

Abstract
Bicuspid aortic valve (BAV) is the most common congenital heart defect, affecting 1-2% of the population. It is generally diagnosed late in adulthood when deterioration of the abnormal leaflet becomes clinically evident. BAV patients have an increased risk of developing serious complications, including stenosis, regurgitation, endocarditis, dilation of the aorta, aortic dissection, and aneurysm. BAV is a heritable trait, but the genetic basis underlying this cardiac malformation remains poorly understood. In the last decade, thanks to studies in animal models as well as genetic and biochemical approaches, a large number of genes that play important roles in heart development have been identified. These discoveries provided valuable insight into cardiac morphogenesis and uncovered congenital-heart-disease-causing genes. This paper will summarize the current knowledge of valve morphogenesis as well as our current understanding of the genetic pathways involved in BAV formation. The impact of these advances on human health including diagnosis of BAV and prevention of cardiovascular complications in individuals with BAV or with a family history of BAV is also discussed.
1. Overview of Valve Development
In human and other mammals, cardiac valves are essential for unidirectional blood flow which is crucial for proper functioning and survival of the organism. Defects in valve structure or function can profoundly alter cardiovascular homeostasis and are among the leading causes of human morbidity and mortality. Thus, understanding the molecular basis of normal and pathologic valve development is of utmost importance and clinical relevance. The valves of the four chambered heart can be classified in 2 groups: atrioventricular valves (mitral and tricuspid) separate the atria from the ventricles while semilunar valves (aortic and pulmonary) divide the ventricles from the great arteries. Normal aortic valve is tricuspid, meaning it possesses three leaflets (or cusps) but defective development involving fusion of two of these produces bicuspid aortic valve (BAV). At 1-2% prevalence in the human population, BAV is the most common form of congenital heart disease and a significant risk factor for premature cardiovascular complications and valve replacement in young adults.
In the embryo, the heart is the first organ to develop starting with the specification and migration of the anterior lateral plate mesoderm cells to form the cardiac crescent [1]. This is closely followed (embryonic day (E) 8.5 in the mouse and 3 weeks gestation in human) by migration of the cardiac progenitors along the ventral midline where they fuse to form a beating linear heart tube. This primitive tube is composed of an inner endocardial cell layer separated by the extracellular matrix (ECM), also known as the cardiac jelly from the outer myocardial lining. At around E9.5 in the mouse and 4-5 weeks in human, cardiac looping occurs and brings the atrial region of the linear tube into a posterior position to the common ventricle. At the same time, increased production of ECM will cause the tissue to swell at localized regions of the heart, initiating the formation of the endocardial cushions at the atrioventricular (AV) canal and outflow tract (OFT) [2, 3]. Endocardial cushion formation is initiated by signals emanating from the AV canal and OFT myocardium that will diffuse into the ECM and reach the adjacent endocardial cells to undergo EMT. A large number of transcription factors and signaling pathways have been implicated in EMT and cushion morphogenesis, including members of the TGF-β superfamily, VEGF, ErbB, NFATc1, Notch, Wnt/β-catenin, Twist-1, Sox9, Tbx20, and Gata4, and their function has been nicely described in a recent review [4].
Endocardial cushion formation is a complex process that relies on the transformation of a subpopulation of inner endocardial cells into mesenchymal cells; this process termed epithelial-to-mesenchymal transformation (EMT) occurs at the AV canal boundary to initiate mitral and tricuspid valve formation and, later, in the OFT to initiate aortic and pulmonary valve formation. As development progresses, these cushions will undergo exhaustive cell proliferation and grow towards each other until they touch initiating a process of fusion between the two cushions. Further remodeling of the endocardial cushions results in the formation of thin protruding leaflets composed of endocardial cells and ECM that go on to form the valves. These last steps are dependent on cell differentiation, apoptosis and ECM remodeling. In the AV canal, EMT-derived mesenchymal cells are the sole contributor to the mitral and tricuspid valves. In the OFT, mesenchymal cells originating from migrating cardiac neural crest cells will reach the OFT cushions and together with endocardially derived mesenchymal cells will contribute to the formation of the aortic and pulmonary valves. It is of no surprise then that subtle perturbations in endocardial cushion development can lead to heart valve diseases, including bicuspid aortic valve (BAV). Of note, valve defects are among the most frequent cardiovascular malformations accounting for 25–30% [5].
In normal individuals, the aortic valve possesses three leaflets, namely, the left coronary, the right coronary, and noncoronary cusps, named after their relationship to the coronary arteries (Figure 1). The morphology of the BAV usually includes leaflets of unequal size as the result of the fusion of two cusps, leaving one leaflet larger than the other [6]. In humans, the larger leaflet is characterized by a raphe, which is a thin ridge of tissue that represents the location where the two cusps fused during valve development. In addition, leaflet orientation has been shown to vary widely among patients. The most frequent BAV subtypes are those with fusion of the right and left (R-L) coronary leaflet, representing 59% of cases and those with union of the right and noncoronary (R-N) leaflet occurring in 37% of BAV cases [7]. Insight into the etiology of the R-N and R-L BAVs was further gained from studies of Nos3 null mice as well as the inbred Syrian hamsters strain which selectively display R-N and R-L subtypes, respectively [8, 9]. These studies suggested that the R-N BAV is caused by defective formation of the OFT cushion whereas the R-L BAV is likely the result of defective OFT septation. Thus, the two BAV subtypes appear to have distinct etiologies. In addition, it has been shown that the R-N fusion is associated with increased dimensions of the aortic root as well as development of aortic insufficiency later in life [10, 11]. The R-L fusion, on the other hand, is associated with a more rapid progression to valve dysfunction and increased dimension of the aortic arch. A new technique, called 3D time resolved phase contrast cardiac magnetic resonance (4D Flow), has been described recently as a powerful tool to allow a better characterization of aortic-valve-related flow dynamics as well as progression of aortic dilation in patients with BAVs [11]. In this study, Hope et al. demonstrated a significant elevation of ascending aortic wall shear stress in a subgroup of patients with BAVs, the majority of whom had R-L fusion. Shear stress to the aortic wall has been linked to aortic aneurysm formation, thus this technique provides a noninvasive mean to quantify the increased blood flow through the ascending aorta in patients with BAVs even before there is a clear manifestation of aortic aneurysms.
Figure 1.
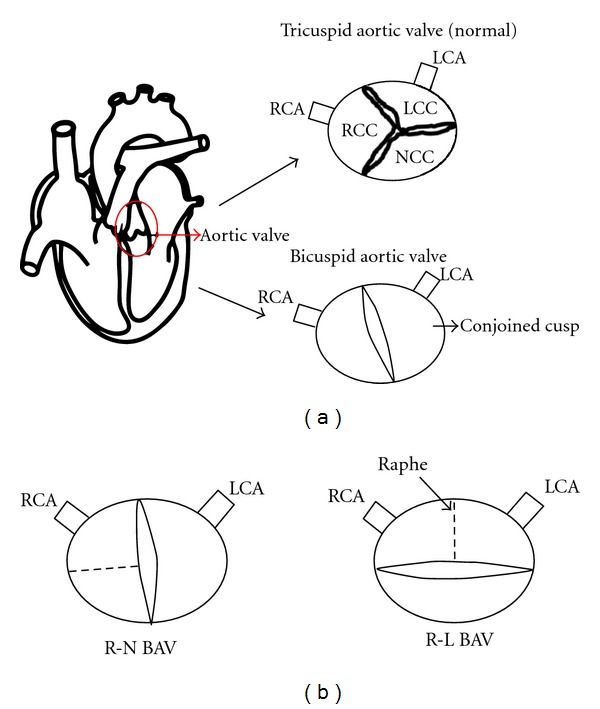
Schematic representation of a bicuspid aortic valve. (a) Transverse section through the aorta showing a normal aortic valve with three leaflets and their corresponding points of attachment to the aortic wall. A bicuspid aortic valve arises from fusion of two different cusps, resulting in the formation of a conjoined leaflet. (b) Schematic representation of the BAV subtypes. The R-N BAV arises from fusion of the right coronary cusp and noncoronary cusp whereas the R-L BAV results from the fusion of the right coronary cusp and left coronary cusp. LCA: left coronary artery; LCC: left coronary cusp; NCC: noncoronary cusp; RCA: right coronary artery; RCC: right coronary cusp.
2. Insights from Human Genetics
Information regarding prevalence of human BAVs has been acquired through autopsy and echocardiographic studies. BAV is now believed to be the most common cardiovascular malformation with a prevalence of 1-2% in the general population [12]. Even though BAV is usually an isolated defect, it may be associated with other cardiovascular syndromes or malformations with at least one-third of patients likely to develop serious complications that will require valve surgery [13]. It is now clear that BAV is a risk factor for adult cardiovascular events. These include aortic stenosis, regurgitation, dissection, dilation/aneurysm as well as valve calcification and infective endocarditis. In the last decade, conflicting results have been obtained regarding the presence of thoracic aortic aneurysms in the setting of BAVs [14]. Some groups have shown that there is a higher rate of aortic root dilatation in individuals with BAVs while others found thoracic aortic aneurysms in first-degree relatives with normal tricuspid aortic valves. Thus, aortic aneurysms in the context of a BAV may not exclusively be due to disturbed blood flow through the aortic valve. For example, Bunton et al. have demonstrated that the fibrilin-1 gene, which is involved in Marfan syndrome, plays an important role in maintaining the integrity of elastic fibers and that mutations in FIBRILIN-1 could decrease the elastin content in the aorta, providing a new mechanism for the presence of aneurysms in patients with Marfan syndrome [15]. A better understanding of the molecular pathways that maintain the integrity of the aorta is warranted in order to elucidate the pathogenesis of aortic aneurysms in BAV patients.
Several studies addressing the heritability of BAV revealed high incidence of familial clustering [16–18]. Glick and Roberts noted that 17 patients out of 71 family members (24%) had aortic valve disease likely secondary to a BAV, with 2 or more family members affected. Using echocardiography screening of 190 first-degree relatives, Huntington and colleagues reported a prevalence of BAV of 9.1% among the first-degree relatives, suggesting that the distribution of BAV follows an autosomal dominant inheritance with reduced penetrance [16]. Using a variance component methodology, Cripe et al. found that, using their mathematical model, the heritability of BAV was 89% suggesting that BAV is almost entirely genetically determined and that mutations in diverse genes with divergent inheritance pattern may be responsible for BAV formation in different families [19]. In conclusion, epidemiologic studies suggest that BAV is heritable and follows an autosomal dominant mode of transmission with reduced penetrance and variable expressivity.
While the heritability of BAV is now well established, genes linked to the defect remain largely unknown. A family-based linkage analysis using microsatellite markers revealed linkage to three loci on chromosome 18q, 5q, and 13q, but the precise gene(s) within these regions were not defined [20]. To date, only mutations in NOTCH1, a gene that maps to 9q34-35, have been associated with BAV in a small number of families (Table 1). Initially, a nonsense and a frameshift mutation in two family members with BAV and valve calcification were identified in a large family with 11 cases of congenital heart disease, four of which required aortic valve replacement [21]. Shortly thereafter, new undescribed missense mutations were identified in patients with BAV and/or aortic aneurysms in two independent studies [22, 23]. These observations provided strong evidence that NOTCH1 haploinsufficiency can cause aortic valve disease.
Table 1.
Human gene mutation and phenotype associated with bicuspid aortic valve formation.
| Gene affected | Human syndrome | Human cardiac phenotype | Mutation and/or expression in BAV patients | Mouse valve phenotype | Reference |
|---|---|---|---|---|---|
| NOTCH1 | BAV, calcification, BAV with aortic aneurysm | R1108X, H1505del, T596M, P179H, A1343V, P1390T | Thick valve leaflets, calcification | [21–23] | |
| GATA6 | ASD, TOF | Mother of the ASD patient had a BAV | No valve phenotype reported | [26] | |
| FBN1 | Marfan syndrome | Mitral valve prolapse, Aortic aneurysm | Reduced expression of FBN1 | Thick and long valve leaflets, mitral valve prolapse | [28–31] |
| TGFBR2 | Loeys-Dielz syndrome, Marfan syndrome | Mitral valve prolapse, aortic aneurysm, BAV | V387M | Defective remodelling of the AV cushion, lethality at E11.5 precluding analysis of valves | [36] |
| ACTA2 | Thoracic aortic aneurysm and dissection | Three family members had a BAV | No valve phenotype | [38] | |
| HOXA1 | Bosley-Salih-Alorainy syndrome, athabascan brainstem dysgenesis syndrome | Interrupted aortic arch type B, aberrant subclavian artery, VSD, TOF, BAV | Not reported | BAV | [39] |
| KCNJ2 | Andersen syndrome | BAV, BAV with coarctation of the aorta, pulmonary stenosis | R67W | Not reported | [41] |
ASD: atrial septal defect; BAV: bicuspid aortic valve; OFT: outflow tract; TOF: tetralogy of Fallot; VSD: ventricular septal defect.
Mutations in transcription factor GATA6 have been associated with congenital heart defects, including tetralogy of Fallot, persistent truncus arteriosus, and atrial septal defects [24–26]. GATA6 is involved in OFT morphogenesis and its deletion from neural crest cells in mice causes perinatal lethality due to a spectrum of aortic arch patterning and cardiac OFT septation defects [27]. Recently, Lin et al. discovered that one of the parents of a patient with ASD had a BAV suggesting a possible relationship between GATA6 and BAV (Table 1). Interestingly, GATA6 maps to chromosome 18q, which is linked to BAV, but no mutations in the GATA6 gene have been reported yet in individuals with BAV [20]. Of note, examination of the aortic valve of Gata6 heterozygote mice revealed partial penetrance of BAVs (our unpublished data) supporting a role for GATA6 as a candidate BAV-causing gene.
Human genetics have also revealed that perturbations in the expression of ECM components (elastin, collagen, and proteoglycans) can lead to cardiac defects (Table 1). For example, mutations in FBN1 (an extracellular glycoprotein of the ECM that acts to maintain tissue elasticity of the valve leaflets and aortic wall by linking smooth muscle cells to adjacent elastin fibrils) have been observed in patients with Marfan syndrome (MFS), a genetic disorder of the connective tissue characterized by mitral valve prolapse, BAV, and/or aortic dissection and aneurysm [28–30]. Moreover, reduced expression of FBN1 in the aorta of patients with BAVs was reported [31]. Of note, targeted deletion of Fbn1 in mice phenocopies the defects observed in Marfan syndrome indicative of a causal relationship between FBN1 mutations and valve malformation [32, 33]. These mice have upregulated TGF-β signalling, mitral valve prolapse and die shortly after birth due to aortic dissection caused by weakening of the aortic wall [34]. Interestingly, mutations in the TGF-β receptors, TGFBR1 and TGFBR2, have been associated with Marfan and Loeys-Dietz syndromes [35, 36]. Recently, a missense mutation in TGFβR2 identical to the one found in MFS patients who tested negative for mutation in FBN1 was found in a patient with BAV and aortic aneurysm [37] but an earlier study found no mutation in either TGFBR1 or TGFBR2 in patients with isolated BAV.
Aortic dilation, dissection, and/or aortic aneurysm are the most common vascular complications in patients with BAV. Linkage analysis of 7 family members with aortic aneurysms and dissection, of whom three had BAVs identified ACTA2 which encodes smooth muscle α-actin [38]. Analysis of aortic tissue from these patients showed increased proteoglycans accumulation, fragmentation, loss of elastic fibers, and decreased numbers of smooth muscle cells, consistent with aortic wall degeneration. However, whether ACTA2 mutations cause BAV remains uncertain.
Other gene mutations have been linked to syndromic disease that include aortic valve abnormalities. For example, homozygous truncating mutations in HOXA1 have been associated to Bosley-Salih-Alorainy syndrome and Athabascan Brainstem Dysgenesis syndrome [39]. Interestingly, severe cardiovascular malformations, including interrupted aortic arch type B, aberrant subclavian artery, ventricular septal defect, tetralogy of Fallot, and BAV, are observed in these syndromes. Targeted inactivation of Hoxa1 in mice was shown to recapitulate these defects [40]. Among cardiac malformations, BAVs were obtained with a prevalence of 24%. It remains to be seen whether mutations in HOXA1 will be discovered in BAV patients.
Similarly, heterozygous missense mutations in the KCNJ2 potassium channel have been linked to Andersen syndrome (Table 1) [41]. This rare disorder is characterized by prolongation of the QT interval with ventricular arrhythmias, periodic paralysis, dysmorphic facies, cleft palate, and scoliosis. Additional features seen in the pedigree were cardiovascular malformations, including BAV, BAV with coarctation of the aorta, or pulmonary stenosis, which had never been associated with this disease before. Whether this or another as yet unidentified mutation causes BAV is unclear and the link between defective potassium current and abnormal aortic valve formation has not been investigated.
In summary, human genetics support the involvement of multiple causative and modifier genes in BAV inheritance. The majority of these genes remain to be identified.
3. Insights from Animal Models
Over the past decade, analysis of aortic valves of genetically engineered mice have provided important insight into valve development and identified potential candidate genes that could underlie BAV formation (Table 2). One of the first mouse models of BAV was reported in Nos3 null mice who had a broad spectrum of CHD including BAV all from the fusion of the right and noncoronary leaflet (R-N) [9, 42]. Nos3 is expressed in endocardial cells of the heart and is shear-stress-dependent [43]. Since formation of the endocardial cushions depends on the EMT, a shear-stress dependent process, Nos3 deficiency might alter endocardial cell migration during EMT, causing abnormal development of the valve cushion. Consistent with this, NOS3 expression was found to be significantly lower in patients with BAV [44], providing further support for the relevance of NOS3 and possibly its regulators in BAV formation.
Table 2.
Mouse models of bicuspid aortic valve formation.
| Gene affected | Human cardiac phenotype | Mouse model | Mouse valve phenotype | Cardiovascular expression | Reference |
|---|---|---|---|---|---|
| Hoxa1 | IAAB, ASC, VSD, BAV, TOF | Hoxa1−/− | BAV | Neural crest cells, OFT endocardium and myocardium, Secondary heart field | [40] |
| NOS3 | Not reported | Nos3−/− | BAV | Endocardium, myocardium | [42] |
| Gata5 | Not reported | Gata5−/− and Tie2Cre; Gata5F/F | BAV | Endocardium, AV and OFT endocardial cushions, subset of endocardial cells, epicardium | [47] |
| Nkx2.5 | ASD, AV block, VSD, TOF, HCM | Nkx2.5GFP het, Nkx2.5HDneo het | BAV, aortic aneurysm | Myocardium | [53] |
ASC: aberrant subclavian artery; ASD: atrial septal defect; AV: atrioventricular; BAV: bicuspid aortic valve; HCM: hypertrophic cardiomyopathy; IAAB: interrupted aortic arch type B; VSD: ventricular septal defect; TOF: tetralogy of Fallot.
Members of the GATA family of transcription factors such as the endothelially expressed GATA2 are transcription activators of NOS3 [45]. Another member, GATA5, has been shown to be restricted to endocardial cells and the endocardial cushions of the AV canal and OFT [46]. Recently, we generated a Gata5 null mouse model and found partial penetrance of BAVs with a prevalence of 26% [47]. To gain more insight into the role of Gata5 in endocardial development including the cell type contributing to these defects, we generated a conditional mouse model lacking Gata5 only in endothelial cells (eGata5−/−). The eGata5 mutant mice had a similar prevalence of BAVs as the Gata5 null mice suggesting a cell autonomous function for Gata5 in endocardial cushion development. Remarkably, in both mouse models, the BAV was due to fusion of the right and noncoronary leaflets (R-N BAV), identical to what was seen in the Nos3 null mice. Reduced expression of Nos3 was observed in the endocardial cushions of the OFT in the Gata5 null mice. Together these observations point to exquisite NOS3 dosage sensitivity in aortic leaflet formation. In vitro studies demonstrated that Gata5 is a potent activator of the Nos3 promoter suggesting that NOS3 may be the downstream effector of GATA5 in endocardial cushion cells. Our studies also revealed that the Notch pathway is significantly downregulated in Gata5 null and eGata5−/− mice. Of note, decreased expression of Jag1 the Notch ligand was observed in Gata5 null and eGata5−/− mice with a concomitant upregulation of Rbpj-κ, the Notch pathway repressor. Downregulation of the Notch pathway in embryonic hearts was confirmed by decreased immunostaining for the Notch1 intracellular domain (NCID). Together, these observations suggest that reduced Notch1 activation and subsequent Notch signaling in the outflow tract could contribute to abnormal endocardial cushion formation and fusion of the aortic valve leaflets. The Notch pathway plays major roles in multiple developmental processes, including cardiovascular development. Among others, it has been shown to be critical for EMT, a key process for valve formation. In mammals, the Notch family consists of 4 type I transmembrane receptors (Notch1 to 4) and 5 type I transmembrane ligands, Jagged1, Jagged2, Delta-like (Dll) 1, Dll3, and Dll4 [48]. Upon ligand binding, a protease complex containing gamma secretase cleaves the intracellular domain of Notch, which enters the nucleus and regulates gene expression through binding to the transcription factor RBP-Jκ. It has been observed that Notch1 plays key roles during valve development and EMT, consistent with its expression pattern in the endocardium and OFT cushion mesenchyme. Of note, Notch1 null mice die early due to severe cardiac defects, including defective EMT [49]. In the last couple of years, different groups have tried to shed light on the molecular mechanism of aortic valve calcification. Nigam and Srivastava reported that inhibition of Bmp2 blocked calcification of murine aortic valves in vivo and in vitro, suggesting that Notch1 represses Bmp2 within the aortic valve [50]. In addition, Acharya et al. discovered that inhibition of Notch1 in an in vitro model of aortic valve calcification was prevented by the addition of Sox9, indicating that Notch1 regulates aortic valve calcification through a Sox-9-dependent pathway [51]. In conclusion, all of these findings demonstrate that NOTCH1 haploinsufficiency plays a key role in valvulogenesis as well as the maintenance of normal valve function in the adult heart. Expression of the three cardiac GATA factors partially overlaps during cardiac development. Our studies revealed that Gata4+/−Gata5+/− and Gata5+/−Gata6+/− die embryonically or perinataly due to profound cardiac defects including double outlet right ventricles and ventricular septal defects [52]. Interestingly, nearly half (3/7) Gata4+/−Gata5+/− mice had very high pressure gradient through the aortic valve and examination of the aortic valve of these mice revealed the presence of BAVs. Similarly, we observed BAV in 25% (1/4) Gata5+/−Gata6+/− embryos at E18.5 (unpublished data). These observations are indicative of dosage sensitivity for cardiac GATA factors in aortic valve formation. They also suggest that CHD-causing genes might interact to influence CHD penetrance and expressivity including BAV. These results have important implications for human genetic studies.
BAVs have been reported in a small proportion (11%) of mice haploinsufficient for Nkx2.5 [53], a gene that has been associated with several human CHDs (Table 2). NKX2.5 maps to chromosome 5q34, which has been linked to BAVs, and although one study did not find any polymorphisms in the coding region of Nkx2.5 in BAV patients [54], it still remains to be determined whether mutations in the NKX2.5 gene will be discovered in future studies of human BAV. Nkx2.5 is a critical regulator of cardiac morphogenesis and was shown to modulate ECM of the aorta through regulation of collagen type I [55]. Interestingly, mutations in collagen type I have been linked to Ehlers-Danlos syndrome, which is characterized by skin and bone abnormalities as well as mitral and aortic valve dysfunction [56].
In conclusion, while the molecular basis of BAV is incompletely understood, at least 2 pathways seem critical for normal tricuspid formation, namely, Notch and Nos3. This information, together with the availability of mouse models of BAV, represents an important step in understanding the pathogenesis of BAV and for studies aimed at the prevention of BAV associated cardiovascular events.
4. Is BAV Related to Other Congenital Heart Defects?
In general most inherited congenital heart defects (CHDs) show variable expressivity and penetrance including many autosomal dominant syndromes. As stated earlier, BAVs have been identified in family members whose parents or siblings had other CHDs. The availability of the Gata5 null mice which do not have other structural heart defects besides BAV offers the unique possibility to genetically test the link between BAV and other heart defects. This is easily achieved by crossing with specific mouse strains. The first set of experiments aimed to explore the consequences of combinatorial heterozygosity of Gata5 and the 2 other cardiac GATA factors, GATA4 and GATA6, both of which have been linked to human CHD including septal and valve defects [24, 26, 57–59].
The resulting double hets offsprings had multiple CHDs and reduced survival suggesting that genes linked to BAVs contribute to other cardiac defects. This information is important for understanding the incidence of BAVs in conjunction with other CHDs in families and for future human genetic studies.
5. Where Do We Go from Here?
BAVs as well as other CHDs are complex multifactorial and multigenetic diseases with variable expressivity and penetrance. Thus, not all family members with a similar gene mutation will have the same heart disease and the same mutation can cause different CHDs in different individuals. These observations are consistent with the presence of modifying factors including genetic and environmental ones that influence the phenotype [60]. Consistent with this, influences of the genetic background on the phenotype is now well documented in experimental animal models and in humans [59, 61, 62]. Thus, analysis of upstream regulators, interacting proteins as well as downstream targets of genes known to be linked to BAV will likely identify new CHD-causing or modifier genes.
Moreover, understanding the progression of the disease starting in childhood would help to slow down disease progression and improve the timing of intervention of each patient. In addition, BAV-linked genes have also been shown to be involved in other adaptive responses of the heart. For example, Notch1-deficient mice subjected to pressure overload develop increased hypertrophy, fibrosis and mortality rate is increased, suggesting that Notch1 is important for survival [63]. Similarly, Gata5 null mice subjected to pressure overload develop more cardiac conduction defects, have increased fibrosis, cardiac hypertrophy, and heart failure (unpublished data).
Such studies combined with our ability to delete genes in specific cells and at specific developmental states in animal models will undoubtedly unravel numerous candidate CHD-causing genes that can be directly tested in human genetic studies. Conversely loci identified through linkage analysis in human cohorts can be tested in animal models to confirm (or not) their causative link to disease.
Additionally, the availability of animal models of BAV and other valve disease will allow direct testing and identification of genetic modifiers as well as the molecular basis of gene-environment interaction in BAV formation.
Lastly, BAV is a risk factor for early onset of serious cardiovascular complications but the molecular basis and tools (such as biomarkers) to monitor disease progression remain largely undefined.
The existing (and future) animal models of disease will hugely facilitate such important analysis by allowing manipulations of diets, of cardiovascular parameters such as volume and pressure overload, and of other aging-related alterations. The results of such studies will undoubtedly be translated into better diagnosis followup and prevention of premature complications in individuals with family history of valve disease.
Conflict of Interests
The authors have no conflict of interests to declare.
Acknowledgments
The authors are grateful to present and past members of the lab for their contributions over the years. Special thanks to Hélène Touchette for excellent secretarial support. Work in the Nemer lab is supported by grants from the Canadian Institute of Health Research (CIHR), the Heart and Stroke Foundation of Ontario, and the McCain Foundation.
References
- 1.Abu-Issa R, Kirby ML. Heart field: from mesoderm to heart tube. Annual Review of Cell and Developmental Biology. 2007;23:45–68. doi: 10.1146/annurev.cellbio.23.090506.123331. [DOI] [PubMed] [Google Scholar]
- 2.Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. American Journal of Anatomy. 1977;148(1):85–119. doi: 10.1002/aja.1001480108. [DOI] [PubMed] [Google Scholar]
- 3.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. International Review of Cytology. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- 4.Chakraborty S, Combs MD, Yutzey KE. Transcriptional regulation of heart valve progenitor cells. Pediatric Cardiology. 2010;31(3):414–421. doi: 10.1007/s00246-009-9616-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loffredo CA. Epidemiology of cardiovascular malformations: prevalence and risk factors. American Journal of Medical Genetics. 2000;97(4):319–325. doi: 10.1002/1096-8628(200024)97:4<319::aid-ajmg1283>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 6.Braverman AC, Guven H, Beardslee MA, Makan M, Kates AM, Moon MR. The bicuspid aortic valve. Current Problems in Cardiology. 2005;30(9):470–522. doi: 10.1016/j.cpcardiol.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Friedman T, Mani A, Elefteriades JA. Bicuspid aortic valve: clinical approach and scientific review of a common clinical entity. Expert Review of Cardiovascular Therapy. 2008;6(2):235–248. doi: 10.1586/14779072.6.2.235. [DOI] [PubMed] [Google Scholar]
- 8.Sans-Coma V, Fernández B, Duran AC, et al. Fusion of valve cushions as a key factor in the formation of congenital bicuspid aortic valves in Syrian hamsters. Anatomical Record. 1996;244(4):490–498. doi: 10.1002/(SICI)1097-0185(199604)244:4<490::AID-AR7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez B, Duran AC, Fernandez-Gallego T, et al. Bicuspid aortic valves with different spatial orientations of the leaflets are distinct etiological entities. Journal of the American College of Cardiology. 2009;54(24):2312–2318. doi: 10.1016/j.jacc.2009.07.044. [DOI] [PubMed] [Google Scholar]
- 10.Fernandes SM, Khairy P, Sanders SP, Colan SD. Bicuspid aortic valve morphology and interventions in the young. Journal of the American College of Cardiology. 2007;49(22):2211–2214. doi: 10.1016/j.jacc.2007.01.090. [DOI] [PubMed] [Google Scholar]
- 11.Hope MD, Hope TA, Crook SES, et al. 4D flow CMR in assessment of valve-related ascending aortic disease. Journal of the American College of Cardiology. 2011;4(7):781–787. doi: 10.1016/j.jcmg.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Hoffman JIE, Kaplan S. The incidence of congenital heart disease. Journal of the American College of Cardiology. 2002;39(12):1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 13.De Mozzi P, Longo UG, Galanti G, Maffulli N. Bicuspid aortic valve: a literature review and its impact on sport activity. British Medical Bulletin. 2008;85(1):63–85. doi: 10.1093/bmb/ldn002. [DOI] [PubMed] [Google Scholar]
- 14.Silberbach M. Bicuspid aortic valve and thoracic aortic aneurysm: toward a unified theory. Journal of the American College of Cardiology. 2009;53(24):2296–2297. doi: 10.1016/j.jacc.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 15.Bunton TE, Jensen Biery N, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circulation Research. 2001;88(1):37–43. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- 16.Huntington K, Hunter AGW, Chan KL. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. Journal of the American College of Cardiology. 1997;30(7):1809–1812. doi: 10.1016/s0735-1097(97)00372-0. [DOI] [PubMed] [Google Scholar]
- 17.Clementi M, Notari L, Borghi A, Tenconi R. Familial congenital bicuspid aortic valve: a disorder of uncertain inheritance. American Journal of Medical Genetics. 1996;62(4):336–338. doi: 10.1002/(SICI)1096-8628(19960424)62:4<336::AID-AJMG2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 18.Glick BN, Roberts WC. Congenitally bicuspid aortic valve in multiple family members. American Journal of Cardiology. 1994;73(5):400–404. doi: 10.1016/0002-9149(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 19.Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. Journal of the American College of Cardiology. 2004;44(1):138–143. doi: 10.1016/j.jacc.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 20.Martin LJ, Ramachandran V, Cripe LH, et al. Evidence in favor of linkage to human chromosomal regions 18q, 5q and 13q for bicuspid aortic valve and associated cardiovascular malformations. Human Genetics. 2007;121(2):275–284. doi: 10.1007/s00439-006-0316-9. [DOI] [PubMed] [Google Scholar]
- 21.Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 22.Mohamed SA, Aherrahrou Z, Liptau H, et al. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochemical and Biophysical Research Communications. 2006;345(4):1460–1465. doi: 10.1016/j.bbrc.2006.05.046. [DOI] [PubMed] [Google Scholar]
- 23.McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ, Sundt TM. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. Journal of Thoracic and Cardiovascular Surgery. 2007;134(2):290–296. doi: 10.1016/j.jtcvs.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 24.Lin X, Huo Z, Liu X, et al. A novel GATA6 mutation in patients with tetralogy of Fallot or atrial septal defect. Journal of Human Genetics. 2010;55(10):662–667. doi: 10.1038/jhg.2010.84. [DOI] [PubMed] [Google Scholar]
- 25.Kodo K, Nishizawa T, Furutani M, et al. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(33):13933–13938. doi: 10.1073/pnas.0904744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maitra M, Koenig SN, Srivastava D, Garg V. Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatric Research. 2010;68(4):281–285. doi: 10.1203/PDR.0b013e3181ed17e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lepore JJ, Mericko PA, Cheng L, Lu MM, Morrisey EE, Parmacek MS. GATA-6 regulates semaphorin 3C and is required in cardiac neural crest for cardiovascular morphogenesis. Journal of Clinical Investigation. 2006;116(4):929–939. doi: 10.1172/JCI27363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 29.Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Human Molecular Genetics. 1995;4:1799–1809. doi: 10.1093/hmg/4.suppl_1.1799. [DOI] [PubMed] [Google Scholar]
- 30.Robinson PN, Booms P, Katzke S, et al. Mutations of FBN1 and genotype-phenotype correlations in Marfan syndrome and related fibrillinopathies. Human Mutation. 2002;20(3):153–161. doi: 10.1002/humu.10113. [DOI] [PubMed] [Google Scholar]
- 31.Fedak PWM, De Sa MPL, Verma S, et al. Vascular matrix remodeling in patients with bicuspid aortic valve malformations: implications for aortic dilatation. Journal of Thoracic and Cardiovascular Surgery. 2003;126(3):797–806. doi: 10.1016/s0022-5223(03)00398-2. [DOI] [PubMed] [Google Scholar]
- 32.Pereira L, Andrikopoulos K, Tian J, et al. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nature Genetics. 1997;17(2):218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- 33.Pereira L, Lee SY, Gayraud B, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(7):3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng CM, Cheng A, Myers LA, et al. TGF-β-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. Journal of Clinical Investigation. 2004;114(11):1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matyas G, Arnold E, Carrel T, et al. Identification and in silico analyses of novel TGFBR1 and TGFBR2 mutations in Marfan syndrome-related disorders. Human Mutation. 2006;27(8):760–769. doi: 10.1002/humu.20353. [DOI] [PubMed] [Google Scholar]
- 36.Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nature Genetics. 2005;37(3):275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 37.Girdauskas E, Schulz S, Borger MA, Mierzwa M, Kuntze T. Transforming growth factor-beta receptor type II mutation in a patient with bicuspid aortic valve disease and intraoperative aortic Dissection. Annals of Thoracic Surgery. 2011;91(5):e70–e71. doi: 10.1016/j.athoracsur.2010.12.060. [DOI] [PubMed] [Google Scholar]
- 38.Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nature Genetics. 2007;39(12):1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 39.Tischfield MA, Bosley TM, Salih MAM, et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nature Genetics. 2005;37(10):1035–1037. doi: 10.1038/ng1636. [DOI] [PubMed] [Google Scholar]
- 40.Makki N, Capecchi MR. Cardiovascular defects in a mouse model of Hoxa1 syndrome. Human Molecular Genetics. 2012;21(1):26–31. doi: 10.1093/hmg/ddr434. Article ID ddr434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL, Jr, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. American Journal of Human Genetics. 2002;71(3):663–668. doi: 10.1086/342360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation. 2000;101(20):2345–2348. doi: 10.1161/01.cir.101.20.2345. [DOI] [PubMed] [Google Scholar]
- 43.Groenendijk BCW, Hierck BP, Gittenberger-De Groot AC, Poelmann RE. Development-related changes in the expression of shear stress responsive genes KLF-2, ET-1, and NOS-3 in the developing cardiovascular system of chicken embryos. Developmental Dynamics. 2004;230(1):57–68. doi: 10.1002/dvdy.20029. [DOI] [PubMed] [Google Scholar]
- 44.Aicher D, Urbich C, Zeiher A, Dimmeler S, Schäfers HJ. Endothelial nitric oxide synthase in bicuspid aortic valve disease. Annals of Thoracic Surgery. 2007;83(4):1290–1294. doi: 10.1016/j.athoracsur.2006.11.086. [DOI] [PubMed] [Google Scholar]
- 45.German Z, Chambliss KL, Pace MC, Arnet UA, Lowenstein CJ, Shaul PW. Molecular basis of cell-specific endothelial nitric-oxide synthase expression in airway epithelium. Journal of Biological Chemistry. 2000;275(11):8183–8189. doi: 10.1074/jbc.275.11.8183. [DOI] [PubMed] [Google Scholar]
- 46.Nemer G, Nemer M. Cooperative interaction between GATA5 and NF-ATc regulates endothelial-endocardial differentiation of cardiogenic cells. Development. 2002;129(17):4045–4055. doi: 10.1242/dev.129.17.4045. [DOI] [PubMed] [Google Scholar]
- 47.Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. Journal of Clinical Investigation. 2011;121(7):2876–2887. doi: 10.1172/JCI44555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niessen K, Karsan A. Notch signaling in cardiac development. Circulation Research. 2008;102(10):1169–1181. doi: 10.1161/CIRCRESAHA.108.174318. [DOI] [PubMed] [Google Scholar]
- 49.Timmerman LA, Grego-Bessa J, Raya A, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes and Development. 2004;18(1):99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. Journal of Molecular and Cellular Cardiology. 2009;47(6):828–834. doi: 10.1016/j.yjmcc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Acharya A, Hans CP, Koenig SN, et al. Inhibitory role of Notch1 in calcific aortic valve disease. PLoS ONE. 2011;6(11) doi: 10.1371/journal.pone.0027743. Article ID e27743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laforest B, Nemer M. GATA5 interacts with GATA4 and GATA6 in outflow tract development. Developmental Biology. 2011;358(2):368–378. doi: 10.1016/j.ydbio.2011.07.037. [DOI] [PubMed] [Google Scholar]
- 53.Biben C, Weber R, Kesteven S, et al. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circulation Research. 2000;87(10):888–895. doi: 10.1161/01.res.87.10.888. [DOI] [PubMed] [Google Scholar]
- 54.Majumdar R, Yagubyan M, Sarkar G, Bolander ME, Sundt TM. Bicuspid aortic valve and ascending aortic aneurysm are not associated with germline or somatic homeobox NKX2-5 gene polymorphism in 19 patients. Journal of Thoracic and Cardiovascular Surgery. 2006;131(6):1301–1305. doi: 10.1016/j.jtcvs.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 55.Ponticos M, Partridge T, Black CM, Abraham DJ, Bou-Gharios G. Regulation of collagen type I in vascular smooth muscle cells by competition between Nkx2.5 and δEF1/ZEB1. Molecular and Cellular Biology. 2004;24(14):6151–6161. doi: 10.1128/MCB.24.14.6151-6161.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malfait F, Symoens S, Coucke P, Nunes L, De Almeida S, De Paepe A. Total absence of the alpha2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity to cardiac valvular problems. Journal of Medical Genetics. 2006;43(7):p. e36. doi: 10.1136/jmg.2005.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garg V, Kathiriya IS, Barnes R, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424(6947):443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 58.Hamanoue H, Rahayuningsih SE, Hirahara Y, et al. Genetic screening of 104 patients with congenitally malformed hearts revealed a fresh mutation of GATA4 in those with atrial septal defects. Cardiology in the Young. 2009;19(5):482–485. doi: 10.1017/S1047951109990813. [DOI] [PubMed] [Google Scholar]
- 59.Rajagopal SK, Ma Q, Obler D, et al. Spectrum of heart disease associated with murine and human GATA4 mutation. Journal of Molecular and Cellular Cardiology. 2007;43(6):677–685. doi: 10.1016/j.yjmcc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gill HK, Splitt M, Sharland GK, Simpson JM. Patterns of recurrence of congenital heart disease: an analysis of 6,640 consecutive pregnancies evaluated by detailed fetal echocardiography. Journal of the American College of Cardiology. 2003;42(5):923–929. doi: 10.1016/s0735-1097(03)00853-2. [DOI] [PubMed] [Google Scholar]
- 61.Bruneau BG, Nemer G, Schmitt JP, et al. A murine model of Holt-Oram syndrome defines roles of the T-Box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106(6):709–721. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 62.Winston JB, Erlich JM, Green CA, et al. Heterogeneity of genetic modifiers ensures normal cardiac development. Circulation. 2010;121(11):1313–1321. doi: 10.1161/CIRCULATIONAHA.109.887687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Croquelois A, Domenighetti AA, Nemir M, et al. Control of the adaptive response of the heart to stress via the Notchl receptor pathway. Journal of Experimental Medicine. 2008;205(13):3173–3185. doi: 10.1084/jem.20081427. [DOI] [PMC free article] [PubMed] [Google Scholar]