

Abstract
Posttranscriptional and posttranslational modification of macromolecules is known to fine-tune their functions. Trm112 is unique, acting as an activator of both tRNA and protein methyltransferases. Here we report that in Saccharomyces cerevisiae, Trm112 is required for efficient ribosome synthesis and progression through mitosis. Trm112 copurifies with pre-rRNAs and with multiple ribosome synthesis trans-acting factors, including the 18S rRNA methyltransferase Bud23. Consistent with the known mechanisms of activation of methyltransferases by Trm112, we found that Trm112 interacts directly with Bud23 in vitro and that it is required for its stability in vivo. Consequently, trm112Δ cells are deficient for Bud23-mediated 18S rRNA methylation at position G1575 and for small ribosome subunit formation. Bud23 failure to bind nascent preribosomes activates a nucleolar surveillance pathway involving the TRAMP complexes, leading to preribosome degradation. Trm112 is thus active in rRNA, tRNA, and translation factor modification, ideally placing it at the interface between ribosome synthesis and function.
INTRODUCTION
Actively growing budding yeast cells produce an average of 33 ribosomes per second, which is considerable (61). Besides the synthesis of its constituents, i.e., 79 ribosomal proteins and 4 rRNAs, ribogenesis involves a large number of so-called trans-acting factors, including proteins and small nucleolar RNAs (13, 56). These interact only transiently with preribosomes and are required for pre-rRNA processing (i.e., cleavages), pre-rRNA modification, and preribosome assembly and transport.
Ribosome synthesis is initiated in the nucleolus, a highly dynamic specialized subcompartment of the nucleus organized around clusters of actively transcribed rRNA genes (60). There, RNA polymerase I produces a 35S/47S (in yeasts and humans, respectively) primary transcript which is processed through a complex succession of endo- and exoribonucleolytic cleavages into three of the four mature rRNAs, the 18S, 5.8S, and 25S/28S rRNAs. The fourth rRNA, 5S, is independently transcribed by RNA polymerase III. Ribosome maturation starts cotranscriptionally in the nucleolus, progresses in the nucleoplasm until preribosomes reach the nuclear pore complex, and is finalized in the cytoplasm. Cytoplasmic maturation steps consisting of pre-rRNA processing and structural reorganization result in the assembly of prominent ribosomal structures, such as the “beak” of the small subunit (SSU) and the “stalk” of the large subunit, and are a prerequisite to the acquisition of functionality (43).
It is not clear how the various facets of ribosome synthesis are integrated with ribosome function. However, there is growing evidence that such coordination exists. For the small subunit, recent cryoelectron microscopy (cryo-EM) maps of late pre-40S subunits indicate that the binding of trans-acting factors literally mask functional sites, preventing premature translation initiation (57). For the large subunit, trans-acting factors that resemble ribosomal proteins (such as Imp3, Rlp7, Rlp24, and Mrt4, discussed in references 9, 33, and 50), and translation elongation factor-like proteins (such as Efl1 [55]), have been suggested to act as “placeholders” or “body doubles” and to have a role in probing functional sites on preribosomes.
Ribosomal components are targets of posttranscriptional and posttranslational modifications. The most frequent rRNA modifications are pseudouridylation and 2′-O ribose methylation, catalyzed, respectively, by box H/ACA and box C/D snoRNA-guided enzymes (5, 23). In addition, several bases are modified by specific protein methyltransferases (MTases), such as Dim1 and Bud23 (28, 64). Within pre-rRNAs, modifications always target residues in the mature rRNA sequences, and, quite suggestively, these often cluster in functional centers of the ribosome (see, e.g., references 22 and 31). However, the exact functions of rRNA modifications remain largely unknown. In the case of Dim1 and Bud23, it was shown, against all odds, that it is the actual presence of the protein rather than its catalytic activity in rRNA modification that is required for pre-40S ribosome synthesis (28, 64). To date, much less is known about the posttranslational modification of ribosomal proteins and trans-acting factors. However, phosphorylation has been shown to control key assembly steps, such as the formation of the “ribosomal beak” on pre-40S (52), and sumoylation to regulate intranucleolar transport reactions, snoRNP assembly, and preribosome export (8, 44, 63).
Ribogenesis is a complex error-prone process which is actively monitored by quality control mechanisms (26). One such mechanism is a 3′→5′ nucleolar surveillance pathway in which the RNA component of defective preribosomes is tagged for degradation by the RNA exosome by the iterative addition of short poly(A) tails by TRAMP complexes (6, 62). Recently, the exosome cofactors and RNA-binding complex Nrd1-Nab3 were suggested to contribute to the identification of defective preribosomes, and to the cotranscriptional recruitment of the exosome, through interactions with the RNA polymerase I elongation complex Spt4-Spt5 (29).
Trm112 is a small zinc finger protein of 15 kDa which acts as a coactivator of several class I S-adenosyl-l-methionine (SAM)-dependent MTases (15, 32, 36, 49, 58). To date, Trm112 is known to interact with, and to activate, three MTases, namely, Mtq2, Trm9, and Trm11, all related to ribosome function. The Mtq2-Trm112 complex methylates the translation termination factor eRF1 on the Gln side chain of its universally conserved GGQ motif (14, 15). Trm11-Trm112 forms 2-methylguanosine at position 10 on tRNAs (49). The Trm9-Trm112 complex catalyzes the addition of a methyl group to uridine at the wobble position (U34) of specific tRNAs (20, 36, 58).
High-throughput affinity purifications indicated that Trm112 interacts with the SSU-processome-associated DEAH box RNA helicase Dhr1 (4, 24). The SSU-processome is a macromolecular complex corresponding to nascent pre-40S ribosomes where the initial U3-dependent pre-rRNA cleavages take place (7, 21, 35, 46). The interaction between Trm112 and Dhr1 prompted us to test the requirement for Trm112 in ribosome synthesis. Here, we report that Trm112 is a bona fide preribosome constituent that interacts, both in vivo and in vitro, with the 18S rRNA MTase Bud23. We further show that Trm112 is required for Bud23 metabolic stability and therefore for Bud23-mediated methylation of the 18S rRNA in vivo and for ribosome assembly.
MATERIALS AND METHODS
Bacterial and yeast strains.
Escherichia coli DH5α was used for DNA amplification and cloning purposes. E. coli SoluBL21 (Amsbio) was used to express the His6-Bud23-Trm112 complex. Cells were grown in complete Luria-Bertani broth (LB). Antibiotics were added, when required, at the final concentrations of 50 μg/ml for kanamycin, 200 μg/ml for ampicillin, and 15 μg/ml for chloramphenicol. Yeast strains used in this study are listed in the supplemental material. Cells were grown at 30°C unless otherwise stated. Cells were grown either in complete yeast extract-peptone-dextrose (2% each) (YPDA) or in synthetic minimal medium, supplemented with 2% sugar (either galactose or glucose). Paromomycin was added at a final concentration of 800 μg/ml and cycloheximide at a final concentration of 100 μg/ml.
Plasmids, protein expression, and purification.
pACDuet(H6Bud23), a derivative of pACYC-Duet1 (Novagen) expressing Bud23 with a His6 tag on its N terminus, was constructed as described in the supplemental material.
The His6-Bud23 and Trm112 proteins were expressed from SoluBL21 strain transformed by pACDuet(His6Bud23) and pVH451 after induction with isopropyl-β-d-thiogalactopyranoside (IPTG) (0.1 mM) at 23°C at an optical density at 600 nm (OD600) of 0.4 in LB medium containing ZnCl2 (100 μM final), ampicillin, kanamycin, and chloramphenicol overnight. The complex was purified as previously described for Mtq2-Trm112 (15).
Western blot analysis.
Proteins were separated on 12% SDS-polyacrylamide gels and transferred to nitrocellulose membranes, and Western blotting was performed with specific primary antibodies, peroxidase-coupled secondary antibodies with SuperSignal chemiluminescent substrate (Pierce), and a Bio-Rad Chemidoc XRS camera. We used anti-Mtq2-Trm112 and anti-Bud23-Trm112 antibodies raised in rabbit against purified recombinant yeast complexes, mouse monoclonal anti-His6 antibody (Roche), and mouse monoclonal anti-PGK1 (Invitrogen). In Western blotting of sucrose gradient fractions, Bud23 and Trm112 were detected by the anti-Bud23-Trm112 complex antibody. Rps8 was detected with a specific antibody raised in rabbit and used at 1:1,000 (a gift from Giorgio Dieci, Università degli studi di Parma), Rpl3 with a specific antibody raised in mouse and used at 1:2,000 (a gift from Jonathan Warner, Albert Einstein College, New York, NY), and Nog1 with an antibody raised in rabbit and used at 1:500 (a gift from Micheline Fromont-Racine, Institut Pasteur, Paris, France). Samples were resolved on Criterion 4 to 12% bis-tris gels (Bio-Rad).
RNA extraction, Northern blotting, pulse-chase labeling, quantitative real-time PCR (qRT-PCR), and primer extension.
RNA extraction from yeast cells and Northern blotting were performed as described previously (3). Oligonucleotides used in the hybridizations are listed in the supplemental material. For the analysis of high-molecular-weight species, 10 μg of total RNA was separated on a 1.2% agarose-6% formaldehyde gel; for low-molecular-weight species, 5 μg of total RNA was separated on an 8% urea-acrylamide gel. Phosphorimager quantification used a Fuji FLA-7000 and the native Multi Gauge software (version 3.1). The pulse-chase labeling was performed as described in reference 28. The gels was transferred to a GeneScreen Plus membrane (NEF-976; Dupont De Nemours), sprayed with tritium enhancer (NEF-970G; Dupont De Nemours), and exposed to autoradiographic film.
For mapping the m7G1575 modification, primer extension reactions were carried out with a molar excess of oligonucleotide LD2092 and 10 μg of total RNA, essentially as described in reference 64. The regular concentration of deoxynucleoside triphosphates (dNTPs) in such primer extension reactions is 1.2 mM (see Fig. 7B). The dNTP concentration was reduced to 0.04 mM to improve detection of the modification (see Fig. 7A). The primer extension stops were mapped with a DNA sequencing ladder generated with the same oligonucleotide as the one used in the primer extension reaction. The primer used in the sequencing reaction was phosphorylated with cold ATP to correct for migration differences. After extension with avian myeloblastosis virus (AMV) reverse transcriptase (Promega), RNA was hydrolyzed and the samples precipitated and migrated on 6% sequencing gels. Alternatively, reverse transcription was performed on RNAs that were first specifically cleaved at N7-methylated guanosine positions (according to reference 39). To break the phosphodiester backbone at the m7G position, total RNA was treated with NaBH4 and aniline and dimethyl sulfate-modified tRNA were added in the reaction to enhance cleavage (39). qRT-PCRs were performed as described in reference 53. An amplicon for yeast actin 1 was used as an endogenous control for the qRT-PCR analysis.
Fig 7.

Trm112 is required for Bud23-mediated formation of the N7-methylguanosine at position G1575 in 18S rRNA. Total RNA extracted from bud23Δ and trm112Δ cells and an isogenic wild-type strain (WT, BY4741) grown to mid-log phase at 30°C in YPDA was analyzed by primer extension from oligonucleotide LD2092, hybridizing 140 nucleotides downstream of the site of modification. (A) Reverse transcription on total RNA. The reverse transcriptase is blocked at the site of modification G1575. The G1575 modification is best detected at the low dNTP concentration of 0.04 mM, which was used here. (B) Reverse transcription on RNAs that have specifically been cleaved at N7-methylated guanosine positions. The reverse transcriptase stops at the site of RNA cleavage, revealing position G1575. The control consisted of untreated RNAs; in the case of chemically treated RNA, total RNA was treated with NaBH4 and aniline to break the phosphodiester backbone at the m7G position (see Materials and Methods), and dimethyl sulfate-modified tRNA was added in the reaction to enhance cleavage at m7G methylation sites. In panel B, the regular dNTP concentration of 1.20 mM was used. Note that by comparison to panel A, which used the lower dNTP concentration of 0.04 mM, a stronger extension ladder was seen in the background in panel B. In panels A and B, the asterisk denotes a structural stop in the substrate and indicates that the same amount of reverse-transcribed RNA was loaded in each lane. In both panels A and B, a 2′-O methylation at position G1572, directed by snoRNA57, was detected 3 nucleotides above G1575 (discussed in reference 64). The detection of this modification was not affected by deletion of either BUD23 or TRM112. (C) Trm112 is required for Bud23 accumulation in vivo. Total protein was extracted from the strain indicated grown to mid-log phase in complete medium. Proteins were analyzed by SDS-PAGE and Western blotting with an antibody specific to the Bud23-Trm112 complex. Lane 1, 100 ng of recombinant His6-Bud23-Trm112 complex purified from E coli was loaded as a control; lanes 2 to 4, 50 μg of total protein. The asterisk denotes a cross-hybridizing band and provides a loading control.
Fluorescence microscopy.
Yeast cells were observed with a Zeiss Axio Imager Z1 microscope, equipped with a 100× objective (numerical aperture [NA] of 1.46), and standard filter sets. Images were captured with a Zeiss HRm charge-coupled-device (CCD) camera and the native Axiovision software (version 4.5) and transferred to Photoshop and Illustrator (CS3; Adobe).
Sucrose gradient sedimentation analysis.
Separation by sucrose gradient was performed exactly as described in reference 53.
Polysome analysis.
Yeast cells were grown to an OD600 of ∼0.5 in YPDA (or 2% sugar-based selective synthetic minimal medium). Cycloheximide was then added to a final concentration of 100 μg/ml. Extracts were prepared in 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 30 mM MgCl2, and 50 μg/ml cycloheximide by vortexing in the presence of glass beads. Extracts were fractionated by ultracentrifugation on a sucrose gradient (10% to 50%) in a buffer containing 50 mM Tris-HCl (pH 7.4), 12 mM MgCl2, 50 mM NH4Cl, and 1 mM dithiothreitol (DTT) for 2 h 45 min at 39,000 rpm and 4°C (SW41 rotor). The fractions were recovered with an ISCO fractionator, and the absorbance at 254 nm was measured. The ratio between the small and the large subunits was obtained by analyzing absorbance profiles of whole cellular extracts prepared in the absence of MgCl2.
Tandem affinity purification.
An affinity purification protocol optimized to detect transient interactions was used (see references 40 and 66). Briefly, yeast cells were collected, resuspended in resuspension buffer, frozen in liquid nitrogen, and lysed in solid phase by cryo-milling on a planetary mill (Retsch) and stored at −80°C. The resuspension buffer consisted of polyvinylpyrrolidone 40 (PVP-40) at 1.2%, HEPES (pH 7.4) at 20 mM, Sigma protease inhibitor cocktail at 1:100, solution P at 1:100 (pepstatin A at 2 mg, phenylmethylsulfonyl fluoride [PMSF] at 90 mg, and ethanol at 5 ml), and DTT (1 M) at 1:1,000. The grindate was resuspended in RNP buffer (HEPES [pH 7.4] at 20 mM, potassium acetate [KOAc] at 110 mM, Triton X-100 at 0.5%, Tween 20 at 0.1%, Ambion SuperRNAsin at 1:5,000, Sigma antifoam at 1:5,000, solution P at 1:100, and NaCl at 150 mM) and incubated with rabbit IgG-conjugated magnetic beads (Dynal) for 1 h at 4°C. The beads were collected using a magnet, washed four times, and eluted twice (once for 20 min and once for 2 min) at room temperature (RT) under denaturing conditions (500 mM NH4OH–0.5 mM EDTA). The pooled eluates were separated on 4 to 12% NuPAGE Novex bis-tris precast gels (Invitrogen) and visualized by Coomassie blue staining. Duplicate experiments were analyzed by liquid chromatography-mass spectrometry (LC/MS-MS).
For the TAP-alone control strain, a tandem affinity purification (TAP) cassette was integrated at the BAT2 locus by homologous recombination, resulting in the expression of a stable full-length TAP polypeptide directly under the control of the BAT2 constitutive promoter (strain YDL2618). BAT2 is a metabolic gene that encodes an enzyme involved in the last step of leucine formation. Genome-wide studies estimated Bat2 to be expressed at around 25,900 copies/cell (11), which is far more than most ribosome synthesis factors, making it a suitable control for unspecific interactions. For Fig. 5B and C (see below), coprecipitating RNAs were identified by primer extension as described in reference 53.
Fig 5.
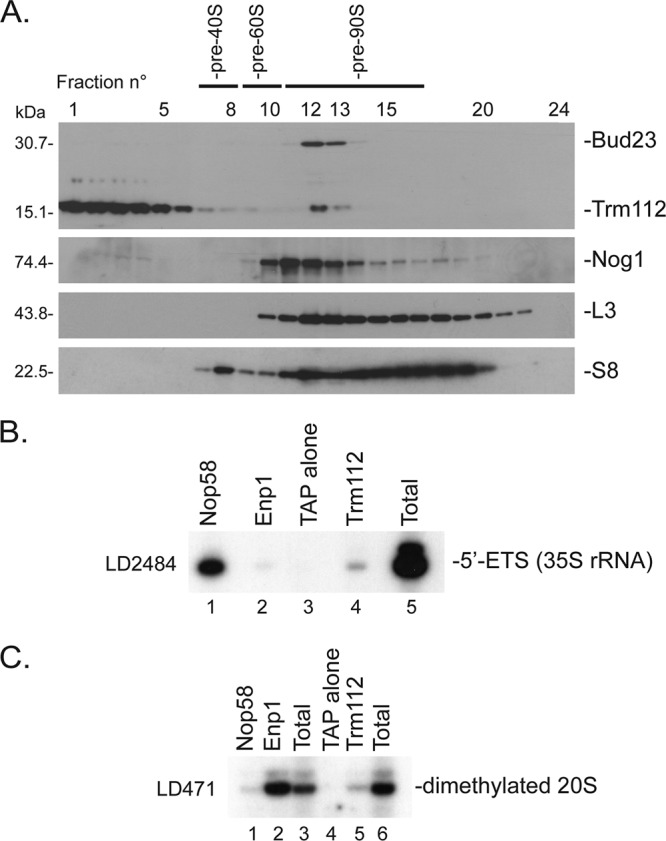
Trm112 interacts with pre-90S and pre-40S ribosomes. (A) Velocity gradient analysis. A total protein extract from wild-type cells was fractionated on a 10 to 50% sucrose gradient. A total of 24 fractions were collected and analyzed by Western blotting with an antibody raised against the recombinant Bud23-Trm112 complex or with antibodies specific to Nog1 or ribosomal proteins S8 and L3. (B and C) Primer extension analysis of RNA coprecipitating with Trm112. Total extracts from yeast cells expressing Trm112-TAP, Nop58-TAP, Enp1-TAP, or TAP alone were engaged in affinity purification. Coprecipitating RNAs were analyzed by primer extension with oligonucleotide LD2484 (B) or LD471 (C). (B) Lanes 1 to 4, pellets from the indicated strains; lane 5, total RNA from Trm112-TAP. The total and pellet fractions were loaded in a 1:9 ratio. (C) Lanes 1, 2, 4, and 5, pellets from the indicated strains; lanes 3, total RNA from cells expressing the TAP alone; lane 6, total RNA from Trm112-TAP.
Generation of Saccharomyces cerevisiae Bud23-Trm112 model.
Previous bioinformatic analyses identified Bud23 as a member of the class I SAM-dependent MTases (64). Members of this family share a well-conserved fold despite little sequence identity; this fold consists of a seven-stranded β-sheet surrounded by helices on each side (54; also see Fig. 7). In Bud23, this MTase domain is predicted to be located within the 200 N-terminal residues, while the C-terminal region (75 residues, with strong enrichment in lysine and arginine residues) is predicted to be poorly folded (64). We have therefore modeled only the Bud23 MTase domain which is predicted to interact with Trm112.
A search within the Protein Data Bank for protein structures displaying significant sequence similarity with S. cerevisiae Bud23 (residues 1 to 205) identified a putative MTase from Anabaena variabilis (PDB code 3CCF; 31% sequence identity over Bud23 residues 40 to 163) as well as human glycine N-methyltransferase (PDB code 2AZT; 26% sequence identity over Bud23 residues 14 to 150 [34]). A chimeric three-dimensional (3D) model for the S. cerevisiae Bud23 MTase domain was then generated by merging regions from both MTases that display the higher similarity with Bud23. This template was used to generate the S. cerevisiae Bud23 model using the I-TASSER server as well as the alignment obtained from analysis of sequence similarity (51). The model's accuracy has been assessed using the MetaMQAPII metaserver (45), yielding a GDT_TS (for global distance test, total score) value of 55.5 and predicting a root-mean square deviation of 3.5 Å. The mapping of the predicted accuracy (estimated agreement with the true but unknown structure) is available upon request.
The structure of the S. cerevisiae Bud23-Trm112 model was obtained by superimposing the coordinates of the S. cerevisiae Trm112 crystal structure (15) and the Bud23 model onto the corresponding parts from our recently solved Encephalitozoon cuniculi Mtq2-Trm112 crystal structure (32). The stereochemistry of this Bud23-Trm112 model was further optimized using energy minimization as implemented in the CNS software so as to avoid steric clashes between the two partners (1). The coordinates of the S. cerevisiae Bud23 model and of the Bud23-Trm112 model are available in the supplemental material.
RESULTS
Trm112 is required for efficient ribosome synthesis in budding yeast.
Trm112 was shown to interact with the U3-specific DEAH RNA helicase Dhr1 (4, 25), suggesting that, in addition to its known functions in tRNA and translation termination factor methylation, the protein might be involved in ribosome synthesis. The subcellular localization of Trm112 in the nucleus and cytoplasm (17) is compatible with this possibility. Since Trm112 is encoded by a nonessential gene, this was directly tested in trm112Δ cells.
S. cerevisiae trm112Δ cells are hypersensitive to paromomycin, which is compatible with a defect in ribosome synthesis and/or function, and they are cryosensitive for growth, which has often been associated with ribosomal assembly defects (Fig. 1A). Polysome analysis indeed revealed a striking ribosomal subunit imbalance in trm112Δ cells, with no 40S subunits detectable, a concomitant elevated peak of 60S, and reduced amounts of polysomes (Fig. 1B). Analysis under dissociation conditions indicated that the reduction in 40S subunits is 66%, and that in 60S subunits is 30% (mean of 4 experiments, with standard deviations [SD] of 6% and 4%, respectively) (Fig. 1C).
Fig 1.

Trm112 is required for small ribosomal subunit accumulation, efficient preribosome export of both ribosomal subunits, and progression through mitosis. (A) (Left) trm112Δ cells are hypersensitive to paromomycin. Serial dilutions of trm112Δ (yVH90) yeast cells and the isogenic wild type (WT) were spotted on complete medium (YPDA), supplemented or not with paromomycin (800 μg/ml). (Right) trm112Δ cells are cryosensitive for growth, and the TRM112-TAP allele is fully functional (no growth defect). Cells were grown for 2 days at 30°C and 37°C and for 3 days at 23°C. (B) trm112Δ cells are characterized by a ribosomal subunit imbalance. Shown is a polysome profile (absorbance at 254 nm) analysis of a trm112Δ strain and the wild-type isogenic control. The positions of 40S, 60S, 80S, and polysomes are indicated. (C) The amount of both the small and the large ribosomal subunits is reduced in trm112Δ cells. Analysis was conducted under dissociation conditions (absence of MgCl2) for determination of the amount of small and large ribosomal subunits. This experiment was repeated 4 times. (D) Preribosome export assay. trm112Δ cells and the isogenic wild type (BY4741) were transformed with a construct expressing either Rps2-GFP or Rpl25-GFP, grown to mid-log phase, and analyzed by fluorescence under a microscope. The arrow points to a nucleolar focus. Arrowheads point to nucleolar foci asymmetrically localized in the mother cell. The insets highlight cells blocked in mitosis with an asymmetrically localized nucleolar focus. DIC, differential interference contrast; DAPI, 4′,6-diamidino-2-phenylindole.
To test whether the loss of ribosomal subunits might result from a preribosome export defect, trm112Δ cells were transformed with suitable reporter constructs expressing a ribosomal protein from either the small or the large subunit (Rps2 or Rpl25, respectively) in fusion with a green fluorescent protein (GFP). These reporters have previously been used to address ribosome export and been shown to assemble properly into preribosomes (18, 37). In wild-type cells, both Rps2-GFP and Rpl25-GFP reporter constructs mostly labeled the cytoplasm, the site of mature ribosome accumulation, as expected. In addition, some cells showed a more intense nuclear signal in the form of a focus that largely colocalized with the nucleolus, as established by counterstaining with a DNA stain (Fig. 1D and Table S1 in the supplemental material). Among trm112Δ cells, the frequency of cells accumulating the GFP-fusions in nucleolar foci increased ∼3-fold (see Table S1 in the supplemental material). This effect on preribosome export was seen both with the small and with the large subunit reporter constructs. We also noted that in trm112Δ populations, a fraction of cells, between 7 and 10%, showed an abnormally elongated morphology, with the daughter cells remaining attached to the mother, reminiscent of cells failing to progress through mitosis. Consistently, in these cells, the GFP fusions always concentrated in a nucleolar focus adjacent to the DNA and asymmetrically distributed in the mother cell. Again, this was seen with both reporters. In budding yeast, the nucleolus is the last part of the nucleus known to partition between the mother and the daughter cells (59). We conclude that trm112Δ cells are characterized by a cell cycle defect and that Trm112 is required for efficient ribosome export.
Most ribosome export defects arise not as a direct consequence of ribosome export inhibition per se but simply as an indirect effect of nuclear ribosome assembly blockage, and this is likely the case for Trm112 since it affects similarly the export of both ribosomal subunits. Therefore, we were interested in finding out whether Trm112 is required for pre-rRNA processing. Total RNA was extracted, separated on denaturing agarose and acrylamide gels, and analyzed by Northern blotting (Fig. 2A; see schematics for a description of the pre-rRNA intermediates detected and the probes used). The analysis was conducted in parallel at the physiological temperature of 30°C and at 23°C, since we found that the growth defect of trm112Δ cells is exacerbated at this temperature (see Fig. 1A). trm112Δ cells showed reduced amounts of both large rRNAs, the 18S and 25S rRNAs (Fig. 2A, gel IV, lanes 3 and 4). The reduction was between 30% and 40%. The synthesis of the 18S and 25S rRNAs was affected to the same extent, as shown by the 25S/18S ratio, which remained constant at ∼1 in both trm112Δ and wild-type cells (see Table S2 in the supplemental material). This is consistent with the ribosomal subunit analysis indicating that the amount of both the small and large subunits is reduced (Fig. 1C). trm112Δ cells accumulated ∼2.5-fold the primary transcript, the 35S pre-rRNA (Fig. 2A, gels I to III, lanes 3 and 4, and inset, gel VIII, lanes 3 and 4; see also Table S2), indicating inhibition of the early nucleolar cleavages, at sites A0, A1, and A2 (Fig. 2A, schematics; also, see reference 38 for a recent review). Inhibition at sites A0 to A2 was confirmed by a weak accumulation of the 23S rRNA (Fig. 2A, gels I and II, and data not shown) that extends between the transcriptional start site and A3 in ITS1. In addition, precursors of the 5.8S and 25S rRNAs, the 7S and 27S pre-rRNAs, were significantly reduced in trm112Δ cells; this reduction was more pronounced at 23°C than at 30°C (Fig. 2A, gels III and V, lanes 3 and 4, and inset, gels VIII and IX, lanes 3 and 4). The reduction in 7S pre-rRNA, however, only had moderate impact on the steady-state accumulation of the 5.8S rRNA (Fig. 2A, gel VI). Note that hybridization with a probe specific to the RNA component of the signal recognition particle, SCR1, was used as a loading control (Fig. 2A, gel VII).
Fig 2.

Trm112 is required for efficient pre-rRNA processing, and the absence of Trm112 activates a 3′→5′ nucleolar surveillance pathway which targets preribosomes for degradation. (A) In trm112Δ cells, early pre-rRNA processing is inhibited and precursors to large subunit rRNA are unstable. (Left) Schematics depicting the pre-rRNA intermediates and probes used. (Right) Total RNA extracted from the indicated strains grown to mid-log phase in YPDA at 30°C (gels I to VII) or 23°C (gels VIII and IX) was separated on agarose-formaldehyde gels, transferred to a nylon membrane, and hybridized with a set of oligonucleotide probes in Northern blots, as indicated on the gels. Gel VI was hybridized with LD915 and gel VII with LD1290. (B) In trm112Δ cells, pre-rRNA precursors are turned over by TRAMP-dependent nucleolar surveillance. Information is the same as for panel A. Cells were grown at 30°C. Gel I, ethidium bromide staining.
The drastic reduction in 27S and 7S precursors suggested that these RNAs are particularly unstable in the absence of Trm112. This might be because in trm112Δ cells, preribosomes are actively targeted by nucleolar surveillance mechanisms. Defective preribosomes are known to be targeted for degradation by polyadenylation of the RNA at its 3′ end by TRAMP complexes, followed by exosome-mediated degradation (see the introduction). An important element of this nucleolar surveillance is the poly(A) polymerase subunit residing within TRAMP complexes and encoded by the related TRF4 and TRF5 genes.
To test whether precursor ribosomes are targeted for degradation in the absence of Trm112, either TRF4 or TRF5 was deleted in trm112Δ cells. trm112Δ trf4Δ and trm112Δ trf5Δ cells were then grown to mid-log phase at 23°C, and total RNA was extracted and analyzed by Northern blotting as described above (Fig. 2B). Normal precursors rRNAs, such as the 35S, 33S/32S, 27S, and the 20S pre-rRNAs, detected in trm112Δ cells were all substantially stabilized upon trf4 or trf5 deletion (Fig. 2B, gels II and III, lanes 5 and 6), with the exception of the 7S pre-rRNA corresponding to 3′-extended forms of 5.8S rRNA, which was not stabilized (see Discussion). This analysis was repeated at 30°C, leading to overall similar conclusions (data not shown). Quite importantly, despite the increased steady-state accumulation of most pre-rRNA precursors and the partial restoration in the steady-state levels of mature rRNAs (Fig. 2B, gel I, lanes 5 and 6), trm112Δ trf4Δ and trm112Δ trf5Δ cells were as defective for growth as trm112Δ cells (data not shown). This is similar to what was observed for SSU-processome mutants under conditions where the nucleolar surveillance is inactivated (see reference 62). The SSU-processome is a macromolecular complex required for A0-A1 cleavages (21). This indicates that encroaching the assembly of defective ribosomal precursors otherwise destined for degradation does not allow generating functional ribosomes.
We conclude that Trm112 is required for early nucleolar pre-rRNA cleavages and that in trm112Δ cells, a significant fraction of large subunit pre-rRNAs are turned over by nucleolar surveillance. The 20S pre-rRNA was also stabilized upon nucleolar surveillance inactivation, indicating that a fraction of 20S pre-rRNA is constitutively degraded in the nucleus (Fig. 2B, gel II, lanes 5 and 6).
The polysome (Fig. 1B) and ribosomal subunit (Fig. 1C) analyses indicated that trm112Δ cells are primarily defective for small ribosomal subunit accumulation. Consistently, the pre-rRNA processing analysis revealed that the early nucleolar cleavages, at sites A0 to A2, are inhibited in the absence of Trm112 (see 35S pre-rRNA accumulation in Fig. 2A). However, and quite surprisingly, the 20S pre-rRNA, the immediate precursor to the small subunit 18S rRNA, which results from cleavage of the 35S pre-rRNA at sites A0 to A2, accumulated stably in trm112Δ cells (Fig. 2A, gel I; compare lanes 1 and 2 with lanes 3 and 4). In order to clarify this observation, and to further characterize the ribosome synthesis defect in trm112Δ cells, we performed a metabolic labeling (Fig. 3). Wild-type and trm112Δ cells were grown to mid-log phase in minimal medium lacking uracil, pulse-labeled with [H3]uracil for 1 min, and then chased with an excess of cold uracil. Total RNA was extracted from samples collected in a time course (1 to 20 min) and analyzed by denaturing agarose gel electrophoresis. Three conclusions could be drawn from the pulse-chase analysis. First, it confirmed the inhibition in early nucleolar cleavages. Indeed, the 35S pre-rRNA, which is not normally detected in wild-type cells because it is normally processed very rapidly, became detectable in trm112Δ cells. Second, it informed us that small subunit synthesis is strongly impaired in the absence of Trm112: in wild-type cells, the conversion of the 20S pre-rRNA into 18S initiated as soon as 3 min following the chase (Fig. 3, lane 3), while in trm112Δ cells, this conversion started only after 10 min of chase (Fig. 3, lane 11). Third, it revealed that the kinetics of large subunit pre-rRNA processing is only mildly affected in trm112Δ cells, which further validated our hypothesis, discussed above, that for the large subunit rRNA precursors it is RNA degradation which is activated rather than RNA synthesis being inhibited.
Fig 3.
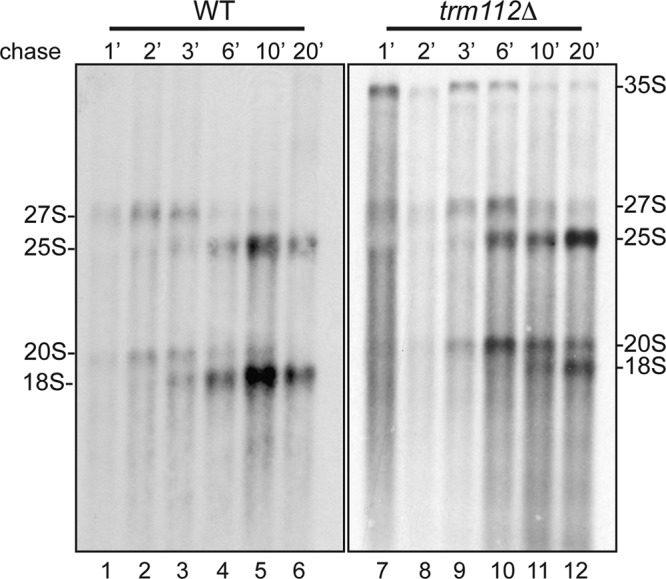
Trm112 is required for efficient small ribosomal subunit rRNA processing. trm112Δ and isogenic wild-type (WT) cells, transformed with a plasmid expressing the URA3 gene, were grown at 23°C in minimal medium lacking uracil, pulse-labeled with tritiated uracil for 1 min, and chased with an excess of cold uracil for the indicated times. Total RNA was extracted, separated on denaturing agarose gels, and transferred to a GeneScreen membrane. The membrane was sprayed with tritium enhancer and exposed to film. Lanes corresponding to trm112Δ cells were exposed 4 times longer than those of the WT control.
Trm112 interacts with the 18S rRNA methyltransferase Bud23 in vivo.
To get further insight into the involvement of Trm112 in ribogenesis, we undertook to identify its protein partners, which we did by affinity purification followed by mass spectrometry analysis in cells expressing as their unique source of the protein a Trm112-TAP fusion from its endogenous promoter. We confirmed that the Trm112-TAP fusion is functional by showing that its expression did not lead to any growth or pre-rRNA processing defects (Fig. 1A and 2A).
Total cellular extracts were prepared in solid phase in a planetary ball mill and engaged in a revised affinity purification protocol optimized to efficiently capture transient interactions (40). Interactions were carefully curated with those identified with a control strain expressing the TAP tag alone, and with an unrelated bait. This analysis confirmed that Trm112 copurifies with the MTases Mtq2, Trm9, and Trm11 (see the introduction) and indicated association with the 18S rRNA MTase Bud23 (peptide ion scores of >95%), as well as numerous ribosome synthesis trans-acting factors involved in the synthesis of the small, the large, or both subunits (Fig. 4; see also Table S3 in the supplemental material). We also further confirmed by a targeted coprecipitation analysis in cells coexpressing as their sole source of Trm112 and Dhr1 a Trm112-HA and a Dhr1-TAP fusion that these two proteins interact in vivo (data not shown).
Fig 4.

Trm112 copurifies with preribosomes that also contain the 18S rRNA methyltransferase Bud23. Partners of Trm112 were identified by affinity purification followed by mass spectrometry analysis. Yeast cells expressing a functional Trm112-TAP fusion (see Fig. 1A) were grown exponentially to mid-log phase at 30°C in YPDA. Total lysates were prepared in solid phase (see Materials and Methods) and engaged in a revised affinity purification protocol (40). As a control, we used a strain expressing the TAP tag alone (data not shown). Interactions were carefully curated with those identified in the TAP-alone control and with an unrelated bait. All interactions are listed in Table S3 in the supplemental material. Interactants were identified by LC-MS/MS. (Left) Major Trm112 interactants sorted and color-coded by known or putative functions. RPS and RPL, ribosomal proteins from the small and large subunit, respectively. (Right) Eluate from a representative Trm112-TAP purification analyzed by 4 to 12% NuPage Novex bis-tris (Invitrogen) followed by Coomassie blue staining. Trm112-TAP is highlighted with an asterisk.
We independently confirmed, by velocity gradient centrifugation, that Trm112 copurifies with preribosomes, including with preribosome species that also contain Bud23 (Fig. 5A). A total extract from wild-type yeast cells was fractionated on 10 to 50% sucrose gradients, and the 24 fractions collected were analyzed by Western blotting. Trm112 showed three peaks, localizing in light fractions (Fig. 5A, fractions 1 to 5), with pre-40S ribosomes (Fig. 5A, fractions 7 and 8), and with pre-90S ribosomes (Fig. 5A, fractions 12 and 13). Bud23 was also detected in the “90S-sized” fractions. As controls, duplicated membranes were probed for Nog1 (a known pre-60S component), as well as for a ribosomal protein of the small and large subunits (RpS8 and RpL3).
To further demonstrate that Trm112 is a bona fide component of preribosomes, we characterized its RNA partners by affinity purification, as described above, followed by primer extension analysis (Fig. 5B and C). We found that Trm112-TAP copurifies low but significant amounts of both early nucleolar and late cytoplasmic pre-rRNAs (35S pre-rRNA and dimethylated 20S pre-rRNA, respectively) (Fig. 5B, lane 4, and Fig. 5C, lane 5). Note that the 20S pre-rRNA (Fig. 2A, schematics) is dimethylated on two conserved adjacent adenines located at the 3′ end of the 18S rRNA coding sequence (28) and that this modification occurs in the cytoplasm. As a control for specificity, we used cells expressing the TAP epitope alone. We also used cells expressing TAP-tagged epitope versions of early- and late-acting assembly factors (Nop58 and Enp1, respectively). Nop58 interacted strongly with the 35S pre-rRNA and only marginally with the dimethylated 20S pre-rRNA, and Enp1 interacted strongly with dimethylated 20S but only very weakly with 35S (Fig. 5B and C, lanes 1 and 2; see also reference 12).
Trm112 interacts with the 18S rRNA MTase Bud23 in vitro.
The presence of the 18S rRNA MTase Bud23 in the Trm112-TAP eluate raised the possibility that the two proteins interact directly. To test this idea, we coexpressed in bacteria a polyhistidine-tagged version of Bud23 with untagged Trm112 and performed purification on nickel agarose resin. We indeed found that Trm112 copurifies with His6-Bud23 (Fig. 6A, lanes 4 to 7) and that formation of the Bud23-Trm112 complex is resistant to high NaCl concentrations (up to 1 M). As a control, the identity of both copurifying proteins was confirmed by Western blotting (Fig. 6B and C). We also checked that untagged Trm112 expressed alone did not interact with nickel agarose resin (see Fig. S1 in the supplemental material). Bud23 had previously been shown to accumulate in inclusion bodies when expressed alone in E. coli (2), similarly to Mtq2 and Trm9 (15, 36). Strikingly, we observed that the coexpression of Trm112 and His6-Bud23 increased the solubility of Bud23 by a factor of ∼5 (Fig. 6D; compare the relative amount of protein in the pellet and supernatant fractions in lanes 3 and 4, respectively, with that in lanes 6 and 7). Further, when expressed alone under the same conditions, the soluble fraction of His6-Bud23 was unable to bind nickel resin, probably because of inappropriate folding and/or protein aggregation (see Discussion).
Fig 6.
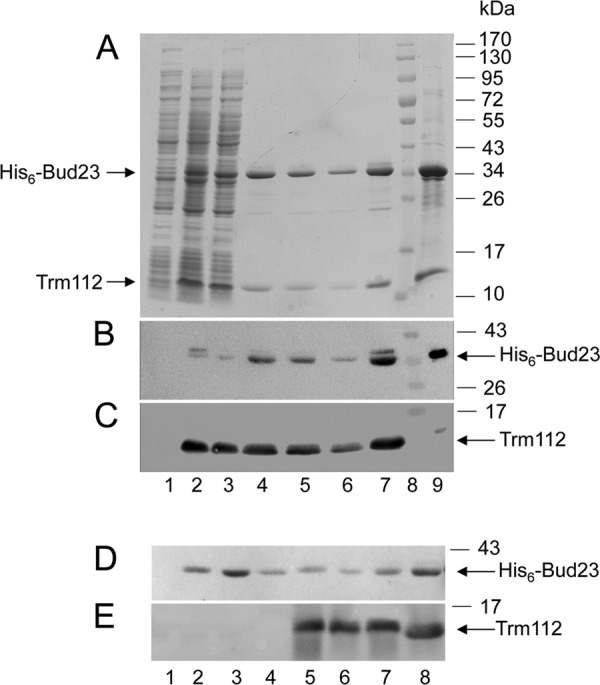
Trm112 interacts directly with the 18S rRNA methyltransferase Bud23 in vitro, and Trm112 increases the solubility of Bud23. (A) Coomassie blue staining of an SDS-PAGE gel showing coelution with imidazole of a poly-His-tagged Bud23 with untagged Trm112 from a nickel affinity matrix. Lane 1, uninduced culture; lane 2, total bacterial extract from cells coexpressing His6-Bud23 and untagged Trm112, respectively, from pACDuet(His6Bud23) and pET11a(Trm112); lane 3, flowthrough; lanes 4 to 7, coelution in a 50 mM imidazole-containing buffer (20 μl of eluted fractions in each lane); lane 8, molecular mass marker; lane 9, 10 μl of purified His6-Bud23-Trm112 complex following a 5× concentration through Amicon 30 (Millipore). (B) Western blot identification of His6-Bud23. The SDS-PAGE gel shown in panel A was duplicated, transferred to nitrocellulose, and probed in a Western blot with an anti-His6 antibody. (C) Information same as for panel B. Trm112 was detected with an antibody specific to the Mtq2-Trm112 complex (that only reveals Trm112 here since these cells do not express Mtq2). (D and E) The coexpression of Trm112 and Bud23 increases the solubility of recombinant Bud23. Lanes 1 to 4, E. coli cells expressing only His6-Bud23; lanes 5 to 7, E. coli cells coexpressing His6-Bud23 and Trm112; lane 8, control purified His6-Bud23-Trm112 complex. Lane 1 contains extract from noninduced cells, lanes 2 and 5 contain total extract from induced cells, lanes 3 and 6 contain pellets from induced cells, and lanes 4 and 7 contain supernatant from induced cells. (D) Western blot detection of His6-Bud23 with an anti-His antibody. (E) Western blot detection of Trm112 with a specific antibody.
Trm112 is required for Bud23-mediated 18S rRNA methylation at G1575 in vivo.
In budding yeast, Bud23 is known to methylate 18S rRNA at position G1575, a highly conserved residue interacting with the anticodon stem of the P-site tRNA within the 40S subunit (47, 64). The observations that Trm112 and Bud23 copurify in vivo and that they directly interact in vitro strongly suggested that Trm112 might act as a coactivator of Bud23.
Whether Trm112 is required for the methylase activity of Bud23 was tested in vivo by primer extension through the methylation site. m7G1575 is known to severely hinder the progression of the reverse transcriptase, leading to the formation of a transcription stop product (64). Total RNA extracted from trm112Δ cells, and as controls from bud23Δ and isogenic wild-type cells, was used as the template for cDNA synthesis (Fig. 7A). Deletion of trm112 abrogated formation of m7G1575, as did the deletion of BUD23.
To further confirm this conclusion, we used an alternative mapping strategy in which the RNAs are specifically cleaved at N7-methylguanosine positions, prior to reverse transcription (Fig. 7B; see also reference 39). In this approach, total RNA was cleaved at m7G by reduction with NaBH4, followed by β-elimination with acetic acid-aniline. Cleavage at position G1575 was detected only on rRNA from wild-type cells, demonstrating unambiguously that in trm112Δ cells the 18S rRNA modification carried out by Bud23 is lost (Fig. 7B).
The loss of rRNA modification prompted us to test whether Trm112 is required for the metabolic stability of Bud23 (Fig. 7C). In trm112Δ cells, and by comparison to wild-type cells, only a minute amount of Bud23 could be detected, indicating that Trm112 is indispensable for Bud23 accumulation (Fig. 7C; compare lanes 2 and 3). In contrast, in the absence of Bud23, the stability of Trm112 was not affected (Fig. 7C, lane 4). To rule out the possibility that the absence of Bud23 in trm112Δ cells simply results from a reduced expression at the mRNA level, total RNA from the samples analyzed in Fig. 7A and B was analyzed by qRT-PCR (standardized against the mRNA encoding actin 1). This analysis indicated that in trm112Δ cells, the BUD23 mRNA accumulated stably ∼2.5-fold (data not shown). Whether this reflects a regulatory loop will require further investigation. Hence, in trm112Δ cells, the loss of 18S rRNA methylation at position G1575 results from a strongly affected level of Bud23 protein but not mRNA. Since Trm112 and Bud23 interact directly in vitro, and since Trm112 substantially increases the solubility of Bud23, we suggest that Trm112 is required to stabilize Bud23 (see Discussion).
Trm112 is a coactivator of four methyltransferases with diverse functions in ribosome metabolism.
Trm112 is truly unique, acting as an activating platform of four MTases, specific to rRNA (Bud23), tRNA (Trm9 and Trm11), and even a translation factor (Mtq2). This ideally positions Trm112 at the interface between ribosome synthesis and function, suggesting that it might possibly coregulate these processes. If so, one might assume that the cellular level of Trm112 is tightly regulated, such that only the correct amount is delivered to each MTase with which it interacts in order to stabilize and/or activate only the amount needed. Quantification of the cellular copy number of Trm112 and its known partners indeed strongly suggests that Trm112 is limiting (the estimated copy number of the four MTases is about twice that of Trm112 (11).
To test the idea that the overexpression of any one of Trm112 partners might sequester it away from its other partners, we overexpressed Mtq2 from a strong inducible pGAL promoter and tested the effects on growth and polysome accumulation (Fig. 8). Overexpression of Mtq2 inhibited growth substantially (Fig. 8A) and led to a strong accumulation of 60S subunits, the absence of 40S, and reduced polysomes (Fig. 8C), a phenotype reminiscent to that observed in trm112Δ cells (Fig. 1B) and in bud23Δ cells (64). Strikingly, coexpression of Trm112 restored growth partially, reduced the amount of free 60S, and somehow restored polysome accumulation. We reached the same conclusion when we overexpressed Trm9 or Trm11 in place of Mtq2 (data not shown). We conclude that the cellular concentration of Trm112 is limiting and that the overexpression of any one of its partners sequesters it away, preventing it from interacting with its other substrates. Consistently, we also found that upon Mtq2 overexpression, the steady-state accumulation of Bud23 was strikingly reduced (up to 5 times [Fig. 8B; compare lanes 1 and 3]).
Fig 8.

Several RNA and protein methyltransferases compete to interact with their cofactor Trm112.(A) Growth plate assay. Serial dilutions (10×) of wild-type cells (BY4741) overexpressing Mtq2, Trm112, or both from pGAL inducible promoters, and an isogenic control, were grown on glucose or galactose-based medium at 30°C for 2 days. +++, overexpression. (B) Western blot analysis of protein steady-state accumulation in strains presented in panel A. Bud23 and Mtq2-Trm112 were detected, respectively, with an antibody raised against the Bud23-Trm112 and Mtq2-Trm112 complexes (see Materials and Methods). Pgk1 (loading control) was detected with a specific commercial antibody. The overexpression of Mtq2 leads to an ∼5-fold reduction in the steady-state accumulation of Bud23 (average calculated on three independent experiments; SD, 0.7). (C) Polysome analysis (254-nm absorbance readings) of strains described for panel A. The arrow points to the 60S peak. The 40S/60S ratio in each panel was determined under dissociation conditions (see Fig. 1C). This experiment was repeated twice. WT, wild type.
DISCUSSION
Trm112 is required for Bud23-mediated methylation of the 18S rRNA at position G1575 and for efficient small ribosome subunit synthesis.
Methylation of proteins and RNAs is a widespread modification known to modulate their functions (42, 48, 65). Trm112 activates several class I SAM-dependent MTases with substrates implicated in translation, such as several tRNAs (modified by Trm9 and Trm11), and the eukaryotic release factor 1 (by Mtq2). Here, we present evidence that Trm112 interacts with and is required for the metabolic stability of Bud23, another class I SAM-dependent MTase, which is specific for rRNA, i.e., the 18S rRNA.
We have shown that Trm112 copurifies with several proteins associated with nuclear preribosomes (Fig. 4), that it colocalizes on velocity gradients with pre-90S and pre-40S ribosomes (Fig. 5A), and that it associates in vivo with early nucleolar and late cytoplasmic pre-rRNAs (Fig. 5B and C). We also demonstrated that Trm112 is required for efficient pre-rRNA processing and for preribosome export (Fig. 1 to 3). Among the numerous ribosome synthesis trans-acting factors with which Trm112 copurifies, we found the 18S rRNA MTase Bud23. We have shown in vitro that Trm112 and Bud23 interact directly (Fig. 6), that coexpression of recombinant Trm112 and Bud23 substantially increases the solubility of Bud23 (Fig. 6), and in vivo that Trm112 is required for Bud23 accumulation and consequently for Bud23-mediated modification of the 18S rRNA at position G1575 (Fig. 7). Bud23 was shown to be important for small ribosome subunit synthesis (64). The effects on ribosome synthesis in trm112Δ cells that we report here are nearly indistinguishable from those previously described for bud23Δ cells. A natural explanation for the effects of Trm112 on ribosome synthesis is thus provided by the observation that Trm112 is required for the metabolic stability of Bud23.
Prior to this work, Trm112 was shown, in high-throughput analysis, to copurify with preribosomes that also contain the box C/D-associated DEAH helicase Dhr1 and several SSU-processome components, all required for the early nucleolar cleavages at sites A0 to A2. We confirmed the association with early preribosomes (presence in 90S-sized fractions and copurification with 35S pre-rRNA [Fig. 5]) and demonstrated the requirement for the early nucleolar cleavages (trm112Δ cells accumulate ∼2.5-fold the primary transcript [Fig. 2]). In addition, we found that Trm112 is required for small ribosome subunit synthesis (Fig. 2 and 3) and that in the absence of Trm112, pre-rRNAs otherwise destined to be incorporated into large subunits are massively turned over by nucleolar surveillance (Fig. 2B). We have demonstrated that the binding of Trm112 to nascent preribosomes occurs at an early nucleolar stage on 35S pre-rRNA. This likely explains that it has consequences for the synthesis not only of the small but also of the large ribosomal subunit. Trm112 interacts with trans-acting factors involved in the synthesis of both the small and the large subunits, such as Rrp5 and Rrp12, providing further support for this notion (Fig. 4). We have further reported that Trm112, or at least a fraction of it, escorts pre-40S ribosomes into the cytoplasm, as shown by its interaction with dimethylated 20S pre-rRNA (Fig. 5C). We do not believe that Trm112 plays any direct role in preribosome transport because it does not interact with large ribosome subunit precursor rRNAs (data not shown) while equally affecting both pre-40S and pre-60S export.
Trm112 is required for progression through mitosis.
Ribogenesis and the cell cycle have long been connected (reviewed in reference 19). It has been known for a very long time that in order to commit to division, cells have to achieve a certain size, for which they require a sufficient amount of functional ribosomes. The G1/S transition, known as START in yeast and restriction point in mammals, is a step in the cell cycle where, unsurprisingly, important quality controls operate (reviewed in reference 10). Recent analyses have indicated that it is not only protein synthesis per se that is monitored at the G1/S transition but also ribosome synthesis. Consistently, mutations in many ribosome synthesis trans-acting factors are characterized by a G1/S arrest. In addition, fewer trans-acting factors were shown to carry functions at specific steps other than G1/S, such as in the S phase (Noc3, Nop7, and Rrp12), mitosis (Ebp2, Rrb1, Rrp14, and Utp7), exit from mitosis (MRP), and cytokinesis (Nop15) in yeasts and centrosome duplication (B23, C23, and Misu) in humans (discussed in reference 10). Trm112 appears to be a novel member of this singular group, since about 10% of trm112Δ cells fail to go through mitosis (Fig. 1). Trm112 is not essential for growth; however, for the 10% of cells for which it is required to get through mitosis, it has become indispensable for cell survival. Interestingly, in plants, trm112 mutants are characterized by cell division and organogenesis defects (16), indicating that this function might have been evolutionarily conserved.
Trm112 is a coactivator of several protein and RNA MTases.
The recent determination of the crystal structure of the Mtq2-Trm112 complex from E. cuniculi has shed important light on the mechanisms by which Trm112 activates its class I MTase partners (Fig. 9; see also reference 32). At the interface between Trm112 and Mtq2 lies a “β-zipper” comprised of two parallel β-strands, one provided by each partner (β4 on Trm112 and β3 on Mtq2 [Fig. 9A, inset]), that interact by four hydrogen bonds formed between main-chain atoms. This affords a communal mode of interaction between proteins differing in primary sequences but sharing a similar fold. In addition, Trm112 interacts with a loop connecting two β-strands in Mtq2 (connecting β3 and β4) and stabilizes it in its interaction with the MTase SAM cofactor. The interaction with SAM, in turn, stabilizes the complex.
Fig 9.

Trm112 interaction with MTases. (A) Ribbon representation of the E. cuniculi Mtq2 (pink)-Trm112 complex. The Trm112 zinc-binding and central domains are shown in green and brown, respectively. The zinc atom is shown as a gray sphere. The SAM cofactor is depicted in sticks and its methyl group by a purple sphere. Inset to the right, enlarged view of the structural elements at the interface. (B) Ribbon representation of the S. cerevisiae Bud23 (orange)-Trm112 complex model. The Trm112 color code and orientation are the same as in panel A. The secondary structure elements predicted for Bud23 are indicated. (C) Overlay of the Mtq2-Trm112 complex structure (gray) and Bud23-Trm112 complex model (same color code as in panel B).
Based on our observation that Trm112 interacts directly with Bud23, another class I MTase, we have generated in silico a three-dimensional model for the yeast Bud23-Trm112 complex (Fig. 9B). We notably found that all the structural elements involved in the catalytic activation of Mtq2 by Trm112 are present in Bud23, suggesting that its mode of activation is largely conserved (Fig. 9C, inset).
Trm112 increases the solubility of several of its partners, including Trm9 and Mtq2 (32, 36) and, as established here, Bud23, suggesting that it is required for optimal folding of this group of class I MTase apoenzymes. Trm112 interaction with Mtq2, and most probably Trm9, buries a large hydrophobic solvent-accessible region, providing a rationale for the increased solubility observed upon complex formation and the resistance of complexes to high-ionic-strength treatment (32). These properties are shared by the Bud23-Trm112 complex, which is resistant to high salt washes in vitro. In addition, our model of the S. cerevisiae Bud23-Trm112 complex shows that the Bud23 region interacting with Trm112 is rich in hydrophobic residues.
Rid2, the Bud23 ortholog from Arabidopsis thaliana, has been involved in the reactivation of cell division during the process of dedifferentiation, which is essential for organ regeneration and wounding in plants (41). A thermosensitive rid2-1 allele, defective for pre-rRNA processing, carries a single amino acid substitution that, quite strikingly, according to our 3D model, is predicted to lie at the Bud23-Trm112 interface (data not shown).
Since the methylation function of Bud23 is dispensable for efficient pre-rRNA processing (64), it was quite unlikely that it is the involvement of Trm112 in Bud23's catalytic activation that underlies the pre-rRNA processing phenotype observed in trm112Δ cells. We found that in trm112Δ cells, the stability of Bud23 is dramatically affected (Fig. 7C). We therefore propose that Trm112 stabilizes Bud23 and that in its absence, Bud23 is unstable and cannot interact productively with preribosomes, which consequently are stalled in their maturation. Previous functional analysis of several rRNA base MTases, including Bud23, Dim1, and Emg1, indicated that it is not the modification per se that is important for ribosome synthesis but, rather, the presence of the MTase itself (27, 28, 30, 64). This led to the proposal that in the course of ribosome assembly, preribosomes that have failed to bind the MTase in a timely fashion, ultimately preventing pre-rRNAs from being modified, are aborted (discussed in references 27 and 28). We now show that these preribosomes are indeed recognized as defective and targeted for degradation by TRAMP complexes (Fig. 2). Indeed, the inactivation of TRF4 or TRF5, which are key elements of a 3′→5′ nucleolar surveillance pathway, stabilizes pre-rRNAs in trm112Δ cells (Fig. 2). It is quite striking that the 7S pre-rRNA, a precursor to the 5.8S rRNA, was not stabilized upon TRAMP complex inactivation (Fig. 2). We suggest that this is because this RNA species is not a substrate for this nucleolar surveillance pathway.
Does Trm112 integrate several aspects of ribosome synthesis and function?
From this work and previous analysis, we conclude that Trm112 acts as a platform that activates multiple proteins and RNA MTases, all involved in ribosomal processes. The observation that the cellular copy number of Trm112 is limited, by comparison to that of its known partners, is compatible with a function as a regulator.
There are several lines of evidence that strongly support the view that at least four MTases (Bud23, Mtq2, Trm9, and Trm11) compete with each other to interact with Trm112. First, overexpression of Mtq2 mimics the deletion of bud23, as revealed by polysome profiles and growth assays, and this is partially suppressed by the concurrent overexpression of Trm112 (Fig. 8). The overexpression of Trm9 or Trm11 led to overall similar conclusions. Second, the overexpression of Trm11 or Mtq2 affects interaction of Trm9 with Trm112 (58). Third, all Trm112 partners are members of the well-defined class I SAM-dependent MTases with the prediction that they share a similar fold and thus interact with Trm112 through the same binding site (e.g., see Fig. 9 for Mtq2 and Bud23). Fourth, our crystal structure of Mtq2-Trm112 revealed that in addition to a large hydrophobic interaction surface, both proteins interact via a β-zipper involving mostly main-chain atoms. Finally, based on the Mtq2-Trm112 structure, we generated a model for the S. cerevisiae Trm9-Trm112 complex and validated directly by site-directed mutagenesis the prediction that specific Trm9 residues lie at the interface with Trm112 (32). Trm112 is thus ideally positioned to integrate various aspects of ribosome synthesis (through Bud23) and function (via Trm9, Trm11, and Mtq2). Future work is now needed to establish the exact nature of this putative regulatory function.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to E. Hurt (University of Heidelberg) for plasmids expressing Rps2-GFP and Rpl25-GFP (pRS316-Rps2p-eGFP and pRS316-RPL25-eGFP) and to B. Séraphin (CNRS, Strasbourg, France) for plasmid pBS1762. We thank Jaunius Urbonavicius (CMMI, Gosselies, Charleroi, Belgium) for suggesting to us the use of an alternative mapping strategy to detect the m7G1575 modification. We are grateful to Stéphanie Kervestin and Marc Dreyfus (IBPC, Paris, France) for critical reading of the manuscript.
Work in the lab of V.H.-H. was supported by the Agence Nationale pour la Recherche (grant ANR-07-JCJC-0105) and the CNRS. During this work, S.F. held a predoctoral grant from the Université Pierre et Marie Curie. Work in the lab of D.L.J.L. was funded by the FRIA, the FRS-FNRS, the Communauté Française de Belgique (ARC), and the Région Wallone (Cibles). M.G. is indebted to the CNRS, the Université Paris-Sud 11, and the Human Frontier Science Program (grant number RGP0018/2009-C) for financial support.
Footnotes
Published ahead of print 9 April 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Brunger AT. 2007. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2: 2728– 2733 [DOI] [PubMed] [Google Scholar]
- 2. Chern MK, et al. 2009. Single-step protein purification by back flush in ion exchange chromatography. Anal. Biochem. 392: 174– 176 [DOI] [PubMed] [Google Scholar]
- 3. Colau G, Thiry M, Leduc V, Bordonné R, Lafontaine DLJ. 2004. The small nucle(ol)ar RNA cap trimethyltransferase is required for ribosome synthesis and intact nucleolar morphology. Mol. Cell. Biol. 24: 7976– 7986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Colley A, Beggs J, Tollervey D, Lafontaine DLJ. 2000. Dhr1p, a putative DEAH-box RNA helicase, is associated with the box C+D snoRNA U3. Mol. Cell. Biol. 20: 7238– 7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Decatur WA, Fournier MJ. 2002. rRNA modifications and ribosome function. Trends Biochem. Sci. 27: 344– 351 [DOI] [PubMed] [Google Scholar]
- 6. Dez C, Dlakic M, Tollervey D. 2007. Roles of the HEAT repeat proteins Utp10 and Utp20 in 40S ribosome maturation. RNA 13: 1516– 1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dutca LM, Gallagher JE, Baserga SJ. 23 February 2011. The initial U3 snoRNA:pre-rRNA base pairing interaction required for pre-18S rRNA folding revealed by in vivo chemical probing. Nucleic Acids Res. doi:10.1093/nar/gkr044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Finkbeiner E, Haindl M, Muller S. 2011. The SUMO system controls nucleolar partitioning of a novel mammalian ribosome biogenesis complex. EMBO J. 30: 1067– 1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gadal O, et al. 2002. Rlp7p is associated with 60S preribosomes, restricted to the granular component of the nucleolus, and required for pre-rRNA processing. J. Cell Biol. 157: 941– 951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gérus M, Caizergues-Ferrer M, Henry Y, Henras AK. Crosstalk between ribosome synthesis and cell cycle and its potential implications in human diseases. In Olson MO. (ed), The nucleolus, protein reviews, in press Springer; [Google Scholar]
- 11. Ghaemmaghami S, et al. 2003. Global analysis of protein expression in yeast. Nature 425: 737– 741 [DOI] [PubMed] [Google Scholar]
- 12. Grandi P, et al. 2002. 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol. Cell 10: 105– 115 [DOI] [PubMed] [Google Scholar]
- 13. Henras AK, et al. 2008. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell. Mol. Life Sci. 65: 2334– 2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heurgué-Hamard V, et al. 2005. The glutamine residue of the conserved GGQ motif in Saccharomyces cerevisiae release factor eRF1 is methylated by the product of the YDR140w gene. J. Biol. Chem. 280: 2439– 2445 [DOI] [PubMed] [Google Scholar]
- 15. Heurgué-Hamard V, et al. 2006. The zinc finger protein Ynr046w is plurifunctional and a component of the eRF1 methyltransferase in yeast. J. Biol. Chem. 281: 36140– 36148 [DOI] [PubMed] [Google Scholar]
- 16. Hu Z, et al. 2010. The Arabidopsis SMO2, a homologue of yeast TRM112, modulates progression of cell division during organ growth. Plant J. 61: 600– 610 [DOI] [PubMed] [Google Scholar]
- 17. Huh WK, et al. 2003. Global analysis of protein localization in budding yeast. Nature 425: 686– 691 [DOI] [PubMed] [Google Scholar]
- 18. Hurt E, et al. 1999. A novel in vivo assay reveals inhibition of ribosomal nuclear export in ran-cycle and nucleoporin mutants. J. Cell Biol. 144: 389– 401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jorgensen P, Tyers M, Warner JR. 2004. Forging the factory: ribosome synthesis and growth control in budding yeast, p 329– 370 In Hall MN, Raff M, Thomas G. (ed), Cell growth: control of cell size. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [Google Scholar]
- 20. Kalhor HR, Clarke S. 2003. Novel methyltransferase for modified uridine residues at the wobble position of tRNA. Mol. Cell. Biol. 23: 9283– 9292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karbstein K. 2011. Inside the 40S ribosome assembly machinery. Curr. Opin. Chem. Biol. 15: 657– 663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. King TH, Liu B, McCully RR, Fournier MJ. 2003. Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol. Cell 11: 425– 435 [DOI] [PubMed] [Google Scholar]
- 23. Kiss T, Fayet-Lebaron E, Jady BE. 2010. Box H/ACA small ribonucleoproteins. Mol. Cell 37: 597– 606 [DOI] [PubMed] [Google Scholar]
- 24. Krogan NJ, et al. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637– 643 [DOI] [PubMed] [Google Scholar]
- 25. Krogan NJ, et al. 2004. High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell 13: 225– 239 [DOI] [PubMed] [Google Scholar]
- 26. Lafontaine DLJ. 2010. A ‘garbage can’ for ribosomes: how eukaryotes degrade their ribosomes? Trends Biochem. Sci. 35: 267– 277 [DOI] [PubMed] [Google Scholar]
- 27. Lafontaine DLJ, Preiss T, Tollervey D. 1998. Yeast 18S rRNA dimethylase Dim1p: a quality control mechanism in ribosome synthesis? Mol. Cell. Biol. 18: 2360– 2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lafontaine DLJ, Vandenhaute J, Tollervey D. 1995. The 18S rRNA dimethylase Dim1p is required for pre-ribosomal RNA processing in yeast. Genes Dev. 9: 2470– 2481 [DOI] [PubMed] [Google Scholar]
- 29. Leporé L, Lafontaine DLJ. 2011. A functional interface at the rDNA connects rRNA synthesis, pre-rRNA processing and nucleolar surveillance in budding yeast. PLoS One 6: e24962 doi:10.1371/journal.pone.0024962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leulliot N, Bohnsack MT, Graille M, Tollervey D, Van Tilbeurgh H. 2008. The yeast ribosome synthesis factor Emg1 is a novel member of the superfamily of alpha/beta knot fold methyltransferases. Nucleic Acids Res. 36: 629– 639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liang XH, Liu Q, Fournier MJ. 2007. rRNA modifications in an intersubunit bridge of the ribosome strongly affect both ribosome biogenesis and activity. Mol. Cell 28: 965– 977 [DOI] [PubMed] [Google Scholar]
- 32. Liger D, et al. 7 April 2011. Mechanism of activation of methyltransferases involved in translation by the Trm112 ‘hub’ protein. Nucleic Acids Res. doi:10.1093/nar/gkr176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lo KY, Li Z, Wang F, Marcotte EM, Johnson AW. 2009. Ribosome stalk assembly requires the dual-specificity phosphatase Yvh1 for the exchange of Mrt4 with P0. J. Cell Biol. 186: 849– 862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luka Z, Pakhomova S, Luka Y, Newcomer ME, Wagner C. 2007. Destabilization of human glycine N-methyltransferase by H176N mutation. Protein Sci. 16: 1957– 1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marmier-Gourrier N, Clery A, Schlotter F, Senty-Segault V, Branlant C. 2011. A second base pair interaction between U3 small nucleolar RNA and the 5′-ETS region is required for early cleavage of the yeast pre-ribosomal RNA. Nucleic Acids Res. 39: 9731– 9745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mazauric MH, Dirick L, Purushothaman SK, Bjork GR, Lapeyre B. 2010. Trm112p is a 15-kDa zinc finger protein essential for the activity of two tRNA and one protein methyltransferases in yeast. J. Biol. Chem. 285: 18505– 18515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Milkereit P, et al. 2003. A Noc complex specifically involved in the formation and nuclear export of ribosomal 40 S subunits. J. Biol. Chem. 278: 4072– 4081 [DOI] [PubMed] [Google Scholar]
- 38.Mullineux ST, Lafontaine DL.Mapping the cleavage sites on mammalian pre-rRNAs: where do we stand? Biochimie. 2012. http://dx.doi.org/10.1016/j.biochi.2012.02.001. [DOI] [PubMed]
- 39. Nishimura K, et al. 2007. Identification of the RsmG methyltransferase target as 16S rRNA nucleotide G527 and characterization of Bacillus subtilis rsmG mutants. J. Bacteriol. 189: 6068– 6073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oeffinger M, et al. 2007. Comprehensive analysis of diverse ribonucleoprotein complexes. Nat. Methods 4: 951– 956 [DOI] [PubMed] [Google Scholar]
- 41. Ohbayashi I, Konishi M, Ebine K, Sugiyama M. 2011. Genetic identification of Arabidopsis RID2 as an essential factor involved in pre-rRNA processing. Plant J. 67: 49– 60 [DOI] [PubMed] [Google Scholar]
- 42. Pang CN, Gasteiger E, Wilkins MR. 2010. Identification of arginine- and lysine-methylation in the proteome of Saccharomyces cerevisiae and its functional implications. BMC Genomics 11: 92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Panse VG, Johnson AW. 2010. Maturation of eukaryotic ribosomes: acquisition of functionality. Trends Biochem. Sci. 35: 260– 266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Panse VG, et al. 2006. Formation and nuclear export of preribosomes are functionally linked to the small-ubiquitin-related modifier pathway. Traffic 7: 1311– 1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pawlowski M, Gajda MJ, Matlak R, Bujnicki JM. 2008. MetaMQAP: a meta-server for the quality assessment of protein models. BMC Bioinformatics 9: 403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Phipps KR, Charette JM, Baserga SJ. 2011. The SSU processome in ribosome biogenesis—progress and prospects. Wiley Interdiscip. Rev. RNA 2: 1– 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Piekna-Przybylska D, Decatur WA, Fournier MJ. 2008. The 3D rRNA modification maps database: with interactive tools for ribosome analysis. Nucleic Acids Res. 36: D178– D183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Polevoda B, Sherman F. 2007. Methylation of proteins involved in translation. Mol. Microbiol. 65: 590– 606 [DOI] [PubMed] [Google Scholar]
- 49. Purushothaman SK, Bujnicki JM, Grosjean H, Lapeyre B. 2005. Trm11p and Trm112p are both required for the formation of 2-methylguanosine at position 10 in yeast tRNA. Mol. Cell. Biol. 25: 4359– 4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rodríguez-Mateos M, et al. 2009. Role and dynamics of the ribosomal protein P0 and its related trans-acting factor Mrt4 during ribosome assembly in Saccharomyces cerevisiae. Nucleic Acids Res. 37: 7519– 7532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5: 725– 738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schafer T, et al. 2006. Hrr25-dependent phosphorylation state regulates organization of the pre-40S subunit. Nature 441: 651– 655 [DOI] [PubMed] [Google Scholar]
- 53. Schillewaert S, Wacheul L, Lhomme F, Lafontaine DLJ. 2012. The evolutionarily conserved protein LAS1 is required for pre-rRNA processing at both ends of ITS2. Mol. Cell. Biol. 32: 430– 444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schubert HL, Blumenthal RM, Cheng X. 2003. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28: 329– 335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Senger B, et al. 2001. The nucle(ol)ar Tif6p and Efl1p are required for a late cytoplasmic step of ribosome synthesis. Mol. Cell 8: 1363– 1373 [DOI] [PubMed] [Google Scholar]
- 56. Strunk BS, Karbstein K. 2009. Powering through ribosome assembly. RNA 15: 2083– 2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Strunk BS, et al. 2011. Ribosome assembly factors prevent premature translation initiation by 40S assembly intermediates. Science 333: 1449– 1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Studte P, et al. 2008. tRNA and protein methylase complexes mediate zymocin toxicity in yeast. Mol. Microbiol. 69: 1266– 1277 [DOI] [PubMed] [Google Scholar]
- 59. Taddei A, Schober H, Gasser SM. 16 June 2010. The budding yeast nucleus. Cold Spring Harb. Perspect. Biol. 2: a000612 doi:10.1101/cshperspect.a000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Thiry M, Lamaye F, Lafontaine DLJ. 2011. The nucleolus: when two became three. Nucleus 2: 289– 293 [DOI] [PubMed] [Google Scholar]
- 61. Warner JR. 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24: 437– 440 [DOI] [PubMed] [Google Scholar]
- 62. Wery M, Ruidant S, Schillewaert S, Lepore N, Lafontaine DLJ. 2009. The nuclear poly(A) polymerase and Exosome cofactor Trf5 is recruited cotranscriptionally to nucleolar surveillance. RNA 15: 406– 419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Westman BJ, et al. 2010. A proteomic screen for nucleolar SUMO targets shows SUMOylation modulates the function of Nop5/Nop58. Mol. Cell 39: 618– 631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. White J, et al. 2008. Bud23 methylates G1575 of 18S rRNA and is required for efficient nuclear export of pre-40S subunits. Mol. Cell. Biol. 28: 3151– 3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yi C, Pan T. 2011. Cellular dynamics of RNA modification. Acc. Chem. Res. 44: 1380– 1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang J, et al. 2007. Assembly factors Rpf2 and Rrs1 recruit 5S rRNA and ribosomal proteins rpL5 and rpL11 into nascent ribosomes. Genes Dev. 21: 2580– 2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.