

Abstract
Herpesviruses morphogenesis occurs stepwise both temporally and spatially, beginning in the nucleus and concluding with the emergence of an extracellular virion. The mechanisms by which these viruses interact with and penetrate the nuclear envelope and subsequent compartments of the secretory pathway remain poorly defined. In this report, a conserved viral protein (VP1/2; pUL36) that directs cytoplasmic stages of egress is identified to have multiple isoforms. Of these, a novel truncated VP1/2 species translocates to the nucleus and assists the transfer of DNA-containing capsids to the cytoplasm. The capsids are handed off to full-length VP1/2, which replaces the nuclear isoform on the capsids and is required for the final cytoplasmic stages of viral particle maturation. These results document that distinct VP1/2 protein species serve as effectors of nuclear and cytoplasmic egress.
INTRODUCTION
Herpesviruses, such as herpes simplex virus type I (HSV-1) and pseudorabies virus (PRV), begin their morphogenesis in the nucleus where 125-nm-diameter capsid shells are assembled and packaged with double-stranded DNA genomes (26, 69). These structures are too large to pass through the channel of a nuclear pore complex, indicating that the biology underlying the nuclear egress of these viruses occurs by a novel export mechanism that is probably not active in healthy cells (55, 75). Consistent with this view, a complex of two virally encoded proteins, pUL31 and pUL34, is critical for the efficient nuclear egress of capsids and is often referred to as the nuclear egress complex (35, 63, 66).
The nuclear egress process is made more remarkable by its inherent selectivity. Herpesvirus capsids assemble around a protein scaffold, which is replaced by DNA during encapsidation (4, 51, 58, 79). However, the fidelity of DNA packaging is somewhat low, with many capsids failing to stably encapsidate viral genomes. Failed capsids accumulate in at least two forms that are readily isolated from infected cell nuclei: those that never expel the protein scaffold (also known as B capsids) and those that expel the scaffold but do not retain full-length genomes (also known as A capsids) (8, 25, 57). Nevertheless, packaged nucleocapsids (also known as C capsids) predominate in extracellular viral particles, and this selectivity is imparted by an inability of A and B capsids to efficiently egress from the nucleus (13, 25, 52, 62, 65, 76, 89). The mechanism of nuclear egress selectivity is a topic of much discussion given that the obvious difference between the three capsid species is hidden inside the capsid shell. The HSV-1 and PRV pUL31 component of the nuclear egress complex binds to all three capsid species indiscriminately, further raising the question of how C capsids are selected for egress (40, 92). The prevailing model is that a C capsid-specific component must tag the exterior of capsids that are ready for egress (86).
Upon exiting the nucleus, viral tegument proteins are acquired onto cytosolic capsids and the resulting capsid/tegument complexes subsequently bud through modified intracellular membranes that contain viral membrane proteins and additional tegument proteins (27, 45). One protein bound to the capsid surface prior to envelopment is the large tegument protein, VP1/2 (pUL36) (31, 37). Recombinant HSV-1 or PRV lacking VP1/2 fails to undergo cytoplasmic envelopment, resulting in the accumulation of capsids in the cytosol (20, 22). These findings document the importance of VP1/2 in cytoplasmic envelopment, but they have also led to the interpretation that VP1/2-null viruses are not impaired in nuclear egress (20, 65, 67). While it is uncontested that VP1/2 is nonessential for nuclear egress, measuring nuclear egress efficiency in the absence of VP1/2 is complicated by the pleiotropic effect of the deletion: the accumulation of cytosolic capsids that cannot proceed to final envelopment could obscure a reduced rate of egress from the nucleus. Consistent with this, the most pronounced nuclear egress defect reported for a VP1/2-null virus was observed under conditions where the nuclear egress of PRV capsids had not yet become extensive (43). Similarly, although HSV-1 capsids were reported to egress from the nucleus unimpeded in the absence of VP1/2 based on accumulations of cytoplasmic capsids by 12 h postinfection (hpi), a decrease in the rate of capsid egress from the nucleus may have been evident at 8 hpi (20).
Here we report that the PRV VP1/2, which is critical for the budding of cytosolic capsids into membranes of the exocytic pathway, is also expressed as a second isoform that enters the nucleus, selectively binds C capsids, and enhances egress to the cytosol. Upon completion of nuclear egress a switch between the nuclear and cytoplasmic forms of capsid-bound VP1/2 occurs, consistent with the requirement for domains in the full-length protein in virion morphogenesis and release from the cell (9, 22, 39, 48).
MATERIALS AND METHODS
Cells.
Viruses were propagated in the pig kidney epithelial cell line PK15. PK15 cell lines stably expressing PRV pUL25 (PK15-UL25) or VP1/2 (PK15-UL36) were used to propagate recombinants of PRV deleted for the respective genes. Both cell lines were previously described (16, 43). Vero cells were used for isolation of intranuclear capsids, live-cell imaging, and quantitation of single-step virus propagation kinetics. PK15 and Vero cells were maintained in Dulbecco modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% bovine growth supplement (BGS) (HyClone); during infection, BGS levels were reduced to 2%. Embryonic chicken (E8-E10) dorsal root ganglion sensory neurons were used for time-lapse imaging of retrograde axon transport and were cultured as previously described (73).
Virus construction.
All viruses were derived from the pBecker3 infectious clone of PRV (strain Becker) (71). Fluorescent derivatives of pBecker3 that encode either green fluorescent protein (GFP) (PRV-GS443) or mRFP1 (PRV-GS847) fused to the VP26 capsid protein were previously described (73, 74). A derivative of PRV-GS443 deleted for the UL36 gene, PRV-GS678, was also described previously (43). Two dually fluorescent derivatives of pBecker3 were produced that encode both mRFP1-VP26 and GFP fused to the carboxyl terminus of VP1/2 (UL36-GFP). A two-step recombination strategy was used to insert the GFP coding sequence in frame at the 3′ end of UL36 (82). The primers used were 5′CTTCAACGTGGACCTATTTCAGGTCCGCCTGATTCTCGGTGTGAGCAAGGGCGAGGAGC and 5′ACCGCAGAGGCAAACAAGTTGGGTAATAAACAATTTATTACTTGTACAGCTCGTCCATGCCG. Following amplification of the pEP-EGFP-in plasmid (underlined sequences provided homology to the template) the PCR product was recombined into pGS847 and pGS2168, the latter of which is a previously described pBecker3 derivative that encodes monomeric RFP1 (mRFP1)-VP26 and contains a deletion of the UL25 open reading frame (ORF) (16). The 5′ portion of the primers directed recombination to the UL36 3′ coding sequence, and a second recombination event was directed by repeated sequences encoded in the EP-EGFP-in cassette (83). The recombinant of pGS847 encoding UL36-GFP was designated pGS1903, which produced PRV-GS1903 upon transfection of PK15 cells. The equivalent recombinant of pGS2168 was pGS3278, which produced PRV-GS3278 upon transfection of PK15-UL25 cells. A dually fluorescent derivative of PRV-GS847 encoding GFP fused to the amino terminus of the VP1/2 tegument protein (PRV-GS909) was described previously (42). To produce a virus encoding mCherry fused to the amino terminus of VP1/2 and GFP simultaneously fused to the carboxyl terminus (mCherry-VP1/2-GFP), the mCherry coding sequence was first inserted in frame into pBecker3 at the 5′ end of UL36 by two-step recombination using the pEP-mCherry-in template with primers 5′AAAGATTTTTCCCCCACGCGCGTGTGTTATTTCAGCCATGGTGAGCAAGGGCGAG and 5′CATACTGATTACGATAGCCGACGACCACCGCGTCGGCCGTCTTGTACAGCTCGTC (underlined sequence provided homology to pEP-mCherry-in) (5). The resulting bacterial artificial chromosome (BAC) infectious clone, pGS1662, was further modified by inserting the GFP coding sequence in frame at the 3′ end of UL36 as described above. These sequential recombination events resulted in pGS1919, which was subsequently transfected into PK15 cells to produce the virus PRV-GS1919. A summary of the recombinant strains of PRV used in this study is provided in Table 1.
Table 1.
Recombinant PRV strains
| Virus strain | Capsid reporter | VP1/2 allelea | Other mutation | Reference |
|---|---|---|---|---|
| PRV-GS443 | GFP-VP26 | WT | 73 | |
| PRV-GS678 | GFP-VP26 | Null (ΔUL36) | 43 | |
| PRV-GS847 | mRFP1-VP26 | WT | 74 | |
| PRV-GS909 | mRFP1-VP26 | GFP-VP1/2 | 42 | |
| PRV-GS1903 | mRFP1-VP26 | VP1/2-GFP | This study | |
| PRV-GS3278 | mRFP1-VP26 | VP1/2-GFP | ΔUL25 | This study |
| PRV-GS1919 | mCherry-VP1/2-GFP | This study |
WT, wild type.
Plasmids.
Two fragments of VP1/2 (encoded by the UL36 ORF; 3,095 codons) were isolated from PRV infectious clones and subcloned into mammalian expression plasmids as fusions to GFP. VP1/2N encoded GFP fused to the amino terminus of UL36 codons 2 to 2018, and VP1/2C encoded UL36 codons 2018 to 3095 fused to a carboxyl-terminal GFP (codon numbering was based on the PRV-Becker sequence; GenBank JF797219.1) (78). To construct the VP1/2N expression plasmid, the R6K plasmid pGS1292 was amplified with the primers 5′CCAAATAAAAAGATTTTTCCCCCACGCGCGTGTGTTATTTCAGCCGATTTTTATCGAATTCGTCATCCATATCACCACG and 5′ACTGATTACGATAGCCGACGACCACCGCGTCGGCCGTCAAAGCTTCCACATGTGGAATTCCCAT. Using RED recombination, the PCR product was inserted in front of the second codon of UL36 in a derivative of pBecker3 (pGS685) that was previously modified to encode a stop codon and a flippase recognition target (FRT) site as a combined 37-nucleotide (nt) insertion after codon 2018 of UL36 (38, 39). The UL36 allele was liberated by digestion with BsaBI, which cut the sequence derived from the forward primer (in italics in the primer sequence) upstream of UL36 and an endogenous site downstream of UL36. The released R6K-containing fragment was circularized with T4 DNA ligase, cloned in S17-1 λpir Escherichia coli following selection with ampicillin, and saved as pGS1756 (19). The UL36 allele was subcloned from pGS1756 into pEGFP-C1 (Clontech) as an in-frame fusion using HindIII and EcoRI sites (underlined in the primer sequences), resulting in pGS1766.
The VP1/2C expression plasmid was derived using a procedure analogous to that described above for VP1/2N with the following changes. The pGS1292 plasmid was amplified using primers 5′CTGGTGGACTCGCAGCTGGCGGTGTCGCGCGTGCTCTGGTGTACACCACATGTGGAATTCCCAT and 5′GAGCGTGAAGCCCGGGCGTCGCGGCGGCTCGGGCGGCGCCATAAGCTTGTCATCCATATCACCACG. The PCR product was recombined into pGS1903 (pBecker3 derivative that encodes a C-terminal GFP fusion to VP1/2; see above) in front of codon 2018 of UL36. A R6K fragment encoding codons 2018 to 3095 of UL36 fused to GFP was liberated by digestion with BsrGI, which cut the sequence derived from the forward primer (in italics) and a site endogenous to the 3′ GFP coding sequence. The circularized fragment was recovered as pGS1946. The VP1/2 region was subcloned from pGS1946 into pEGFP-N1 (Clontech) by digestion with HindIII (underlined) and BsrGI, resulting in pGS1952. A mCherry version of VP1/2C was constructed by the same procedure but using a recombinant BAC encoding mCherry fused to the 3′ end of UL36, resulting in expression plasmid pGS2213.
Single-step growth curves.
Quantitation of viral propagation kinetics by single-step growth curve analysis was conducted as previously described, with the exception that Vero cells were substituted for PK15 cells (81). Vero cells were infected at a multiplicity of infection (MOI) of 5. Viral titers from cells or medium supernatants harvested at 2, 5, 8, 12, or 24 hpi were determined in duplicate by plaque assay.
Fluorescence microscopy and image analysis.
Microscopy was performed on a Nikon TE2000-U inverted wide-field microscope with a 60× 1.4-numerical aperture (NA) oil objective (Nikon) and a Cascade:650 charge-coupled device (Photometrics). The microscope was housed in a 37°C environmental box (Life Imaging Services). Samples were maintained in coverslip chambers sealed with VALAB (Vaseline, lanolin, and beeswax) as previously described (73). MetaMorph software (Molecular Devices) was used for image acquisition and analysis. Time-lapse recordings of retrograde capsid motion in sensory axons within the first hour postinfection were captured by automated sequential imaging of mRFP1 and GFP emissions using continuous 200-ms exposures. Nuclear egress in Vero cells was analyzed by acquiring both fluorescence and differential interference contrast (DIC) still images of infected cells. The DIC images were used to identify the location of nuclei. Because fluorescent capsids in proximity to the nuclear membrane cannot be spatially resolved as cytoplasmic or nuclear, cells were scored as positive only if five or more fluorescent capsids were recognizably outside the nucleus (43).
Released particle assay.
The structural incorporation of viral proteins fused to GFP was measured in extracellular viral particles that emerged from infected cells and were imaged in situ, as described previously (42). Briefly, Vero cells seeded at low density were grown on coverslips and infected with dually fluorescent viral strains PRV-GS909 or PRV-GS1903. Three days postinfection, newly released extracellular viral particles were imaged on areas of the coverslip devoid of cells. The MetaMorph software package was used with a custom automated algorithm to identify capsids tagged with mRFP1-VP26 as diffraction-limited red fluorescent punctae, and the emissions of the coincident GFP signal were scored. A minimum of 1,000 viral particles were analyzed per sample. The percent incorporation was recorded as the proportion of extracellular viral particles that produced detectable GFP emissions.
Isolation of intranuclear capsids.
Intranuclear A, B, and C capsids were isolated from PK15 or Vero cells at 18 hpi following infection at an MOI of 10, as previously described (40). The capsid gradients were collected from top to bottom either as 18 continuous fractions or as 3 fractions specific to the A, B, and C light-scattering bands. In both cases, fractions were collected with a gradient fractionator (BioComp Instruments). For fluorescence microscopy experiments, the isolated capsids were spotted on glass coverslips, imaged, and analyzed for coincident RFP and GFP emissions as described for the released particle assay. For Western blot analysis, the samples were precipitated in a final trichloroacetic acid (TCA) concentration of 10% for 1 h on ice. The samples were pelleted in a refrigerated microcentrifuge for 10 min at 18,000 × g and resuspended in loading buffer (31.25 mM Tris [pH 6.8], 5% glycerol, 0.005% bromophenol blue, 1% sodium dodecyl sulfate, 5% β-mercaptoethanol) supplemented with 0.1 N NaOH.
Purification of extracellular virions.
Extracellular virions were purified from the supernatant of PK15 cells infected in one 850-cm2 roller bottle. Virions were pelleted through a 10% Nycodenz cushion made in phosphate-buffered saline by centrifuging for 1 h at 13,000 rpm at 4°C in a SW28 rotor. The pellet was resuspended in TNE buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 10 mM EDTA) and gently dispersed using a cup-horn sonicator. The material was then transferred to a 12 to 32% continuous dextran gradient made in TNE buffer with a gradient master (BioComp Instruments). After a 1-h centrifugation at 20,000 rpm at 4°C in a SW41 rotor, either the heavy particle (virion-containing) band was selectively extracted or the gradient was fractionated into 0.58-ml volumes using a gradient fractionator (BioComp Instruments). Fractions were precipitated with TCA and resuspended in loading buffer as described above.
Transient transfection.
To transfect Vero cells, 2 μg of plasmid DNA and 10 μl of Lipofectamine 2000 (Invitrogen) were incubated in 400 μl of DMEM for 20 min. The mixture was added to cells in a 10-cm plate. After 24 h, the cells were either harvested for Western blot analysis or split onto glass coverslips for imaging experiments. In a subset of experiments, the transfected cells were infected 16 h after the cell split.
Western blot analysis.
GFP-fusion proteins were detected in either Vero cells infected with PRV-GS847, PRV-GS909, or PRV-GS1903 or HEK293 cells transfected with pGS1766 or pGS1952. Cellular lysates were prepared by harvesting cells in 2× final sample buffer (62.5 mM Tris [pH 6.8], 10% glycerol, 0.01% bromophenol blue, 2% sodium dodecyl sulfate, 10% β-mercaptoethanol). The samples were boiled for 5 min and separated on 4 to 15% SDS-polyacrylamide gels. Two sets of antibodies were used to detect GFP. A mixture of clones 7.1 and 13.1 (Roche) was used for detection of GFP on Western blots of intranuclear capsids, and clone B-2 (Santa Cruz) was used to detect GFP-tagged proteins in purified virions and infected cell lysates. The monoclonal mouse antibody directed against PRV VP5 (clone 3C10) was provided by Lynn Enquist. Rabbit antiserum raised against a UL37 peptide has been previously described (39). To detect VP1/2 antigen, a rabbit polyclonal antibody was generated by immunization with the PRV VP1/2 peptide sequence CQPSQQRPPEAPWTWPEPRD. Secondary goat antibodies conjugated to horseradish peroxidase were acquired from Jackson ImmunoResearch. Horseradish peroxidase was detected with a luminol-coumaric acid-H2O2 chemiluminescence solution and exposed to film. Densitometry analysis was performed with ImageJ (3).
RESULTS
Initial characterization of viruses expressing fluorescent protein fusions to the large tegument protein.
VP1/2 homologs are essential for the propagation of alphaherpesviruses such as HSV-1 and PRV (20, 22, 36, 72). We previously reported that a recombinant of PRV (PRV-GS909) expressing RFP fused to the capsid surface (mRFP1-VP26) and GFP fused to the amino terminus of VP1/2 (GFP-VP1/2) possessed wild-type propagation kinetics and, therefore, was a good reagent for studying VP1/2 dynamics during infection. Using this virus, time-lapse fluorescence imaging of infected sensory neurons documented that the VP1/2 large tegument protein remains capsid associated during the long-distance transit from the distal axon to the nuclear membrane (42). There is increasing evidence that VP1/2 is proteolyzed in cells during infection (28, 30, 46, 61). To examine the localization of the carboxyl-terminal end of VP1/2 during infection, a recombinant of PRV was made that encodes mRFP1-VP26 and GFP fused in frame to the VP1/2 carboxyl terminus (VP1/2-GFP). The resulting virus, PRV-GS1903, propagated with wild-type kinetics and produced extracellular viral particles that had green emissions coincident with RFP-tagged capsids (RFP-capsids) (Fig. 1A and B and Table 1). Following infection of cultured primary sensory neurons with either reporter virus, RFP-tagged capsids transported retrograde in axons in association with GFP emissions (Fig. 1C). Because fusions of GFP to either end of VP1/2 were tolerated remarkably well in PRV, a recombinant virus was made that encoded two fluorescent tags on VP1/2: mCherry (RFP) on the amino terminus and GFP on the carboxyl terminus (PRV-GS1919). Like the viruses encoding either GFP-VP1/2 (PRV-GS909) or VP1/2-GFP (PRV-GS1903) fusions, PRV-GS1919 propagation did not substantially deviate from wild-type kinetics (Fig. 1A). Although capsids were not fluorescently tagged in PRV-GS1919 and could not be observed directly, both ends of VP1/2 cotransported in axons following infection of sensory neurons (Fig. 1C). These initial findings indicated that if proteolysis of VP1/2 occurs at these stages of infection, it does not result in a notable loss of either VP1/2 terminus from capsids or virions.
Fig 1.

Characterization of viruses encoding fluorescent proteins fused to VP1/2. (A) Single-step growth kinetics of PRV-GS847 (R-capsid), PRV-GS1903 (R-capsid + VP1/2-G) and PRV-GS1919 (R-VP1/2-G). Culture media (open symbols) or adherent cells (filled symbols) were harvested at the times indicated, and the titers were determined by plaque assay. PRV-GS847 was previously documented to propagate with wild-type kinetics (7, 42). (B) Percent incorporation of GFP fusions into released extracellular particles. Vero cells were infected with PRV-GS909 (R-capsid + G-VP1/2) or PRV-GS1903 (R-capsid + VP1/2-G). Released extracellular particles were detected as a homogenous population of diffraction-limited red fluorescent punctae and individually scored for the presence of a corresponding GFP signal. (C) Axon transport of PRV-GS909 (R-capsid + G-VP1/2), PRV-GS1903 (R-capsid + VP1/2-G), and PRV-GS1919 (R-VP1/2-G) in sensory neurons cultured from dorsal root ganglia. Green (G) and red (R) emissions were captured in alternating succession. All frames are 2.52 by 9.12 μm and are oriented with the axon terminal above the field.
Differential expression of VP1/2 fragments during infection.
Because GFP fusion to either end of VP1/2 did not alter PRV propagation kinetics, the recombinant PRV strains were used to identify VP1/2 species produced during infection. Lysates from cells infected with recombinant viruses were probed with antibodies against GFP. A high-molecular-weight band common to both lysates was consistent with full-length PRV VP1/2, which normally runs as a 330-kDa species (31, 46, 47). In addition, each virus produced a distinct profile of smaller bands (Fig. 2A). The ∼100-kDa band present in both samples was nonspecific, inasmuch that its presence was inconsistent and it was also sometimes observed from infections with PRV that did not express GFP (data not shown). Extracellular viral particles showed a dramatic enrichment for the full-length form of VP1/2, indicating that the smaller N- and C-terminal species that were abundant in infected cells were not efficiently packaged into virions (Fig. 2A).
Fig 2.

Distinct profiles of VP1/2 amino- and carboxyl-terminal GFP fusions. (A) Western blots of total cell lysates from Vero cells infected with PRV-GS909 (G-VP1/2) or PRV-GS1903 (VP1/2-G) were probed with an antibody directed against GFP. An antiserum directed against virally encoded pUL37 served as a loading control. Extracellular virions were probed to document the size of structurally incorporated VP1/2. Molecular size markers (in kDa) are indicated to the left of each blot. (B) Representative fluorescence images of live Vero cells infected with either PRV-GS909 (G-VP1/2) or PRV-GS1903 (VP1/2-G) at 7 to 8 hpi. Capsids and VP1/2 were imaged by the intrinsic fluorescence from the fusion proteins encoded by the viruses. DIC, differential interference contrast. (C) Example of a fluorescence profile from a live Vero cell nucleus following infection with PRV-GS1919 (R-VP1/2-G). The image is a color merge of GFP and mCherry (RFP) emissions. The yellow horizontal line overlaid on the image indicates a line scan used to measure pixel intensity values that are plotted below the image. Asterisks on the plot mark pixels intersecting the nuclear envelope.
To determine if the different species of VP1/2 observed by Western blotting were present in intact cells, infected cells were examined by live-cell imaging. During late infection (7 to 8 hpi), newly assembled mRFP1-capsids and mRFP1-capsid clusters were observed in the nuclei of cells. GFP-VP1/2 emissions from PRV-GS909 were diffusely distributed throughout the cytoplasm but were less evident in nuclei, consistent with previous reports (Fig. 2B) (39, 49). In contrast, VP1/2-GFP emissions from PRV-GS1903 were enriched at the nuclear membrane. The prevalence of the VP1/2 fusions at the nuclear envelope of infected cells was quantitated as part of a followup analysis (see below). The difference in localization between the VP1/2 terminal ends was also seen in cells infected with PRV-GS1919; the GFP-tagged C terminus of VP1/2 was observed at the nuclear envelope in the absence of the RFP-tagged amino terminus (Fig. 2C). These findings provided supporting evidence that the small forms of VP1/2 seen in cell lysates were present in intact cells and possessed distinct properties.
Carboxyl-terminal fragments of VP1/2 uniquely associate with nuclear capsids.
Intranuclear GFP emissions from cells infected with either GFP-VP1/2 or VP1/2-GFP viruses were typically not observed (Fig. 2BC). However, in cells infected with PRV-GS1903 (mRFP1-VP26 + VP1/2-GFP) in which several RFP-capsid assemblies aligned in a single focal plane, dim coincident emissions from VP1/2-GFP were often noted. Like nuclear envelope localization, GFP emissions from intranuclear capsids were evident with the VP1/2-GFP fusion expressed by PRV-GS1903 but not the GFP-VP1/2 fusion expressed by PRV-GS909 (Fig. 3A). Quantitative analysis of VP1/2 localization indicated that nearly half of all cells infected with PRV-GS1903 produced RFP-capsid assemblies with coincident VP1/2-GFP emissions (Fig. 3B). We cannot currently discriminate if the absence of VP1/2-GFP emissions from capsid assemblies in the majority of cells is a naturally occurring variable between infected cells or a technical limitation of detecting the dim VP1/2-GFP emissions from intranuclear capsid assemblies. Support for the latter theory comes from the difficulty in detecting the dim VP1/2-GFP emissions from intranuclear capsid assemblies; while slight adjustments in focus did not impact imaging of the bright RFP-capsid assemblies, emissions from coincident VP1/2-GFP could become easily overlooked. Cells that scored positive for intranuclear VP1/2-GFP emissions typically had several capsid assemblies in a single focal plane (Fig. 3A).
Fig 3.
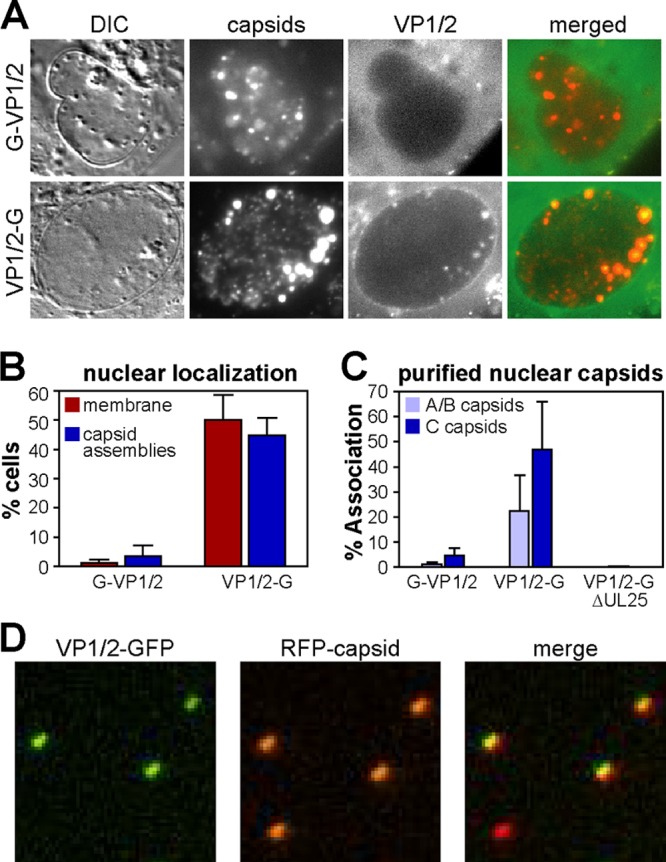
Nuclear localization of the VP1/2 carboxyl terminus. (A) Vero cells were infected with PRV-GS909 (G-VP1/2) or PRV-GS1903 (VP1/2-G). Images of infected Vero cell nuclei document the selective colocalization of VP1/2-G emissions with intranuclear capsid assemblies at 7 to 8 hpi. (B) Summary of GFP fluorescence localization at the nuclear membrane or with capsid assemblies in infected cells, as documented in panel A and in Fig. 2B (n = 202 for PRV-GS909; n = 332 for PRV-GS1903). The error bars represent the standard errors of the means. (C) Percent of RFP-capsids emitting GFP fluorescence following isolation of A/B and C capsids from the nuclei of infected Vero cells by rate zonal centrifugation. Only A/B capsid data are provided for the VP1/2-G + ΔUL25 sample, as C capsids were not recovered in the absence of the UL25 gene product. A minimum of 400 capsids were analyzed per sample. Each experiment was performed a minimum of three times. The error bars represent the standard errors of the means between experiments. (D) Subregion of an example image used to measure the frequency of VP1/2 association with isolated C capsids. Three of the four capsids shown were scored positive for GFP emissions, which in this example (PRV-GS1903) indicated the presence of the VP1/2 carboxyl terminus.
Whether VP1/2 interactions with capsids occur in the nucleus prior to egress to the cytosol is controversial (see Discussion). Therefore, capsids were purified from the nuclei of cells infected with PRV-GS909 (mRFP1-VP26 + GFP-VP1/2) and PRV-GS1903 (mRFP1-VP26 + VP1/2-GFP) by rate zonal centrifugation. Using this method, three types of capsids are isolated: A (empty), B (scaffold containing), and C (DNA containing) (8, 25, 57). A and B capsids sediment closely together from PRV-infected cells, and in these initial experiments the A and B capsids were harvested together (40). The isolated A/B and C capsids were each spotted onto coverslips and imaged by fluorescence microscopy (Fig. 3C). Because herpesvirus capsids are 125 nm in diameter, the RFP-tagged capsids produce diffraction-limited fluorescent punctae (Fig. 3D). Using an automated fluorescence detection algorithm, individual RFP-capsids were identified and the RFP and GFP emission profiles were recorded. Using a simple threshold to score RFP-capsids that were coincident with a GFP signal, the percentage of capsids associated with GFP was tallied for each sample. Although GFP-VP1/2 produced emissions from extracellular viral particles (Fig. 1B), the GFP signal was generally absent from isolated nuclear capsids (Fig. 3C). In contrast, the VP1/2-GFP fusion produced robust fluorescence from a subset of nuclear C capsids and was present to a lesser extent on A/B capsids. When fluorescence was present above the threshold, VP1/2-GFP emissions from individual A/B capsids were on average 40% the level of C capsids (n > 1,700 per sample). The relative occupancy of VP1/2 species on nuclear capsids is further examined below. The carboxyl terminus of VP1/2 contains a capsid-binding domain (CBD) that binds to the pUL25 capsid protein (16, 56). To determine if pUL25 was required for the interaction between the VP1/2 carboxyl terminus and capsids, intranuclear capsid isolation was repeated using a derivative of PRV-GS1903 (mRFP1-VP26 + VP1/2-GFP) that has a deletion of the UL25 gene. UL25 is required for stable DNA encapsidation, and as expected, C capsids were absent from the UL25-null sample (15, 44). Of greater relevance to the current study, the A/B capsids lacked VP1/2-GFP signal in the absence of UL25 (Fig. 3C).
To further examine the association of VP1/2 with nuclear capsids, capsid purification was repeated but the resulting sucrose gradients were separated into 18 0.58-ml fractions that were subsequently precipitated with trichloroacetic acid and assayed by Western blotting. Using an antibody directed against GFP, no reactivity was detected with nuclear capsids of the GFP-VP1/2 virus; however, a VP1/2-GFP species migrating at ≅85 kDa was associated with A/B and C capsid-containing fractions (Fig. 4A and B). Because GFP emissions were specifically associated with isolated capsids (Fig. 3D), we conclude that the cosedimentation is not coincidental but rather the result of an association of a carboxyl-terminal VP1/2 species with capsids isolated from infected cell nuclei. The capsid-associated species was smaller than the predominant carboxyl fragments observed in total cell lysates (Fig. 2A), indicating that either further processing had occurred or the fragment was labile from the physical stresses imposed during capsid isolation (50).
Fig 4.

Association of the VP1/2 carboxyl terminus with intranuclear capsids. Intranuclear capsids were harvested from infected Vero cells (A to C) or PK15 cells (D) and separated by rate zonal centrifugation. Fractions collected from the resulting sucrose gradient were precipitated with TCA and analyzed by Western blot. Fraction 1 represents the top of the gradient, and fraction 18 represents the bottom. Membranes were probed with antibodies directed against GFP, VP1/2, and the VP5 capsid protein. (A) PRV-GS909 (G-VP1/2). (B) PRV-GS1903 (VP1/2-G). (C and D) PRV-Becker (wild type). Peak fractions containing A/B or C capsids are indicated. (E) Extracellular PRV particles purified from supernatants of infected Vero cells were reacted with the VP1/2 antiserum. Molecular mass markers (kDa) are illustrated to the left of each panel.
The presence of a carboxyl-terminal fragment of VP1/2 on nuclear capsids conflicts with immunofluorescence data previously obtained with cells infected with PRV (49). Because the above-described experiments were carried out with recombinant PRV that expressed fluorescent proteins fused to the capsid and VP1/2, the presence of VP1/2 on nuclear capsids was reexamined in cells infected with wild-type PRV. For these experiments, an antibody was raised against a synthetic peptide derived from VP1/2 amino acids 2845 to 2863 (of 3095 amino acids total; GenBank no. JF797219.1). Consistent with the results obtained from cells infected with the fluorescent recombinants of PRV, a carboxyl-terminal species of VP1/2 cosedimented with capsids. Two bands between 50 and 75 kDa were detected (Fig. 4C). A reduction in the size of these bands relative to those seen with PRV-GS1903 was expected due to the absence of the GFP fusion. The presence of a carboxyl-terminal species was confirmed in two cell lines: Vero (Fig. 4C) and PK15 (Fig. 4D). Notably, in addition to the carboxyl-terminal species, a large VP1/2 species consistent with full-length protein was present throughout the gradient from infected PK15 cells. An equivalent high-molecular-weight reactive species was also observed throughout the Vero cell gradient on overexposed film, indicating its presence to a lesser degree (data not shown). Unlike the C-terminal nuclear isoform, the larger VP1/2 species was not restricted to capsid-containing fractions and enrichment in capsid-containing fractions appeared minimal. Taken together, these results show that the large VP1/2 species does not have the property of capsid-specific fractionation, but importantly its variable presence may underlie some of the mixed reports regarding if VP1/2 is present in the nucleus (see Discussion). Finally, to confirm that the antibody reacted with VP1/2, extracellular viral particles were isolated. As expected, the antibody detected a large VP1/2 species that was consistent with full-length VP1/2 (Fig. 4E).
Characterization of endogenous VP1/2 fragments.
To investigate the functional significance of the nuclear carboxyl-terminal VP1/2 fragments, subclones of VP1/2 fused to GFP were prepared and examined for migration in SDS-PAGE (data not shown). Two subclones produced VP1/2 fragments with apparent molecular weights equivalent to those of the endogenous fragments from total cell lysates of cells infected with PRV-GS909 (mRFP1-VP26 + GFP-VP1/2) and PRV-GS1903 (mRFP1-VP26 + VP1/2-GFP), respectively (Fig. 5A and B). The predominant amino-terminal subspecies seen in PRV-GS909-infected cells was approximated by a fragment, VP1/2N, that extended up to the beginning of the second proline-rich region of VP1/2 (amino acids 2 to 2018). Likewise, the predominant carboxyl-terminal subspecies produced during PRV-GS1903 infection was approximated by the counterpart fragment, VP1/2C, beginning at the second proline-rich region (amino acids 2018 to 3095). Taken together, these findings are consistent with a cleavage site(s) near the beginning of the second proline-rich region in the full-length protein resulting in the VP1/2 subspecies. Because multiple closely migrating VP1/2 subspecies can sometimes fail to resolve in denaturing gels (Fig. 2A), several closely juxtaposed cleavage sites may exist; further testing of this hypothesis will be necessary to better understand the genesis of the VP1/2 fragments.
Fig 5.

Identification of a recombinant VP1/2 fragment sharing properties with the native carboxyl-terminal isoform. (A) Illustration of full-length VP1/2 (top) and two cloned fragments (VP1/2N and VP1/2C) fused to GFP. DUB, deubiquitinase; PRR, proline-rich region; ARR, alanine-rich region; CBD, capsid binding domain; G, green fluorescent protein. (B) Vero cells were transiently transfected with VP1/2N or VP1/2C expression constructs, and total cell lysates were separated by SDS-PAGE. Protein migration was compared to that of endogenous VP1/2 species from Vero cells infected with PRV-GS909 (G-VP1/2) or PRV-GS1903 (VP1/2-G). Molecular size markers (in kDa) are indicated to the left of each blot. (C) Localization of GFP emissions from a Vero cell transiently expressing VP1/2C. (D) Nuclear GFP emissions from a Vero cell transiently expressing VP1/2C and subsequently infected with the RFP-capsid virus, PRV-GS847.
Transient expression of VP1/2C reproduced the nuclear envelope fluorescence previously seen in cells infected with PRV-GS1903 (Fig. 5C). Therefore, nuclear membrane localization is an intrinsic feature of the C-terminal fragment of PRV VP1/2 that does not require other viral proteins. Nuclear localization was restricted to the envelope in transfected cells, and no intranuclear punctae were observed. To determine if VP1/2C was capable of relocalizing to capsid assemblies, cells transiently transfected with VP1/2C were infected with a recombinant of PRV that expresses RFP-capsids (PRV-GS847) (74). At 8 to 9 h postinfection, intranuclear capsid assemblies associated with VP1/2C fluorescence became evident (Fig. 5D). These results indicated that VP1/2C recapitulated the behavior of the endogenous VP1/2 C-terminal fragments.
VP1/2C enhances nuclear egress.
We previously documented that PRV lacking VP1/2 is less effective at capsid egress from the nucleus than wild-type virus (43). To measure this deficit, a recombinant of PRV-expressing capsids fused to GFP (GFP-VP26; PRV-GS443) was scored for the presence of capsids in the cytoplasm of living cells by fluorescence microscopy at various times postinfection. This assay was reapplied here to test the hypothesis that association of the C-terminal VP1/2 species with intranuclear capsids was responsible for nuclear egress enhancement. For these experiments, Vero cells were transiently transfected with a plasmid encoding VP1/2C fused to the mCherry red fluorescent protein, which allowed for monitoring egress in transfected cells using the previously described GFP-capsid viruses encoding either wild-type VP1/2 or VP1/2 null. Cells were tallied for nuclear egress manually by scoring cells with five or more cytoplasmic capsids as positive, which reduced spurious counts from the final data sets. Cytoplasmic GFP-capsid emissions were diffraction-limited punctae, and no evidence of capsid clumping in the cytoplasm was observed. At 8 to 11 h postinfection, 16% of cells infected with the VP1/2 null mutant had cytoplasmic capsids compared with 81% for the wild-type virus. However, the percentage of the VP1/2 null mutant increased to 55% in cells expressing VP1/2C. Complementation became more effective at later times postinfection (11 to 14 h) although baseline egress of the null virus also increased (Fig. 6). These data demonstrate that VP1/2C functions as an enhancer of nuclear egress, and this activity functions independently of the first two-thirds of the protein. More broadly, VP1/2 is an accessory component of the herpesvirus nuclear egress machinery.
Fig 6.

Nuclear egress complementation. Vero cells were mock transfected or transfected with a plasmid encoding VP1/2C fused to the mCherry red fluorescent protein (indicated by row labeled VP1/2C). Cells were subsequently infected with recombinant PRV expressing GFP-tagged capsids and encoding either wild-type VP1/2 (indicated by row labeled WT VP1/2) or a deletion allele (indicated by row labeled VP1/2-null). Capsid nuclear egress was scored based on the presence of cytoplasmic capsids that were identified as fluorescent punctae. Cells were scored as positive if five or more capsids were visible within the cytoplasm. Two separate time periods were examined, 8 to 11 and 11 to 14 h postinfection.
A switch between VP1/2 isoforms occurs during virion morphogenesis.
Herpesvirus morphogenesis consists of an unusual biphasic egress pathway in which capsids interact with successive membrane-delineated compartments in the nucleus and the cytoplasm (45). While these studies demonstrate that the VP1/2 C-terminal isoform performs a supportive role during nuclear egress, the isoform lacks regions that are required for continued virion morphogenesis in the cytoplasm (1, 2, 39, 48). Because VP1/2 is essential for the cytoplasmic envelopment of capsids that have egressed from the nucleus, the C-terminal capsid-bound fragment presumably must be replaced by the full-length protein prior to cytoplasmic envelopment and exocytosis from the cell (20, 22, 65). The absence of the nuclear VP1/2 isoforms in virions is documented in Fig. 4E. To determine the relative levels of VP1/2 incorporation onto nuclear capsids and fully assembled virions, each type of viral particle was purified from PK15 cells infected with wild-type PRV. PK15 cells were used in place of Vero cells for these experiments because yields of extracellular viral particles from Vero cells were inadequate for the analysis. Extracellular viral particles were separated in a continuous dextran gradient and fractionated (Fig. 7A). Infectious virions sediment as capsid-containing heavy particles and were concentrated in fraction 11 based on the location of the VP5 major capsid protein. Light particles, which are devoid of capsids, were observed as a broad light-scattering band in the gradient above the heavy particles (77). Full-length VP1/2 was the sole species throughout the gradient, confirming that the C-terminal isoform of VP1/2 was removed from capsids prior to the cytoplasmic step of envelopment that gave rise to the extracellular particles. The material in fraction 11 was next used for comparison with nuclear capsid composition.
Fig 7.
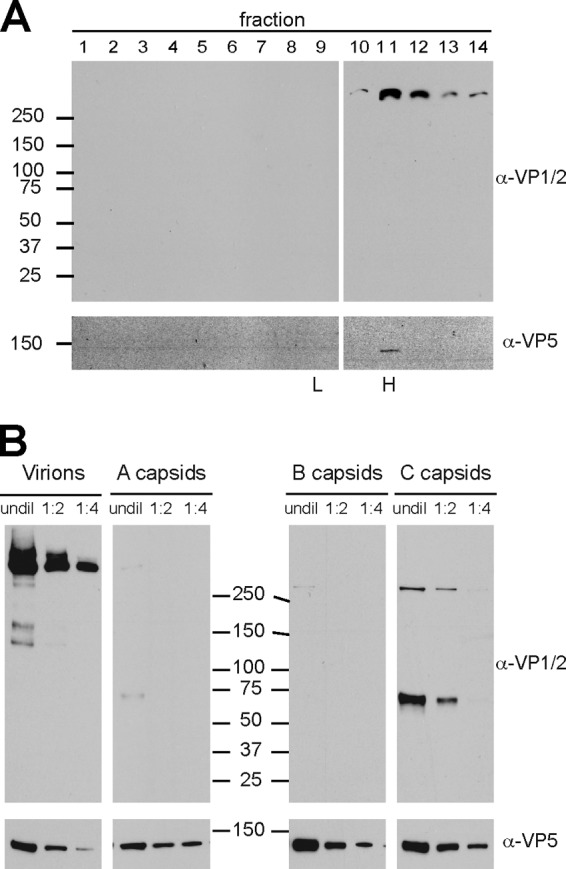
Relative incorporation levels of VP1/2 isoforms onto nuclear capsids and extracellular viral particles. (A) Extracellular viral particles from PK15 cells infected with wild-type PRV were separated through a 12 to 32% dextran gradient. Fractions were collected from the top, TCA precipitated, and blotted with antibodies directed against VP1/2 and the VP5 major capsid protein. Molecular size markers (in kDa) are indicated at left; peak fractions containing heavy (H) and light (L) viral particles observed as light-scattering bands in the gradient are indicated at bottom. (B) Heavy particles (virions) from fraction 11 of panel A were serially diluted 2-fold alongside preparations of intranuclear A, B, and C capsids from PK15 cells infected with wild-type PRV. The A, B, and C capsid light-scattering bands were harvested as three discrete fractions from a 20 to 50% sucrose gradient. The membranes were probed with antibodies directed against VP1/2 and the VP5 capsid protein, the latter serving as the loading control. Molecular size markers (in kDa) are shown down the middle to indicate the relative migration of the samples across two gels. undil, undiluted.
For these experiments, nuclear capsids were isolated following rate zonal centrifugation by selectively harvesting the A, B, and C light-scattering capsid bands, rather than fractionating the entire sucrose gradient. This afforded the discrete isolation of all three capsid species. Because these experiments were carried out with PK15 cells, full-length VP1/2 was present in these samples due to its diffuse distribution throughout the initial density gradient (Fig. 4D). Samples of each preparation were separated on denaturing gels and probed with the VP1/2 and VP5 antibodies, the latter serving as a loading control (Fig. 7B). The nuclear VP1/2 isoform was highly enriched on C capsids relative to the A and B capsid populations (Table 2). Despite the dramatic C capsid enrichment, full-length VP1/2 was present in extracellular H particles at an approximately 5-fold-greater copy number than the C capsid nuclear isoform. Based on cryoelectron microscopy (cryoEM) reconstructions of the herpesvirus capsid, there are five copies of pUL25 surrounding each vertex (60 copies total), and quantitative fluorescence microscopy indicates that the copy numbers of pUL25 and VP1/2 are closely matched in extracellular heavy particles that include virions (6, 17, 86). Given these observations, the nuclear VP1/2 isoform appears to occupy a subset of the available pUL25 binding sites present on C capsids, averaging approximately one copy per capsid vertex.
Table 2.
VP1/2:VP5 ratios based on densitometry of bands from Fig. 7B
| Virion or capsid group | Ratio at dilutiona |
||
|---|---|---|---|
| Undiluted | 1:2 | 1:4 | |
| Virions | 1.014 | 0.944 | 1.281 |
| A capsids | 0.012 | ND | ND |
| B capsids | 0.004 | ND | ND |
| C capsids | 0.400 | 0.210 | 0.011 |
ND, not detected.
DISCUSSION
Very little is known regarding how viruses that assemble inside the nucleus escape to the cytoplasm. Adenoviruses and the polyomavirus simian virus 40 (SV40) each encode proteins important for nuclear egress of viral particles that are suspected to function by lysing the nucleus and possibly the plasma membrane (18, 84, 85). The herpesviridae are large DNA viruses that assemble 125-nm icosahedral capsids in the nucleus (69). The capsids are exported to the cytosol, where they bud into membranes of the secretory pathway and nonlytically exocytose from the cell. Like adenovirus and SV40, herpesviruses encode proteins critical for nuclear egress. These include a capsid protein, pUL25, and a nuclear membrane complex consisting of the viral proteins pUL31 and pUL34 (35, 54, 63, 66). How these proteins coordinate nuclear egress of capsids is also poorly understood. Although lytic events and distortions of nuclear pores have been documented, the nuclear release of these viruses appears to predominantly occur by a budding mechanism (34, 41, 76). In support of the budding egress model, cooverexpression of nuclear egress proteins pUL31 and pUL34 results in the accumulation of vesicles between the inner and outer nuclear membranes, which may reflect an activity associated with budding during infection (21, 32).
Here we report that a protein previously recognized for its essential role in capsid envelopment in the cytoplasm, VP1/2, also participates in the nuclear egress of capsids and thereby coordinates the transition between nuclear and cytoplasmic steps in herpesvirus morphogenesis. Key to these findings was the observation that VP1/2 is expressed as multiple isoforms during late stages of infection, one of which functions in the nucleus. Although expression of VP1/2 fragments during infection has previously been observed under differing circumstances, a nuclear-specific isoform expressed as a consequence of normal infection had not been previously documented (30, 46, 61). The nuclear VP1/2 isoform was initially detected using a recombinant strain of PRV that encoded RFP-tagged capsids and GFP fused to the carboxyl end of VP1/2. This virus, PRV-GS1903, produced green fluorescence at the nuclear envelope upon infection. Closer examination of infected cells also revealed dim intranuclear GFP emissions that were localized to capsid clusters referred to as assemblons (90). The latter observation encouraged us to isolate nuclear capsids to look for the presence of a VP1/2 peptide. Rate zonal centrifugation was combined with fluorescence microscopy to confirm that a truncated carboxyl-terminal species of VP1/2 sedimented specifically as a capsid-bound protein.
The production of amino- and carboxyl-terminal fragments of VP1/2 during infection was consistent with proteolysis at sites near the beginning of the second proline-rich region (PRR2). Although proteolysis of VP1/2 was not examined as part of this report, the protein is cleaved at nuclear pores during early infection and cleavage in PRR2 can also occur (28, 46). Cleavage events may contribute to the fragments seen here during late infection given that the amino and carboxyl fragments were approximated by nonoverlapping contiguous portions of the protein that we referred to as VP1/2N and VP1/2C. VP1/2 itself encodes a cysteine protease in its amino terminus that functions as a deubiquitinase and deneddylase; however, a catalytically inactivating point mutation in PRV (C26A) did not prevent expression of the amino and carboxyl fragments during infection (data not shown) (24, 30, 68).
The amino-terminal VP1/2 fragments described here are likely related to a previously reported 220-kDa fragment that can assemble into viral particles to a small degree (46, 61). The amino-terminal half of VP1/2 includes binding sites for the VP16 and pUL37 tegument proteins, which is consistent with structural incorporation of the fragment into viral particles by coassembly with the tegument and envelope components of virions (31, 88). In contrast, the carboxyl-terminal isoform lacks these sites but includes a binding site for the pUL25 capsid protein (16, 56). The carboxyl-terminal isoforms had the novel property of localizing to the nuclear membrane and intranuclear capsid assemblies. Nuclear membrane localization was intrinsic to the VP1/2C fragment, whereas assembly on intranuclear capsids was dependent upon pUL25.
The interaction of the carboxyl VP1/2 fragment with pUL25 is noteworthy. DNA-containing C capsids preferentially egress from nuclei, indicating that nuclear egress and DNA encapsidation are coupled (13, 52, 62, 65, 76, 89). The pUL25 minor capsid protein is enriched on C capsids and may therefore contribute to C capsid selectivity (14, 29, 53, 80, 86). In yeast two-hybrid analysis pUL25 interacts with pUL15, pUL31, and VP1/2 (87). pUL15 is a component of the viral terminase that packages DNA into capsids; this potentially interesting interaction has not been pursued (59). The pUL25-pUL31 interaction has been reciprocated by coimmunoprecipitation, but mapping of the pUL25 binding site in pUL31 proved challenging as multiple nonoverlapping fragments bind pUL25, calling into question the specificity of the interaction (40). In parallel HSV-1 and PRV studies, pUL31 was determined to bind capsids, but this interaction was reported to be pUL25 dependent in HSV-1 and pUL25 independent in PRV-infected cells (40, 92). While the role of pUL25 in pUL31 binding to capsids will require further investigation, the studies were consistent in the finding that pUL31 association with capsids was not C capsid enriched. The absence of C capsid-selective binding is consistent with reports that pUL31 contributes to DNA encapsidation, which begins prior to pUL25 capsid enrichment (12, 60, 70, 80). In sharp contrast to pUL31, the carboxyl VP1/2 fragment exhibited dramatic C capsid-selective binding. pUL25 is essential for the nuclear egress of capsids, and based on the current finding that VP1/2C is sufficient to complement the reduction in nuclear egress efficiency associated with the VP1/2-null virus, a portion of the pUL25 activity appears to be exerted through its interaction with the nuclear VP1/2 isoform (43). Taken together, these findings support a role for pUL31 in initial anchoring of capsids to the nuclear egress complex in the inner nuclear membrane and assisting with encapsidation, followed by pUL25 addition to the capsid, recruitment of the VP1/2 carboxyl fragment, and C capsid-selective egress from the nucleus.
Once C capsids leave the nucleus, domains of VP1/2 that are absent in the carboxyl fragment are essential to continue viral assembly in the cytoplasm and produce infectious virions (1, 2, 39, 48). We infer that a near total replacement of carboxyl fragments with full-length VP1/2 occurs prior to cytoplasmic envelopment based on the VP1/2 composition of extracellular viral particles. Given that both full-length and carboxyl fragments of VP1/2 share the same pUL25 capsid binding domain, the mechanism by which the exchange occurs is not immediately obvious; however, the completeness of the exchange is evidence of a directed process (16). Full-length VP1/2 has a second pUL25 binding site which could assist in the exchange process (56). The noted ≅5-fold-higher copy number of VP1/2 in virions than on C capsids may also provide an indication of how the swap occurs.
Prior to this report, the majority of available evidence indicated that VP1/2 functions as a cytoplasmic molecule. VP1/2 is required for delivery of viral genomes into the nucleus during initial infection and for cytoplasmic envelopment postreplication; both of these events are essential for viral propagation (1, 6, 20, 22, 36, 65). Reports of VP1/2 on nuclear capsids have differed. Studies of HSV-1 have mostly failed to detect full-length VP1/2 on nuclear capsids (50, 86, 91). In some studies, the antibodies used were directed against amino-terminal fragments of VP1/2 that would not be expected to detect the nuclear VP1/2 isoform described here (10, 31, 65, 67, 91). However, evidence of HSV-1 VP1/2 in the nucleus and on C capsids was noted in a subset of studies (2, 11). For studies employing nuclear fractionation, the detection of full-length VP1/2 may be confounded by the propensity for VP1/2 to smear through density gradients, including, but not specific to, fractions containing capsids (Fig. 4D). The amino terminus of VP1/2 can be observed associated with the nuclear membrane in a manner that is dependent upon the time postinfection, which may be a source of its presence in density gradients of nuclear extracts (7). Three studies have used antibodies raised against peptides derived from sequences that reside within the VP1/2C fragment. Interestingly, two of these studies examined HSV-1 and reported the detection of small C-terminal fragments that may be consistent with those reported here (28, 61). However, the third study, which examined PRV, failed to detect nuclear reactivity by immunofluorescence, and by immunoelectron microscopy no VP1/2 was detected on capsids resident in the lumen of the nuclear envelope (49). The antibody used in the latter study was raised against the pUL25 binding sequence at the extreme tail end of the carboxyl terminus and reacted with extracellular virions by immunoelectron microscopy. Based on our finding that virions contain ≅5-fold more VP1/2 than do nuclear C capsids, one possible explanation is that the nuclear signal was below the detection threshold for these experiments. Detection of the VP1/2 pUL25 binding domain may have also been obscured if the protein was bound to capsids, and it would be of interest to see if the antibody recognized the VP1/2 nuclear isoform on isolated capsids when assayed by Western blotting.
The most common approach to assessing defects in the nuclear egress step of herpesvirus morphogenesis has been to examine cell sections by transmission electron microscopy. In the case of viruses deleted for the genes encoding pUL25, pUL31, and pUL34, which are critical for the nuclear egress of capsids, the defect is immediately evident (23, 33, 35, 54, 64, 66). However, the defect observed in the absence of VP1/2 is not absolute (43). Furthermore, because VP1/2 performs multiple functions during infection, a null mutation will exert pleiotropic effects during infection. Postreplication, capsids egress from the nucleus with decreased kinetics in the absence of VP1/2, but the capsids nevertheless accumulate to unusually large numbers in the cytoplasm due to the absolute loss of secondary envelopment. A pleiotropic egress effect in HSV-1- and PRV-infected cells may have been previously documented but overlooked (20, 22). At late times postinfection, finding the cell cytoplasm filled with capsids is not unexpected but could lead to the premature conclusion that there is no nuclear egress defect.
In summary, we have identified a novel VP1/2 isoform that functions as an accessory nuclear egress factor. To our knowledge, the carboxyl VP1/2 isoform is the first protein found with the property of selective C capsid enrichment that is not itself a capsid component. VP1/2 is the first viral protein found to actively contribute to both nuclear and cytoplasmic stages of egress, and further dissection of the molecular interactions between VP1/2, capsids, and the nuclear egress machinery will undoubtedly shed light on the process of C capsid selectivity and the coordination between nuclear and cytoplasmic stages of herpesvirus morphogenesis.
ACKNOWLEDGMENTS
We thank Jenifer Klabis for contributions toward the production of recombinant PRV strains and Kevin Bohannon and Oana Maier for their critical feedback on the manuscript. The PRV VP5 antibody was a gift from Lynn Enquist.
This work was funded by NIH grant R01 AI080658 to G.A.S.
Footnotes
Published ahead of print 21 March 2012
REFERENCES
- 1. Abaitua F, Daikoku T, Crump CM, Bolstad M, O'Hare P. 2011. A single mutation responsible for temperature-sensitive entry and assembly defects in the VP1-2 protein of herpes simplex virus. J. Virol. 85:2024–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abaitua F, O'Hare P. 2008. Identification of a highly conserved, functional nuclear localization signal within the N-terminal region of herpes simplex virus type 1 VP1-2 tegument protein. J. Virol. 82:5234–5244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abramoff MD, Magalhaes PJ. 2004. Image processing with ImageJ. Biophotonics Int. 11:36–42 [Google Scholar]
- 4. Addison C, Rixon FJ, Preston VG. 1990. Herpes simplex virus type 1 UL28 gene product is important for the formation of mature capsids. J. Gen. Virol. 71(Pt 10):2377–2384 [DOI] [PubMed] [Google Scholar]
- 5. Antinone SE, Smith GA. 2010. Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J. Virol. 84:1504–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Batterson W, Furlong D, Roizman B. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol. 45:397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bohannon KP, Sollars PJ, Pickard GE, Smith GA. 2012. Fusion of a fluorescent protein to the pUL25 minor capsid protein of pseudorabies virus allows live-cell capsid imaging with negligible impact on infection. J. Gen. Virol. 93:124–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Booy FP, et al. 1991. Liquid-crystalline, phage-like packing of encapsidated DNA in herpes simplex virus. Cell 64:1007–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Böttcher S, et al. 2007. Identification of functional domains within the essential large tegument protein pUL36 of pseudorabies virus. J. Virol. 81:13403–13411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bottcher S, et al. 2006. Identification of a 709-amino-acid internal nonessential region within the essential conserved tegument protein (p)UL36 of pseudorabies virus. J. Virol. 80:9910–9915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bucks MA, O'Regan KJ, Murphy MA, Wills JW, Courtney RJ. 2007. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361:316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang YE, Van Sant C, Krug PW, Sears AE, Roizman B. 1997. The null mutant of the U(L)31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J. Virol. 71:8307–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Church GA, Wilson DW. 1997. Study of herpes simplex virus maturation during a synchronous wave of assembly. J. Virol. 71:3603–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cockrell SK, Huffman JB, Toropova K, Conway JF, Homa FL. 2011. Residues of the UL25 protein of herpes simplex virus that are required for its stable interaction with capsids. J. Virol. 85:4875–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cockrell SK, Sanchez ME, Erazo A, Homa FL. 2009. Role of the UL25 protein in herpes simplex virus DNA encapsidation. J. Virol. 83:47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coller KE, Lee JI, Ueda A, Smith GA. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 81:11790–11797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conway JF, et al. 2010. Labeling and localization of the herpes simplex virus capsid protein UL25 and its interaction with the two triplexes closest to the penton. J. Mol. Biol. 397:575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Daniels R, Sadowicz D, Hebert DN. 2007. A very late viral protein triggers the lytic release of SV40. PLoS Pathog. 3:e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Lorenzo V, Timmis KN. 1994. Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 235:386–405 [DOI] [PubMed] [Google Scholar]
- 20. Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608–11618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Desai PJ, Pryce EN, Henson BW, Luitweiler E, Cothran J. 2012. Reconstitution of the Kaposi's sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by co-expression of ORF67 and ORF69 gene products. J. Virol. 86:594–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fuchs W, Klupp BG, Granzow H, Mettenleiter TC. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 78:11879–11889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76:364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gastaldello S, et al. 2010. A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-RING ligases. Nat. Cell Biol. 12:351–361 [DOI] [PubMed] [Google Scholar]
- 25. Gibson W, Roizman B. 1972. Proteins specified by herpes simplex virus. 8. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 10:1044–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Granzow H, et al. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 75:3675–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 28. Jovasevic V, Liang L, Roizman B. 2008. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 82:3311–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaelin K, Dezelee S, Masse MJ, Bras F, Flamand A. 2000. The UL25 protein of pseudorabies virus associates with capsids and localizes to the nucleus and to microtubules. J. Virol. 74:474–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, Ploegh HL. 2005. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 19:547–557 [DOI] [PubMed] [Google Scholar]
- 31. Klupp BG, Fuchs W, Granzow H, Nixdorf R, Mettenleiter TC. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 76:3065–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klupp BG, et al. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. U. S. A. 104:7241–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klupp BG, Granzow H, Keil GM, Mettenleiter TC. 2006. The capsid-associated UL25 protein of the alphaherpesvirus pseudorabies virus is nonessential for cleavage and encapsidation of genomic DNA but is required for nuclear egress of capsids. J. Virol. 80:6235–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klupp BG, Granzow H, Mettenleiter TC. 2011. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J. Virol. 85:8285–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klupp BG, Granzow H, Mettenleiter TC. 2000. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 74:10063–10073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knipe DM, Batterson W, Nosal C, Roizman B, Buchan A. 1981. Molecular genetics of herpes simplex virus. VI. Characterization of a temperature-sensitive mutant defective in the expression of all early viral gene products. J. Virol. 38:539–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ko DH, Cunningham AL, Diefenbach RJ. 2010. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J. Virol. 84:1397–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee EC, et al. 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56–65 [DOI] [PubMed] [Google Scholar]
- 39. Lee JI, Luxton GW, Smith GA. 2006. Identification of an essential domain in the herpesvirus VP1/2 tegument protein: the carboxy terminus directs incorporation into capsid assemblons. J. Virol. 80:12086–12094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leelawong M, Guo D, Smith GA. 2011. A physical link between the pseudorabies virus capsid and the nuclear egress complex. J. Virol. 85:11675–11684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leuzinger H, et al. 2005. Herpes simplex virus 1 envelopment follows two diverse pathways. J. Virol. 79:13047–13059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Luxton GW, et al. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U. S. A. 102:5832–5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80:201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McNab AR, et al. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J. Virol. 72:1060–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 46. Michael K, Bottcher S, Klupp BG, Karger A, Mettenleiter TC. 2006. Pseudorabies virus particles lacking tegument proteins pUL11 or pUL16 incorporate less full-length pUL36 than wild-type virus, but specifically accumulate a pUL36 N-terminal fragment. J. Gen. Virol. 87:3503–3507 [DOI] [PubMed] [Google Scholar]
- 47. Michael K, Klupp BG, Mettenleiter TC, Karger A. 2006. Composition of pseudorabies virus particles lacking tegument protein US3, UL47, or UL49 or envelope glycoprotein E. J. Virol. 80:1332–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mohl BS, et al. 2010. Random transposon-mediated mutagenesis of the essential large tegument protein pUL36 of pseudorabies virus. J. Virol. 84:8153–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mohl BS, et al. 2009. Intracellular localization of the pseudorabies virus large tegument protein pUL36. J. Virol. 83:9641–9651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Newcomb WW, Brown JC. 2010. Structure and capsid association of the herpesvirus large tegument protein UL36. J. Virol. 84:9408–9414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Newcomb WW, Brown JC. 1991. Structure of the herpes simplex virus capsid: effects of extraction with guanidine hydrochloride and partial reconstitution of extracted capsids. J. Virol. 65:613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Newcomb WW, Brown JC. 2009. Time-dependent transformation of the herpesvirus tegument. J. Virol. 83:8082–8089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ogasawara M, Suzutani T, Yoshida I, Azuma M. 2001. Role of the UL25 gene product in packaging DNA into the herpes simplex virus capsid: location of UL25 product in the capsid and demonstration that it binds DNA. J. Virol. 75:1427–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. O'Hara M, et al. 2010. Mutational analysis of the herpes simplex virus type 1 UL25 DNA packaging protein reveals regions that are important after the viral DNA has been packaged. J. Virol. 84:4252–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pante N, Kann M. 2002. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol. Biol. Cell 13:425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 83:6610–6623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Perdue ML, Cohen JC, Kemp MC, Randall CC, O'Callaghan DJ. 1975. Characterization of three species of nucleocapsids of equine herpesvirus type-1 (EHV-1). Virology 64:187–204 [DOI] [PubMed] [Google Scholar]
- 58. Perdue ML, Cohen JC, Randall CC, O'Callaghan DJ. 1976. Biochemical studies of the maturation of herpesvirus nucleocapsid species. Virology 74:194–208 [PubMed] [Google Scholar]
- 59. Poon AP, Roizman B. 1993. Characterization of a temperature-sensitive mutant of the UL15 open reading frame of herpes simplex virus 1. J. Virol. 67:4497–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Popa M, et al. 2010. Dominant negative mutants of the murine cytomegalovirus M53 gene block nuclear egress and inhibit capsid maturation. J. Virol. 84:9035–9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Radtke K, et al. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 6:e1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Remillard-Labrosse G, Guay G, Lippe R. 2006. Reconstitution of herpes simplex virus type 1 nuclear capsid egress in vitro. J. Virol. 80:9741–9753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Reynolds AE, et al. 2001. UL31 and UL34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 75:8803–8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Roberts AP, et al. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 83:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. 2000. Herpes simplex virus type 1 UL34 gene product is required for viral envelopment. J. Virol. 74:117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schipke J, et al. 2012. The C-terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 86:3682–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schlieker C, Korbel GA, Kattenhorn LM, Ploegh HL. 2005. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J. Virol. 79:15582–15585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schrag JD, Prasad BV, Rixon FJ, Chiu W. 1989. Three-dimensional structure of the HSV1 nucleocapsid. Cell 56:651–660 [DOI] [PubMed] [Google Scholar]
- 70. Sheaffer AK, et al. 2001. Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J. Virol. 75:687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Smith GA, Enquist LW. 2000. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 97:4873–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Smith GA, Enquist LW. 1999. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J. Virol. 73:6405–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Smith GA, Gross SP, Enquist LW. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. U. S. A. 98:3466–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Smith GA, Pomeranz L, Gross SP, Enquist LW. 2004. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. U. S. A. 101:16034–16039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Solmaz SR, Chauhan R, Blobel G, Melcak I. 2011. Molecular architecture of the transport channel of the nuclear pore complex. Cell 147:590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stackpole CW. 1969. Herpes-type virus of the frog renal adenocarcinoma. I. Virus development in tumor transplants maintained at low temperature. J. Virol. 4:75–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Szilagyi JF, Cunningham C. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 72(Pt 3):661–668 [DOI] [PubMed] [Google Scholar]
- 78. Szpara ML, et al. 2011. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 7:e1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Thomsen DR, Roof LL, Homa FL. 1994. Assembly of herpes simplex virus (HSV) intermediate capsids in insect cells infected with recombinant baculoviruses expressing HSV capsid proteins. J. Virol. 68:2442–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Thurlow JK, et al. 2005. The herpes simplex virus type 1 DNA packaging protein UL17 is a virion protein that is present in both the capsid and the tegument compartments. J. Virol. 79:150–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tirabassi RS, Enquist LW. 1998. Role of envelope protein gE endocytosis in the pseudorabies virus life cycle. J. Virol. 72:4571–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430 [DOI] [PubMed] [Google Scholar]
- 83. Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 84. Tollefson AE, Ryerse JS, Scaria A, Hermiston TW, Wold WS. 1996. The E3-11.6-kDa adenovirus death protein (ADP) is required for efficient cell death: characterization of cells infected with adp mutants. Virology 220:152–162 [DOI] [PubMed] [Google Scholar]
- 85. Tollefson AE, et al. 1996. The adenovirus death protein (E3-11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J. Virol. 70:2296–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Trus BL, et al. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol. Cell 26:479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Uetz P, et al. 2006. Herpesviral protein networks and their interaction with the human proteome. Science 311:239–242 [DOI] [PubMed] [Google Scholar]
- 88. Vittone V, et al. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566–9571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Vlazny DA, Kwong A, Frenkel N. 1982. Site-specific cleavage/packaging of herpes simplex virus DNA and the selective maturation of nucleocapsids containing full-length viral DNA. Proc. Natl. Acad. Sci. U. S. A. 79:1423–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ward PL, Ogle WO, Roizman B. 1996. Assemblons: nuclear structures defined by aggregation of immature capsids and some tegument proteins of herpes simplex virus 1. J. Virol. 70:4623–4631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wolfstein A, et al. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227–237 [DOI] [PubMed] [Google Scholar]
- 92. Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc. Natl. Acad. Sci. U. S. A. 108:14276–14281 [DOI] [PMC free article] [PubMed] [Google Scholar]