

Introduction
Cystic Fibrosis is an autosomal recessive inheritable condition principally involving the lungs, pancreas, liver and intestines. Pulmonary involvement is characterized by airway inflammation and infection starting at an early age1,2 and is the primary cause of premature death.3 This paper will review the current situation in terms of prevalence of bacterial pathogens causing pulmonary infections and the strategies currently employed to treat these infections, focusing primarily on inhaled antibiotic therapy. This will be followed by a review of newly introduced inhaled antibiotics and of those in development.
Current situation
Prevalence data from Europe and North America have identified that younger children are most frequently infected with Staphylococcus aureus and Haemophilus influenzae though gram negative pathogens such as Pseudomonas aeruginosa, Stenotrophomonas maltophilia and Burkholderia cepacia may all occur even in young infants.4 With increasing age, Pseudomonas becomes the dominant pathogen and it's chronic infection is associated with a decline in lung function, chest X-ray score, quality of life and with increased requirement for hospitalization and for antibiotic therapy.3,5,6 Early infection with Pseudomonas is treated aggressively with the intention and expectation that eradication will be successful and so prevent/delay the acquisition of chronic infection.7,8 Chronic infection with Pseudomonas cannot usually be eradicated and therapy is directed towards suppression and control of the infection with intermittent intensive therapy according to symptoms. Antibiotic therapy against Pseudomonas may be delivered by the oral, intravenous or inhaled route. The principle advantages of inhaled therapy are:
Increased antibiotic concentration at the site of infection;
Enhanced bacterial killing;
Reduction in need for intravenous and prolonged oral therapy so reduced systemic toxicity.
The two most commonly used inhaled antibiotics in the UK are nebulized colistin and tobramycin. Colistin is usually the drug of first choice and is recommended as a part of eradication therapy for early Pseudomonas infection in conjunction with either oral therapy (usually ciprofloxacin) or intravenous therapy (usually a combination of a beta-lactam antibiotic such as ceftazidime with a once daily aminoglycoside such as tobramycin). Nebulized colistin is also widely used as a long term treatment for patients chronically infected with Pseudomonas aeruginosa.9 Nebulized tobramycin solution for inhalation is the only other antibiotic licensed in the UK for inhalation for the treatment of chronic infection with Pseudomonas aeruginosa and has been used in clinical trials in the eradication of early infection with Pseudomonas aeruginosa.7,10–12
Other intravenous antibiotics have been delivered by the nebulized route to cystic fibrosis patients but are not licensed for this method of administration. These include gentamicin, amikacin, ceftazidime, meropenem and imipenem. There are no published randomized controlled trials of these treatments in cystic fibrosis patients.
Colistin and tobramycin may be delivered by several nebulizer delivery devices. Conventionally this has been one such as a Pari LC Sprint, using a compressor producing a flow rate of 6 litres per minute. Systems faster than conventional jet nebulizer have been developed using vibrating mesh technology to produce a fine, dense aerosol cloud of low velocity, such as the eFlow rapid and the I-neb. Both of these systems deliver the antibiotic dose over a shorter treatment time and are small, lightweight, very quiet and can be driven by battery without the need for a mains electricity supply.
Despite these advances in nebulizer technology and with the reduction in the amount of time taken with taking the nebulizer, they remain unpopular with many patients for several reasons: the nebulized medication often does not taste great; they still take time to deliver; it's a hassle; it can cause wheeze/cough; there is a possibility of emergence of bacterial resistance.
Many studies have been done in which compliance with medication in CF has been studied. A recent study of adult patients from Leeds, UK investigated three month adherence to nebulized treatment and was able to compare self-reported and clinician-reported levels of compliance with that downloaded from data stored by the I-neb.13 Whilst self-reported adherence was 80%, the nebuliser download indicated adherence levels of 36% with an interquartile range of 5–84%.13 A 2010 audit of Bristol inpatient paediatric nebulizer compliance found that the drug chart and nebulizer (I-neb) download correlated in only 36% of days; with only 341 of 668 doses signed in the drug chart correlating with activation of the I-neb (51%) (data prepared for publication). Even with rapid nebulizers, well motivated patients and normal levels of inpatient supervision, nebulized doses are received by patients at a far lower level than the rate at which they are prescribed.
New kids on the block
Over the last ten years there have been two new inhaled antibiotics licensed for use in cystic fibrosis: Aztreonam lysine (AZLI with the trademark name Cayston) and Tobramycin Inhalation Powder (TIP).
Aztreonam lysine (Cayston)
The intravenous preparation of aztreonam is a dry mixture of aztreonam and arginine, and has been available for CF patients as an anti-Pseudomonas antibiotic for many years. Aztreonam is a monocyclic beta-lactam antibiotic with good gram-negative cover whose side effects include allergy, rash, nausea, vomiting, diarrhoea and elevated liver function tests. The arginine salt is a substrate for nitric oxide production which has been associated with airway inflammation in asthma. Uncertainty about the nitric oxide balance in individual CF patients led to the lysine salt being chosen as a potentially safer formulation of aztreonam for aerosolized use.14
The pharmaceutical company, Gilead, developed AZLI which was approved in the US and UK in 2010 for use as a nebulized antibiotic in CF patients with Pseudomonas aeruginosa. It has been developed for use specifically with it's own nebulizer, the Altera, which is based on the e-Flow rapid compressor system (Figure 1). The mean particle size is 3.6 micrometres (1–5 µm is the optimal size for deposition in the small airways) and a standard 1 ml dosage is delivered over 2–3 minutes.15
Figure 1.

Altera nebulizer for administration of AZLI
Following a 2006 phase 1 dose escalation trial in 35 older patients with CF, AZLI was found to be well tolerated with high sputum antibiotic levels.15 Phase II and phase III clinical trials followed and have been published. The chosen dose of AZLI is 75 mg delivered three times daily. Randomized controlled trials in CF have shown improvements seen in FEV1, symptom score and bacterial sputum density. A summary of AZLI controlled trials is shown in Table 1 (adapted from Kirkby et al.14).
Table 1.
Summary characteristics of published evidence relating to AZLI in CF patients
| Author | Pts | Design | Outcome |
|---|---|---|---|
| Gibson 200615 | 35 adults/adol FEV1 >40% | DB, placebo-controlled daily dose escalation study | Well tolerated, sputum levels >> MIC |
| Retsch-Bogart 200816 | 105 pts >13y, FEV1 >40 | DBRPCT 14d AZLI 75 or 225 mg vs placebo | No change FEV1 at d14 75 mg seems favoured |
| McCoy 200817 AIR-CF1 |
246 pts >5y, FEV1 25–75 Mean age 26.2y |
RDBPCT 28d AZLI 75 mg, bd or tds v placebo | Increased time to more antiPa abx, FEV1, symptom scores, Pa density |
| Retsch-Bogart 200918 AIR-CF2 |
164 pts >6y, FEV1 25–75 | RDBPCT 28d AZLI tds v placebo | Improved symptom scores CFQR, Pa density |
| Oermann 201019 | 274 pts >8y, from previous 2 studies | 18 m open label cyclical treatment bd or tds | Long term use well tolerated, cyclical improved FEV1, symptom scores, Pa density, wt |
| Wainwright 201120 | 157 pts, >5y, FEV1 >75% | RDBPCT 28d AZLI tds v placebo | No change symptom score, improved FEV1, Pa density |
RDBPCT – randomized double blind placebo controlled trial
Further open studies are underway investigating the use of AZLI in chronic Burkholderia cepacia infection in CF. AZLI is not yet licensed below the age of 18 years.
In conclusion, AZLI is a new inhaled antibiotic for use in adult CF patients with Pseudomonas aeruginosa infection. Its place in relation to tobramycin inhalation solution is not yet established but may be an alternative therapy in patients who do not tolerate TIS or in whom there is unfavourable response despite TIS. The UK cost of 28 days of therapy is currently set at £2566.50 or £15,399 per annum (based on alternating cycles of 28 days on and 28 days off treatment).21
Tobramycin inhalation powder
Nebulized tobramycin inhalation solution (TIS) improves lung function and quality of life and reduces hospitalization in patients with chronic Pseudomonas aeruginosa infection but must be delivered by a nebulizer.22–24 Tobramycin Inhalation Powder (TIP) has been developed by Novartis Pharmaceuticals to offer an alternative preparation which may advantage some patients.
TIP in a dosage of 112 mg is administered as four powder capsules used twice daily and was licensed for UK use, 28 days on and 28 days off, from September 2011 for patients aged 6 years and over. The capsules are delivered using a dry powder inhaler developed specifically for TIP known as a Podhaler (Figure 2). Once the capsule has been inserted, the Podhaler is manually activated. The patient then inhales and breath-holds before repeating the cycle for all 4 capsules. Once pierced, the capsules release spherical light porous particles which are manufactured using an emulsion-based spray drying process. These hollow particles have a high surface area and low density, creating a powder that is highly dispersible even in young children and those with reduced lung function.25
Figure 2.
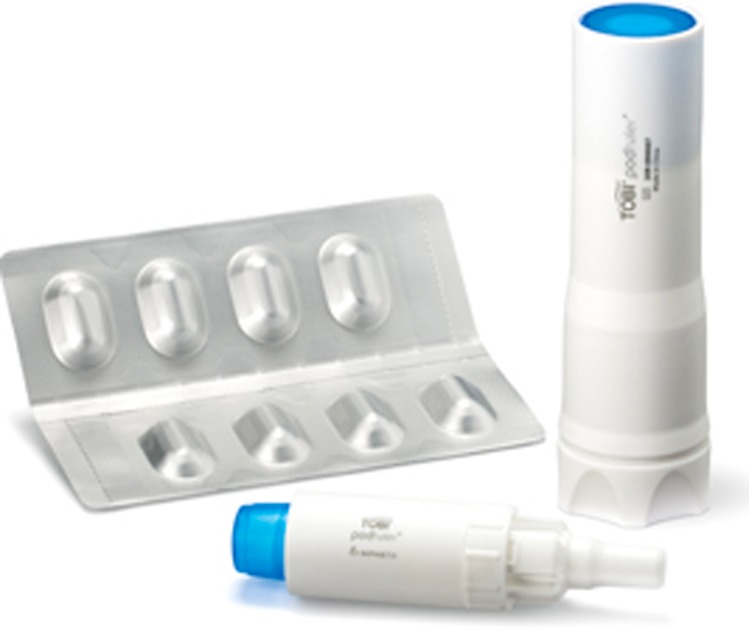
Podhaler for inhalation of Tobramycin Inhalation Powder
Table 2 shows the results of published trials to date.26,27 TIP via the Podhaler has been shown to be safe, similarly effective to Tobramycin inhalation solution (TIS) and faster to deliver if compared to TIS delivered through the PARI LC PLUS jet nebulizer (mean time for delivery 5.7 versus 19.7 minutes respectively) though delivery would have been faster through other commonly used vibrating mesh nebulizers such as the eFlow. Patient-rated satisfaction scores favour TIP over TIS, though there was a higher rate of cough, dysphonia and taste disturbance and of discontinuation of TIP.
Table 2.
Summary characteristics of clinical trials with Tobramycin Inhalation Powder
| Author | Pts | Design | Outcome |
|---|---|---|---|
| Konstan 201027 | 95 patients aged 6–21yrs, FEV1 25–80% predicted | RDBPCT for first cycle then open for two further cycles, TIP versus placebo, 24 week study | Improvement in FEV1 at d28 in favour of TIP. Also reduced sputum density, respiratory hospitalization and anti-pseudomonas antibiotic usage |
| Konstan 201126 | 553 adults and children with CF, FEV1 25–75% predicted | Open label randomized study, 3:2 to TIP or TIS via Pari LC Plus | Increased cough, dysphonia and taste disturbance in TIP-treated group, shorted time for administration and higher overall treatment-specific satisfaction scores favouring TIP |
In summary, TIP is now licensed and available for children and adults with CF, and offers an alternative delivery method to TIS which is portable, more convenient than a nebulizer and not dependant on an electrical power source. Some patients will prefer this though should be warned of the possibility of cough, dysphonia and taste disturbance after inhalation. The UK cost of a 28 day cycle will be £2148, or £12,888 per annum.21
What is in the pipeline?
There are no other licensed inhaled antibiotics at present for cystic fibrosis. Other antibiotics are in earlier stages of development and license application. These include:28
Liposomal amikacin – Arikace
In phase III clinical trials as an anti-Pseudomonas antibiotic under development with Insmed. In the US, clinical trials are on hold following a request by FDA for further animal studies to be performed to investigate toxicity. Unlicensed and no clear launch date available.
Liposomal ciprofloxacin
Undergoing phase II clinical trials in development with Aradigm. Trials are focusing on once daily usage as an anti-Pseudomonas antibiotic in CF and non-CF bronchiectasis. No UK launch date.
Colistin – Colobreathe
Dry powder formulation of colistin (already in use as an intravenous and nebulized antibiotic) developed by Forest. Phase III studies have been performed but not yet published so Colobreathe is currently unlicensed and with a launch date yet to be confirmed.
Levofloxacin – Aeroquin
Another quinolone antibiotic undergoing phase III clinical trials with Mpex. A multicentre multinational trial in 267 stable CF patients over 12 years of age with Pseudomonas aeruginosa infection is due to complete during 2012.
Fosfomycin/tobramycin combination
Phase II safety and efficacy trials are underway in the United States in adult patients with chronic Pseudomonas aeruginosa infection and preliminary data have been released but not yet published.
Summary and Conclusion
The last 30 years have seen progressive improvement in CF survival, even without new treatments directed at the specific molecular targets unique to CF. These improvements have been brought about by good multidisciplinary care, attention to nutrition, chest physiotherapy and aggressive appropriate use of antibiotics. The paper by Dodge et al. (2007)29 demonstrated very well these improvements and act as a reminder that CF teams and their patients should remain optimistic about further improvements in survival. Dodge et al. suggest that median survival beyond 50 years should be expected for children born in 2000 (Figure 3).
Figure 3.

UK cystic fibrosis population. Proportion of a) males and b) females of each 3-yr cohort surviving until 2003. ▪: 1968–1970; ○: 1971–1973; ▴: 1974–1976; □: 1977–1979; ♦: 1980–1982; ◊: 1983–1985; ▵: 1986–1988; •: 1989–1991; ▿: 1992–1994
Development of intravenous, oral and inhaled antibiotics have played a part in this improvement and will continue to do so in the future. The two new inhaled antibiotics, Cayston (aztreonam) and TIP (Tobramycin Inhalation Powder) represent an important development and will offer greater choice for CF physicians and patients. Their place in the antibiotic formulary is established in so far as achieving licensing but not yet their place in relation to other commonly used antibiotics. Our patients should be encouraged to participate in randomized controlled trials of antibiotics so that the prescribing clinicians are best informed about their use. As physicians, we should contribute to the design of these trials so that we are able to give evidence-based advice to our patients.
DECLARATIONS
Competing interests
None declared
Funding
None
Ethical approval
Not required
Guarantor
SLH
Contributorship
Simon Lagton Hewer is the sole contributor
Acknowledgements
I am very grateful to Kirsten Thomson, pharmacist at Bristol Royal Hospital for Children, for information that has contributed to this manuscript
References
- 1.Armstrong DS, Grimwood K, Carlin JB, Carzino R, Olinsky A, Phelan PD Bronchoalveolar lavage or oropharyngeal cultures to identify lower respiratory pathogens in infants with cystic fibrosis. Pediatric Pulmonology 1996;21:267–75 [DOI] [PubMed] [Google Scholar]
- 2.Ranganathan SC, Dezateux C, Bush A, et al. Airway function in infants newly diagnosed with cystic fibrosis. Lancet 2001;358:1964–5 [DOI] [PubMed] [Google Scholar]
- 3.Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatric Pulmonology 2002;34:91–100 [DOI] [PubMed] [Google Scholar]
- 4.Cystic Fibrosis Foundation. 2009. Patient Registry Annual Report.
- 5.Kosorok MR, Zeng L, West SE, et al. Acceleration of lung disease in children with cystic fibrosis after Pseudomonas aeruginosa acquisition. Pediatric Pulmonology 2001;32:277–87 [DOI] [PubMed] [Google Scholar]
- 6.Nixon GM, Armstrong DS, Carzino R, et al. Clinical outcome after early Pseudomonas aeruginosa infection in cystic fibrosis. The Journal of Pediatrics 2001;138:699–704 [DOI] [PubMed] [Google Scholar]
- 7.Langton Hewer SC, Smyth AR Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database Syst Rev 2009:CD004197 [DOI] [PubMed] [Google Scholar]
- 8.Li Z, Kosorok MR, Farrell PM, et al. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA: the Journal of the American Medical Association 2005;293:581–8 [DOI] [PubMed] [Google Scholar]
- 9.UK Cystic Fibrosis Trust. 2009. Antibiotic Treatment for Cystic Fibrosis.
- 10.Ratjen F, Munck A, Kho P, Angyalosi G Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis: the ELITE trial. Thorax 2010;65:286–91 [DOI] [PubMed] [Google Scholar]
- 11.Rosenfeld M, Emerson J, McNamara S, et al. Baseline characteristics and factors associated with nutritional and pulmonary status at enrollment in the cystic fibrosis EPIC observational cohort. Pediatric Pulmonology 2010;45:934–44 [DOI] [PubMed] [Google Scholar]
- 12.Treggiari MM, Rosenfeld M, Mayer-Hamblett N, et al. Early anti-pseudomonal acquisition in young patients with cystic fibrosis: rationale and design of the EPIC clinical trial and observational study’. Contemp Clin Trials 2009;30:256–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daniels T, Goodacre L, Sutton C, Pollard K, Conway S, Peckham D Accurate assessment of adherence: self-report and clinician report vs electronic monitoring of nebulizers. Chest 2011;140:425–32 [DOI] [PubMed] [Google Scholar]
- 14.Kirkby S, Novak K, McCoy K Aztreonam (for inhalation solution) for the treatment of chronic lung infections in patients with cystic fibrosis: an evidence-based review. Core Evid 2011;6:59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibson RL, Retsch-Bogart GZ, Oermann C, et al. Microbiology, safety, and pharmacokinetics of aztreonam lysinate for inhalation in patients with cystic fibrosis. Pediatric Pulmonology 2006;41:656–65 [DOI] [PubMed] [Google Scholar]
- 16.Retsch-Bogart GZ, Burns JL, Otto KL, et al. A phase 2 study of aztreonam lysine for inhalation to treat patients with cystic fibrosis and Pseudomonas aeruginosa infection. Pediatric Pulmonology 2008;43:47–58 [DOI] [PubMed] [Google Scholar]
- 17.McCoy KS, Quittner AL, Oermann CM, Gibson RL, Retsch-Bogart GZ, Montgomery AB Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2008;178:921–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Retsch-Bogart GZ, Quittner AL, Gibson RL, et al. Efficacy and safety of inhaled aztreonam lysine for airway pseudomonas in cystic fibrosis. Chest 2009;135:1223–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oermann CM, Retsch-Bogart GZ, Quittner AL, et al. An 18-month study of the safety and efficacy of repeated courses of inhaled aztreonam lysine in cystic fibrosis. Pediatric Pulmonology 2010;45:1121–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wainwright CE, Quittner AL, Geller DE, et al. Aztreonam for inhalation solution (AZLI) in patients with cystic fibrosis, mild lung impairment, and P. aeruginosa. Journal of Cystic Fibrosis: Official Journal of the European Cystic Fibrosis Society 2011;10:234–42 [DOI] [PubMed] [Google Scholar]
- 21.Joint Formulary Committee British National Formulary. 62 ed. London: BMJ Group and Pharmaceutical Press; September 2011. [Google Scholar]
- 22.Ramsey BW, Pepe MS, Quan JM, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 1999;340:23–30 [DOI] [PubMed] [Google Scholar]
- 23.Murphy TD, Anbar RD, Lester LA, et al. Treatment with tobramycin solution for inhalation reduces hospitalizations in young CF subjects with mild lung disease. Pediatric Pulmonology 2004;38:314–20 [DOI] [PubMed] [Google Scholar]
- 24.Quittner AL, Buu A Effects of tobramycin solution for inhalation on global ratings of quality of life in patients with cystic fibrosis and Pseudomonas aeruginosa infection. Pediatric Pulmonology 2002;33:269–76 [DOI] [PubMed] [Google Scholar]
- 25.Geller DE, Konstan MW, Smith J, Noonberg SB, Conrad C Novel tobramycin inhalation powder in cystic fibrosis subjects: pharmacokinetics and safety. Pediatric Pulmonology 2007;42:307–13 [DOI] [PubMed] [Google Scholar]
- 26.Konstan MW, Flume PA, Kappler M, et al. Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: The EAGER trial. Journal of Cystic Fibrosis: Official Journal of the European Cystic Fibrosis Society 2011;10:54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konstan MW, Geller DE, Minic P, Brockhaus F, Zhang J, Angyalosi G Tobramycin inhalation powder for P. aeruginosa infection in cystic fibrosis: The EVOLVE trial. Pediatric Pulmonology 2010;46:230–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.http://www.ukmi.nhs.uk/. New Drugs Online, 2011. (last accessed 14 February 2012)
- 29.Dodge JA, Lewis PA, Stanton M, Wilsher J Cystic fibrosis mortality and survival in the UK: 1947–2003. The European Respiratory Journal: Official Journal of the European Society for Clinical Respiratory Physiology 2007;29:522–6 [DOI] [PubMed] [Google Scholar]