

Abstract
The endoplasmic reticulum (ER) resident PKR-like kinase (PERK) is necessary for Akt activation in response to ER stress. We demonstrate that PERK harbors intrinsic lipid kinase, favoring diacylglycerol (DAG) as a substrate and generating phosphatidic acid (PA). This activity of PERK correlates with activation of mTOR and phosphorylation of Akt on Ser473. PERK lipid kinase activity is regulated in a phosphatidylinositol 3-kinase (PI3K) p85α-dependent manner. Moreover, PERK activity is essential during adipocyte differentiation. Because PA and Akt regulate many cellular functions, including cellular survival, proliferation, migratory responses, and metabolic adaptation, our findings suggest that PERK has a more extensive role in insulin signaling, insulin resistance, obesity, and tumorigenesis than previously thought.
INTRODUCTION
The cellular membrane phospholipid phosphatidylinositol (PtdIns) and its metabolites are critical signaling molecules. PtdIns is synthesized at the endoplasmic reticulum (ER) membrane (1) and can then be phosphorylated at the 3, 4, and 5 positions of the inositol ring, generating a variety of monophosphorylated metabolites. These metabolites serve as precursors for additional phosphorylation events that result in the generation of PtdInsP2 and PtdInsP3 (10, 42, 48). PtdIns(4,5)P2 is in turn hydrolyzed by phospholipase C (PLC), generating diacylglycerol (DAG) and inositol-1,4,5-triphosphate, resulting in the formation of additional molecules capable of intracellular signaling (38). PtdIns(3,4,5)P3 (PIP3) is generated by the PtdIns3-kinase (PI3K) superfamily of lipid kinases (16). PI3K activity and PIP3 production are regulated by growth factors and chemokines, leading to the activation of Akt, one of the key growth and survival pathways in the cell. Additionally, generation of phosphatidic acid via the mitogen-stimulated activation of phospholipase D (PLD) provides another signal promoting Akt activation due to the phosphatidic acid-dependent assembly of the mTORC2 complex (18, 49).
PI3K class IA is composed of a 110-kDa catalytic subunit (p110) and an 85-kDa adaptor/regulatory subunit (p85). Mammalian cells have three p110 isoforms (p110α, -β, and -δ), encoded by three separate genes and at least seven adaptor proteins that are generated through alternative splicing of transcripts encoded by three distinct genes, p85α, p85β, and p55γ. The p85 subunit has two Src homology 2 (SH2) domains that dock with phosphorylated tyrosine residues generated by activated tyrosine kinases (3, 11). The p85 SH2 domain mediates recruitment of the cytosolic PI3Ks to cellular membranes where their lipid substrates reside. The p110 subunit-binding site within p85 is located between the two SH2 domains (inter-SH2 domain) (13, 21).
The ER transmembrane serine/threonine kinase PERK, or EIF2AK3 (26, 45), is activated under conditions of physiological ER stress such as low carbon source (glucose deprivation), low oxygen (hypoxia), or increased synthetic demand in secretory tissues, as well as by chemical inducers of ER stress (tunicamycin, thapsigargin). PERK-dependent phosphorylation of eIF2α triggers decreased protein synthesis to alleviate ER client protein load while simultaneously increasing production of key protein substrates necessary for cell adaptation (26). PERK also maintains cellular redox homeostasis via direct phosphorylation of the Nrf2 transcription factor (12). Moreover, PERK has been linked to the activation of PI3K signaling and Akt during conditions of ER stress (25, 29, 31). PKR, another eIF2α kinase, can also stimulate Akt phosphorylation (31). Since these two kinases share eIF2α as their substrate, it was proposed that PI3K activation is dependent on the effect of PERK/PKR on translation attenuation.
Here, we present evidence that PERK possesses an intrinsic lipid kinase activity toward diacylglycerol (DAG), generating phosphatidic acid (PA) as a major product. Additional evidence is provided demonstrating that PERK lipid kinase activity is regulated in a PI3K regulatory subunit p85α-dependent manner. The lipid kinase function of PERK mediates mTOR, Akt, and Erk1/2 activation during ER stress. Critically, the activity of PERK is necessary for the activation of anabolic pathways downstream of Akt in a physiologically relevant setting.
MATERIALS AND METHODS
Cell culture conditions, transfections, and infections.
Mouse embryo fibroblasts (MEFs) were grown in Dulbecco's modified Eagle's medium (DMEM) (high glucose formulation) with 4 mM l-glutamine, 10% (vol/vol) fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 55 mM β-mercaptoethanol. H1299 cells were grown in RPMI 1640 medium with 10% (vol/vol) fetal bovine serum. PERKLoxP/LoxP MEFs were derived as previously described (7) and immortalized using 3T9 protocol. Transient expression of plasmids encoding Myc-PERK and Myc-K618APERK, HA-p85α and FLAG-p110α, was achieved using Lipofectamine Plus (Invitrogen). For MEF in vitro differentiation, cells were grown to confluence in 60-cm2 dishes and then treated with adipocyte differentiation cocktail as described previously (8). Small molecule inhibitors used were as follows: FIPI (Cayman Chemical); rapamycin (Calbiochem); PIK-294, IC-87114, and MK-2206 (Selleck Chem Company); and wortmannin (Millipore). Compounds were added directly to cell culture media at the concentrations indicated.
Construction of PERK mutants.
PERK deletion mutants were generated by site-directed mutagenesis with the QuikChange mutagenesis kit (Stratagene) according to the manufacturer's instructions and confirmed by sequencing.
Immunoprecipitation and Western blot analysis.
Cells were lysed in EBC buffer (50 mM Tris, pH 8.0, 120 mM NaCl, 0.5% NP-40) supplemented with protease and phosphatase inhibitors. The following antibodies were used: PERK (Rockland Immunochemicals); phospho-eIF2α, phospho-Ser473 Akt, Akt, eIF4E, phospho-S6 ribosomal protein (Ser235/236), S6 ribosomal protein, phospho-p44/42 MAPK (Thr202/Tyr204 of Erk1 and Thr185/Tyr187 of Erk2), and p44/42 MAPK (Erk1/2), (Cell Signaling); CHOP (Santa Cruz Biotechnology); p110α (BD Biosciences); and p85 and IRS1 (Millipore).
In vitro lipid kinase assays.
PERK was isolated from cells by immunoprecipitation, or recombinant GST-tagged ΔN-PERK or ΔN-K618A PERK was purified from bacteria, using glutathione (GSH)-Sepharose. Commercial recombinant PERK was obtained from Invitrogen. Recombinant PKR was purchased from Millipore. Recombinant p38α was obtained from Upstate. Recombinant GSK3β and PKA C-α were purchased from Cell Signaling. Recombinant DGK was purchased from Sigma. 1,2-Dilauroyl-sn-glycerol (DAGC12), 1,2-dimyristoyl-sn-glycerol (DAGC14), 1,2-dipalmitoyl-sn-glycerol (DAG16), and 1,2-distearoyl-sn-glycerol (DAG18) were from Echelon Biosciences. A lipid kinase assay was performed as described previously (50). 1,2-Dihexanoyl-sn-glycero-3-phosphate (06:0 PA) was purchased from Avanti Polar Lipids.
In vitro transcription and translation.
In vitro transcription and translation were performed with expression plasmid-encoding p85α using a coupled transcription and translation system (Promega) according to the manufacturer's instructions.
Oil Red O staining.
Lipids on cultured cells were visualized after cells were fixed for 2 min in 3.7% formaldehyde, washed with deionized water, and stained with Oil Red O solution.
Cellular PA measurement.
Total cellular phosphatidic acid content was determined using either the total phosphatidic acid kit (Cayman Chemical) or fluorescent monitors of PA in living cells (36). In brief, cells were transfected with a PA biosensor expression construct (wild-type [WT] biosensor; Pii-DK) or control plasmid with mutated PA binding domain (mutant [MUT] biosensor; Pii-DK-9A). Twenty-four hours after transfection, cells were serum-starved (0.1% fetal bovine serum [FBS]) for 16 h and then treated with 500 nM thapsigargin. Cyan fluorescent protein (CFP) (excitation [ex] of 440 nm; emission [em] of 480 nm) and fluorescence resonance energy transfer (FRET) (ex of 440 nm; em of 550 nm) values were obtained using fluorescence microscopy. FRET data represent results from three independent experiments where data were acquired from a minimum of three cells. For data acquisition, a Leica AF6000LX system equipped with a Hamamatsu ORCA-R2 charge-coupled-device (CCD) camera using a 20× objective at 37°C was used. FRET/CFP ratios, the corresponding fold change in PA production, and P values were calculated using Sigma Stat software from results of a minimum of three independent experiments.
RESULTS
Activation of Akt during ER stress is independent of eIF2α.
Many PERK-dependent biological effects result from reduced translation following direct phosphorylation of eIF2α (26, 30, 43). It was therefore important to establish whether there was a role for eIF2α in PERK-mediated activation of Akt following ER stress. For these experiments, we used tunicamycin, an inhibitor of N-linked glycosylation and a commonly used inducer of protein misfolding in the endoplasmic reticulum. As previously noted, induction of CHOP (C/EBP homologous protein) was ablated in eIF2αS51A knock-in cells (Fig. 1a) (43). In contrast, the ER stress-dependent increases in Akt phosphorylation on Ser473 as well as mTOR-dependent ribosomal S6 phosphorylation were preserved in eIF2αS51A knock-in cells (Fig. 1a), demonstrating that neither was a feature of altered eIF2α-dependent translation initiation. Because the PI3K p110α catalytic subunit is responsible for the majority of class IA PI3K activity in fibroblasts, p110α−/− cells were utilized to determine its contribution to ER stress-dependent Akt phosphorylation (57). Akt phosphorylation on Ser473 was still induced in p110α−/− cells (data not shown), suggesting that it is a p110-independent activity. However, because fibroblasts also express p110β, we used pharmacological inhibition as a mechanism to ablate all p110-related PI3K activity. PERK-proficient fibroblasts were treated with one of three distinct chemical inhibitors of the catalytic p110 subunit of PI3K: wortmannin, PIK-294, and IC-87114. Treatment of cells with these inhibitors did not prevent ER stress-induced AKT phosphorylation (Fig. 1b), whereas they did inhibit serum-induced Akt phosphorylation (data not shown). Finally, we also used a p110α dominant negative allele as an independent method to reduce endogenous total PI3K function. Expression of dominant negative p110α in H1299 cells did not significantly diminish Akt phosphorylation in response to ER stress (Fig. 1c; compare lanes 2 and 6). Increased phosphorylation of 4E-BP1, a substrate of mTOR, was also observed in PERK+/+ but not PERK−/− MEFs, revealing PERK-dependent activation of mTOR signaling (Fig. 1d; third panel). This was paralleled by reduced affinity of 4E-BP1 for the 7-methyl-GTP-Sepharose-bound complex of the mRNA Cap-binding proteins (Fig. 1d, upper panel).
Fig 1.

Activation of Akt during ER stress is independent of eIF2α phosphorylation and is correlated with activation of mTOR. (a) Western blot analysis for phospho-Ser473 Akt (pAkt) in eIF2α wild-type (wt) and eIF2αS51A knock-in fibroblasts following exposure of cells to tunicamycin (Tunic). (b) Activation of Akt in response to ER stress in NIH 3T3 cells coadministered with 10 nM wortmannin, 10 μM PIK-294, or 10 μM IC-87114 to inhibit endogenous PI3K activity. (c) Activation of Akt during ER stress in H1299 cells overexpressing p85α wt and p110α, p85α wt, and dominant negative (DN) p110α. (d) PERK+/+ or PERK−/− cells were treated with tunicamycin for the times indicated, and lysates were resolved by SDS-PAGE or utilized to pull down eIF4E complex with 7-Met-GTP-Sepharose. Arrows indicate relative positions of phosphorylated 4E-PB1 and CHOP.
PERK possesses intrinsic lipid kinase activity.
We next interrogated the mechanism whereby PERK regulates mTOR. Surprisingly, we observed that PERK immunopurified from either p110+/+ or p110α−/− cells treated with thapsigargin, which induces protein misfolding due to calcium depletion in the ER as a consequence of SERCA pump inhibition, exhibited an ability to phosphorylate lipids present in a preparation of phosphatidylinositol (PI) (Fig. 2a). For direct assessment, we performed an in vitro lipid kinase assay utilizing the recombinant catalytic domain of PERK and PI. The catalytic domain of PERK, but not kinase-dead PERK-K618A, generated a phosphorylated lipid product in vitro (Fig. 2b). No significant lipid kinase activity was detected when other recombinant active kinases (p38, GSK3β, PKA Cα) were tested (Fig. 2c). To identify the phospholipid product, in vitro lipid kinase assays were carried out using recombinant PERK and various phosphoinositide substrates. Products were resolved by thin-layer chromatography (TLC) (Fig. 2d) and subsequently excised and subjected to high-performance liquid chromatography (HPLC) for identification (Fig. 2e). PERK-dependent phosphorylation of phosphoinositides resulted in the production of phosphatidic acid (PA), as evidenced by the mobility of this metabolite on TLC plates (Fig. 2d) and the elution profile of deacylated product on HPLC (Fig. 2e). We hypothesized that PERK was preferentially phosphorylating a small pool of diacylglycerol (DAG) present in PI preparation. Consistent with this hypothesis, PERK phosphorylated purified DAG in vitro, demonstrating a preference for shorter-fatty-acid side chains (Fig. 2f). The identity of the PERK-generated metabolite as PA was also confirmed by using recombinant diacylglycerol kinase (DGK) (Fig. 2g) as a control. Thus, the serine/threonine kinase PERK is a bifunctional enzyme possessing both protein kinase and lipid kinase activity toward DAG generating PA as a product in vitro.
Fig 2.

The ER stress sensor PERK demonstrates lipid kinase activity in vitro and in vivo. (a) PERK was immunopurified from p110+/+ or p110α−/− MEFs, and lipid kinase activity was determined in an in vitro assay with phosphatidylinositol as a substrate. NRS, nonspecific rabbit serum. Thaps, thapsigargin. (b) In vitro lipid kinase assay with phosphatidylinositol as a substrate and recombinant catalytic domain of PERK (GST-ΔNPERK) or kinase-dead PERK (K618A). (c) In vitro lipid kinase assay with phosphatidylinositol as a substrate and recombinant catalytic domain of PERK (GST-ΔNPERK), kinase-dead PERK (K618A), p38, GSK3β, PKA C (500 ng of each kinase), and PI3K p85/p110 complex. (d) In vitro lipid kinase assay with phosphatidylinositol as a substrate and recombinant catalytic domain of PERK (GST-ΔNPERK) or recombinant PI3K and various phosphoinositide substrates. CBE, crude brain extract. Products were resolved by chromatography with propanol and acetic acid as the mobile phase. (e) The radioactive spot from the TLC plate in panel d (with CBE as a substrate) was excised, and PERK product was deacylated and then resolved by HPLC with 3H-labeled PI, PI4P, and PI4,5P2 as standards. (f) In vitro lipid kinase assay with diacylglycerol (DAG) as a substrate and recombinant catalytic domain of PERK (GST-ΔNPERK). DAGC12, 1,2-dilauroyl-sn-glycerol. (g) In vitro lipid kinase assay with phosphatidylinositol (PI) or diacylglycerol (DAG) as a substrate and the recombinant catalytic domain of PERK in the presence or absence of recombinant PI3K p85α or with recombinant diacylglycerol kinase (DGK). DAGC14, 1,2-dimyristoyl-sn-glycerol; DAGC16, 1,2-dipalmitoyl-sn-glycerol; DAGC18, 1,2-distearoyl-sn-glycerol.
We next assessed PERK-dependent generation of PA in vivo. We detected a significant increase in intracellular levels of total PA in PERK+/+ cells treated with thapsigargin but not in PERK−/− cells (Fig. 3a). PA production in response to thapsigargin was retained in eIF2αS51A knock-in fibroblasts (Fig. 3b), consistent with this being a translation-independent activity of PERK. To address the role of endogenous DGK in the generation of PA during an ER stress response, we treated cells concurrently with thapsigargin and a pharmacological inhibitor of DGK R59949; no significant inhibition of ER stress-dependent PA production was noted (Fig. 3c), suggesting that DGK does not contribute significantly under these conditions. We also addressed the potential contribution of PLD to stress-dependent PA production. Treatment of cells with the PLD inhibitor, FIPI, failed to reduce stress-dependent PA generation (Fig. 3c). Finally, we also evaluated stress-dependent PA generation following inhibition of PI3K activity (wortmannin), mTORC1 (rapamycin), and Akt (MK-2206). Consistent with the ability of PERK to directly generate PA, inhibition of these kinases failed to significantly inhibit ER stress-dependent PA induction (Fig. 3d).
Fig 3.

PERK-dependent generation of PA in vivo. (a) Total phosphatidic acid (PA) content in lysates from PERK+/+ and PERK−/− MEFs that were treated with 500 nM thapsigargin (Thaps) for the times indicated. (b) Total phosphatidic acid (PA) content in lysates from wild-type (wt) and eIF2αS51A knock-in cells treated with 500 nM thapsigargin for the times indicated. (c) PA formation in MEFs treated with 500 nM thapsigargin and either DGK inhibitor R59949 (20 μM) or PLD inhibitor FIPI (1.5 μM). (d) Total PA content in lysates from wild-type MEFs treated with 10 nM wortmannin (Wort), 25 nM rapamycin-specific inhibitor against mTOR (Rap), and 1 mM MK-2206-specific inhibitor against Akt. Thaps (500 nM) was used to induce ER stress. (e) FRET-based PA biosensors were used to measure PERK-dependent PA production in live cells: wild-type and PERK−/− MEFs were transfected with Pii-DK (wild type) or Pii-DK-9A (PA binding domain mutant) constructs. CFP and FRET (red, high FRET; blue, no/low FRET; see scale on right) images were obtained. Schematic representation of the FRET biosensor is provided. FRET signal decreases when the wild-type biosensor recognizes PA. (f) Values for FRET and CFP levels were obtained using fluorescence microscopy before and after treatment with 500 nM thapsigargin. FRET/CFP ratios were calculated for both wild-type (WT) and mutant (MUT) biosensors. (g) Fold change in PA measured 4 h after thapsigargin treatment (500 nM). Significant changes were observed between PERK+/+ MEFs transfected with WT versus MUT biosensor and PERK−/− cells expressing WT versus MUT biosensor (*, P < 0.001, determined by Student t test).
As an independent test of PA production, we utilized a FRET biosensor (Pii-DK) (36) to measure PA production in live cells (Fig. 3e). Consistent with PA production, the FRET signal (as represented by FRET/CFP) decreased in a time-dependent manner in PERK+/+ cells treated with thapsigargin; in contrast, high FRET levels were sustained in PERK+/+ cells expressing a mutant biosensor (Pii-DK-9A); a high FRET level was also maintained with the wild-type or mutant biosensors in PERK−/− cells (Fig. 3e and f). A significant increase in PA production was measured in PERK+/+ cells but not in PERK−/− cells in response to thapsigargin treatment (Fig. 3g). Thus, PERK lipid kinase activity can directly generate PA in vitro and contribute to PA production in vivo. However, because more than one product migrated on a TLC plate when PI was used as a substrate in an in vitro assay (Fig. 2a and b), we cannot exclude at this point that PERK initially mediates PI phosphorylation followed by lipase-like cleavage of PIP2 generating DAG substrate.
The PI3K class IA p85α subunit binds to and stimulates PERK-dependent lipid kinase activity.
We next determined whether PI3K class IA p85 regulatory subunits contribute to PERK-dependent Akt activation. Indeed, ER stress-dependent Akt phosphorylation was significantly reduced in p85α/β double-knockout (DKO) MEFs (Fig. 4a and 5a) (9). This result suggests a potential functional regulatory relationship between PERK and p85α. The most direct scenario would be one mediated by direct association. Indeed, purified GST-tagged catalytic domain of PERK and K618A PERK associated with in vitro transcribed and translated p85α (Fig. 4b). Supporting this in vitro interaction, PERK was also recovered in p85 immune complexes collected from 293T cells expressing exogenous PERK, p85α, and p110α (Fig. 4c and d). This interaction was diminished when the C terminus of PERK was deleted (Fig. 4c). Finally, endogenous p85 exhibited stress-dependent association with PERK in wild-type murine fibroblasts (Fig. 4e).
Fig 4.

PERK directly binds the regulatory subunit of PI3K in vitro and in vivo. (a) Activation of Akt in response to ER stress or in p85α/β double-knockout MEFs. (b) In vitro binding assay using GST-ΔNPERK and in vitro transcribed and translated p85α. (c) Coimmunoprecipitation of Myc-tagged PERKK618A and HA-p85α/FLAG-p110 in 293T cells. The diagram depicts deletion mutants of PERK cytosolic kinase domain. SP, signal peptide; TM, transmembrane domain. (d) Control for the experiment shown in panel c. Immunoprecipitation and Western blot with anti-myc antibody are shown. (e) Immunoprecipitation and Western blot for PERK and p85α from PERK+/+ MEFs treated with 50 nM thapsigargin (Thaps) for 2 or 4 h or stimulated with 100 nM insulin (Ins) for 15 or 30 min.
Fig 5.
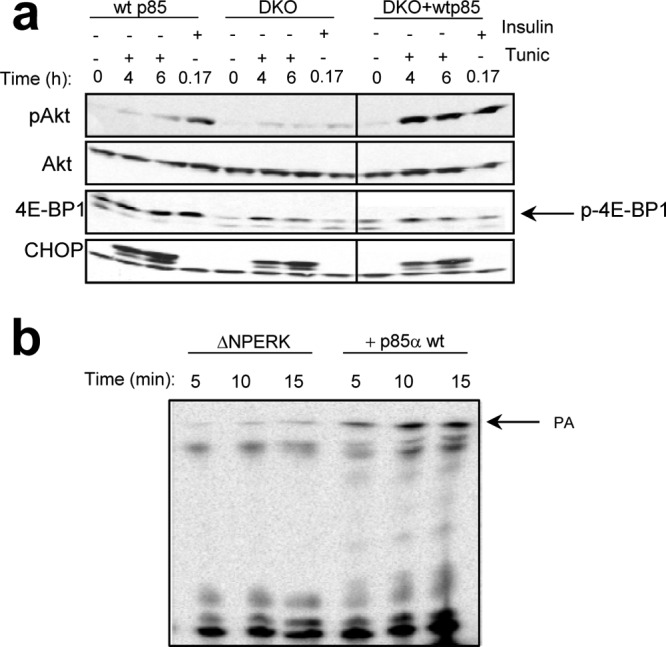
p85 increases PERK lipid kinase activity and Akt activation. (a) Activation of Akt in response to ER stress in wild-type, p85/p85 double-knockout (DKO), or DKO MEFs reconstituted with wild-type p85α (+wt p85). Phosphorylated p-4EBP1 is indicated by the arrow. (b) In vitro lipid kinase with DAGC12 as a substrate and recombinant catalytic domain of PERK (GST-ΔNPERK) alone or in the presence of recombinant p85α wild type. The intensity of phosphorylated lipid product signal was measured using phosphor imaging screen and STORM scanner (graph shown).
To establish the functional relationship between p85 and PERK, we utilized p85α/p85β double-knockout fibroblasts or those reconstituted with wild-type p85α. While ER stress-dependent activation of Akt was greatly diminished in knockout cells, reconstitution with p85 restored Akt activation (Fig. 5a). Finally, in an in vitro lipid kinase assay, addition of purified p85α stimulated PERK-dependent lipid kinase activity (Fig. 5b) revealing a regulatory role for p85.
PERK lipid kinase activity promotes mitogenic signaling.
Increased PA generation can directly promote mTORC1 and mTORC2 complex assembly and Akt Ser473 phosphorylation (49). PA also contributes to Ras activation via recruitment of the nucleotide exchange factor Son of Sevenless (SOS) to membranes (35, 56) and through recruitment of cRaf-1 to the plasma membrane followed by its interaction with Ras (39); both events contribute to activation of Erk1/2. We thus hypothesized that PERK-dependent generation of PA should link the unfolded protein response (UPR) with the induction of pathways downstream of Ras, such as MAPK, Akt, and mTOR. Indeed, we observed PERK-dependent activation of Akt (25, 29), as well as increased phosphorylation of S6, an event dependent upon mTOR, and Erk1/2 phosphorylation (Fig. 6a). Activation of mTOR and Ras pathways in response to ER stress was lost in PERK−/− MEFs; importantly, this could be restored by addition of PA to the growth medium in the presence or absence of tunicamycin (Fig. 6a). These data reveal that PA generation is sufficient to trigger signaling downstream of Ras in cells experiencing a robust ER stress response. Thus, PERK-dependent generation of PA is necessary and sufficient for mTOR-Akt and Ras-MEK-Erk1/2 signaling following initiation of ER stress.
Fig 6.

PERK promotes mitogenic signaling. (a) PI3K-mTOR-Akt and Ras-MEK-Erk1/2 pathway activation downstream of PERK was measured in PERK+/+/PERK−/− MEFs treated with tunicamycin (Tunic) and assessed by Western blotting using phospho-Ser473 Akt (pSer473), phospho-Erk1/2 (p-Erk1/2), and phospho-S6 kinase (p-S6) antibodies. PA was added to PERK−/− MEF cell culture media where indicated at 100 μM. (b) PERK+/+ and PERK−/− cells were serum starved overnight and then treated with 100 nM insulin for 10 or 20 min. Akt activation and levels were subsequently determined. (c) The indicated cells were serum deprived for 16 h, followed by addition of medium containing 10% FBS where indicated. Pathway activation was monitored by Western blot analysis with phospho-specific antibodies as indicated. (d) Lipid kinase activity in PERK immune complexes. The graph represents average activity, and the error bars are standard deviations from results of three independent experiments. (e) PERK was immunopurified from cells treated with 100 nM insulin, and the levels of associated p85 were assessed by Western blot analysis. (f) PERK+/+ or PERK−/− cells were serum starved overnight and then left untreated or pretreated with 500 nM thapsigargin for 4 h followed by 100 nM insulin as indicated. Lysates were probed for phospho-Ser473 Akt and total Akt levels. NRS, nonspecific rabbit serum.
To further test the impact of PERK lipid kinase activity on growth factor receptor signaling, we measured Akt activation in response to insulin stimulation in the context of PERK wild-type and PERK-deficient cellular environment. Indeed, insulin-dependent Akt phosphorylation was attenuated in PERK−/− cells (Fig. 6b). In contrast, PERK−/− cells remained responsive to serum stimulation (Fig. 6c), demonstrating that while PERK function contributes to insulin signaling, it is not required for generalized growth factor signaling. Assessment of PERK lipid kinase activity in response to insulin treatment revealed an insulin-dependent 2-fold increase in PERK-lipid kinase activity (Fig. 6d). We also detected recruitment of p85α in PERK immune complexes upon insulin treatment (Fig. 6e). Finally, thapsigargin pretreatment of PERK+/+ cells enhanced the amplitude of PERK-dependent Akt phosphorylation elicited by insulin treatment (Fig. 6f). Thus, PERK-dependent generation of PA promotes mitogenic signaling in the setting of ER stress.
Role of PERK lipid kinase activity in adipocyte differentiation in vitro.
To query the physiological significance of PERK lipid kinase activity, we utilized an in vitro adipocyte differentiation assay. We previously reported that differentiation of MEFs transduced with Myc-SREBP1 was attenuated in the absence of PERK (8). We measured levels of Akt phosphorylation in PERK wild-type and PERK-null MEFs during in vitro differentiation. Akt phosphorylation was severely attenuated in the absence of PERK (Fig. 7a). This correlated with reduced accumulation of lipid droplets in PERK-null MEFs as evidenced by the Oil Red O staining (Fig. 7b). We isolated p85α, PERK, and the insulin receptor substrate 1 (IRS1) by immunoprecipitation from wild-type MEFs at different stages of differentiation (Fig. 7c). Recruitment of the p85α into both IRS1 and PERK immune complexes was observed by day 7 of differentiation (Fig. 7c). p110α was detected in p85α and IRS1 but not in PERK immune complexes even though total levels of p85α were similar in PERK and IRS1 immune complexes (Fig. 7c). Finally, we measured PERK lipid kinase activity and detected an increase in the ability of immunopurified PERK to phosphorylate phosphatidylinositol in vitro on day 7 and day 10 of differentiation (Fig. 7d), consistent with recruitment of p85α into PERK immune complexes at this time (Fig. 7c). The increase in PERK activity does not reflect changes in PERK expression, as PERK levels remain constant through the first 7 days of differentiation followed by a modest decline at day 10 (8). These data provide proof of principle for the importance of PERK lipid kinase activity in a physiologically relevant model.
Fig 7.

Role of PERK lipid kinase activity in adipocyte differentiation in vitro. (a) Levels of phospho-Ser473 Akt in adipocytes differentiating from PERK+/+ or PERK−/− MEFs. (b) Oil Red O staining of adipocytes differentiating from PERK+/+ or PERK−/− MEFs on day 13 of treatment. (c) PERK, p85α, or IRS1 was immunopurified from MEFs on day 0, 7, 10, or 15 of differentiation, and the levels of PERK, p85α, p110, and IRS1 were determined. (d) PERK was immunopurified from differentiating adipocytes, and PERK-dependent lipid kinase activity was assessed.
DISCUSSION
The ER can be viewed as a sensor of metabolic status in the cell. Imbalance between available intracellular resources relative to the functional demand of the ER results in protein misfolding and activation of the adaptive response pathway referred to as the unfolded protein response (UPR) and sometimes as the integrated stress response (ISR) due to the contribution of cytosolic signal transducers that regulate common downstream pathways. Activation of the UPR is mediated by three major proximal sensor molecules: PERK (26, 45), inositol-requiring enzyme 1 (Ire 1α/β) (47, 51), and transmembrane transcription factor ATF6 (27, 33, 52). The transcriptional programs activated by Ire1, ATF6, and PERK either adjust ER functional capacity and alleviate ER client protein load or trigger apoptosis if nutrient/energy availability is severely compromised. Accumulating evidence demonstrates that UPR signaling molecules regulate both ER functional capacity and cellular metabolic pathways. PERK (8) and Xbp1 (32, 46) have been implicated in anabolic regulation of lipid synthesis. The intersection with lipid synthesis likely reflects a need for ER membrane expansion necessary for adaptation. Another connection between UPR and cellular metabolic networks is provided by the PERK-dependent activation of the Akt pathway (25, 29, 31). Akt is the primary regulator of anabolic metabolism in the cell via its effects on glucose transport (14, 53), capture by hexokinase, and utilization (15, 23, 40).
Consequences of PERK-dependent regulation of Akt during ER stress.
Initial work suggested that ER stress-dependent Akt activation depends on phosphorylation of eIF2α and PI3K catalytic function (31). However, our data demonstrate that Akt phosphorylation is independent of eIF2α phosphorylation and p110 catalytic function. The discrepancy likely arises from use of LY294002 (29), which at the dose used also inhibits mTOR activity and, thus, Akt Ser-473 phosphorylation. Our data support a model where Akt activation is mediated by PERK-dependent generation of PA which can, in turn, either directly promote assembly of the mTORC2 complex (49) or function through SOS-Ras activation (35, 56).
Our data demonstrate that PERK can generate PA via direct phosphorylation of DAG and that PERK is required for PA generation in cells exposed to ER stress-inducing agents. Canonically, PA is generated via pathways wherein phospholipase D (PLD) utilizes phosphatidylcholine (PC) as a substrate (20) or where DAG kinases (DGKs) directly phosphorylate cellular DAG (34). The use of inhibitors that directly target each molecule failed to inhibit ER stress-dependent PA generation, suggesting that these molecules do not significantly contribute to PA generation under these conditions.
A question that arises from PERK-dependent generation of PA concerns the source of the DAG substrate. As an ER transmembrane protein kinase, PERK localization should facilitate access to phosphoinositides. Indeed, the ER is a major compartment of lipid and phospholipid generation in cells (17). DAG can be generated via phospholipase C-mediated hydrolysis of phosphoinositides. Environmental growth factors can trigger the utilization of PC at least in part via activation of protein kinase C (PKC) (4). There is little direct evidence to support an active role for PKC in ER stress signaling. However, PKCδ activation has been observed in steatohepatitis-associated ER stress (24). While we cannot rule out this mechanism, there are few data to support ER stress-dependent activation of PKC in other systems. Finally, studies utilizing labeled glucose suggest that the majority of DAG comes from de novo synthesis from glycolytic precursors and thus ultimately requires the action of phosphatidic acid phosphatase, which utilizes PA as a substrate (41). As this pathway would oppose PA accumulation, it seems unlikely to play a major role during ER stress.
Akt activation promotes anabolic metabolism, which may seem counterintuitive with regard to cell adaptation to stress, as the cell needs to be reprogrammed to preserve energy and halt anabolic reactions. However, increased activity of Akt and the subsequent increase in intracellular glucose availability would help to generate additional energy via glycolysis and/or oxidative phosphorylation as well as maximize glucose-derived cellular biosynthetic precursors necessary to synthesize ER membrane lipids, thereby accelerating recovery from stress. Critically, the accumulation of long-chain fatty acids and PA at the ER acts as a signal for increased association of the phosphatidate phosphatase 1 with the ER membrane, thereby increasing synthesis of triacylglycerols (TAG) (28).
The cellular function of PA may be dependent upon the structure of the PA molecule generated. With regard to de novo synthesis from glycolytic precursors, the newly generated PA is associated with decreased mTORC2 activity. This is achieved through PA-dependent dissociation of the mTORC2-specific component Rictor (55). Thus, these data are in direct contrast with our results and those of other groups (2, 18, 49) demonstrating that PA stimulates mTOR function. A resolution to this seeming paradox likely stems from functionally and structurally distinct PA. Our data reveal that PERK favors short-chain DAG as a substrate (C12), while the chain length of PA molecules generated from the biosynthetic pools is unknown (55). The possibility that structurally distinct PA molecules could have opposing effects on cellular signaling is an exciting notion that will require additional investigation.
PERK lipid kinase activity and implications for cellular physiology.
Although lipid synthesis and storage in adipocytes are attenuated in p110α-null MEFs, deletion of p110 was also accompanied by decreased levels of two major adipogenic transcription factors, PPARγ and C/EBPα, as well as p85α (57). Because p85α deficiency results in reduced PERK-dependent lipid kinase activity, the reduced lipid production during adipogenesis in p110α-null MEFs could also reflect reduced PERK function. Indeed, a significant increase in PERK lipid kinase activity is apparent upon p85α recruitment to PERK. Our data support a model wherein activation of the lipogenic program in adipocytes is coordinately regulated by PERK and PI3K p110α for the optimal induction of lipid synthesis.
PERK lipid kinase function is not only essential for engaging mitogenic and prosurvival pathways during a bona fide ER stress response; it also synergizes with the insulin receptor signaling. Since PERK is not generally required for growth factor (e.g., serum)-dependent activation of mitogenic pathways, it is not as yet clear how PERK synergizes with insulin. One possibility is that in addition to engaging Ras, insulin also triggers ER stress, which engages PERK, and together these signals contribute to maximum activation of relevant pathways.
In addition to connecting with PERK, monomeric p85α and p85β subunits regulate nuclear transport of the ATF6/Ire1-regulated transcription factor Xbp1 (37, 54), and this mechanism appears to be downstream of insulin receptor signaling. Critically, this novel regulatory mechanism is lost in ob/ob mice, leading to insulin resistance that could be rescued by adenoviral delivery of p85. Thus, p85-dependent regulation of at least two UPR transducers may provide a mechanism for increasing insulin sensitivity with regard to PERK and preventing development of insulin resistance due to the unresolved stress in the case of Xbp1.
Finally, the third major UPR transducer, ATF6α, was directly implicated in transcriptional upregulation of the mTOR activator Ras homolog enriched in brain (RheB), which correlated with increased levels of phospho-S6 ribosomal protein in ATF6α-positive cells, consistent with higher mTOR activity (44). Thus, the UPR/ISR/PI3K/mTOR signaling pathways appear to be hyper-connected and may cooperate to promote survival of the cell under conditions of stress.
PERK lipid kinase activity and PI3K/mTOR pathway inhibition.
The PI3K-mTOR pathway represents an attractive pharmacological target in cancer. We have observed that while insulin- and PI3K p110α-dependent activation of Akt is impaired in response to growth factors, its activation is essentially unaffected in response to ER stress. This may have significant implications for the development of successful treatment protocols. For instance, increased expression of ER chaperone and the ER stress marker BiP was observed in human breast tumors and cancer-derived cell lines (19, 22), suggesting that the UPR and PERK play active roles in promoting tumorigenesis. Given recent work demonstrating a role for PERK in tumor progression (5–7), it is tempting to speculate that PERK may directly contribute to tumorigenesis by promoting PA-dependent regulation of mTOR, thereby triggering Akt activation. Critically, PERK lipid kinase activity may also influence the efficacy of strategies that target PI3K/mTOR.
ACKNOWLEDGMENTS
We thank Lucia Rameh for assistance in conduction of HPLC characterization of lipid metabolites, Craig B. Thompson for insightful comments, Lewis Cantley, Jean Zhao, Morris Birnbaum, Randal Kaufman, and Etsuko Kiyokawa for sharing cell lines and reagents, and Margarita Romero for outstanding technical assistance.
This work was supported by National Institutes of Health grants F32CA1238252 (E.B.-M.) and P01 CA104838 and a Leukemia & Lymphoma Scholar award (J.A.D.).
Footnotes
Published ahead of print 9 April 2012
E.B.-M. and D.P. contributed equally to the manuscript.
REFERENCES
- 1. Antonsson B. 1997. Phosphatidylinositol synthase from mammalian tissues. Biochim. Biophys. Acta 1348: 179– 186 [DOI] [PubMed] [Google Scholar]
- 2. Avila-Flores A, Santos T, Rincon E, Merida I. 2005. Modulation of the mammalian target of rapamycin pathway by diacylglycerol kinase-produced phosphatidic acid. J. Biol. Chem. 280: 10091– 10099 [DOI] [PubMed] [Google Scholar]
- 3. Backer JM, et al. 1992. Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J. 11: 3469– 3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Besterman JM, Duronio V, Cuatrecasas P. 1986. Rapid formation of diacylglycerol from phosphatidylcholine: a pathway for generation of a second messenger. Proc. Natl. Acad. Sci. U. S. A. 83: 6785– 6789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bi M, et al. 2005. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 24: 3470– 3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blais JD, et al. 2006. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 26: 9517– 9532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bobrovnikova-Marjon E, et al. 2010. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 29: 3881– 3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bobrovnikova-Marjon E, et al. 2008. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc. Natl. Acad. Sci. U. S. A. 105: 16314– 16319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brachmann SM, et al. 2005. Role of phosphoinositide 3-kinase regulatory isoforms in development and actin rearrangement. Mol. Cell. Biol. 25: 2593– 2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bunney TD, Katan M. 2010. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat. Rev. Cancer 10: 342– 352 [DOI] [PubMed] [Google Scholar]
- 11. Carpenter CL, et al. 1993. Phosphoinositide 3-kinase is activated by phosphopeptides that bind to the SH2 domains of the 85-kDa subunit. J. Biol. Chem. 268: 9478– 9483 [PubMed] [Google Scholar]
- 12. Cullinan SB, et al. 2003. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23: 7198– 7209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dhand R, et al. 1994. PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J. 13: 511– 521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Edinger AL, Thompson CB. 2002. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell 13: 2276– 2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elstrom RL, et al. 2004. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 64: 3892– 3899 [DOI] [PubMed] [Google Scholar]
- 16. Engelman JA, Luo J, Cantley LC. 2006. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7: 606– 619 [DOI] [PubMed] [Google Scholar]
- 17. Fagone P, Jackowski S. 2009. Membrane phospholipid synthesis and endoplasmic reticulum function. J. Lipid Res. 50 (Suppl.):S311–S316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. 2001. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294: 1942– 1945 [DOI] [PubMed] [Google Scholar]
- 19. Fernandez PM, et al. 2000. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 59: 15– 26 [DOI] [PubMed] [Google Scholar]
- 20. Foster DA, Xu L. 2003. Phospholipase D in cell proliferation and cancer. Mol. Cancer Res. 1: 789– 800 [PubMed] [Google Scholar]
- 21. Fu Z, Aronoff-Spencer E, Wu H, Gerfen GJ, Backer JM. 2004. The iSH2 domain of PI 3-kinase is a rigid tether for p110 and not a conformational switch. Arch. Biochem. Biophys. 432: 244– 251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gazit G, Lu J, Lee AS. 1999. De-regulation of GRP stress protein expression in human breast cancer cell lines. Breast Cancer Res. Treat. 54: 135– 146 [DOI] [PubMed] [Google Scholar]
- 23. Gottlob K, et al. 2001. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 15: 1406– 1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Greene MW, et al. 2010. PKC{delta} is activated in a dietary model of steatohepatitis and regulates endoplasmic reticulum stress and cell death. J. Biol. Chem. 285: 42115– 42129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hamanaka RB, Bobrovnikova-Marjon E, Ji X, Liebhaber SA, Diehl JA. 2009. PERK-dependent regulation of IAP translation during ER stress. Oncogene 28: 910– 920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harding HP, Zhang Y, Ron D. 1999. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397: 271– 274 [DOI] [PubMed] [Google Scholar]
- 27. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. 1999. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10: 3787– 3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hopewell R, Martin-Sanz P, Martin A, Saxton J, Brindley DN. 1985. Regulation of the translocation of phosphatidate phosphohydrolase between the cytosol and the endoplasmic reticulum of rat liver. Effects of unsaturated fatty acids, spermine, nucleotides, albumin and chlorpromazine. Biochem. J. 232: 485– 491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu P, Han Z, Couvillon AD, Exton JH. 2004. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J. Biol. Chem. 279: 49420– 49429 [DOI] [PubMed] [Google Scholar]
- 30. Reference deleted.
- 31. Kazemi S, et al. 2007. A novel function of eIF2alpha kinases as inducers of the phosphoinositide-3 kinase signaling pathway. Mol. Biol. Cell 18: 3635– 3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee AH, Scapa EF, Cohen DE, Glimcher LH. 2008. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320: 1492– 1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li M, et al. 2000. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell. Biol. 20: 5096– 5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Merida I, Avila-Flores A, Merino E. 2008. Diacylglycerol kinases: at the hub of cell signalling. Biochem. J. 409: 1– 18 [DOI] [PubMed] [Google Scholar]
- 35. Mor A, et al. 2007. The lymphocyte function-associated antigen-1 receptor costimulates plasma membrane Ras via phospholipase D2. Nat. Cell Biol. 9: 713– 719 [DOI] [PubMed] [Google Scholar]
- 36. Nishioka T, Frohman MA, Matsuda M, Kiyokawa E. 2010. Heterogeneity of phosphatidic acid levels and distribution at the plasma membrane in living cells as visualized by a Foster resonance energy transfer (FRET) biosensor. J. Biol. Chem. 285: 35979– 35987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Park SW, et al. 2010. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 16: 429– 437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Payrastre B, et al. 2001. Phosphoinositides: key players in cell signalling, in time and space. Cell Signal. 13: 377– 387 [DOI] [PubMed] [Google Scholar]
- 39. Rizzo MA, et al. 1999. Phospholipase D and its product, phosphatidic acid, mediate agonist-dependent raf-1 translocation to the plasma membrane and the activation of the mitogen-activated protein kinase pathway. J. Biol. Chem. 274: 1131– 1139 [DOI] [PubMed] [Google Scholar]
- 40. Robey RB, Hay N. 2009. Is Akt the “Warburg kinase”?—Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 19: 25– 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rossi F, Grzeskowiak M, Della Bianca V, Sbarbati A. 1991. De novo synthesis of diacylglycerol from glucose. A new pathway of signal transduction in human neutrophils stimulated during phagocytosis of beta-glucan particles. J. Biol. Chem. 266: 8034– 8038 [PubMed] [Google Scholar]
- 42. Sasaki T, et al. 2009. Mammalian phosphoinositide kinases and phosphatases. Prog. Lipid Res. 48: 307– 343 [DOI] [PubMed] [Google Scholar]
- 43. Scheuner D, et al. 2001. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7: 1165– 1176 [DOI] [PubMed] [Google Scholar]
- 44. Schewe DM, Aguirre-Ghiso JA. 2008. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. U. S. A. 105: 10519– 10524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shi Y, et al. 1998. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 18: 7499– 7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sriburi R, Jackowski S, Mori K, Brewer JW. 2004. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 167: 35– 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tirasophon W, Welihinda AA, Kaufman RJ. 1998. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12: 1812– 1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tolias KF, Cantley LC. 1999. Pathways for phosphoinositide synthesis. Chem. Phys. Lipids 98: 69– 77 [DOI] [PubMed] [Google Scholar]
- 49. Toschi A, et al. 2009. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol. Cell. Biol. 29: 1411– 1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang L-P, Summers SA. 2003. Diabetes mellitus. Methods Mol. Med. 83: 127– 136 [DOI] [PubMed] [Google Scholar]
- 51. Wang XZ, et al. 1998. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 17: 5708– 5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang Y, et al. 2000. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 275: 27013– 27020 [DOI] [PubMed] [Google Scholar]
- 53. Wieman HL, Wofford JA, Rathmell JC. 2007. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 18: 1437– 1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Winnay JN, Boucher J, Mori MA, Ueki K, Kahn CR. 2010. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 16: 438– 445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang C, et al. 2012. Glycerolipid signals alter mTOR complex 2 (mTORC2) to diminish insulin signaling. Proc. Natl. Acad. Sci. U. S. A. 109: 1667– 1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D. 2007. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat. Cell Biol. 9: 706– 712 [DOI] [PubMed] [Google Scholar]
- 57. Zhao JJ, et al. 2006. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc. Natl. Acad. Sci. U. S. A. 103: 16296– 16300 [DOI] [PMC free article] [PubMed] [Google Scholar]