Xpos, and Eva) and PBMCs from a healthy donor. The first-round reaction parameters were 95°C for 3 min, followed by 35 cycles (95°C for 30 s, 55.3°C for 30 s, and 72°C for 1 min), and 5 min at 72°C. Second-round 3DPCR was performed using 0.5 μl of the first-round PCR products as input using a denaturing temperature gradient between 88°C and 84°C. The reaction parameters were 95°C for 1 min, followed by 40 cycles (1 cycle consists of 88 to 84°C for 30 s, 61.5°C for 30 s, and 72°C for 1 min), and 5 min at 72°C. Both rounds were performed using recombinant Taq polymerase. The primers used were described by Suspène et al. (79).
Statistical analysis.
Paired t tests were performed using GraphPad Prism 5 software. The values were determined to be statistically significant at 0.05 and 0.01 levels (P values of 0.05 and 0.01).
RESULTS
A3A, A3B, and A3H hapII restrict HTLV-1.
To test the hypothesis that several A3 proteins target HTLV-1, we first compared the ability of A3A, A3B, A3C, A3D, A3F, A3G, and two A3H haplotypes (hapI and hapII) to restrict HTLV-1 using a previously established HTLV-1 infectivity assay (13). Briefly, A3 expression plasmids were cotransfected with an HTLV helper construct, which carries genes that encode all HTLV-1 proteins except Env, a packageable luciferase reporter construct, and a VSV-G expression plasmid. Transfected HEK-293T cells were subsequently cocultured with Jurkat T cells, and luciferase activity was measured in the cell lysates. The luciferase reporter contains a reversed CMV promoter-driven luciferase gene with an internal intron preventing expression in transfected cells. The functional luciferase protein is expressed only upon successful splicing and packaging (in the producer cells) as well as completion of reverse transcription and integration in the target cell (the assay is explained in detail in reference 48). A3A, A3B, and A3H hapII inhibited HTLV-1 infectivity up to 10-fold compared to the no-A3 controls (Fig. 1A). The restriction by the deaminases was dose dependent, whereas the other A3 proteins poorly restricted even at higher DNA levels (Fig. 1B). In contrast, HTLV-1 infectivity was largely resistant to human A3C, A3D, A3F, and A3G action (Fig. 1A and B). The observed lack of A3G activity is in agreement with previous reports (14, 41, 56, 59). No luciferase was detected when either HTLV-1 helper construct, luciferase reporter, or the VSV-G plasmid was omitted from the transfection (Fig. 1A).
Fig 1.

APOBEC3 restriction of HTLV-1 and HIV-1. (A) The indicated A3 expression plasmids were cotransfected with HTLV-1 helper (GagPol and nonstructural proteins), luciferase reporter, and VSV-G plasmids in HEK-293T cells. The cells were overlaid with Jurkat cells, and luciferase (relative light units [RLU]) was measured after 3 days. The GFP control is set at 100% infectivity. Values are means plus standard deviations (error bars) for three independent experiments. (B) Similar to panel A but with increasing amounts of A3 expression plasmids. (C) HIV-1 WT and HIV-1 ΔVif expression plasmids were cotransfected with the indicated A3 plasmids in HEK-293T cells. Two days posttransfection, supernatants were used to infect TZM-bl reporter cells, and β-galactosidase activity (RLU) was measured 2 days postinfection. Infectivity without A3 is set at 100%. Values are means plus standard deviations (error bars) of a representative experiment performed in triplicate. Unpaired t tests were computed to determine whether differences between GFP and each A3 protein reach the level of statistical significance (P < 0.05 [*] and P < 0.01[**], using GraphPad Prism 5 software).
High A3A expression has been reported to degrade plasmid DNA, which could have a negative effect on the HTLV plasmids and, thus, on luciferase expression (78). To exclude this possibility, we included LacZ expression plasmids in the assay. Equal β-galactosidase expression was observed, suggesting that, in our experimental system, expression was not affected by A3A or other A3 proteins (data not shown).
To provide a framework for the activity spectrum of human A3 proteins against HTLV-1, we additionally assessed the level of restriction of wild-type HIV-1 (WT) and an HIV-1 lacking a functional Vif (ΔVif) for each of the A3s. The A3 expression plasmids were transfected with HIV molecular clones NL4-3 WT and ΔVif in HEK-293T cells, and the culture supernatants were used to infect TZM-bl reporter cells. The Vif-sensitive A3D, A3F, and A3G restricted only HIV-1 ΔVif, an observation which is in good agreement with numerous other reports (reviewed in reference 2). HIV-1 WT and HIV-1 ΔVif were both restricted by A3B and A3H hapII to similar levels, indicating their lack of sensitivity to Vif degradation (Fig. 1C). Interestingly, A3A, which potently restricted HTLV-1, did not affect HIV-1 WT and HIV-1 ΔVif infectivity in our experimental assay system. In summary, both HTLV-1 and HIV-1 WT are restricted by A3B and A3H hapII, whereas A3A exclusively restricts HTLV-1. Of note, this finding makes HTLV-1, next to avian Rous sarcoma virus (91), the only retrovirus that is sensitive to human A3A restriction.
Packaging of A3 into HTLV-1 virions.
A3 proteins need to be packaged into the virion before they can exert their antiviral activity in the target cell during reverse transcription. We therefore analyzed A3 expression and incorporation into HTLV-1 virions by cotransfecting A3 plasmids with HTLV-1 or an irrelevant plasmid in HEK-293T cells. The APOBEC3 expression levels were not affected by HTLV expression, indicating that, unlike WT HIV, HTLV does not encode a protein that degrades A3 proteins in the producer cells (Fig. 2A). Supernatants were concentrated through a 20% sucrose cushion and analyzed by Western blotting (Fig. 2B, left panels). We observed that A3 proteins were present at very low levels in the concentrated pellets compared to HIV-1 (data not shown), but also that A3 levels were similar for the concentrated mock-transfected and HTLV-transfected cell culture supernatants (Fig. 2B, left panels). Although some A3 members, like A3A, A3B, A3G, and A3H hapII, pelleted slightly more efficiently in the presence of HTLV-1, the majority of the concentrated A3 proteins were concentrated unspecifically, likely due to association with exosomes and/or microvesicles. To remove these nonviral particles, we incubated the concentrated supernatants with the protease subtilisin A, which digests exosomes and microvesicles, but leaves enveloped virions intact (62). Subtilisin A digestion removed all non-HTLV-1-specific packaging (Fig. 2B, right panels) and showed HTLV-1-specific packaging of A3 proteins. Although all A3 members were specifically packaged into HTLV-1 virions, the active A3A, A3B, A3H hapII, and inactive A3G are packaged more efficiently than the other inactive A3C, A3D, A3F, and A3H hapI proteins. Taken together, HTLV-1 fails to degrade APOBEC3 proteins in the producer cells, but several APOBEC3 members are specifically packaged with low efficiencies into HTLV-1 virions.
Fig 2.

Packaging of APOBEC3 into HTLV-1 virions. (A) The indicated A3 expression plasmids were cotransfected with an irrelevant plasmid (−) or with an HTLV-1 helper plasmid (+ HTLV) (all HTLV proteins except Env) in HEK-293T cells, and the cells were lysed 2 days after transfection. The lysates were analyzed by immunoblotting and probed for A3 expression (HA), and tubulin served as a loading control. α-HA, anti-HA antibody. (B) Filtered supernatants from panel A were concentrated through a 20% sucrose cushion and were either mock treated or treated with subtilisin A to remove exosomes/microvesicles and reconcentrated through a 20% sucrose cushion. Virion lysates were analyzed by immunoblotting. HTLV capsid (CA), matrix (MA), and nucleocapsid (NC) were detected by anti-HTLV-1 antibodies (α-HTLV).
Comparison between human and macaque A3A for HTLV-1 restriction.
Human A3A is highly expressed in monocytes/macrophages, but its expression is very low or even absent in T cells (69). Thus, the in vivo relevance of A3A for HTLV-1 infection in humans is currently undefined. Rhesus macaques are natural hosts of simian T-lymphotropic virus type 1 (STLV-1), a retrovirus closely related to HTLV-1 (35, 40, 73). Activated T cells from rhesus macaques express high levels of A3A in contrast to humans (75). Moreover, rhesus A3A strongly inhibits HIV-1 ΔVif but is inactive against adeno-associated virus type 2 (AAV-2) and L1 retrotransposons (75). In contrast, human A3A is inactive against HIV-1 ΔVif but represses AAV-2 and L1 replication (5, 7, 9, 55). A recent study showed that hominids acquired a three-amino-acid deletion in A3A (ΔSVR at positions 27 to 29 [75] [Fig. 3A, see alignment]). The deletion of these three amino acids in rhesus A3A abrogated its activity against HIV-1 ΔVif yet failed to bring back the inhibitory capacity against L1, indicating that the difference between human and macaque A3A is not dictated by the indel (75).
Fig 3.

Comparison between human and macaque A3A restriction of HTLV-1. (A) Protein sequence alignment of human A3A and Macaca mulatta A3A. Human A3A contains a specific deletion of 27S, 28V, and 29R. (B) The indicated A3A WT and mutant expression plasmids were cotransfected with HTLV-1 helper (GagPol and nonstructural proteins), luciferase reporter, and VSV-G plasmids in HEK-293T cells. The cells were overlaid with Jurkat cells, and luciferase (RLU) was measured after 3 days. GFP is set at 100%. Values are means plus standard deviations (error bars) for three independent experiments. P values were computed to determine whether differences between GFP and each A3 protein or between A3A mutants reach significance (P < 0.05 [*] and P < 0.01[**] by unpaired t test using GraphPad Prism 5 software). no luc, no luciferase. (C) The indicated A3A WT and mutant expression plasmids were cotransfected with HTLV-1 helper (GagPol and nonstructural proteins), luciferase reporter, and VSV-G plasmids in HEK-293T cells, and the cells were lysed 2 days after transfection. Supernatants were cleared and concentrated through a 20% sucrose cushion. Cell and virion lysates were analyzed by Western blotting. Of note, human A3A is 3× HA tagged and is therefore larger than the 1× HA tagged macaque A3A. Tubulin serves as a loading control.
Because our data identified human A3A as a potent inhibitor of HTLV-1, we analyzed the effect of these three amino acids in rhesus and human A3A proteins on HTLV-1 restriction. Using the described luciferase reporter virus, we show that both human and rhesus A3A displayed a potent inhibition of HTLV-1 (Fig. 3B). Moreover, inserting two or three amino acids in the human A3A (+SV and +SVR) did not affect its antiviral activity against HTLV-1. Interestingly, deletion of two and three amino acids in rhesus A3A (ΔSV and ΔSVR A3A) reduced its anti-HTLV-1 activity (Fig. 3B). We also observed that WT and mutant A3A proteins were expressed equally in the producer cells, but rhesus A3A was packaged less efficiently than human A3A was (Fig. 3C). We conclude that both rhesus and human A3A proteins share an evolutionarily conserved capacity to inhibit HTLV-1, which indicates that the three-amino-acid deletion in human A3A likely arose because of its beneficial nature in counteracting viruses other than HTLV-1.
A3A and A3B, but not A3H hapII, require deaminase activity for HTLV-1 restriction.
Since A3 proteins can restrict retroviruses by editing and nonediting mechanisms (3, 25, 28, 49, 57), we next determined whether the HTLV-1 restriction exerted by A3A, A3B, and A3H hapII required catalytic deaminase activity. The deaminase activity of each deaminase domain can be abolished by mutating the essential glutamic acid in the catalytic site (22, 52). We introduced these previously described active site mutations into A3A, A3B, and A3H hapII (Fig. 4A) and tested their ability to block HTLV-1 infectivity as described in Materials and Methods. We show that HTLV-1 infectivity was restricted by WT A3A, but not by the corresponding deaminase active site mutant (Fig. 4B). A3B contains two deaminase domains, and both appear to contribute to efficient HTLV-1 restriction, since only virus produced in the presence of the double deaminase mutant A3B (E68A-E255A) showed infectivity levels comparable to the no-A3 control (Fig. 4B). Surprisingly, the A3H hapII deaminase mutant failed to rescue infectivity, suggesting that A3H hapII restricts HTLV-1 in a deaminase-independent manner.
Fig 4.
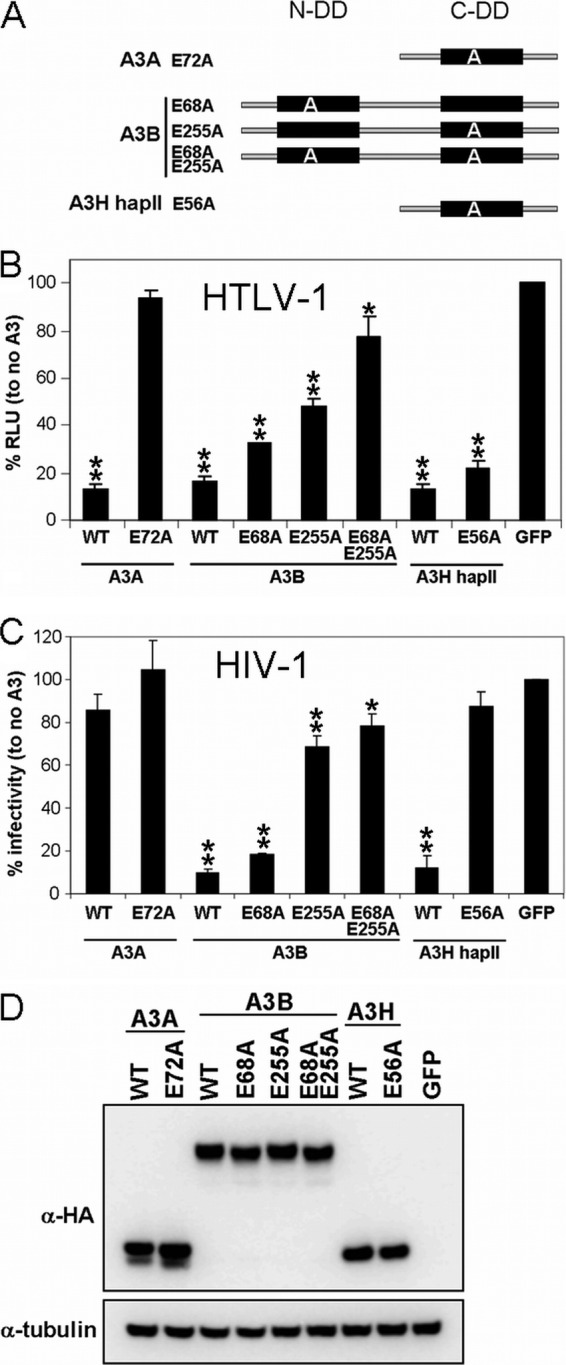
Deaminase activity requirements for restriction of HTLV-1 and HIV-1. (A) Schematic representations of the deaminase domains present in A3A, A3B, and A3H. A3A and A3H hapII contain a single deaminase domain, whereas A3B contains two domains, which are indicated by a black box. The letter “A” denotes the alanine mutations of the essential glutamic acid in the catalytic active site. N-DD and C-DD denote the N-terminal and C-terminal deaminase domains, respectively. (B) The indicated A3 WT and deaminase mutants were cotransfected with HTLV-1 helper (GagPol and nonstructural proteins), luciferase reporter, and VSV-G plasmids in HEK-293T cells. The cells were overlaid with Jurkat cells, and luciferase (RLU) was measured after 3 days. GFP is set at 100%. Values are means plus standard deviations (error bars) for three independent experiments. Unpaired t tests were computed to determine whether differences between GFP and each A3 protein are statistically different (P < 0.05 [*] and P < 0.01[**], using GraphPad Prism 5 software). (C) HIV-1 ΔVif expression plasmids were cotransfected with the indicated A3 WT and deaminase mutants in HEK-293T cells. Two days posttransfection, supernatants were used to infect TZM-bl reporter cells, and β-galactosidase (RLU) was measured 2 days postinfection. Infectivity with GFP is set at 100%. Values are means plus standard deviations (error bars) of a representative experiment performed in triplicate. The values were compared for statistical significance (P < 0.05 [*] and P < 0.01[**] by unpaired t test, using GraphPad Prism 5 software). (D) Immunoblot analysis of A3A, A3B, and A3H hapII and their corresponding deaminase mutants in HEK-293T cells. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) serves as a loading control.
We also tested the deaminase mutants for their ability to inhibit HIV-1 to confirm the activity spectra of the deaminase mutants. As expected, rescue was not observed for either WT A3A or A3A E72A, suggesting that both proteins have no activity against HIV-1 (Fig. 4C). The anti-HIV-1 activity of A3B was determined mainly by the C-terminal deaminase motif, and a complete loss of its antiretroviral activity required mutations in both domains (6). Interestingly, the A3H hapII deaminase mutant failed to restrict HIV-1, while A3H hapII deaminase activity was not required for HTLV-1. We conclude that A3A exclusively restricts HTLV-1 in a deaminase-dependent manner, while A3B requires its catalytic sites for restricting HTLV-1 and HIV-1. In contrast, an intact deaminase domain for A3H hapII is dispensable for HTLV-1 restriction but necessary and required for anti-HIV-1 activity.
Mutagenesis of HTLV-1 by A3.
We next assessed whether reduction of HTLV-1 infectivity correlated with mutagenesis. We used a previously described 3DPCR approach (82) to amplify mutagenized HTLV-1 sequences. The underlying principle of selective 3DPCR is that edited DNA contains fewer GC base pairs, resulting in a lower melting temperature. Therefore, successful PCR amplification at lower denaturing temperatures (88 to 84°C) is indicative of mutagenized sequences. In order to avoid amplifying transfected plasmids, we selected a region in the spliced luciferase gene for amplification. We performed 3DPCR on DNA extracted from cells obtained from the HTLV-1 infectivity assay (Fig. 1A, GFP, A3A, A3B, and A3H hapII). The GFP control did not yield any PCR products amplified at lower denaturing temperatures, suggesting that no editing took place (Fig. 5A). A3A and A3B resulted in efficient amplification at lower temperatures, which was absent for the A3A E72A and the A3B double deaminase mutant E68A-E255A (Fig. 5A). For A3H hapII, no PCR products could be amplified at lower denaturing temperatures, indicating the absence of editing. Amplified PCR products from the lowest denaturing temperature were cloned, and a minimum of eight clones was sequenced. Figure 5B illustrates the extensive mutagenesis observed in the presence of A3A and A3B, predominantly in an A3-specific G-to-A context (17 to 64 G-to-A mutations per 501 bp for A3A and 4 to 45 G-to-A mutations per 501 bp for A3B). G-to-A mutations were not induced by A3H hapII, which was in stark contrast to its restriction of HIV-1, which required deaminase activity and was previously shown to induce editing (22).
Fig 5.

A3-induced editing in HTLV-1 genomes. (A) A region overlapping with the intron in the luciferase gene of the HTLV-1 reporter sequence was PCR amplified from DNA obtained from HTLV-1 infectivity assays (Fig. 1A). PCR was performed using a range of denaturing temperatures (84 to 88°C) and analyzed on agarose gels. The denaturing temperature (Td) (84, 85.3, 86.3, 87, 87.5, and 88°C) is indicated by the height of the black triangle above the gel. (B) PCR products amplified at the lowest denaturing temperature were cloned, and eight individual clones were sequenced (501 bp). The mutations are indicated by color as follows: GA-to-AA mutations (blue lines), GG-to-AG mutations (red lines), GT-to-AT mutations (pink lines), and GC-to-AC mutations (green lines). (C) Pie chart representation of the relative dinucleotide preferences of A3A and A3B.
A3 enzymes favor different dinucleotide contexts, and the analysis of the edited sequences reproduced these specific dinucleotide preferences very well (Fig. 5C). In our experiments, A3A preferred GA-to-AA mutations (56%) followed by GG-to-AG (26%), GT-to-AT (13%), and GC-to-AC (4%) mutations. A3B predominantly edited GA-to-AA (73%) and GG-to-AG (25%) mutations, as previously reported (4, 6, 16). In summary, A3A and A3B restricted HTLV-1 in a deaminase-dependent manner, inducing G-to-A mutations in a GA-to-AA dinucleotide context, while A3H hapII acted independently of catalytic activity, leaving no specific footprints in HTLV-1 proviruses.
A3G editing of HIV-1 and HTLV-1.
In contrast to HIV-1, HTLV-1 editing by A3 proteins is rarely observed in patients. A recent report analyzed the presence of mutations in HTLV-1 proviral DNA from infected individuals and detected only a relatively low number of mutations (17). Most substitutions were G-to-A mutations in a GG dinucleotide context, which is favored by A3G. However, our data suggest that HTLV-1 is poorly restricted by A3G. To test whether A3G is able to edit HTLV-1 proviral DNA under experimental conditions resulting in minimal restriction, we used 3DPCR to seek evidence for editing. We also performed 3DPCR on HIV-1 proviral DNA by amplifying a 1,905-bp fragment of the HIV-1 pol gene (22). Agarose gel analysis of the 3DPCR products show that selective amplification was possible only with HIV-1 produced in the presence of A3G (Fig. 6A). Cloning and sequencing showed a high number of G-to-A mutations (17 to 45 G-to-A mutations per 800 bp), which were predominantly found in the GG dinucleotide context (Fig. 6B and C). A3G also increased the efficiency of amplification of HTLV-1 proviral DNA at lower temperatures, indicating a moderate level of editing (Fig. 6A). Sequencing showed a low level of mutations (1 to 6 G-to-A mutations per 501 bp), which were mostly in the A3G preferred GG dinucleotide context (Fig. 6B and C). Taken together, A3G is able to introduce high levels of GG-to-AG specific mutations in HIV-1 but also appears to be responsible for low levels of GG-to-AG mutations in HTLV-1 genomic DNA, despite its limited level of restriction in our reporter infectivity assay.
Fig 6.

A3G editing of HIV-1 ΔVif and HTLV-1. (A) Supernatants of cells cotransfected with HIV-1 ΔVif and GFP or A3G was used to infect TZM-bl cells. Proviral DNA was extracted, and a 1,905-bp fragment in HIV-1 pol was PCR amplified and purified from gel. PCR fragments were subsequently used as a template in the 3DPCR with a range of denaturing temperatures (80.5 to 83.2°C) and analyzed on agarose gels. The denaturing temperature (Td) (80.5, 80.9, 81.2, 81.5, 81.2, and 83.2°C) is indicated by the height of the black triangle above the gel. HTLV-1 editing was performed as described in the legend to Fig. 5. (B) PCR products amplified at the lowest denaturing temperature were cloned, and eight individual clones were sequenced. Red lines indicate GG-to-AG mutations, blue lines indicate GA-to-AA mutations, pink lines indicate GT-to-AT mutations, green lines indicate GC-to-AC mutations, and black lines indicate non-G-to-A mutations. (C) Pie chart representation of the relative dinucleotide preferences of A3G for HIV-1 and HTLV-1.
HTLV-1 editing in patient-derived T-cell lines.
We speculated that if A3A and A3B target HTLV-1 in vivo, specific deaminase footprints should be present in HTLV-1 proviral DNA. To assess whether A3A and A3B play a role in HTLV-1 replication in vivo, we analyzed five different T-cell lines obtained from HTLV-1-infected individuals (65, 72, 88). Three of these donors were diagnosed with ATLL (StEd, Champ, and PaBe cell lines), and two donors were diagnosed with HAM/TSP (Xpos and Eva). To specifically document independently edited HTLV-1 sequences, which likely represent a small fraction of total proviral sequences, we performed 3DPCR using HTLV-1 tax-specific primers. Nested PCR fragments were amplified using lower denaturing temperatures from all but one cell line (Fig. 7A). DNA extracted from 293T cells transfected with HTLV-1 packaging plasmid served as a negative control and did not show amplification at lower temperatures (Fig. 7A, plasmid). To determine whether editing was specific for HTLV-1, a nested 3DPCR was performed on the cellular myc gene (78), which showed no difference compared to DNA extracted from PBMCs from a healthy donor (Fig. 7B). Taken together, HTLV-1 is a specific target for editing in patient-derived cell lines.
Fig 7.
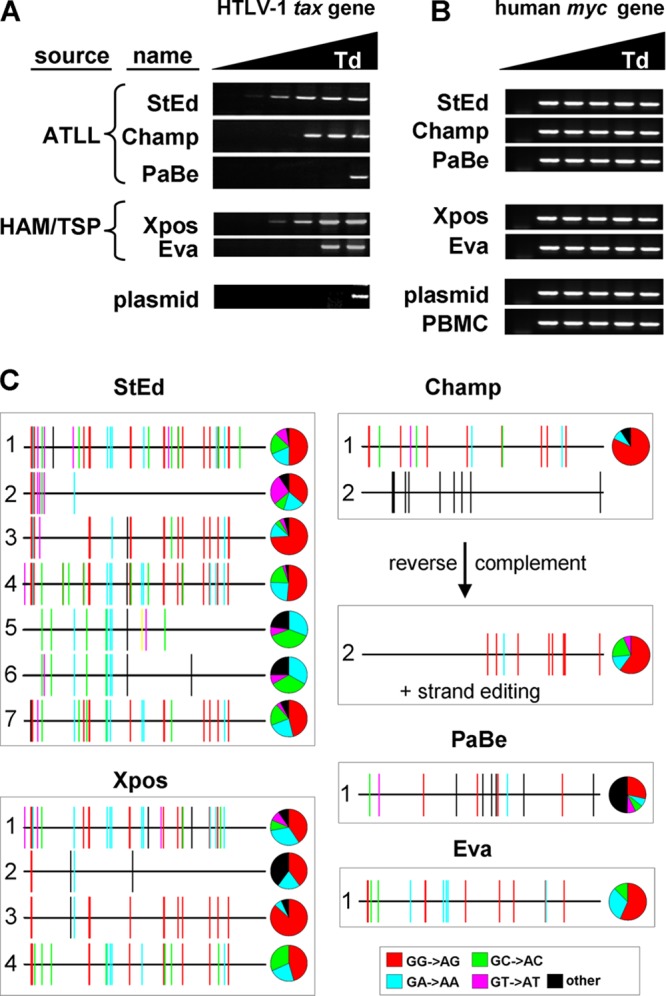
A3 editing of HTLV-1 in vivo. (A) Cellular DNA was extracted from several cell lines obtained from HTLV-1-infected individuals diagnosed with ATLL or HAM/TSP. Nested 3DPCR was performed with HTLV-1 tax-specific primers. DNA extracted from HTLV-1 plasmid-transfected HEK-293T cells served as a negative control for editing (plasmid). (B) Nested 3DPCR was performed on DNA extracted from the different HTLV-1 cell lines using specific primers to amplify a fragment of the cellular myc gene. DNA extracted from a healthy donor and from HTLV-transfected HEK-293T cells were used as controls (PBMC and plasmid). (C) PCR fragments from the 3DPCR were cloned and sequenced. Only unique sequences (419 bp) are shown. A3-specific dinucleotide contexts are shown in the indicated colors. All non-G-to-A mutations are represented in black. + strand, plus strand.
Next, we cloned PCR products from the lowest and highest denaturing temperatures. A minimum of 8 clones from each cell line were sequenced, and only unique tax sequences (419 nucleotides [nt]) were used for mutational analysis. Comparing the edited sequences to their own reference sequence (high denaturing temperature) showed a large number of mutations in each cell line (48 to 78 mutations for StEd cells, 11 to 15 mutations for Champ cells, 14 mutations for PaBe cells, 5 to 44 mutations for Xpos cells, and 16 mutations for Eva cells [all mutations per 419 bp] [Fig. 7C]). The mutations were mostly G-to-A substitutions, implicating A3 action as the underlying mechanism (85% of all mutations for StEd cells, 96% of all mutations for Champ cells, 50% of all mutations for PaBe cells, 96% of all mutations for Xpos cells, and 100% of all mutations for Eva cells). The dinucleotide contexts observed were GG followed by GA and GC (Fig. 7C), indicating that A3G but also other A3 proteins, like A3A and A3B, could have edited HTLV-1 in these patient-derived cell lines. The low number of unique sequences for Champ, PaBe, and Eva cells may reflect their reported lower HTLV-1 proviral loads (66). Interestingly, one clone from Champ cells (sequence 2) showed C-to-T mutations in the positive strand, and only analysis of the negative strand showed A3-specific G-to-A mutations, indicating that the positive strand was edited. Evidence of plus-stranded editing has been reported once for HIV-1, but it is more common for hepatitis B virus A3-induced editing (81, 87). In addition, few C-to-T mutations have been found in HTLV-1 proviral DNA, which may have been induced by activation-induced deaminase (17). In conclusion, multiple independently edited HTLV-1 proviral DNA sequences were detected in HTLV-1-infected cell lines. The pattern of mutagenesis suggests a role for A3G as well as other A3 members in HTLV-1 editing.
DISCUSSION
HTLV-1 is a human retrovirus that infects cell populations that express several A3 proteins, but the full spectrum of their antiviral activity toward HTLV-1 is unknown. We tested all human A3 proteins and show that A3A, A3B, and A3H hapII potently restrict HTLV-1. We confirmed the reported lack of A3G activity and show that A3C, A3D, and A3F also lack anti-HTLV-1 activity. Interestingly, the restriction pattern partially overlaps with the pattern observed for HIV-1 WT, which is also sensitive to A3B and A3H hapII but not to A3A. HTLV-1 does not express a Vif-like protein. The lack of HTLV-1 sensitivity to A3C, A3D, A3F, and A3G does not seem to be mediated by A3 degradation in the producer cells (Fig. 2A), but A3C, A3D, and A3F are packaged with lower efficiency (Fig. 2B) into HTLV-1 particles. It is conceivable that one or more of the many nonstructural HTLV-1 proteins could bind or inactivate packaged A3 proteins. Alternatively, packaged A3 proteins without anti-HTLV-1 activity, like A3G, may not be located within the virion core where reverse transcription takes place. Indeed, intravirion location appeared to be important for A3A and A3H activity against HIV (20, 61).
A3A and A3B both required functional deaminase domains for restriction and introduced extensive G-to-A mutations in HTLV-1, indicating that their restriction is exerted through deamination. However, A3H hapII did not require an intact deaminase domain for restriction nor did it lead to deamination of HTLV-1 proviral DNA. This contrasts with A3H hapII-mediated HIV-1 restriction, which both requires a functional deaminase domain and is accompanied by extensive proviral DNA editing (22). There are numerous reports of APOBEC3 deaminase editing-independent restriction of HIV, including APOBEC3-mediated reduction of reverse transcription activity, strand transfer, or integration (3, 25, 28, 49, 57), and we speculate that one or more of these steps in the HTLV-1 life cycle could be affected by A3H hapII. Elucidating the viral determinants underlying the difference in A3H restriction between HIV-1 and HTLV-1 could lead to an improved understanding of the mechanism of viral A3 restriction.
The entire human A3 locus is highly polymorphic, especially the regions containing A3B and A3H hapII, both of which potently restrict HTLV-1. The A3B coding region is deleted with high frequencies in East Asia, South America, and Oceania (32), while multiple A3H haplotypes have been reported, of which only A3H hapII has strong activity against HIV-1 (12, 22, 58). The frequency of the active A3H hapII is high in African but low in Asian populations (58, 89). If A3B and A3H hapII affect HTLV-1 in vivo, individuals carrying inactive A3H haplotypes (e.g., A3H hapI) combined with a deleted A3B locus would be more susceptible to infection by HTLV-1. Interestingly, HTLV-1 prevalence is extremely high in Japan, which coincides with high A3B deletion and low A3H hapII frequencies at a population level.
We here report that A3A, A3B, and A3H hapII restrict HTLV-1, but the editing profiles observed in the HTLV-1-infected T-cell lines suggest that A3G may also contribute to HTLV-1 proviral DNA editing. The major targets for HTLV-1 are CD4+ T cells (42), and depending on their activation state, these cells express only very limited amounts of A3A and A3B transcripts but considerable amounts of A3G and A3H transcripts (26, 69). A3H, however, does not mutate HTLV-1 DNA and does not contribute to the mutations observed in the cell lines. A3A may restrict HTLV-1 replication in cells in monocytes and macrophages, which express high levels of A3A (34, 64, 69). Although A3B expression is low in CD4+ T cells, mammary epithelial cells express high A3B levels (60), which may affect mother-to-child transmission of HTLV-1 through breast milk.
In conclusion, HTLV-1 is a target for A3A, A3B, and A3H hapII in cell culture. Further studies will elucidate whether and to what extent they curb HTLV-1 replication in vivo. An individual's genetic A3 profile may have major implications on HTLV-1 transmission and disease progression.
ACKNOWLEDGMENTS
We thank Gisela Heidecker and David Derse for the kind gift of the pCMVHT1M-ΔEnv and HTLV1-inLuc plasmids. We thank the NIH AIDS Reagents program for TZM-bl cells, goat anti-HTLV-1 serum, NL43 WT, and NL4-3 ΔVif. We thank all the members of the Simon laboratory for helpful discussions, Wioletta Hörschken for excellent technical assistance, and Dieter Häussinger for constant support.
This research was supported in part by grant AI089246 from NIAID, the National Institutes of Health (V.S.), and a grant (MU 1608/4-1) from the Deutsche Forschungsgesellschaft (C.M.). C.M. is supported by the Heinz Ansmann Foundation for AIDS research. A.K. is supported by the Jürgen Manchot Foundation, Molecules of Infection Graduate School.
Footnotes
Published ahead of print 28 March 2012
REFERENCES
- 1. Adachi A, et al. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Albin JS, Harris RS. 2010. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev. Mol. Med. 12:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bishop KN, Holmes RK, Malim MH. 2006. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 80:8450–8458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bishop KN, et al. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14:1392–1396 [DOI] [PubMed] [Google Scholar]
- 5. Bogerd H, Wiegand H, Doehle B, Lueders K, Cullen B. 2006. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 34:89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bogerd HP, Wiegand HL, Doehle BP, Cullen BR. 2007. The intrinsic antiretroviral factor APOBEC3B contains two enzymatically active cytidine deaminase domains. Virology 364:486–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bogerd HP, et al. 2006. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. U. S. A. 103:8780–8785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boross P, Bagossi P, Weber IT, Tozser J. 2009. Drug targets in human T-lymphotropic virus type 1 (HTLV-1) infection. Infect. Disord. Drug Targets 9:159–171 [DOI] [PubMed] [Google Scholar]
- 9. Chen H, et al. 2006. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 16:480–485 [DOI] [PubMed] [Google Scholar]
- 10. Chiu YL, Greene WC. 2008. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 26:317–353 [DOI] [PubMed] [Google Scholar]
- 11. Conticello S, Harris R, Neuberger M. 2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 13:2009–2013 [DOI] [PubMed] [Google Scholar]
- 12. Dang Y, et al. 2008. Human cytidine deaminase APOBEC3H restricts HIV-1 replication. J. Biol. Chem. 283:11606–11614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Derse D, Hill SA, Lloyd PA, Chung H, Morse BA. 2001. Examining human T-lymphotropic virus type 1 infection and replication by cell-free infection with recombinant virus vectors. J. Virol. 75:8461–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Derse D, Hill SA, Princler G, Lloyd P, Heidecker G. 2007. Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. Proc. Natl. Acad. Sci. U. S. A. 104:2915–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de The G, Kazanji M. 1996. An HTLV-I/II vaccine: from animal models to clinical trials? J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 13(Suppl. 1):S191–S198 [DOI] [PubMed] [Google Scholar]
- 16. Doehle BP, Schafer A, Cullen BR. 2005. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology 339:281–288 [DOI] [PubMed] [Google Scholar]
- 17. Fan J, et al. 2010. APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J. Virol. 84:7278–7287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gessain A, et al. 1996. Virological aspects of tropical spastic paraparesis/HTLV-I associated myelopathy and HTLV-I infection. J. Neurovirol. 2:299–306 [DOI] [PubMed] [Google Scholar]
- 19. Gibbs JS, Regier DA, Desrosiers RC. 1994. Construction and in vitro properties of HIV-1 mutants with deletions in “nonessential” genes. AIDS Res. Hum. Retroviruses 10:343–350 [DOI] [PubMed] [Google Scholar]
- 20. Goila-Gaur R, Khan MA, Miyagi E, Kao S, Strebel K. 2007. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology 4:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goila-Gaur R, Strebel K. 2008. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harari A, Ooms M, Mulder LC, Simon V. 2009. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J. Virol. 83:295–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harris RS, et al. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809 [DOI] [PubMed] [Google Scholar]
- 24. Hlela C, Shepperd S, Khumalo NP, Taylor GP. 2009. The prevalence of human T-cell lymphotropic virus type 1 in the general population is unknown. AIDS Rev. 11:205–214 [PubMed] [Google Scholar]
- 25. Holmes RK, Koning FA, Bishop KN, Malim MH. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 282:2587–2595 [DOI] [PubMed] [Google Scholar]
- 26. Hultquist JF, et al. 2011. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85:11220–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Igakura T, et al. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716 [DOI] [PubMed] [Google Scholar]
- 28. Iwatani Y, et al. 2007. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 35:7096–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Izumo S, Umehara F, Osame M. 2000. HTLV-I-associated myelopathy. Neuropathology 20(Suppl.):S65–S68 [DOI] [PubMed] [Google Scholar]
- 30. Janini M, et al. 2001. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4+ T cells. J. Virol. 75:7973–7986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kao S, et al. 2004. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology 1:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE. 2007. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 3:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kijak GH, et al. 2008. Variable contexts and levels of hypermutation in HIV-1 proviral genomes recovered from primary peripheral blood mononuclear cells. Virology 376:101–111 [DOI] [PubMed] [Google Scholar]
- 34. Koning FA, et al. 2009. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 83:9474–9485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koralnik IJ, et al. 1994. Phylogenetic associations of human and simian T-cell leukemia/lymphotropic virus type I strains: evidence for interspecies transmission. J. Virol. 68:2693–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lecossier D, et al. 2003. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300:1112. [DOI] [PubMed] [Google Scholar]
- 37. Lee HH, et al. 1990. Patterns of HIV-1 and HTLV-I/II in intravenous drug abusers from the Middle Atlantic and central regions of the USA. J. Infect. Dis. 162:347–352 [DOI] [PubMed] [Google Scholar]
- 38. Li HC, et al. 2004. Provirus load in breast milk and risk of mother-to-child transmission of human T lymphotropic virus type I. J. Infect. Dis. 190:1275–1278 [DOI] [PubMed] [Google Scholar]
- 39. Li HC, et al. 1999. The presence of ancient human T-cell lymphotropic virus type I provirus DNA in an Andean mummy. Nat. Med. 5:1428–1432 [DOI] [PubMed] [Google Scholar]
- 40. Mahieux R, Pecon-Slattery J, Gessain A. 1997. Molecular characterization and phylogenetic analyses of a new, highly divergent simian T-cell lymphotropic virus type 1 (STLV-1marc1) in Macaca arctoides. J. Virol. 71:6253–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mahieux R, et al. 2005. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J. Gen. Virol. 86:2489–2494 [DOI] [PubMed] [Google Scholar]
- 42. Manel N, Battini JL, Taylor N, Sitbon M. 2005. HTLV-1 tropism and envelope receptor. Oncogene 24:6016–6025 [DOI] [PubMed] [Google Scholar]
- 43. Mangeat B, et al. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99–103 [DOI] [PubMed] [Google Scholar]
- 44. Mansky LM, et al. 2000. In vivo analysis of human T-cell leukemia virus type 1 reverse transcription accuracy. J. Virol. 74:9525–9531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mariani R, et al. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21–31 [DOI] [PubMed] [Google Scholar]
- 46. Marin M, et al. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9:1398–1403 [DOI] [PubMed] [Google Scholar]
- 47. Matsuoka M, Jeang KT. 2007. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 7:270–280 [DOI] [PubMed] [Google Scholar]
- 48. Mazurov D, Ilinskaya A, Heidecker G, Lloyd P, Derse D. 2010. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 6:e1000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mbisa JL, et al. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 81:7099–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mbisa JL, Bu W, Pathak VK. 2010. APOBEC3F and APOBEC3G inhibit HIV-1 DNA integration by different mechanisms. J. Virol. 84:5250–5259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mehle A, et al. 2004. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 279:7792–7798 [DOI] [PubMed] [Google Scholar]
- 52. Muckenfuss H, et al. 2006. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J. Biol. Chem. 281:22161–22172 [DOI] [PubMed] [Google Scholar]
- 53. Nagai M, Brennan MB, Sakai JA, Mora CA, Jacobson S. 2001. CD8(+) T cells are an in vivo reservoir for human T-cell lymphotropic virus type I. Blood 98:1858–1861 [DOI] [PubMed] [Google Scholar]
- 54. Naldini L, et al. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263–267 [DOI] [PubMed] [Google Scholar]
- 55. Narvaiza I, et al. 2009. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 5:e1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Navarro F, et al. 2005. Complementary function of the two catalytic domains of APOBEC3G. Virology 333:374–386 [DOI] [PubMed] [Google Scholar]
- 57. Newman EN, et al. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15:166–170 [DOI] [PubMed] [Google Scholar]
- 58. OhAinle M, Kerns JA, Li MMH, Malik HS, Emerman M. 2008. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4:249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ohsugi T, Koito A. 2007. Human T cell leukemia virus type I is resistant to the antiviral effects of APOBEC3. J. Virol. Methods 139:93–96 [DOI] [PubMed] [Google Scholar]
- 60. Okeoma CM, Huegel AL, Lingappa J, Feldman MD, Ross SR. 2010. APOBEC3 proteins expressed in mammary epithelial cells are packaged into retroviruses and can restrict transmission of milk-borne virions. Cell Host Microbe 8:534–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ooms M, Majdak S, Seibert CW, Harari A, Simon V. 2010. The localization of APOBEC3H variants in HIV-1 virions determines their antiviral activity. J. Virol. 84:7961–7969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ott DE. 2009. Purification of HIV-1 virions by subtilisin digestion or CD45 immunoaffinity depletion for biochemical studies. Methods Mol. Biol. 485:15–25 [DOI] [PubMed] [Google Scholar]
- 63. Pais-Correia AM, et al. 2010. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 16:83–89 [DOI] [PubMed] [Google Scholar]
- 64. Peng G, et al. 2007. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 110:393–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pichler K, et al. 2008. Strong induction of 4-1BB, a growth and survival promoting costimulatory receptor, in HTLV-1-infected cultured and patients' T cells by the viral Tax oncoprotein. Blood 111:4741–4751 [DOI] [PubMed] [Google Scholar]
- 66. Pichler K, Schneider G, Grassmann R. 2008. MicroRNA miR-146a and further oncogenesis-related cellular microRNAs are dysregulated in HTLV-1-transformed T lymphocytes. Retrovirology 5:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Poiesz BJ, Ruscetti FW, Reitz MS, Kalyanaraman VS, Gallo RC. 1981. Isolation of a new type C retrovirus (HTLV) in primary uncultured cells of a patient with Sezary T-cell leukaemia. Nature 294:268–271 [DOI] [PubMed] [Google Scholar]
- 68. Proietti FA, Carneiro-Proietti AB, Catalan-Soares BC, Murphy EL. 2005. Global epidemiology of HTLV-I infection and associated diseases. Oncogene 24:6058–6068 [DOI] [PubMed] [Google Scholar]
- 69. Refsland EW, et al. 2010. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38:4274–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ross SR. 2009. Are viruses inhibited by APOBEC3 molecules from their host species? PLoS Pathog. 5:e1000347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Roucoux DF, et al. 2005. A prospective study of sexual transmission of human T lymphotropic virus (HTLV)-I and HTLV-II. J. Infect. Dis. 191:1490–1497 [DOI] [PubMed] [Google Scholar]
- 72. Ruckes T, Saul D, Van Snick J, Hermine O, Grassmann R. 2001. Autocrine antiapoptotic stimulation of cultured adult T-cell leukemia cells by overexpression of the chemokine I-309. Blood 98:1150–1159 [DOI] [PubMed] [Google Scholar]
- 73. Rudolph DL, Yee J, Palker T, Coligan JE, Lal RB. 1993. Antibody responses to the env epitopes of human T-lymphotropic virus type I in rhesus macaques' naturally infected with simian T-lymphotropic virus type I. Res. Virol. 144:193–199 [DOI] [PubMed] [Google Scholar]
- 74. Sasada A, et al. 2005. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology 2:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schmitt K, et al. 2011. Differential virus restriction patterns of rhesus macaque and human APOBEC3A: implications for lentivirus evolution. Virology 419:24–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sheehy A, Gaddis N, Choi J, Malim M. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650 [DOI] [PubMed] [Google Scholar]
- 77. Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9:1404–1407 [DOI] [PubMed] [Google Scholar]
- 78. Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. 2010. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 17:222–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Suspène R, et al. 2011. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc. Natl. Acad. Sci. U. S. A. 108:4858–4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Suspène R, et al. 2011. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 85:7594–7602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Suspène R, et al. 2005. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 102:8321–8326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Suspène R, Henry M, Guillot S, Wain-Hobson S, Vartanian JP. 2005. Recovery of APOBEC3-edited human immunodeficiency virus G→A hypermutants by differential DNA denaturation PCR. J. Gen. Virol. 86:125–129 [DOI] [PubMed] [Google Scholar]
- 83. Tanaka G, et al. 2005. The clonal expansion of human T lymphotropic virus type 1-infected T cells: a comparison between seroconverters and long-term carriers. J. Infect. Dis. 191:1140–1147 [DOI] [PubMed] [Google Scholar]
- 84. Van Dooren S, Salemi M, Vandamme AM. 2001. Dating the origin of the African human T-cell lymphotropic virus type-I (HTLV-I) subtypes. Mol. Biol. Evol. 18:661–671 [DOI] [PubMed] [Google Scholar]
- 85. Van Prooyen N, et al. 2010. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc. Natl. Acad. Sci. U. S. A. 107:20738–20743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Vartanian JP, Guetard D, Henry M, Wain-Hobson S. 2008. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 320:230–233 [DOI] [PubMed] [Google Scholar]
- 87. Vartanian JP, Meyerhans A, Asjo B, Wain-Hobson S. 1991. Selection, recombination, and G→A hypermutation of human immunodeficiency virus type 1 genomes. J. Virol. 65:1779–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Waldele K, et al. 2006. Requirement of the human T-cell leukemia virus (HTLV-1) tax-stimulated HIAP-1 gene for the survival of transformed lymphocytes. Blood 107:4491–4499 [DOI] [PubMed] [Google Scholar]
- 89. Wang X, et al. 2011. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J. Virol. 85:3142–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wei X, et al. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wiegand HL, Cullen BR. 2007. Inhibition of alpharetrovirus replication by a range of human APOBEC3 proteins. J. Virol. 81:13694–13699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Worobey M, et al. 2008. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature 455:661–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yu X, et al. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060 [DOI] [PubMed] [Google Scholar]