

Abstract
Signaling of transforming growth factor β (TGF-β) is redirected in cancer to promote malignancy, but how TGF-β function is altered in a transformed cell is not fully understood. We investigated TGF-β signaling by profiling proteins that differentially bound to type I TGF-β receptor (TβRI) in nontransformed, HER2-transformed, and HER2-negative breast cancer cells using immunoprecipitation followed by protein identification. Interestingly, several nuclear proteins implicated in posttranscriptional RNA processing were uniquely identified in the TβRI coprecipitates from HER2-transformed cells. Ligand-inducible nuclear translocation of TβRI was observed only in transformed cells, and the translocation required importin β1, nucleolin, and Smad2/3. This trafficking was dependent on the high Ran GTPase activity resulting from oncogenic transformation. In the nucleus, TβRI associated with purine-rich RNA sequences in a synergistic manner with the RNA-binding factor hnRNP A1. We further found that nuclear translocation of TβRI specifically induced epidermal growth factor receptor (EGFR) transcript isoform c, which encodes a soluble EGFR protein, through alternative splicing or 3′-end processing. Our study confirms a cancer-specific nuclear translocation of TβRI and demonstrates its potential function in regulating nuclear RNA processing, as well as a novel gain-of-function mechanism of TGF-β signaling in cancer.
INTRODUCTION
Signaling initiated by transforming growth factor β (TGF-β) and the corresponding physiological consequences are highly context dependent, resulting in different or even opposite TGF-β functions in cancerous and normal cells. TGF-β family members signal through heteromeric complex of transmembrane serine/threonine kinases, the type I and type II receptors (TβRI and TβRII), which subsequently phosphorylate receptor-regulated Smad proteins (R-Smads). R-Smads usually translocate to the nucleus together with common mediator Smad4, where they regulate gene transcription via binding the promoter of target genes (17, 28). Smad-independent pathways, including phosphatidylinositol-3 kinase (PI3K), extracellular signal-regulated kinase (ERK; mitogen-activated protein kinase [MAPK]), c-Jun NH2-terminal kinase (JNK), p38MAPK, and Rho GTPases, have also been implicated in TGF-β action (12, 14). TGF-β acts as a tumor suppressor in normal epithelia by inhibiting cell proliferation and inducing apoptosis, but it accelerates progression of established cancers by autocrine and paracrine mechanisms (11, 13, 14). We and others have previously reported that transforming oncogenes such as HER2 (ERBB2; Neu, the rat/mouse homologue of HER2), a proto-oncogene frequently activated by gene amplification or overexpression in human breast cancer, contribute to the transition of TGF-β function during cellular transformation and cancer progression (7). In transformed cells, signaling of TGF-β loses its tumor-suppressive effects observed in normal cells and begins to function as a cancer-promoting agent that synergizes with transforming oncogenes (29).
As a well-studied example of context-dependent TGF-β action, synergy between TGF-β and HER2 has been initially demonstrated in crossbred mice simultaneously expressing Neu and TβRIT204D (a constitutively active mutant of TβRI) or TGF-β1S223/225 (a constitutively active mutant of TGFβ1) in the mammary glands (31, 32, 37). In both bitransgenic models, overexpression of activated TGF-β receptor or ligand accelerates metastasis of Neu-driven mammary tumors. Compared to tumors that only express Neu, the Neu/TβRIT204D and Neu/TGF-β1S223/225 bigenic tumors exhibit less apoptosis and are more locally invasive and display higher histological grades (31, 32). Conversely, mice expressing a dominant negative TβRII exhibit delayed formation of Neu-driven mammary tumors and reduced metastasis (37). These data suggest that TGF-β can accelerate metastasis of Neu-induced mammary tumors and that Neu, in turn, requires TGF-β signaling to maximally drive cancer progression.
The functional synergy between TGF-β and HER2 has been investigated using the MCF10A nontransformed human mammary epithelial cell model. A genetic modifier screen in MCF10A cells stably overexpressing transfected HER2 (MCF10A/HER2) showed that TGF-β1 and TGF-β3 cDNAs cooperate with HER2 in inducing cell motility and invasion (36). Subsequent studies from several groups, including ours, revealed that the cross talk between TGF-β and HER2 occurs at multiple levels and includes suppression of Smad-dependent transcriptional regulation and induction of ErbB ligands through a Smad-independent mechanism (7). A gene expression signature induced by TGF-β activation is frequently found in primary human breast tumors with a HER2-positive or basal-like molecular profile and is associated with shorter survival in a cohort of 295 breast cancer patients (48). In addition, TGF-β is also implicated in resistance to trastuzumab, a HER2-targeted therapeutic agent, by inducing heterodimerization of HER2 and ErbB3 (48). These previous works prelude the understanding of context-dependent TGF-β signaling and its clinical relevance to cancer.
In the present study, we set out to interrogate context-dependent TGF-β signaling by profiling proteins that differentially bound to activated TβRI in MCF10A, MCF10A/HER2, and the HER2-negative MDA-MB-231 breast cancer cells. TβRI immunoprecipitation (IP) coupled with liquid chromatography-tandem mass spectrometry (LC-MS/MS) revealed proteins that associated with TβRI only in a transformed cellular context (MCF10A/HER2 and MDA-MB-231) and not in MCF10A cells. Interestingly, a number of nuclear proteins that are implicated in RNA posttranscriptional regulation were discovered. We further found that in transformed cells, TβRI translocated to the nucleus upon activation by TGF-β and bound to a purine-rich consensus RNA sequence present in transcripts of multiple genes, including EGFR, to exert posttranscriptional regulation. Although the nuclear translocation of several receptor tyrosine kinases, including HER2, has been previously reported (6), our results demonstrated the spatial regulation of full-length TβRI through a cancer-specific nuclear transporting mechanism that requires high Ran GTPase activity. Unlike the reported function of nuclear HER2, which associates with multiple DNA targets to stimulate gene transcription (42), TβRI specifically recognized RNA targets and regulated RNA processing in the nucleus. These novel findings open a new avenue for studying TGF-β signaling mediated by its RNA targets and elucidating the complex functional switch of TGF-β function during cancer initiation and progression.
MATERIALS AND METHODS
Cell lines, plasmids, and viruses.
MCF10A, BT474, and MDA-MB-231 cells were obtained from American Type Culture Collection (Manassas, VA) and cultured in the recommended media in a humidified 5% CO2 incubator at 37°C. MCF10A/HER2 and MCF10A/vec (vector control) cells were generated and grown as described previously (40). To generate stably transduced cells, retroviruses expressing hemagglutinin (HA)-tagged TβRI (wild type), TβRIT204D (48), or the empty pBMN-I-GFP vector were produced by transfecting Ampho-Phoenix cells and then utilized for transduction, followed by green fluorescent protein (GFP) selection. For GFP-fused TβRI constructs, full-length or truncated (1 to 287 or 1 to 205 amino acids [aa]) TβRI coding regions were subcloned from the pBMN-TβRI plasmid to the EcoRI/SacII sites of pEGFP-N3 vector (Clontech; Mountain View, CA). The ALK5(D266A)/HA and ALK5(3A)/HA constructs were kindly provided by Peter ten Dijke (Leiden University Medical Center, Netherlands) and are described elsewhere (23). The Ran(WT), Ran(F35A), and Ran(T24N) constructs (25) were kindly provided by Richard Cerione (Cornell University). Recombinant human TGF-β1 was purchased from R&D Systems (Minneapolis, MN). Lapatinib ditosylate was purchased from LC Laboratories (Woburn, MA). The type I/II TGF-β receptor inhibitor LY2109761 was provided by Eli Lilly and Company (Indianapolis, IN).
RNA extraction, RT, and qPCR.
RNA extraction, reverse transcription (RT), and real-time quantitative PCR (qPCR) were performed as described previously (49). Primer sequences used were as follows: 5′-CCTGTTTCCTTGAGATCAGCTGC-3′ and 5′-GTGAGGGAAGAAAGTTGGGAGCG-3′ for the Chr7-6 TβRI-binding site, 5′-CCCATAACCCCTGAGGGTAG-3′ and 5′-CTCAGGCGGCAGTCATAGA-3′ for the Chr7-193 TβRI-binding site, 5′-GAGCCGCGAGAAGTGCTAGCTCG-3′ and 5′-CTGGAGCACTGTCTGCGCACACC-3′ for the Chr6-6 TβRI-binding site, 5′-GGAAGTGTTGAAGGGAGGTGGCA-3′ and 5′-CAAACCGTGCCTGGAAGTCAACG-3′ for the Chr11-110 TβRI-binding site, 5′-CACTGCAGCACTTGAAGGAGG-3′ and 5′-TGAGGCAGAGGCTGCCATCTA-3′ for the Chr15-7 TβRI-binding site, 5′-TGGAGCCTCTTACACCCAGT-3′ and 5′-GCTTTCGGAGATGTTGCTTC-3′ for human epidermal growth factor receptor (EGFR) isoform a, 5′-AACAACACCCTGGTCTGGAA-3′ and 5′-TGAAGCAAAGGGAGAAATTGA-3′ for human EGFR isoform b, 5′-GGATATTCTGAAAACCGTAAAGGAAA-3′ and 5′-CGAAAAGTTCTCTCTAAAACACTGATT-3′ for human EGFR isoform c, 5′-CCAGTGTGCCCACTACATTG-3′ and 5′-CGCTGCCATCATTACTTTGA-3′ for human EGFR isoform d, 5′-GGCTCTGGAGGAAAAGAAAG-3′ and 5′-CAATGAGGACATAACCAGCCAC-3′ for all EGFR isoforms, 5′-GGCTCTGGAGGAAAAGAAAG-3′ and 5′-AGAACGAAACGTCCCGTTCCTC-3′ for primary EGFR transcripts, and 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (as a control). An annealing temperature of 55°C was used for all primers.
Fractionation, IP, Western blotting, and pulldown assays.
Cell fractionation, IP, and Western blotting were carried out as described previously (44, 46). To pull down GTP-bound (active) Ran GTPase, His-tagged RanBP1 recombinant protein (Cytoskeleton Inc., Denver, CO) precoupled to His Mag Sepharose Ni magnetic beads (GE Healthcare, Piscataway, NJ) was utilized to pull down active Ran from the nuclear and cytoplasmic fractions as described previously (3). Eluted Ran proteins were detected by Western blotting. Pulldown assays using 5′-end biotinylated RNA probes and MCF10A/HER2 nuclear extracts were carried out as described previously using magnetic streptavidin beads (Promega, Madison, WI) (47). Sequences of the RNA probes were as follows: Chr7-6(wt), 5′-GGGCGGAGGAGGAGACGCGU-3′; Chr7-6(mt), 5′-GGGCGUGCACGUCGACGCGU-3′; Chr7-193(wt), 5′-GGUAGAGGGAGGGGACAGGG-3′; and Chr7-193(mt), 5′-GGUAGCGUUAUCCGACAGGG-3′ (mutated nucleotides are underlined.). Primary antibodies used in Western blots included HA tag, GAPDH, histone H3, importin β1, nucleolin, nucleophosmin, Ran, His tag, Smad2/3 (Cell Signaling, Danvers, MA), hnRNP A1, EGFR C-term (Santa Cruz Biotechnology, Santa Cruz, CA), TβRI (Abcam, Cambridge, MA), and EGFR N-term (Abgent, San Diego, CA).
SDS-PAGE, in-gel digestion, and LC-MS/MS analysis.
The eluates from HA antibody-conjugated agarose beads (Sigma-Aldrich, St. Louis, MO) following IP of MCF10A/TβRIT204D, MCF10A/HER2/TβRIT204D, or MDA-MB-231/TβRIT204D lysates were collected and electrophoresed into a 4 to 12% bis-Tris gel and stained with colloidal blue (Invitrogen, Carlsbad, CA). The protein bands were excised and subjected to in-gel trypsin digestion using a standard protocol (19). Peptides were analyzed using a Thermo Finnigan LTQ ion trap instrument equipped with a Thermo MicroAS autosampler and Thermo Surveyor high-performance liquid chromatography (HPLC) pump, nanospray source, and Xcalibur 2.0 SR2 instrument control. Tandem spectra were searched against the Human IPI database (version 3.37) using the Myrimatch algorithm (version 1.2.11) (39). The database was concatenated with the reverse sequences of all proteins in the database to allow for the determination of false-positive rates. The IDPicker algorithm (version 2.1.5) was used to filter the data using a false-positive identification threshold (default is 0.05 or 5% false positives) based on reverse sequence hits in the database (26, 50). Proteins with two or more peptide matches confidently identified from the samples were determined to be TβRI associated and are listed in Table S1 in the supplemental material.
IPA.
The 62 proteins for which the association with TβRI was identified only in MCF10A/HER2 and MDA-MB-231 cells and not MCF10A cells were examined for functional annotation by Ingenuity pathway analysis (IPA; Ingenuity Systems). IPA mapped these genes to their corresponding gene objects in the Ingenuity Pathways Knowledge Base (IPKB). A P value was generated for each mapped network by comparing the number of submitted genes that participated in a given network relative to the total number of occurrences of those genes in all the possible network permutations for all the objects in the IPKB. The network score was assessed for statistical significance and the generated pathway maps examined for biological correlation.
Cell transfection with DNA and siRNAs.
DNA transfection was performed using Lipofectamine 2000 (Invitrogen) by following the manufacturer's protocol, as described previously (43). The FlexiTube GeneSolution small interfering RNAs (siRNAs) for TGFBR1, KPNB1, NCL, SMAD2, SMAD3, and HNRNPA1, as well as AllStars negative-control siRNAs, were purchased from Qiagen (Valencia, CA) and transfected using DharmaFECT Duo transfection reagent (Dharmacon, Lafayette, CO) according to the manufacturer's procedures.
ChIP, RIP, and deep sequencing.
Chromatin IP (ChIP) was performed as previously described (47) in MCF10A and MCF10A/HER2 cells stably expressing HA-tagged TβRIT204D. In brief, DNA-binding proteins were cross-linked by formaldehyde, and nuclear fractions were purified, sonicated to shear DNA, and subjected to IP with HA antibody. Normal mouse IgG was used as a negative control. For RNA IP (RIP), the same cross-linking and IP procedures were performed, followed by RNA purification from the eluate using TRIzol reagent (Invitrogen), treatment with DNase I, and reverse transcription. The first-strand cDNA synthesis was carried out using random primers and SuperScript II reverse transcriptase (Invitrogen), and the second-strand cDNA was synthesized using the mixture of RNase H, T4 DNA ligase, and DNA polymerase I (New England BioLabs, Ipswich, MA). The TβRI-binding DNA fragments obtained from ChIP and the cDNA products of the RNA fragments obtained from RIP were subjected to Solexa deep sequencing at the City of Hope DNA Sequencing Core. The DNA and cDNA ends were repaired using T4 polynucleotide kinase and Klenow enzyme (New England BioLabs), followed by treatment with Taq polymerase to generate a protruding 3′ A base used for adaptor ligation. Following ligation of a pair of Solexa adaptors to the repaired ends, DNA and cDNA were amplified using the adaptor primers for 18 cycles and fragments of ∼250 bp (mononucleosome plu adaptors) were isolated from agarose gels. The purified DNA or cDNA was used directly for cluster generation and sequencing analysis using the Illumina Genome Analyzer II by following manufacturer protocols. Sequence reads were mapped to whole human genome NCBI build 36.1 (ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/) using NovoAlign (22). The aligned results were converted to browser extensible data (BED) format to identify sequences enriched in the ChIP/RIP-seq samples using a binding site detection tool, Hypergeometric Optimization of Motif (HOMER) (21), which uses the direction and density of the aligned reads and the average length of sample fragments to identify the binding sites. The DNA or RNA input purified prior to IP was used as a control for the calculation of fold enrichment. The locations of the binding sites were summarized and analyzed using the genome-wide binding portfolio. MEME (2) with default parameters was used to identify statistically overrepresented consensus motifs from all identified sense RNA sequences that coprecipitated with TβRI. E values, the probabilities of finding equally well-conserved patterns in random sequences, were calculated as previously described (2). These analyses were carried out at the City of Hope Bioinformatics Core.
IFA.
Indirect immunofluorescence assay (IFA) was carried out as described previously (44) using cells grown on coverslips. Fluorescent images were captured using a Princeton Instruments cooled charge-coupled device (CCD) digital camera from a Zeiss upright LSM 510 2-Photon confocal microscope with a 20×/0.6 objective.
RESULTS
Identification of TβRI-associated proteins in various cellular contexts.
To establish our cell line models, a retroviral vector-encoded, HA-tagged constitutively active TβRIT204D was used to transduce MCF10A/HER2 cells that stably overexpressed exogenous HER2, as well as the vector-transduced MCF10A/vec cells that expressed barely detectable level of endogenous HER2 (43). The resultant cell lines, MCF10A/HER2/TβRIT204D and MCF10A/TβRIT204D, were used to investigate alterations of TβRI-initiated signaling upon HER2-mediated cell transformation. MDA-MB-231, a HER2-negative and hormone receptor-negative breast cancer cell line, was also transduced by TβRIT204D to serve as a model for HER2-independent cancerous context. TβRIT204D was immunoprecipitated in these three cell lines, and the protein complexes that coprecipitated with TβRIT204D were subjected to mass spectrometry.
We identified 98 TβRI-associated proteins from MCF10A/TβRIT204D cells, all of them also associated with TβRI in MCF10A/HER2/TβRIT204D cells. This suggests that the TGF-β-initiated signaling events directly mediated by these proteins are retained in HER2-transformed cells. However, 119 unique proteins were also identified in MCF10A/HER2/TβRIT204D cells, representing a gain of novel TGF-β-mediated functions upon transformation by HER2. Among these unique proteins, 62 also associated with TβRI in the HER2-negative MDA-MB-231 cells, suggesting that their association with TβRI was specific to a general transformed/cancerous context but not to HER2 overexpression (see Fig. S1A and Tables S1 and S2 in the supplemental material). These 62 proteins that only associated with TβRI in a transformation-dependent manner were examined for functional annotation using Ingenuity pathway analysis. Interestingly, the most significant molecular function of these proteins was RNA posttranscriptional modification (see Fig. S1B and C and Table S3 in the supplemental material), with 16 proteins that are known to directly participate in this function identified as context-dependent TβRI-binding partners (Table 1). In addition, 31 out of the identified 62 proteins are known to reside in the nucleus (see Table S2 in the supplemental material), including 14 proteins that participate in RNA posttranscriptional modification (Table 1). Therefore, the comparison of the repertoires of TβRI-binding partners in various cellular contexts suggested a previously unrecognized nuclear translocation of TβRI and its potential nuclear function in regulating RNA posttranscriptional modification, which apparently occur specifically in transformed cells.
Table 1.
Proteins associated with TβRI in transformed cells with a known function in RNA posttranscriptional regulationa
| Symbol | Entrez name | Location | Type(s) | Function annotation |
|---|---|---|---|---|
| DDX5 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 5 | Nucleus | Enzyme | rRNA processing |
| DDX17 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 17 | Nucleus | Enzyme | RNA modification, unwinding, and processing |
| DDX21 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 21 | Nucleus | Enzyme | RNA modification and unwinding |
| DDX50 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 50 | Nucleus | Enzyme | RNA modification and unwinding |
| DHX15 | DEAH (Asp-Glu-Ala-His) box polypeptide 15 | Nucleus | Enzyme | RNA modification and processing |
| EIF4A1 | Eukaryotic translation initiation factor 4A, isoform 1 | Cytoplasm | Translation regulator | mRNA binding, unwinding, and modification |
| FBL | Fibrillarin | Nucleus | Other | rRNA processing |
| HNRNPA1 | Heterogeneous nuclear ribonucleoprotein A1 | Nucleus | Other | RNA modification, processing, and splicing |
| HNRNPH3 | Heterogeneous nuclear ribonucleoprotein H3 (2H9) | Nucleus | Other | RNA modification, processing, and splicing |
| HNRNPR | Heterogeneous nuclear ribonucleoprotein R | Nucleus | Other | RNA modification and processing |
| HNRNPL | Heterogeneous nuclear ribonucleoprotein L | Nucleus | Other | RNA modification, processing, and splicing |
| HNRNPM | Heterogeneous nuclear ribonucleoprotein M | Plasma membrane | Transmembrane receptor | RNA splicing |
| NPM1 | Nucleophosmin (nucleolar phosphoprotein B23, numatrin) | Nucleus | Transcription regulator | rRNA processing |
| SF3B3 | Splicing factor 3b, subunit 3, 130 kDa | Nucleus | Other | RNA modification, processing, and splicing |
| SYNCRIP | Synaptotagmin binding, cytoplasmic RNA interacting protein | Nucleus | Other | RNA modification, processing, and splicing |
| WDR43 | WD repeat domain 43 | Nucleus | Other | rRNA processing |
Proteins associate with TβRI only in MCF10A/HER2 and MDA-MB-231 cells and not in MCF10A cells.
Nuclear transport of TβRI is ligand inducible and requires a transformed cellular context.
To seek direct evidence for the nuclear translocation of TβRI and examine the effect of ligand-mediated activation on TβRI trafficking, we purified the nuclear and cytoplasmic fractions from MCF10A and MCF10A/HER2 cells stably expressing the wild-type TβRI gene in a time course after TGF-β treatment. Before TGF-β was added, TβRI was detected at low levels in the nucleus of MCF10A/HER2 but not MCF10A cells. Ligand treatment further induced the nuclear translocation of TβRI in the HER2-overexpressing line, but not the other, starting at 0.5 h after adding TGF-β (Fig. 1A). Inhibition of HER2 kinase activity using lapatinib abolished the TβRI nuclear transport upon TGF-β treatment in MCF10A/HER2 cells (Fig. 1B), suggesting that the translocation event was dependent on the downstream cascades of HER2 signaling. In contrast to the ligand-inducible nuclear translocation of wild-type TβRI, the constitutively active TβRIT204D exhibited high levels of nuclear localization in the absence of TGF-β in both MCF10A/HER2 and MDA-MB-231 cells (Fig. 1C). Using immunofluorescent staining with a TβRI-specific antibody, the ligand-induced nuclear translocation of endogenous TβRI and the inhibitory effect of lapatinib were further confirmed in MCF10A/HER2 cells (Fig. 1D). These results demonstrated a context-dependent, ligand-activated nuclear transport event of TβRI, enabling its interaction with multiple nuclear proteins involved in RNA regulation.
Fig 1.

Nuclear transport of TβRI is ligand inducible and requires a transformed context. (A) Cytoplasmic and nuclear fractions were prepared from MCF10A/TβRI and MCF10A/HER2/TβRI cells that were treated with TGF-β (2 ng/ml) for the indicated times. Western blotting was carried out using the indicated antibodies. Purity of different cellular fractions was confirmed by Western blotting of GAPDH and histone H3, which are exclusively located in the cytoplasm and nucleus, respectively. (B) MCF10A/HER2/TβRI cells were treated with lapatinib (1 μM) for 6 h before TGF-β treatment. Cell fractionation and Western blotting were performed as indicated. (C) Cells were transfected with vector, wild-type (wt) TβRI construct, or a constitutively active TβRI construct (T204D) as indicated. At 24 h after transfection, cells were treated with TGF-β for 0.5 h or left untreated, and they were then subjected to fractionation and Western blotting. (D) IFA images of MCF10A and MCF10A/HER2 cells treated with TGF-β for 0.5 h or left untreated, in the presence or absence of a prior 6-h lapatinib treatment. Cells were stained with TβRI antibody for detection of endogenous TβRI (red) and 4′,6-diamidino-2-phenylindole (DAPI) for nuclei (blue). The bar equals 10 μm. Nuclear TβRI is indicated by arrows. The graph at the bottom shows percentages of cells with nuclear staining of TβRI. Two hundred cells under each treatment were counted, and the average of three independent experiments is shown. *, P < 0.001.
Importin β1 and nucleolin are required for TβRI nuclear transport.
A thorough search of the amino acid sequence of TβRI failed to identify a putative nuclear localization signal (NLS). Therefore, TβRI may enter the nucleus through the interaction with other nuclear proteins carrying an NLS. The nuclear transport receptor importin β1 and nucleolar protein nucleolin have been reported to mediate the nuclear translocation of ErbB receptors (15, 18) and were found to coprecipitate with TβRI in both MCF10A/HER2 and MDA-MB-231 breast cancer cells but not in MCF10A cells (see Table S1 in the supplemental material). We therefore hypothesized that in a permissive cellular context, TβRI was translocated to the nucleus through the interaction with importin β1 and/or nucleolin. We first confirmed the association of TβRI with these two proteins by coimmunoprecipitation and Western blotting. Both importin β1 and nucleolin, as well as another nucleolar protein nucleophosmin, associated with TβRI in both cytoplasmic and nuclear fractions of MCF10A/HER2 cells (Fig. 2A, top). Inhibition of the HER2 kinase activity abolished these associations in the nucleus but not in the cytoplasm (Fig. 2A, bottom). In MCF10A cells, associations of TβRI with importin β1, nucleolin, and nucleophosmin were also detected in the cytoplasm at levels comparable to those in the MCF10A/HER2 cytoplasmic fractions, but there was little nuclear accumulation of TβRI (Fig. 2A, top). Smad2/3, the well-known effectors of TGF-β signaling, associated with TβRI in the cytoplasm, but not the nucleus, in a ligand-inducible manner in both MCF10A and MCF10A/HER2 cells (Fig. 2A, top). Using siRNAs of importin β1 and nucleolin to specifically knock down their respective expression, we determined that both proteins were required for the basal and ligand-induced nuclear transport of TβRI in MCF10A/HER2 cells (Fig. 2B and C). Knockdown of importin β1 also reduced the nuclear levels of nucleolin (Fig. 2B), suggesting that as part of the general nuclear import machinery, importin β1 is also essential for the nuclear transport of nucleolin.
Fig 2.

Importin β1 and nucleolin are required for TβRI nuclear transport. (A) TβRI IP was performed using HA antibody in cells treated and fractionated as indicated. Importin β1 (KPNB1), nucleolin (NCL), nucleophosmin (NPM), Smad2/3, and TβRI (HA tag) were detected by Western blotting. (B) MCF10A/HER2/TβRI cells were transfected with siRNAs targeting importin β1, nucleolin, or a negative-control (Ctrl) siRNA. At 48 h after transfection, cells were treated with TGF-β as indicated, fractionated, and analyzed by Western blotting. (C) IFA of cells transfected with various siRNAs and treated with TGF-β for 0.5 h. HA antibody was used to stain for HA-tagged TβRI.
The context-dependent TβRI nuclear transport is controlled by Ran GTPase activity.
The nuclear import of proteins through nuclear pore complexes involves dynamic interactions between cargoes, carriers, and the Ran GTPase. TβRI interacted with the carrier protein importin β1, possibly through other NLS-carrying proteins such as nucleolin and Smads, in the cytoplasm of both MCF10A and MCF10A/HER2 cells and in the presence of lapatinib (Fig. 2A). However, the nuclear import of TβRI was achieved only in MCF10A/HER2 cells undergoing active HER2 signaling, suggesting a control of the transport event by contextual factors that are altered in HER2-transformed cells. Interestingly, recent studies indicate that Ran GTPase is activated by growth factors, especially heregulin, a ligand that signals through the HER2 receptor (25). In the nucleus, Ran GTPase predominantly exists as the active GTP-bound form, whereas in the cytoplasm, the predominant form is inactive and GDP bound. This results in a gradient of Ran activity between the two cellular compartments which is essential for nuclear protein import (38). Activation of Ran GTPase, represented by elevated levels of RanGTP (active form) in the nucleus, can facilitate cargo release from the importin complex into the nucleoplasm upon nuclear entry, resulting in the accumulation of transported cargo in the nucleus. To examine if Ran is activated in MCF10A/HER2 cells, and if this contributes to the context-dependent nuclear transport of TβRI, we first measured Ran activity by RanBP1 pulldown assay in MCF10A and MCF10A/HER2 cells, and we indeed detected significantly higher activity and nuclear distribution of Ran in the latter (Fig. 3A). Next, we modulated Ran activity by transfecting MCF10A cells with a constitutively active mutant of Ran, Ran(F35A), and MCF10A/HER2 cells with a dominant negative mutant of Ran, Ran(T24N). In the absence of HER2 transformation, activation of Ran enabled the nuclear transport of endogenous TβRI in MCF10A cells, whereas inhibition of Ran disabled this translocation event in MCF10A/HER2 cells (Fig. 3A). The nuclear transport of endogenous TβRI was also observed in two breast cancer cell lines, BT474 (HER2 positive) and MDA-MB-231 (HER2 negative), and was abolished by expression of the dominant negative Ran(T24N) (Fig. 3B). A basal level of TβRI nuclear transport was observed in these cells in the absence of exogenous TGF-β, likely due to the autocrine TGF-β signaling and the relatively high endogenous Ran activity in these cells. In MCF10A cells expressing active Ran(F35A), knockdown of importin β1 and nucleolin both abolished the Ran-induced nuclear import of TβRI (Fig. 3C). In addition, we noted that the nuclear levels of importin β1, nucleolin, and Smad2/3 were not significantly affected by Ran activity (Fig. 3C). These results suggest that although TβRI complexes with importin β1 in the cytoplasm regardless of the cellular context (Fig. 2A), the subsequent release of TβRI from the nuclear import complex upon nuclear entry and the accumulation of TβRI in the nucleus are highly dependent on the Ran activity. This Ran-dependent mechanism seems different from the highly efficient nuclear transports of the NLS-containing proteins nucleolin and Smads, which require only the basal level of Ran activity observed in untransformed MCF10A cells.
Fig 3.

The context-dependent TβRI nuclear transport is controlled by Ran GTPase activity. (A) Cells were transfected as indicated with plasmids encoding the wild-type Ran, constitutively active Ran(F35A), dominant negative Ran(T24N), or the empty vector. At 48 h after transfection, cells were treated with TGF-β for 0.5 h or left untreated, fractionated, or subjected to RanBP1 pulldown assay and analyzed by Western blotting. (B) BT474 and MDA-MB-231 cells were transfected with Ran constructs and treated with TGF-β as described above, followed by RanBP1 pulldown assay and Western blotting. (C) MCF10A cells cotransfected with Ran(F35A) and siRNAs of nucleolin, importin β1, or control sequence, as well as control MCF10A cells, were treated with TGF-β for 0.5 h and subjected to fractionation and Western blotting. Abbreviations are the same as in Fig. 2.
TβRI nuclear transport requires the L45 loop and Smad2/3.
To determine the TβRI motif responsible for its nuclear translocation, we constructed GFP fusion proteins containing the full-length (1 to 503 aa) or C-terminally truncated (1 to 287 aa and 1 to 205 aa) TβRI (Fig. 4A). When expressed in the MDA-MB-231 cells treated with TGF-β, the full-length and 287-aa TβRI constructs exhibited both cytoplasmic and nuclear staining in the majority of cells, whereas the 205-aa TβRI construct was not detected in the nucleus (Fig. 4B). Because the TβRI motif of 205 to 287 aa contains the L45 loop that serves as the docking site for R-Smads (16, 24, 34), we further examined the role of the L45 loop in TβRI nuclear transport using two previously reported full-length TβRI constructs carrying point mutations in the L45 loop, ALK5(D266A) and ALK5(3A) (Fig. 4A). Both mutants retain kinase activity but are unable to activate Smads (23). In MDA-MB-231 cells transfected with ALK5(D266A), ALK5(3A), or wild-type TβRI (Alk5) constructs, ligand-induced nuclear translocation was observed only with the wild-type receptor and not the L45 loop mutants (Fig. 4C). Similar results were observed in MCF10A/HER2 cells transfected with these truncated or mutated TβRI constructs (see Fig. S2 in the supplemental material). We therefore speculated that Smad2/3 could be involved in TβRI translocation and that the inability of ALK5(D266A) and ALK5(3A) to interact with Smad2/3 could be responsible for their impaired nuclear transport. Using a mixture of siRNAs targeting Smad2 and Smad3, we knocked down the Smad2/3 expression in MDA-MB-231/TβRI (wild-type) cells, where the siRNAs reduced the associations of TβRI with importin β1 and nucleolin and abolished the nuclear translocation of TβRI (Fig. 4D). Thus, TGF-β induces transient interaction between TβRI and Smad2/3 (Fig. 2A), followed by the complexation with importin β1 and nucleolin in the cytoplasm to trigger nuclear translocation. In a transformed cellular context with high Ran activity, this event results in accumulation of TβRI in the nucleus.
Fig 4.

TβRI nuclear transport requires the L45 loop and Smad2/3. (A) Schematics of the TβRI constructs and their potential of nuclear translocation. ECD, extracellular domain; GS, glycine-serine-rich domain; TM, transmembrane domain. (B) IFA images of TGF-β-treated MDA-MB-231 cells expressing the GFP-fused TβRI constructs indicated in panel A. (C) MDA-MB-231 cells were transfected with HA-tagged wild-type TβRI, the L45(3A) mutant, or the D266A mutant, with the sequences of the L45 loop indicated in panel A. Transfected cells were treated with TGF-β for 0.5 h or left untreated, and the HA-tagged TβRI levels were analyzed in the nuclear fraction and whole-cell lysates by Western blotting. (D) MDA-MB-231/TβRI cells transfected with siRNAs targeting Smad2/3 or control siRNA were treated with TGF-β and subjected to IP, fractionation, and Western blotting as indicated. Abbreviations are the same as in Fig. 2.
Identification of TβRI-binding RNA sequences.
The context-dependent nuclear translocation of TβRI may enable unique, previously unrecognized functions of the receptor in the nucleus of transformed cells. To explore the potential nuclear function of TβRI, we purified the DNA and RNA sequences that bound to TβRI in MCF10A and MCF10A/HER2 cells stably expressing TβRIT204D by using chromatin immunoprecipitation (ChIP) and RNA IP (RIP), respectively. Consistent with the absence of nuclear TβRI in MCF10A cells, no DNA or RNA was obtained from the TβRI precipitates in these cells. In contrast, detectable amounts of DNA and RNA were coprecipitated with TβRI from equal numbers of MCF10A/HER2 cells. The TβRI-bound DNA sequences and the cDNA products of TβRI-bound RNA sequences were subjected to deep sequencing. We identified 7.5 million reads from the DNA and 17.2 million reads from the RNA/cDNA samples. The reads were aligned to the human genome, and high-frequency binding sites were determined. Most of the aligned DNA tags (99.92%) scattered in a nonspecific pattern, and only 0.08% of total aligned tags could possibly be mapped to 130 particular DNA sites, suggesting that TβRI did not bind to specific DNA sequences. In contrast, a large fraction (13.2%) of aligned RNA tags clustered at 9,094 RNA sites, suggesting that TβRI likely bound to specific RNA sequences (Fig. 5A; see also Tables S4 and S5 in the supplemental material). Most RNA sites were located in exons (53%) or introns (28%), whereas only 3 DNA sites (2%) were located in putative promoter regions (Fig. 5B). In addition, the majority of RNA sites resided within the −1 × 104 to 3 × 104 bp region relative to transcription start sites (TSSs), likely indicating their intragenic locations, whereas most DNA sites were more distal to the TSSs (within −2 × 105 to 2 × 105 bp) (Fig. 5C). Thus, the sequence counts of TβRI-binding DNA and TβRI-binding RNA sites, as well as their genomic distributions, strongly suggested that TβRI specifically bound to RNA, rather than DNA, targets. Among the identified 9,094 TβRI-binding RNA sites that involve genes (see Fig. S3 in the supplemental material), a purine-rich consensus motif of AGGAGGAG was discovered (Fig. 5D). This consensus motif appeared at least once in 2,292 identified TβRI-binding RNA sites (see Table S5 in the supplemental material).
Fig 5.

Identification of TβRI-binding RNA sequences. (A) Summary of the total tags, aligned tags, tags on binding sites, and binding sites detected from ChIP or RIP deep sequencing. (B) Regional distributions of identified TβRI-binding RNA and DNA sequences. (C) Summarized locations of identified TβRI-binding RNA and DNA sequences relative to transcription start sites (TSS). (D) Consensus motif identified from TβRI-binding RNA sequences with the most significant E value, the probability of finding an equally well-conserved pattern in random sequences. (E) Confirmation of selected TβRI-binding RNA sequences using RIP followed by RT-PCR. The nuclear and cytoplasmic fractions of cells that were treated as indicated were subjected to RIP using HA tag antibody or normal IgG (as a negative control). Input RNA was used as a positive control for RT-PCR. Primer pairs specific for each selected TβRI-binding site or GAPDH (as a negative control) were used for fragment detection. (F) Pulldown assays were carried out using biotinylated RNA probes with wild-type (wt) or mutated (mt) sequences as indicated. MCF10A/HER2 cells were transfected with siRNAs of TβRI (RI), hnRNP A1 (A1), or control sequence (C) and treated with TGF-β for 0.5 h before nuclear extracts were prepared and incubated with each RNA probe. After pulldown with streptavidin beads, the eluates as well as the input nuclear extracts were analyzed by Western blotting using the indicated antibodies. (G) Co-IP assay was carried out using TβRI antibody in cells treated with TGF-β for 0.5 h or left untreated. The precipitates as well as the input lysates were analyzed by Western blotting as indicated.
We selected five TβRI-binding RNA sites for which a relatively high abundance of read counts was obtained, and we validated their associations with TβRI by RIP coupled with RT-PCR. These included 2 sites in the EGFR gene, 1 site in the VEGFA gene, 1 site in the CCND1 gene, and 1 site in the PKM2 gene. MCF10A and MCF10A/HER2 cells expressing wild-type TβRI were treated with TGF-β or left untreated before TβRI-RIP was performed using purified nuclear and cytoplasmic fractions. All target RNAs were detected in the TβRI precipitates from the nuclear fractions of MCF10A/HER2 cells following TGF-β-induced TβRI nuclear transport but not detected or detected at a very low level in TGF-β-treated MCF10A cells (Fig. 5E). This suggests that the nuclear transport of TβRI is required for its interaction with these RNA targets and reveals a novel nuclear function of TβRI mediated by these RNA targets.
We further examined the affinity of binding of TβRI to the two validated RNA binding sites in EGFR introns 1 and 10, as well as their mutated versions in which the purine-rich consensus motif had been destroyed, through incubating biotinylated RNA probes with the nuclear extracts of TGF-β-induced MCF10A/HER2 cells. The Chr7-6 probe contained the AGGAGGAG consensus motif, while the Chr7-193 probe contained a G/A-rich region resembling the identified consensus sequence (Fig. 5F). Both probes were able to efficiently pull down TβRI proteins, and their binding to TβRI was significantly reduced by disruption of the G/A-rich region in the mutated probe and by siRNA-mediated knockdown of TβRI expression (Fig. 5F). Heterogeneous ribonucleoprotein A1 (hnRNP A1) is an RNA-binding protein involved in various aspects of RNA processing and was identified as a TβRI-binding partner in transformed cells (Table 1). Interestingly, hnRNP A1 was also pulled down by the two wild-type RNA probes, but at much lower levels by the mutated probes, suggesting that hnRNP A1 can also bind to the purine-rich motif in the target RNA sequences. Moreover, TβRI and hnRNP A1 seem to bind to the RNA targets in a synergistic manner as a complex, as knockdown of either gene resulted in diminished binding of both proteins to the wild-type RNA probes (Fig. 5F). We noted that the weak binding of hnRNP A1 to the two mutated probes was not further diminished by TβRI depletion (Fig. 5F), which suggests that the mutated probes likely contain sites that enable low levels of hnRNP A1 binding and that the purine-rich region is responsible for enhanced binding of hnRNP A1 and TβRI. Consistent with the mass spectrometry result (see Table S1 in the supplemental material), coimmunoprecipitation (co-IP) experiments confirmed that TβRI associated with hnRNP A1 in MCF10A/HER2 and MDA-MB-231, but not MCF10A cells, and that this association was enhanced by TGF-β treatment (Fig. 5G).
Nuclear TβRI selectively induces EGFR splicing isoform c.
Among all genes involved, EGFR harbored the highest number of identified TβRI-binding RNA sites (see Table S5 in the supplemental material) and was therefore selected for further investigation. Within EGFR gene, 17 different RNA sites were identified in TβRI RIP-seq with a peak score of ≥4, including 12 sites located within intron 1 and 1 site in each of introns 7, 10, and 20 and exons 5 and 8 (Table S5). The two sites validated by PCR and RNA probe pulldown assays (Fig. 5E and F), Chr7-6 and Chr7-193, respectively, resided in intron 1 and intron 10 and were enriched 403- and 105-fold in TβRI RIP, compared to the RNA input control (Fig. 6A and Table S5). Human EGFR is encoded by two transcripts of 10.5 kb and 5.8 kb (isoform a), both of which give rise to full-length protein. In addition, three alternative transcripts of 1.8, 2.4, and 3.0 kb, referred to as isoforms c, b, and d, respectively, are also derived from the EGFR gene (1, 35). The 1.8-kb transcript is generated by terminating at intron 10 and contains exon 10/intron 10 boundary, whereas the 2.4- and 3.0-kb transcripts are generated by incorporating alternate exon 15A or 15B, which results in unique C-terminal sequences in the protein products. Interestingly, the TβRI-binding RNA site Chr7-193 was localized to intron 10, a position that could be important to maintain the exon 10/intron 10 boundary that generates the 1.8-kb isoform c. This speculation was further supported by the binding of TβRI to hnRNP A1, a known splicing repressor, on the Chr7-193 site (Fig. 5F).
Fig 6.

Nuclear TβRI selectively induces EGFR splicing isoform c. (A) Schematic of the human EGFR gene and the positions of all identified TβRI-binding RNA sites that were aligned to the EGFR gene. (B) Quantitative RT-PCR of various EGFR-derived RNAs in MCF10A/HER2 cells treated or transfected as indicated. Data were normalized to the level of GAPDH and then compared to that in untreated cells (the first set of bars). Each data point represents the mean ± standard deviation of 3 wells. *, P < 0.001. Abbreviations are the same as in Fig. 2. (C) Quantitative RT-PCR of EGFR-derived RNAs in MCF10A/HER2 cells treated or transfected as indicated. *, P < 0.001. DMSO, dimethyl sulfoxide. (D) Quantitative RT-PCR of EGFR-derived RNAs in cells transfected with various Ran constructs. *, P < 0.001. (E) MCF10A and MCF10A/HER2 cells were treated or transfected as indicated before whole-cell lysates were subjected to Western blotting for detection of various EGFR isoforms. The positions of 60- and 80-kDa sEGFR generated from the splicing isoform c are indicated.
We measured the levels of various EGFR transcript variants as well as the primary RNA transcripts in MCF10A/HER2 cells in response to TGF-β. Within 1 h of treatment, TGF-β significantly increased isoform c, ∼4-fold, did not affect isoform a, modestly increased isoforms b and d, and reduced the primary EGFR transcripts (Fig. 6B; see also Fig. S4 in the supplemental material). The induction of EGFR isoform c by TGF-β was abrogated by conditions that destroyed TβRI nuclear translocation, including treatments with the HER2 kinase inhibitor lapatinib and type I/II TGF-β receptor inhibitor LY2109761, siRNAs of Smad2/3, importin β1, nucleolin, or TβRI and overexpression of the ALK5(3A) mutant that failed to undergo nuclear translocation (Fig. 6B and C). In addition, knockdown of hnRNP A1 expression by siRNA, which diminished TβRI binding to RNA targets (Fig. 5F), reduced the basal level of isoform c and completely abolished its induction by TGF-β (Fig. 6B). Moreover, TGF-β induced isoform c ∼3-fold in MCF10A cells expressing constitutively active Ran(F35A) but failed to do so in the control MCF10A cells that lacked TβRI nuclear transport (Fig. 6D). Consistent with this result, inhibition of Ran activity by dominant negative Ran(T24N) abolished the effect of TGF-β on EGFR splicing in MCF10A/HER2 and MDA-MB-231 breast cancer cells (Fig. 6D). These results suggest that TGF-β promotes the generation of EGFR transcript isoform c through inducing nuclear transport of TβRI and its association with RNA targets in synergy with hnRNP A1.
The 1.8-kb EGFR isoform c encodes a secreted 60- or 80-kDa soluble EGFR (sEGFR) protein, which contains only part of the EGFR extracellular region (35). To determine if TGF-β induces the production of sEGFR in a context-dependent manner, we performed Western blotting in both MCF10A and MCF10A/HER2 cells using two different EGFR antibodies, one recognizing an N-terminal region present in the protein products of all EGFR isoforms and the other recognizing a C-terminal region that is present only in the product of isoform a. In MCF10A/HER2 cells, TGF-β induced both the 60- and 80-kDa bands of EGFR recognized by the N-terminal specific antibody, without affecting the 170-kDa full-length EGFR. This effect was abrogated by lapatinib, Smad2/3 siRNA, or inhibition of Ran activity. In MCF10A cells, TGF-β failed to induce 60- or 80-kDa sEGFR but gained this ability when they underwent activation of Ran GTPase (Fig. 6E). Therefore, the context-dependent, ligand-inducible TβRI nuclear translocation confers a novel function of TGF-β in regulating EGFR RNA processing, resulting in increased production of soluble, but not full-length, EGFR.
DISCUSSION
In this study, we used our previously established model of HER2-transformed MCF10A (MCF10A/HER2) (43) and the HER2-negative breast cancer cell line MDA-MB-231, as well as the untransformed MCF10A model, to investigate how a transformed/cancerous cellular context alters the repertoire of proteins that bind to activated TβRI and possibly mediate its downstream signaling. We found that overexpression of HER2 significantly broadened the spectrum of TβRI-binding proteins, with 119 additional proteins identified in MCF10A/HER2 cells. Although an association between HER2 and TβRI has been reported in our previous study (45) and was demonstrated here again by identification of HER2 in the TβRI coprecipitates from MCF10A/HER2 cells (see Table S1 in the supplemental material), most of the additional TβRI-binding partners identified from MCF10A/HER2 are unlikely to be pulled down due to a direct interaction with HER2 instead of with TβRI, as many of these proteins (62 out of 119) also associated with TβRI in the HER2-negative MDA-MB-231 cells. The fact that half of the context-dependent TβRI-binding partners are located in the nucleus urged us to test the hypothesis that in a permissive cellular context, TβRI is translocated to the nucleus, where it interacts with these nuclear proteins. During the course of this study, Mu et al. reported that TβRI undergoes ubiquitination upon ligand treatment, resulting in cleavage of the receptor by TNF-α converting enzyme (TACE) and nuclear translocation of a 34-kDa TβRI intracellular domain (29a). Here, we determined that full-length TβRI also underwent nuclear translocation, which was specific to transformed cells and enabled the interaction of TβRI with the herein identified RNA targets, through the mechanisms summarized in Fig. 7 and discussed below.
Fig 7.
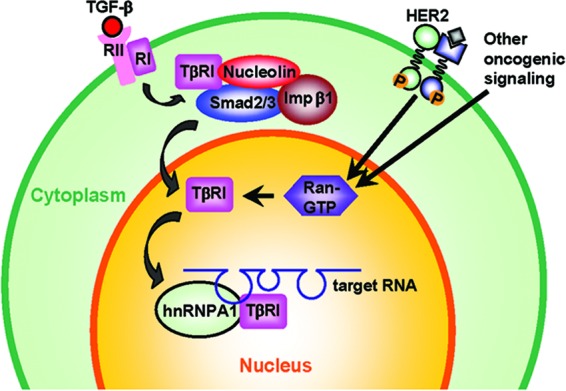
Schema of the context-dependent nuclear translocation of TβRI. In our proposed model, TGF-β induces transient interaction between TβRI and Smad2/3, followed by their complexation with importin β1 and nucleolin in the cytoplasm. Consequently, the nuclear transport of TβRI can be initiated at a certain degree in both normal and cancer cells. However, upon nuclear entry, efficient release of the TβRI cargo into the nucleus requires a high level of RanGTP, which is observed only in HER2-transformed cells and HER2-negative cancer cells and not in untransformed cells. Once accumulated in the nucleus, TβRI dissociates from Smad2/3 and binds to the G/A-rich motif in RNA targets in synergy with hnRNP A1, exerting a regulatory function in RNA processing.
Spatial regulation through either subcellular trafficking or interaction with spatially restricted partners is a key determinant of protein function. Several receptor tyrosine kinases, including the ErbB family members, fibroblast growth factor receptors, insulin receptor, and vascular endothelium growth factor receptor Flk1/KDR, are known to migrate to the nucleus and act as transcription factors for certain target genes (6). Nuclear HER2 has been found to associate with multiple genomic targets, including the cyclooxygenase enzyme COX-2 gene promoter, and stimulate gene transcription (42). The nuclear transport of HER2 involves interaction with the transport receptor importin β1 and other molecules in endocytic internalization (18). Unlike HER2, which contains an NLS that directs its nuclear transport through importin β1 (18), TβRI does not carry a putative NLS and appeared to depend on other proteins, such as nucleolin and Smad2/3, for the nuclear transport. Based on the size of the nuclear TβRI as indicated by Western blotting, nuclear TβRI seemed to be full length and exhibited the same migration speed in the gel as the cytoplasmic TβRI (Fig. 1A). This observation further differentiates the mechanism of full-length TβRI nuclear transport from the reported mechanisms for the translocation of its intracellular domain (29a) and ErbB4 (6), in which the receptor is processed by membrane-localized proteases to produce a soluble cytoplasmic domain fragment that translocates into the nucleus. Our data indicated that the cytoplasmic interactions between TβRI and low levels of importin β1 and nucleolin were also detectable in untransformed MCF10A cells (Fig. 2A). Therefore, nuclear transport of TβRI mediated by importin β1 may be initiated at a certain degree regardless of the cellular context in both normal and cancer cells. However, upon nuclear entry, efficient release of the TβRI cargo into the nucleus requires a high level of RanGTP to dissociate the importin complex (38). Therefore, only transformed cells that express high Ran activity (Fig. 3) exhibit significant nuclear accumulation of TβRI. It is possible that the high Ran activity in cancer cells, possibly as a result of the growth factor-mediated induction (25), has a global effect on the dynamics of nuclear protein import, and perhaps different proteins, due to their unique features, may be affected to various extents. The different features of the proteins that determine Ran dependency are not understood and will require further investigation. Whether the cancer-specific activation of Ran GTPase allows more proteins to shuttle to the nucleus and the novel nuclear functions of these “conditionally” nuclear proteins, as well as their relevance to the cancer disease, are intriguing future research directions.
Using RIP coupled with deep sequencing, we identified a novel nuclear function of TβRI through binding to specific RNA targets. A consensus G/A-rich motif was identified among TβRI-binding RNA sites based on the probability of finding this pattern among random sequences. Although in the case study of EGFR introns 1 and 10 the G/A-rich motif seems to mediate the synergistic binding of TβRI and hnRNP A1 to RNA targets (Fig. 5F), a more specific motif discovery approach, such as CLIP-Seq, would be desired to precisely define the TβRI-binding sequences in future studies. Although recent studies demonstrated the capacity of R-Smads to bind to double-stranded RNA targets (8, 9), TβRI does not interact with Smad2/3 in the nucleus (Fig. 2A), and the previously reported Smad-binding RNA motif (CAGAC or CAGGG) was not overrepresented in the herein-identified TβRI-binding sites, suggesting that TβRI and Smad2/3 act on different RNA targets in the nucleus. Despite their independent roles in the nucleus, the cytoplasmic interaction between TβRI and Smad2/3 triggered by ligand treatment is required for the nuclear entry of TβRI, as knockdown of Smad2/3 abolished TβRI complexation with importin β1 and nucleolin, as well as the nuclear transport of TβRI (Fig. 4D). In addition, the nuclear translocation of Smads and their binding to DNA and RNA targets are not cancer specific, whereas those of TβRI are strictly dependent on a transformed/cancerous cellular context, in particular high Ran GTPase activity. Thus, our study identified a novel RNA-binding activity of the nuclear localized TβRI that is clearly distinct from the previously reported RNA-regulating function of Smads.
We further found that through triggering the nuclear transport of TβRI, TGF-β induced the EGFR transcript isoform c, which is generated by ignoring a splice donor site and use of an alternative polyadenylation signal located in intron 10 (35). The herein-identified TβRI-binding RNA site Chr7-193 resides in intron 10, 184 nucleotides (nt) downstream of the poly(A) addition signal. Interestingly, TβRI and hnRNP A1 complex on the purine-rich region in Chr7-193 in a synergistic manner; depletion of hnRNP A1 reduced the basal level of isoform c and disrupted TβRI binding to Chr7-193 probe as well as isoform c induction by TGF-β (Fig. 5F and 6B). These results suggest that hnRNP A1 is required for the generation of isoform c, possibly through its reported function to suppress splicing (33), allowing the alternative polyadenylation signal in intron 10 to be used. Nuclear TβRI significantly enhanced the binding of hnRNP A1 to intron 10 (Fig. 5F), possibly potentiating the effect of hnRNP A1 on splicing and/or 3′-end RNA processing, which results in increased production of isoform c. Although the herein-identified G/A-rich motif may not resemble the hnRNP A1 high-affinity canonical sequence, UAGRG(A/U), identified by Burd and Dreyfuss (5), there are reports (10, 20) that noncanonical hnRNP A1 binding sites may be used in alternative splicing. In addition, Martinez-Contreras et al. have shown that certain intronic hnRNP A/B-binding sites can stimulate in vitro splicing of pre-mRNAs (27), suggesting a context-dependent function of hnRNPs A and B. Despite the herein-observed effect of TGF-β and hnRNP A1 on EGFR splicing, the mechanism that regulates EGFR splicing and 3′-end RNA processing and the precise role of TβRI/hnRNP A1 complex in this biological event remain unclear and await future investigations.
Through enhancing the generation of EGFR transcript isoform c, TGF-β increased the expression of 60- and 80-kDa sEGFR in transformed but not untransformed cells (Fig. 6E). These truncated receptors are thought to sequester the EGF ligands in the extracellular environment and/or form inactive heterodimers with the cell surface EGFR, resulting in suppression of EGFR-mediated signaling (35). sEGFR is detectable in the serum of cancer patients and appears to be a clinical marker for cancer prognosis. High sEGFR levels in patients with various types of cancer, including breast cancer, are associated with better clinical outcomes, possibly due to the inhibitory effect of sEGFR on EGFR signaling (4, 30). The effects of TGF-β-induced sEGFR production on HER2 signaling and cell response to anti-EGFR/HER2 therapeutics need to be further evaluated using in vitro and in vivo models. Tumorigenesis often involves large-scale alterations in alternative RNA processing (41). It is possible that through the hnRNP A1-mediated mechanism identified in this study, nuclear TβRI regulates the alternative processing of a panel of genes, such as those harboring the TβRI-binding RNA sites, as a novel means to participate in global RNA regulation in cancer cells. Elucidations of these genes and their regulation by TGF-β will open a new avenue for studying redirected TGF-β signaling in cancer.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by grant numbers R00 CA125892 (S.E.W.), R01 GM40639 (R.-J.L.), and P30 CA033572 from the National Institutes of Health.
We thank Kenneth Dery, Shiuan Chen, Susan Kane, Mei Kong, and Yate-Ching Yuan for valuable comments, the DNA Sequencing/Solexa Core, Bioinformatics Core, and Light Microscopy Digital Imaging Core for professional services, and many other colleagues at City of Hope for enthusiastic support and discussion. We acknowledge Peter ten Dijke (Leiden University Medical Center, Netherlands) for kindly providing the ALK5(D266A)/HA and ALK5(3A)/HA constructs and Richard Cerione (Cornell University) for kindly providing the Ran(WT), Ran(F35A), and Ran(T24N) constructs.
Footnotes
Published ahead of print 2 April 2012
Supplemental material can be found at http://mcb.asm.org/.
REFERENCES
- 1.Albitar L, et al. 2010. EGFR isoforms and gene regulation in human endometrial cancer cells. Mol. Cancer 9:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey TL, Williams N, Misleh C, Li WW. 2006. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34:W369–W373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. 2002. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 115:3193–3206 [DOI] [PubMed] [Google Scholar]
- 4.Baron AT, Wilken JA, Haggstrom DE, Goodrich ST, Maihle NJ. 2009. Clinical implementation of soluble EGFR (sEGFR) as a theragnostic serum biomarker of breast, lung and ovarian cancer. IDrugs 12:302–308 [PubMed] [Google Scholar]
- 5.Burd CG, Dreyfuss G. 1994. RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J. 13:1197–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpenter G. 2003. Nuclear localization and possible functions of receptor tyrosine kinases. Curr. Opin. Cell Biol. 15:143–148 [DOI] [PubMed] [Google Scholar]
- 7.Chow A, Arteaga CL, Wang SE. 2011. When tumor suppressor TGFbeta meets the HER2 (ERBB2) oncogene. J. Mammary Gland Biol. Neoplasia 16:81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis BN, Hilyard AC, Lagna G, Hata A. 2008. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 454:56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. 2010. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol. Cell 39:373–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dery KJ, et al. 2011. Mechanistic control of carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM1) splice isoforms by the heterogeneous nuclear ribonuclear proteins hnRNP L, hnRNP A1, and hnRNP M. J. Biol. Chem. 286:16039–16051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Derynck R, Akhurst RJ, Balmain A. 2001. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 29:117–129 [DOI] [PubMed] [Google Scholar]
- 12.Derynck R, Zhang YE. 2003. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425:577–584 [DOI] [PubMed] [Google Scholar]
- 13.Dumont N, Arteaga CL. 2003. Targeting the TGF beta signaling network in human neoplasia. Cancer Cell 3:531–536 [DOI] [PubMed] [Google Scholar]
- 14.Ewan KB, et al. 2002. Latent transforming growth factor-beta activation in mammary gland: regulation by ovarian hormones affects ductal and alveolar proliferation. Am. J. Pathol. 160:2081–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farin K, Di Segni A, Mor A, Pinkas-Kramarski R. 2009. Structure-function analysis of nucleolin and ErbB receptors interactions. PLoS One 4:e6128 doi:10.1371/journal.pone.0006128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng XH, Derynck R. 1997. A kinase subdomain of transforming growth factor-beta (TGF-beta) type I receptor determines the TGF-beta intracellular signaling specificity. EMBO J. 16:3912–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng XH, Derynck R. 2005. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 21:659–693 [DOI] [PubMed] [Google Scholar]
- 18.Giri DK, et al. 2005. Endosomal transport of ErbB-2: mechanism for nuclear entry of the cell surface receptor. Mol. Cell. Biol. 25:11005–11018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ham AJL. 2004. Proteolytic digestion protocols, p 10–17. In Caprioli RM, Gross ML. (ed), The encyclopedia of mass spectrometry, vol 2 Biological applications, part A. Elsevier, Oxford, United Kingdom [Google Scholar]
- 20.Han SP, Tang YH, Smith R. 2010. Functional diversity of the hnRNPs: past, present and perspectives. Biochem. J. 430:379–392 [DOI] [PubMed] [Google Scholar]
- 21.Heinz S, et al. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38:576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hercus C. 2009. Novocraft short read alignment package. http://ryanmlayer.files.wordpress.com/2009/10/novocraftv2-05.pdf
- 23.Itoh S, et al. 2003. Elucidation of Smad requirement in transforming growth factor-beta type I receptor-induced responses. J. Biol. Chem. 278:3751–3761 [DOI] [PubMed] [Google Scholar]
- 24.Liu IM, et al. 2009. TGFbeta-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGFbeta switch. EMBO J. 28:88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ly TK, et al. 2010. Activation of the Ran GTPase is subject to growth factor regulation and can give rise to cellular transformation. J. Biol. Chem. 285:5815–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma ZQ, et al. 2009. IDPicker 2.0: improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 8:3872–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Contreras R, et al. 2006. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 4:e21 doi:10.1371/journal.pbio.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Massagué J, Blain SW, Lo RS. 2000. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103:295–309 [DOI] [PubMed] [Google Scholar]
- 29.Massagué J, Gomis RR. 2006. The logic of TGFbeta signaling. FEBS Lett. 580:2811–2820 [DOI] [PubMed] [Google Scholar]
- 29a.Mu Y, et al. 2011. TRAF6 ubiquitinates TGFbeta type I receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller V, et al. 2006. Prognostic and predictive impact of soluble epidermal growth factor receptor (sEGFR) protein in the serum of patients treated with chemotherapy for metastatic breast cancer. Anticancer Res. 26:1479–1487 [PubMed] [Google Scholar]
- 31.Muraoka-Cook RS, et al. 2006. Activated type I TGFbeta receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene 25:3408–3423 [DOI] [PubMed] [Google Scholar]
- 32.Muraoka RS, et al. 2003. Increased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor beta1. Mol. Cell. Biol. 23:8691–8703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okunola HL, Krainer AR. 2009. Cooperative-binding and splicing-repressive properties of hnRNP A1. Mol. Cell. Biol. 29:5620–5631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Persson U, et al. 1998. The L45 loop in type I receptors for TGF-beta family members is a critical determinant in specifying Smad isoform activation. FEBS Lett. 434:83–87 [DOI] [PubMed] [Google Scholar]
- 35.Reiter JL, Maihle NJ. 1996. A 1.8 kb alternative transcript from the human epidermal growth factor receptor gene encodes a truncated form of the receptor. Nucleic Acids Res. 24:4050–4056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seton-Rogers SE, et al. 2004. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 101:1257–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. 2003. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc. Natl. Acad. Sci. U. S. A. 100:8430–8435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stewart M. 2007. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 8:195–208 [DOI] [PubMed] [Google Scholar]
- 39.Tabb DL, Fernando CG, Chambers MC. 2007. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6:654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ueda Y, et al. 2004. Overexpression of HER2 (erbB2) in human breast epithelial cells unmasks TGF-induced cell motility. J. Biol. Chem. 279:24505–24513 [DOI] [PubMed] [Google Scholar]
- 41.Venables JP, et al. 2009. Cancer-associated regulation of alternative splicing. Nat. Struct. Mol. Biol. 16:670–676 [DOI] [PubMed] [Google Scholar]
- 42.Wang SC, et al. 2004. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell 6:251–261 [DOI] [PubMed] [Google Scholar]
- 43.Wang SE, et al. 2006. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 10:25–38 [DOI] [PubMed] [Google Scholar]
- 44.Wang SE, et al. 2007. Convergence of p53 and transforming growth factor beta (TGFbeta) signaling on activating expression of the tumor suppressor gene maspin in mammary epithelial cells. J. Biol. Chem. 282:5661–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang SE, Shin I, Wu FY, Friedman DB, Arteaga CL. 2006. HER2/Neu (ErbB2) signaling to Rac1-Pak1 is temporally and spatially modulated by transforming growth factor beta. Cancer Res. 66:9591–9600 [DOI] [PubMed] [Google Scholar]
- 46.Wang SE, et al. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 78:4248–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang SE, Wu FY, Shin I, Qu S, Arteaga CL. 2005. Transforming growth factor beta (TGF-beta)-Smad target gene protein tyrosine phosphatase receptor type kappa is required for TGF-beta function. Mol. Cell. Biol. 25:4703–4715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang SE, et al. 2008. Transforming growth factor beta engages TACE and ErbB3 to activate phosphatidylinositol-3 kinase/Akt in ErbB2-overexpressing breast cancer and desensitizes cells to trastuzumab. Mol. Cell. Biol. 28:5605–5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Y, et al. 2010. Context-dependent bidirectional regulation of the MutS homolog 2 by transforming growth factor beta contributes to chemoresistance in breast cancer cells. Mol. Cancer Res. 8:1633–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang B, Chambers MC, Tabb DL. 2007. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 6:3549–3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.