

Abstract
Prokaryotes are in general believed to possess small, compactly organized genomes, with repetitive sequences forming only a small part of them. Nonetheless, many prokaryotic genomes in fact contain species-specific repeats (>85 bp long genomic sequences with less than 60% identity to other species) as we have previously demonstrated. However, it is not known at present how frequent such species-specific repeats are and what their functional roles in bacterial genomes may be. Therefore, we have conducted a comprehensive survey of prokaryotic species-specific repeats and characterized them to examine as to whether there are functional classes among different repeats or not and how they are mutually related to each other. Of the 613 distinct prokaryotic species analyzed, 97% were found to contain at least one species-specific repeats. It seems interesting to note that the species-specific repeats thus identified appear to be functionally variable in different genomes: in some genomes, they are mostly associated with duplicated protein-coding genes, whereas in some other genomes with rRNA and tRNA genes. Contrary to what may be expected, only one-fourth of the species-specific repeats were found to be associated with mobile genetic elements.
Keywords: species-specific, repeat, identification, prokaryote, pathogen
1. Introduction
To date, more than 1000 prokaryotic species have been completely sequenced. These genome data provide an opportunity to conduct large-scale genome-wide studies in comparative genomics. Many analyses are carried out to explore eukaryotic genomes side by side, but not many comparative analyses are performed with prokaryotes. Even though prokaryotic organisms demonstrate recognizable, common architectural principles, they have a great variability in ecology and in metabolic and genomic complexity.1 For example, the lengths of sequenced bacterial genomes range from 180 kb (Carsonella ruddii)2 to 13 Mb (Sorangium cellulosum).3 Although a lot of this variability can be attributed to varying metabolic complexity, it also suggests that some species have more compact genomes than others. Large differences exist in the fraction of intergenic regions.1 The percentage of intergenic regions in the genomic sequence varies from ∼5% (Thermotoga neapolitana, NC_011978) to 50% (Sodalis glossinidius, NC_007712). The median value of the fraction of intergenic regions per genome size is 12%.
Though prokaryotic genomes are known to be compactly organized, there is still room for different repetitive sequences in them.4,5 There are very few comprehensive studies of repetitive sequences; most of the studies focus on specific types of repeats or on a limited number of bacterial species.4–8
The processes generating repeats include duplication, horizontal gene transfer, transposition, and replicon fusion.1,5,9–17 These processes allow bacteria to adapt to environmental changes or to evolve into pathogens (owing to the distribution of genes encoding toxins, effector proteins, cell wall modification enzymes, fitness factors, and antibiotic and heavy metal resistance determinants).10 A large fraction of known types of repetitive sequences belong to integrative and conjugative elements, also known as mobilome.18,9 The mobilome consists of bacteriophages, plasmids, and transposable elements. It mediates the movement of DNA within and between genomes. Integrative elements play a key role in the emergence of infectious diseases, antibiotic resistance and other problems.1,19–21
Thus, different mechanisms give rise to repeats and also different roles are suggested for repeats. Though many papers about completely sequenced prokaryotic genomes also try to analyze repetitive sequences, the characterization is frequently limited to some types of repeats (e.g. CRISPR, MITE, IS) or repeats as whole without classification.22,23 No exhaustive research about the characterization of prokaryotic species-specific repeats is published. However, as further shown, different types of species-specific repeats with different functions are present in prokaryotic genomes. At least some of them are the characteristic of a particular species (e.g. specific protein- biosynthesis-coding genes or specific bacterial toxins).
We have previously reported the existence of species-specific repeats in several bacterial species of medical importance and we have developed a computational method for de novo detection of such repeats previously.24 However, two questions remain unanswered: do all species have species-specific repeats and can we associate these repeats with sequences other than mobile genetic elements?
The first topic we cover in this paper is the general description of prokaryotic repeats. It is important for species characterization and identification to know what fraction of a prokaryotic genome contains repetitive DNA regions, how many of them are species-specific, and what is the common size range of the repetitive sequences. Our main focus is on species-specific repeats, with a few comparisons with universal repeats (definitions of these repeat types are given below, at the beginning of the ‘Results’ section).
Thereafter, we focus on the functional characterization of species-specific repeats. The main interest here is whether species-specific repeats are reliable for use as target sequences in identifying a species or whether they are really only selfish elements or sequences with unknown function according to the common view. For this, different functional classes of repeats are distinguished. In this paper we use the following classes to describe repeats: (I) mobile genetic elements, (II) repeats associated with RNA genes, (III) repeats associated with protein-coding genes, and (IV) non-coding repeats.
Finally, we discuss the different practical aspects of detecting prokaryotic repetitive sequences. The Supplementary material contains a list of all of the detected sequence-specific repeats.
2. Materials and methods
2.1. Genomic sequences used
To identify prokaryotic repeats, DNA sequences for all completely sequenced prokaryotic chromosomes and plasmids were retrieved from the FTP site of the NCBI RefSeq (ftp://ftp.ncbi.nih.gov/genomes/Bacteria). The human genome (version NCBI36) was downloaded from Ensembl (ftp://ensembl.org/pub/). A full list of analyzed genomes and their basic features (genome length, superkingdom, number of chromosomes, number of plasmids, etc.) is provided in the Supplementary Table S3. In total, 876 chromosomal genomes (63 archaeal and 819 eubacterial) and 613 prokaryotic species (containing 54 archaeal and 561 eubacterial genomes) were analyzed. Of the 613 species, 95 are represented with more than one strain of chromosomal genome sequence. For 232 prokaryotic species at least one plasmid genome was available, 733 prokaryotic plasmid genomes were downloaded in total. The RefSeq accession numbers of the virus genomes used in the current work are provided in the additional files (Additional File 1: Supplementary Table S3). To find strain-specific repeats online, the NCBI BLAST database for microbes was used on genomic sequences with sequencing status ‘Complete, in Progress or Assembly’.
2.2. Annotation data
To explore the positions of prokaryotic repeats in relation to genes, gene coordinates were retrieved from the FTP site of the NCBI RefSeq (ftp://ftp.ncbi.nih.gov/genomes/Bacteria) for all analyzed prokaryotic chromosomal genomes.
2.3. Method for identifying microbial repetitive sequences
The method for finding microbial repeats has been described earlier.24 The availability of complete genome sequences for the species of interest is required in order to find repetitive regions. The species of interest is called the ‘target genome’ in this article. In the context of species-specific repetitive sequences, the ‘non-target genome’ is defined as genomic DNA sequence(s) of all other organisms not in the target genome group. In the case of bacterial species-specific repeats, all sequenced chromosomal genomes of prokaryotic species and their plasmid genomes were used as non-target genomes. In this study, the human genome was always used by default as one of the non-target genomes.
To identify repeats, the genomic sequence of the target genome was segmented into consecutive windows. The length of a window was 100 bp and the length of overlap between consecutive windows was 50 bp. We also tested different length for the overlap (75 bp) to find species-specific repeats (50 randomly chosen species was used). However, no additional species-specific repeats could be found. Thus, to achieve smaller search time, we decided to use 50 bp for overlap. Matches of sequence windows inside the target genome were identified using the similarity search software BLAST. A sequence window was classified as a candidate repeat if the following two criteria were met: the length of the BLAST match in target genome was between 85 and 115 bp and the BLAST identity between the matching region and the query window was >80%. Thereafter, in the case of finding species-specific repeats, the specificity of candidate repeats was checked. A candidate repeat was defined as a non-species-specific repeat if the length of any BLAST match in any non-target genome had a length of at least 50 bp and the identity between the match of background sequence and the query window was >60%. Finally, overlapping candidate repeats were joined to form complete (species-specific) repeats. All scripts were written in the Perl programming language.
2.4. Definition of length and number of copies of the repeat
The length of different copies of the same repeat is, to some extent, variable between species or between strains of the same species. In this analysis, the median length of all copies of a given repeat is shown as the length of the repeat. To define the copy number of a given species-specific repeat, the median value of all copy numbers of that repeat over the analyzed strains of that species was used. Similarly, the median copy number of particular repeat over different species where this repeat was present was used as the copy number of a universal repeat.
2.5. Repetitive sequence functionality analysis
First, we identified prophage regions using the Phage_finder program.25 After that, IS elements and transposons were identified from the ISfinder database26 and from RefSeq annotations. Next, a BLAST search against plasmid sequences (2473 completely sequenced plasmid genomes downloaded from the NCBI FTP site ftp://ftp.ncbi.nih.gov/genomes/Plasmids) was conducted to find repeats of possible plasmidic origin. Clustered regularly interspaced short palindromic repeat (CRISPR) sequences were identified within repetitive sequences with CRISPRFinder.27 Intergenic repeat units (IRUs; ERIC/IRU sequences) were retrieved from the collection of short repeated palindromes (http://www.pasteur.fr/recherche/unites/pmtg/repet/intro.IRU.html).
We used RefSeq gene annotation data for calculating how many species-specific and universal repetitive sequences cover genes (RNA genes and protein-coding genes) in more than 50% of the repeat length. Even if only one copy of a repeat contained a gene annotation then the repeat was considered a repeat associated with a gene (we assumed annotation faults are responsible for the missing annotation of other copies). The same logic was used to identify other features of the repeats except intergenic regions. None of the copies of intergenic repeats were allowed to contain any of the aforementioned features by more than 5% of the length of the repeated region.
Finally, Pfam database was used to predict the functions of NCBI RefSeq hypothetical proteins.28
3. Results
3.1. Definition of repeats
Throughout this paper, ‘repeats’ are defined as DNA sequences at least 85 bp long that are present in at least two copies in the genome of each strain of the species. Repeat copies within the same species are required to have at least 80% identity with each other. ‘Species-specific repeat’ is defined as a repeat that is present in only one species and there are no similar sequences (more than 60% identity over 50 bp or more) in chromosomes of other fully sequenced species. If more than one strain of a species has been sequenced, the species-specific repeat must be present in all strains, with at least two copies. The minimum length and homology cutoffs for repeat definition in this paper were chosen for practical reasons—to allow the design of species-specific polymerase chain reaction (PCR) primers for each identified repeat.24 Our methodology is also able to identify ‘strain-specific repeats’, but we deliberately chose not to analyze these in order to maintain the focus of this article on species-specific repeats.
Some repeats are detected in more than one species. These can be further divided into ‘universal repeats’ and ‘intermediate repeats’. Universal repeat is a repeat that is present in at least two species (in both as a repeat) which must belong to different genera. Thus, universal repeats are repeats that are shared between some species of separate genera. For example, widely known universal repeats are ribosomal RNA genes and elongation factor Tu. All the rest of the repeats are defined as intermediate repeats. For example, intermediate repeat can be identified in one species as a repeat and can be present in one strain of another species with only one copy. Also intermediate repeats are repeats present in different species of one genus. In our analysis, we compare species-specific repeats to universal repeats; intermediate repeats are not analyzed further. Generally, intermediate repeats share characteristics of both universal and species-specific repeats.
An additional difficulty in repeat definition comes from the existence of bacterial plasmids. Typically, people search for repeats in bacterial chromosome(s), but some repeat copies can also be located in plasmids. In our data set, 232 of 613 species contained plasmids. Some of these plasmids also contained species-specific repeats. However, the analysis of species-specific plasmidic repeats is unequivocal because plasmid transfer between different bacterial species is a common phenomenon.29,30 It has been shown that at least 51% of sequenced proteobacterial plasmids are transmissible.9 Thus, the plasmid-based repeats are not reliable targets for characterization of bacterial species. For this reason, we have omitted plasmid sequences from our analysis and identified only the repeats that have multiple copies on chromosomes (no repetitive sequences were searched from species plasmid sequences).
As a first step, we analyzed the frequencies and lengths of all types of repeats in prokaryotic genomes. In the following analysis, we show the characteristic features of the species-specific repeats.
3.2. Characterization of repeats
3.2.1. Almost all prokaryotes contain repetitive sequences
In total, 613 microbial species comprising 876 different strains were analyzed. Repetitive DNA was detected in almost all species, with three exceptions: Buchnera aphidicola (six sequenced strains; NC_002528, NC_004061, NC_004545, NC_008513, NC_011833, and NC_011834), Candidatus Carsonella ruddii (NC_008512), and Candidatus Hodgkinia cicadicola (NC_012960). All these bacteria are symbionts with extremely small genomes.
The number of different repeats per genome was highly variable (Fig. 1), ranging from 1 to 690 with the median 32 repeats per species. 95% of species contained fewer than 149 repeats. The highest number of repeats was found in the genome of the myxobacterium S. cellulosum' So ce 56′, which also has the largest size (13 Mb) of all the analyzed genomes.
Figure 1.

The number of repetitive sequences per species. Most of the species contain fewer than 50 repeats.
3.2.2. The fraction of genome sequence covered by repeats
The median value of the repeat coverage was 1.8% of the bacterial genome. In only 12 bacterial species, repetitive sequences made up 7% of the total size of their genome (Fig. 2). Most of these bacteria are pathogens (insect pathogen Wolbachia sp. wRi NC_012416, plant pathogens Phytoplasma mali NC_011047 and Hamiltonella defensa NC_012751, fish pathogen Aliivibrio salmonicida NC_011312 and NC_011313, and mammalian pathogens Mycoplasma mycoides NC_005364 and NC_015431, Bartonella tribocorum NC_010161, Anaplasma phagocytophilum NC_007797, Bartonella grahamii NC_012846, and Bartonella henselae NC_005956). In general, larger genomes tend to contain larger number of repeats and also a higher fraction of repetitive sequences from the genome sequence.
Figure 2.

The repeat coverage in bacterial genomes. In 95% of the species, the fraction of all repeats from the length of the genome is lower than 6%.
The fraction of pathogens among species with elevated percentages of repetitive sequences [9 of 12 species (75%) are pathogenic] is somewhat higher than one would expect. The fraction of pathogenic species among all analyzed genomes (all that are known to be pathogenic or non-pathogenic) is 37.7%. We suggest that pathogenic bacteria have a pressure to adapt to the environment more quickly and repetitive sequences may aid this or, alternatively, be the result of this. In many pathogenic bacteria, a fraction of repetitive sequences are associated with pathogenicity genes. For example, repetitive sequences in A. phagocytophilum (∼9% of the genome comprises repeats) contain genes functionally involved with the type IV secretion system. Also, repetitive sequences in B. tribocorum and B. henselae (∼11 and ∼7% of the genomes are repeats, respectively) contain several genes associated with pathogenicity—the Trw-conjugation system, filamentous hemagglutinin, specific adhesions, and components of the type IV secretion system. As many pathogenic processes remain uncharacterized, the fraction of genes involved in pathogenesis may be larger than we could interpret. An example of a non-pathogenic genome containing a high fraction of repeats is Dehalococcoides ethenogenes. It contains large duplications that include genes involved in the ability of dechlorinate groundwater pollutants. Supplementary Table S1 gives an overview of the numbers of identified repetitive sequences from all analyzed prokaryotic species.
3.2.3. Most of the repeats are species-specific
From a total of 33 921 different repeats found in this work, 29 771 (88%) were species-specific (Fig. 3). The total number of universal repeats (those that are present in two or more different genera) found in this work was 554; approximately half of them were present in only two species. After analysis of the universal repeats, we estimated that only 0.25% (84 repeats) of all repeats were truly universal, present in more than six species. Measured in nucleotides, species-specific repeats constituted more than half of the total length of all repetitive sequences. Perhaps more informative is the number of nucleotides included in either universal or species-specific repeats. In Fig. 3 (second set of three columns), one can see that species-specific repeats constitute ca. 58% of the total nucleotides included in all repeats. In all analyzed genomes, there are three times more nucleotides in species-specific repeats than in universal repeats (from Fig. 3, 58% divided to 19%).
Figure 3.

The frequencies of repeats considered in this study. The first three columns represent the percentage of the number of particular repeats from the total number of repeats found in this work. The second three columns show the percentage of repeats in nucleotides from the total length of repeats found in this work. This figure can be viewed in colour online.
Of the analyzed species, 97% contained at least one species-specific repeat. Species-specific repetitive DNA was not detected in 20 of 613 species (listed in Supplementary Table S1). There are several possible explanations as to why we were not able to detect species-specific repeats in these 20 species. For example, B. aphidicola (six sequenced strains in our data set) is very widely defined and contains many variable genomic sequences, not sharing sufficient similarity over their repeats. Two species (Candidatus Carsonella ruddii and Candidatus Hodgkinia cicadicola) contain no repeats at all, probably because of their small genomes. In the other species (e.g. Brucella ovis, Brucella canis, and Brucella abortus), the existence of closely related fully sequenced species makes it impossible to find truly species-specific repeats.
3.2.4. The majority of species-specific repeats are shorter than 200 bp
In Fig. 4, the length distribution for species-specific repeats is shown. We found that 95% of species-specific repeats were shorter than 1000 bp. Universal repeats had a similar repeat length distribution.
Figure 4.

The length distribution of species-specific and universal repeats. Only repeats shorter than 2000 nucleotides are shown (which constitute ∼98% of all species-specific and ∼98% of all universal repeats). Light blue triangles represent species-specific repeats and dark blue squares represent universal repeats. y-axis represents the fraction of repeats of specific length to the total number of the repeats.
Although most repeats are very short, many long species-specific repeats also exist in prokaryotic genomes. The longest species-specific repeat (36 kb) belongs to Alkaliphilus metalliredigens (NC_009633), which has a genome size of 4.9 Mb. The repeat has three copies overlapping 178 gene sequences with different RefSeq gene functions, mostly annotated as hypothetical proteins. Part of this repeat (23 kb of 36 kb) contains a prophage sequence identified by the computer software Phage_finder.25 The remaining sequences of this repeat may also have a phage origin as the genes present in this region are associated with regulation and initiation of transcription, cell wall component breakdown, and regeneration.
3.2.5. Most of the species-specific repeats have two copies
As the copy number of a species-specific repeat might differ between different strains of the same species, the median value of copies of particular repeat in the analyzed strains of particular species was used as the copy number of a given repeat. Similarly, the median copy number of particular repeat in different species was used as a copy number of a universal repeat.
Approximately 77% of species-specific repeats had two copies per repeat (Fig. 5). Only a small fraction had a large number of copies per repeat. For example, only 0.4% of species-specific repeats had more than 20 copies per repeat. The maximum number of copies of a species-specific repeat was identified in the marine bacterium Hahella chejuensis KCTC 2396 (NC_007645, genome length 7.2 Mb) with 111 copies. The median length of the copies of this repeat was 141 bp. The copies are dispersed over the genome and the origin of this sequence is unclear. The fact that several copies overlap by a few nucleotides at the 5′ ends of different genes indicates that the repeat may play a role in the regulation of transcription. An overview of the copy numbers for all identified repeats is shown in Supplementary Table S2.
Figure 5.

The distribution of the number of copies per species-specific repeat. Although the number of copies per species-specific repeat in different species varies, the median value (rounded down to the nearest integer) over the number of copies per particular repeat is considered. The number above the column shows the percentage of prevalence of particular number of copies per repeat from the total number of repeats. Only the values over 1% are shown. y-axis is on a logarithmic scale.
In general, shorter repeats tend to have more copies (Fig. 6). However, Fig. 6 also shows somewhat higher copy numbers for species-specific repeats with lengths between 1000 and 2000 bp. This is due to species-specific transposons that contribute to the higher copy numbers in the length range between 900 and 1900 nucleotides.
Figure 6.
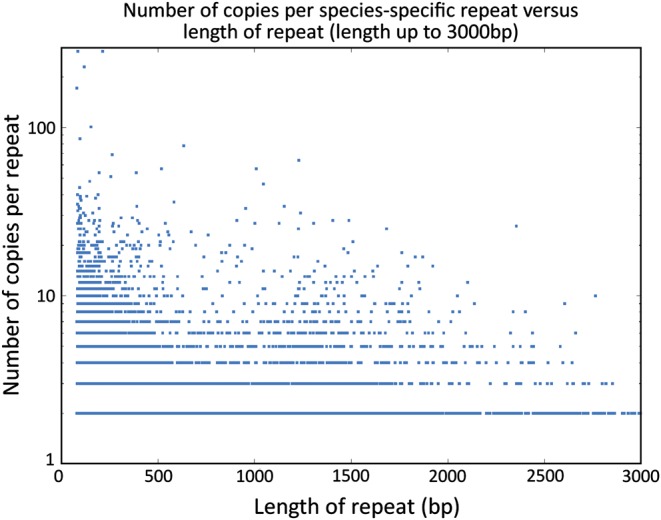
Correlation between the length of species-specific repeat and the number of copies per species-specific repeat. Only repeats up to 3000 nucleotides are shown. y-axis is on a logarithmic scale.
From the repeats analyzed in this study, ∼11% of species-specific repeats were located in the genome in tandem. Interestingly, species-specific repeats were much more frequently located in tandem than universal repeats, ∼2% of which were located in tandem.
3.3. Functional analysis of species-specific repeats
3.3.1. A large fraction of the species-specific repeats are associated with protein-coding genes
We analyzed 29 771 species-specific repeats in order to understand the possible functions of repetitive sequences. We were interested in the general functional categories encoded within species-specific and universal repeats.
Here, we have used the following classification of bacterial repetitive elements: (I) mobile genetic elements such as insertion sequences (ISs), transposons, phage sequences, and plasmid sequences; (II) RNA genes; (III) protein-coding genes; (IV) non-coding short interspersed repeats (ERIC/IRU, CRISPR, intergenic regions). All these repeat classes were represented among the species-specific repeats (Fig. 7). The protein-coding genes (class III) were the most abundant class (64% of all repeats). Mobile genetic elements (class I) and non-coding short interspersed repeats (class IV) both constituted 13–14% of all species-specific repeats. The smallest class (class II) of repeats included tRNA and rRNA genes. Approximately 6% of species-specific repeats could not be classified with our methodology.
Figure 7.

Different classes of species-specific repeats. (A) Repeat frequencies of species-specific repeats. (B) Repeat coverage of species-specific repeats. As ERIC/IRU sequences constitute under 0.03% of all species-specific repeats, they are not shown.
For comparison, the universal repeats (Supplementary Fig. S1) contained mainly mobilome elements (class I), some long regions of universally repeated rRNA genes (class II), and ∼40% were protein-coding genes (class III).
We identified that 19% of species-specific repeats belong to the class of non-coding short intergenic repeats and repeats with unknown origin. A fraction of these repeats may comprise a certain conserved regulatory elements. For example, IRUs and specific intergenic repeat motifs (CIR motifs, >100 bp) are supposedly associated with gene conversion or with regulatory or structural requirements of the bacterial genomes.31–33 In our data set, we found that more than half of the species (365 species) contain at least one species-specific repeat which is located prior to, following to, or between RNA gene sequences (e.g. between 23S rRNA and 5S rRNA gene sequences, between different tRNA sequences, etc). The overlap between the gene sequence and the repeat constitutes a small extent from repeat/gene sequence. Thus, in many cases, sequences between RNA genes may characterize a species uniquely. Still, many intergenic repeats have no associated function and are considered to be predominantly genomic parasites.34,35
An overview of the most frequent functions of species-specific repeats associated with genes (class III) is shown in Table 1. A notable group of species-specific repeated genes contains membrane proteins, transport proteins, and repeat-containing proteins. High sequence variation of these genes between species may be related to the need to adapt to specific environments or the need of pathogenic microbes to vary their immunogenic properties. The largest group, 31% of all repeat-associated genes, is annotated as ‘hypothetical protein’ by NCBI RefSeq. We analyzed the regions of hypothetical proteins containing repeats against Pfam database and found Pfam entries for 28% (2372 repeats) of them. Over the half of the repetitive sequences searched for are found from automatically generated Pfam-B families (866 families) or from the curated Pfam-A collection (471 families) predicted as proteins of unknown function. The rest of the matches represent variable functions of proteins. For example, the highest number of matches (for 48 repeats) was gained from the tetratricopeptide repeat superfamily, which are found from numerous and diverse proteins involved in such functions as cell cycle regulation, transcriptional control, mitochondrial and peroxisomal protein transport, neurogenesis, and protein folding.
Table 1.
Classification of genes containing species-specific repeats
| Gene class | Number of repeats contained in the gene | Gene class | Number of repeats contained in the gene |
|---|---|---|---|
| Hypothetical protein | 8264 | Kinase | 497 |
| Transport protein | 1711 | ATP-dependent protein | 453 |
| Pseudogene | 1212 | Transcriptional regulator | 431 |
| Membrane protein | 656 | Synthetase | 337 |
| Hydrogenase | 603 | Bacterial chemotaxis | 272 |
| Repeat containing protein | 548 | Hydrolase | 254 |
| Synthase | 504 |
The number shows how many repeats are overlapping the gene by at least 50% of the length of a given repeat. The total number of repeats overlapping gene sequences is 18 961, of which 15 742 repeats are shown in this table.
3.3.2. The function of repetitive full-length species-specific genes remains unknown
We analyzed repeats that contain full-length protein-coding gene sequences that are represented in at least two copies of a particular repeat. We found that 1034 repetitive sequences contain full-length species-specific protein-coding genes in at least two copies of that repeat, resulting in 4001 different genes in total. Most of these genes (56.7 versus the 30.6% expected on the basis of RefSeq annotations of prokaryotic protein-coding genes) are hypothetical proteins (statistically over-represented P < 0.0001).
However, several interesting details emerged. For example, we found that pseudogenes are statistically significantly over-represented (P < 0.0001). The RefSeq annotations of prokaryotic protein-coding genes imply that on average ∼1.68% of randomly chosen protein-coding genes are pseudogenes. In contrast, we found that 6.0% of the analyzed genes are pseudogenes. We suggest that this can be explained by gene duplication. It is known that gene duplication is a significant contributor to the evolution of genomes. In some cases, one duplicated copy may become non-functionalized by randomly accumulating degenerative mutations in the absence of selective advantage.36 Another notable group of genes, encoding transport proteins, is under-represented (P < 0.0001) among the analyzed genes (12.6 and 5.8% are the percentages of expected and observed transport proteins, respectively). It is shown that at least some transport proteins are conserved among a wide range of species.37,38 This is also supported by our analysis of universal repeats where transport proteins are over-represented.
3.3.3. Example of species-specific repeats of H. defensa
Hamiltonella defensa (NC_012751) is an endosymbiont found in sap-feeding insects, including aphids, psyllids, and whiteflies. Hamiltonella defensa is an example of species containing relatively various repeat classes, whereas many species contain only one or two different classes of repeats.
The complete list of species-specific repeats in H. defensa contains 95 repeats (plus three universal repeats that contain rRNA and/or tRNA sequences and four repeats that were present in other species but not as repeats). In total, 55 species-specific repetitive sequences are classified as mobile genetic elements; 4 are classified as ISs; 34 are classified as phage-related sequences; and 17 are classified as plasmidic repeats.
The remaining 40 species-specific repeats are associated with protein-coding genes. Sixteen of them are related to proteins of the RTX family, which contains putative virulence factors, RTX toxins (repeats in toxins), which are exported proteins.39 This could explain why there are 10 species-specific repeats associated with different inner and outer membrane proteins, some of which are suggested to be auto-transporters. Also, one repeat contains genes of which the functions involve different type IV pilus biosynthesis proteins (type IV pilus proteins are also essential for virulence).
Furthermore, only two repeats contain hypothetical protein-coding genes and only four species-specific repeats contain pseudogenes.
The final six repetitive sequences are associated with genes with various functions (methionine sulfoxide reductase, phosphomannomutase, heat shock protein, amidase and lipoprotein, serine endoprotease, transcriptional regulator, and GTP cyclohydrolase). Some of these genes could also be involved with pathogenicity but no direct reference can be indicated.
3.4. Firmness of species-specific repeats
One of the major questions that arose during this work was related to the definition of species and the genetic variation within species. Does a species-specific repeat exist as a repeat in every newly sequenced strain? Or, if a new, closely related species is sequenced, does the repeat remain species-specific? In reality, of all the prokaryotic genomes existing on the Earth, only a small part of each has been sequenced; this part may represent distantly related genomes. Because of these questions, we have validated species-specific repeats against a prokaryotic database that consists of ∼1.42 times more species than the database used for finding those repeats (data not shown). Approximately 88% of detected species-specific repeats remained species-specific in this larger data set. Interestingly, closest species where the species-specific repeats could still be found are 99% identical in terms of 16S rRNA (e.g. Shewanella pealeana NC_009901 and Shewanella halifaxensis NC_010334). Nevertheless, some repeats turned out to be non-species-specific in the larger data set, because they existed in another newly sequenced species or the repeat was not present in the genome of a newly sequenced strain of the same species. This raises the question of whether the species-specific repeats we have identified are real species-specific repeats or just data analysis artifacts, dependent on the number of available genomes and the choice of species for analysis.
The possibility of finding, or not finding, a species-specific repeat is partly determined by the controversial definition of bacterial species. Currently, the definition comprises 70% DNA–DNA re-association as a standard.40 As this definition is somewhat arbitrary, the definition of species-specific repeats is also arbitrary. However, the problem of the species concept is not the topic of this article. We use the definition of species as it comes from the NCBI genome database, together with the genomic DNA sequence. In most cases, the repeats we define as species-specific repeats will remain specific to a group of phylogenetically related strains, even if the concept of the species will be changed in future. Yet, we have identified strain-specific repetitive sequences (data not shown) in 50 of 91 species for which 2–26 completely sequenced strains per species were available. Strain-specific repetitive sequences add some confidence to the existence of repeats specific to a small phylogenetically related group.
4. Discussion
Although repetitive sequences are less common in prokaryotes than in eukaryotes, almost all prokaryotic species still have some identifiable repeats. In this work, we have characterized mainly the species-specific repetitive sequences—repeats common to all strains of a species, but not to other species.
In accordance with widely accepted understanding that prokaryotic genomes are compactly organized, we have demonstrated that the median value of the repeat coverage of bacterial genome is 1.8% (species-specific repeats constitute ∼1% from this). Despite of a small fraction of repetitive sequences in prokaryotic genomes, many novel findings have arisen. We have found that not all repetitive sequences in prokaryotic genomes belong to mobile genetic elements. Our results indicate that only ∼14% of the species-specific repetitive sequences and ∼53% of universal repeats are associated with mobile genetic elements. Further, the functions of many intergenic repeats and repeats from the class of unknown origin (in total ∼17% from the species-specific repeats) are not understood. However, a fraction of intergenic repeats may comprise a certain conserved regulatory elements. For example, specific intergenic repeats (CIR motifs, >100 bp) in Caulobacter crescentus may be associated with gene conversion and with gene regulation.33 A large number of various functions of protein-coding genes, reflecting the diversity of sequenced prokaryotic genomes, have emerged from the functional analysis. In different species, genes associated with repeats have variable roles and no common phenomenon can be identified for all species or for groups of particular species. As shown, the function of many protein-coding genes associated with species-specific repeats remains unknown. We rather think that this interesting question cannot be answered by automatic analysis but must be left to biologists who investigate a particular genome in more detail. The functional analysis also revealed that different repeat classes appear in different species. In some species, phage-related sequences are in prevalence; in the other, only rDNA-related repeats appear; or in the third, one large duplication containing different protein-coding genes is found. Every species seems to be somewhat unique regarding to its repeats.
One important application of species-specific repeats is their usage in the identification of prokaryotic species. The repetitive sequences increase the sensitivity of the PCR test simply because of the higher concentration (double or more) of the initial sequence.24 In this work, we have shown that many of the species-specific repetitive sequences are associated with protein-coding genes, so at least these repeats can be reliably used as target sequences in PCR tests. Species-specific or a group of phylogenetically related strains-specific repeats may sometimes not be the preferred target in medical diagnostics. This is in cases where pathogenic genes are known and the objective of the diagnostic test is to detect such a gene rather than to detect the species.41 However, we believe that this does not diminish the value of species-specific repeats as a PCR target region for the detection of species. Pathogenic gene-specific PCR primers can be designed with similar methodology and used in addition to species-specific PCR primers in medical tests.
In conclusion, the current work enhances knowledge about prokaryotic species-specific repeats in many novel aspects. Most prokaryotic species contain several short species-specific repeats, which are represented in the genome with a low copy number. In a typical prokaryotic species, the species-specific repeats cover ∼1% of the genome. Over half of these repeats contain protein-coding genes, approximately one-eighth are associated with mobile genetic elements and one-eighth are associated with short non-coding interspersed elements. Thus, most of the species-specific repeats are associated with protein-coding genes and probably related to the biochemical processes required for adaptation and survival of any given prokaryote. Unfortunately, the functions of many species-specific repeats related to protein-coding genes currently remain unexplained.
Supplementary Data
Supplementary data are available online at www.dnaresearch.oxfordjournal.org.
Funding
This work was funded by the EU European Regional Development Fund, through the Estonian Centre of Excellence in Genomics and grant SF0180026s09 from the Estonian Ministry of Education and Research.
Supplementary Material
Footnotes
Edited by Katsumi Isono
References
- 1.Koonin E. V., Wolf Y. I. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008;36:6688–719. doi: 10.1093/nar/gkn668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakabachi A., Yamashita A., Toh H., et al. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science. 2006;314:267. doi: 10.1126/science.1134196. [DOI] [PubMed] [Google Scholar]
- 3.Schneiker S., Perlova O., Kaiser O., et al. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat. Biotechnol. 2007;25:1281–9. doi: 10.1038/nbt1354. [DOI] [PubMed] [Google Scholar]
- 4.Delihas N. Small mobile sequences in bacteria display diverse structure/function motifs. Mol. Microbiol. 2008;67:475–81. doi: 10.1111/j.1365-2958.2007.06068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Achaz G., Rocha E. P., Netter P., Coissac E. Origin and fate of repeats in bacteria. Nucleic Acids Res. 2002;30:2987–94. doi: 10.1093/nar/gkf391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu M., Sun L. V., Vamathevan J., et al. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004;2:E69. doi: 10.1371/journal.pbio.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rocha E. P. C., Danchin A., Viari A. Functional and evolutionary roles of long repeats in prokaryotes. Res. Microbiol. 1999;150:725–33. doi: 10.1016/s0923-2508(99)00120-5. [DOI] [PubMed] [Google Scholar]
- 8.Rocha E. P., Danchin A., Viari A. Analysis of long repeats in bacterial genomes reveals alternative evolutionary mechanisms in Bacillus subtilis and other competent prokaryotes. Mol. Biol. Evol. 1999;16:1219–30. doi: 10.1093/oxfordjournals.molbev.a026212. [DOI] [PubMed] [Google Scholar]
- 9.Smillie C., Garcillan-Barcia M. P., Francia M. V., Rocha E. P., de la Cruz F. Mobility of plasmids. Microbiol. Mol. Biol. Rev. 2010;74:434–52. doi: 10.1128/MMBR.00020-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mavrodi D. V., Loper J. E., Paulsen I. T., Thomashow L. S. Mobile genetic elements in the genome of the beneficial rhizobacterium Pseudomonas fluorescens Pf-5. BMC Microbiol. 2009;9:8. doi: 10.1186/1471-2180-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toleman M. A., Bennett P. M., Walsh T. R. ISCR elements: novel gene-capturing systems of the 21st century? Microbiol. Mol. Biol. Rev. 2006;70:296–316. doi: 10.1128/MMBR.00048-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nesmelova I. V., Hackett P. B. DDE transposases: Structural similarity and diversity. Adv. Drug Deliv. Rev. 2010;62:1187–1195. doi: 10.1016/j.addr.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brochet M., Da Cunha V., Couve E., Rusniok C., Trieu-Cuot P., Glaser P. Atypical association of DDE transposition with conjugation specifies a new family of mobile elements. Mol. Microbiol. 2009;71:948–59. doi: 10.1111/j.1365-2958.2008.06579.x. [DOI] [PubMed] [Google Scholar]
- 14.Wozniak R. A., Waldor M. K. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 2010;8:552–63. doi: 10.1038/nrmicro2382. [DOI] [PubMed] [Google Scholar]
- 15.Tobes R., Pareja E. Bacterial repetitive extragenic palindromic sequences are DNA targets for insertion sequence elements. BMC Genomics. 2006;7:62. doi: 10.1186/1471-2164-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mes T. H., Doeleman M. Positive selection on transposase genes of insertion sequences in the Crocosphaera watsonii genome. J. Bacteriol. 2006;188:7176–85. doi: 10.1128/JB.01021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nunvar J., Huckova T., Licha I. Identification and characterization of repetitive extragenic palindromes (REP)-associated tyrosine transposases: implications for REP evolution and dynamics in bacterial genomes. BMC Genomics. 2010;11:44. doi: 10.1186/1471-2164-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cortez D., Forterre P., Gribaldo S. A hidden reservoir of integrative elements is the major source of recently acquired foreign genes and ORFans in archaeal and bacterial genomes. Genome Biol. 2009;10:R65. doi: 10.1186/gb-2009-10-6-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gogarten J. P., Townsend J. P. Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 2005;3:679–87. doi: 10.1038/nrmicro1204. [DOI] [PubMed] [Google Scholar]
- 20.Canchaya C., Fournous G., Chibani-Chennoufi S., Dillmann M. L., Brussow H. Phage as agents of lateral gene transfer. Curr. Opin. Microbiol. 2003;6:417–24. doi: 10.1016/s1369-5274(03)00086-9. [DOI] [PubMed] [Google Scholar]
- 21.Frost L. S., Leplae R., Summers A. O., Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 2005;3:722–32. doi: 10.1038/nrmicro1235. [DOI] [PubMed] [Google Scholar]
- 22.Ravin N. V., Mardanov A. V., Beletsky A. V., et al. Complete genome sequence of the anaerobic, protein-degrading hyperthermophilic crenarchaeon Desulfurococcus kamchatkensis. J. Bacteriol. 2009;191:2371–9. doi: 10.1128/JB.01525-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salzberg S. L., Sommer D. D., Schatz M. C., et al. Genome sequence and rapid evolution of the rice pathogen Xanthomonas oryzae pv. oryzae PXO99A. BMC Genomics. 2008;9:204. doi: 10.1186/1471-2164-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koressaar T., Joers K., Remm M. Automatic identification of species-specific repetitive DNA sequences and their utilization for detecting microbial organisms. Bioinformatics. 2009;25:1349–55. doi: 10.1093/bioinformatics/btp241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fouts D. E. Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006;34:5839–51. doi: 10.1093/nar/gkl732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varani A. M., Siguier P., Gourbeyre E., Charneau V., Chandler M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 2011;12:R30. doi: 10.1186/gb-2011-12-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grissa I., Vergnaud G., Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–7. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finn R. D., Mistry J., Tate J., et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–22. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soda S., Otsuki H., Inoue D., Tsutsui H., Sei K., Ike M. Transfer of antibiotic multiresistant plasmid RP4 from Escherichia coli to activated sludge bacteria. J. Biosci. Bioeng. 2008;106:292–6. doi: 10.1263/jbb.106.292. [DOI] [PubMed] [Google Scholar]
- 30.Zhao F., Bai J., Wu J., et al. Sequencing and genetic variation of multidrug resistance plasmids in Klebsiella pneumoniae. PLoS One. 2010;5:e10141. doi: 10.1371/journal.pone.0010141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bachellier S., Clement J. M., Hofnung M., Gilson E. Bacterial interspersed mosaic elements (BIMEs) are a major source of sequence polymorphism in Escherichia coli intergenic regions including specific associations with a new insertion sequence. Genetics. 1997;145:551–62. doi: 10.1093/genetics/145.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharples G. J., Lloyd R. G. A novel repeated DNA sequence located in the intergenic regions of bacterial chromosomes. Nucleic Acids Res. 1990;18:6503–8. doi: 10.1093/nar/18.22.6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S. L., Shapiro L. Identification of long intergenic repeat sequences associated with DNA methylation sites in Caulobacter crescentus and other alpha-proteobacteria. J. Bacteriol. 2003;185:4997–5002. doi: 10.1128/JB.185.16.4997-5002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doolittle W. F., Sapienza C. Selfish genes, the phenotype paradigm and genome evolution. Nature. 1980;284:601–3. doi: 10.1038/284601a0. [DOI] [PubMed] [Google Scholar]
- 35.Orgel L. E., Crick F. H. Selfish DNA: the ultimate parasite. Nature. 1980;284:604–7. doi: 10.1038/284604a0. [DOI] [PubMed] [Google Scholar]
- 36.Lynch M., O'Hely M., Walsh B., Force A. The probability of preservation of a newly arisen gene duplicate. Genetics. 2001;159:1789–804. doi: 10.1093/genetics/159.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolhuis A. The archaeal Sec-dependent protein translocation pathway. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004;359:919–27. doi: 10.1098/rstb.2003.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holland K. A., Holland I. B. Adventures with ABC-proteins: highly conserved ATP-dependent transporters. Acta Microbiol. Immunol. Hung. 2005;52:309–22. doi: 10.1556/AMicr.52.2005.3-4.4. [DOI] [PubMed] [Google Scholar]
- 39.Lally E. T., Hill R. B., Kieba I. R., Korostoff J. The interaction between RTX toxins and target cells. Trends Microbiol. 1999;7:356–61. doi: 10.1016/s0966-842x(99)01530-9. [DOI] [PubMed] [Google Scholar]
- 40.Achtman M., Wagner M. Microbial diversity and the genetic nature of microbial species. Nat. Rev. Microbiol. 2008;6:431–40. doi: 10.1038/nrmicro1872. [DOI] [PubMed] [Google Scholar]
- 41.Belanger S. D., Boissinot M., Menard C., Picard F. J., Bergeron M. G. Rapid detection of Shiga toxin-producing bacteria in feces by multiplex PCR with molecular beacons on the smart cycler. J. Clin. Microbiol. 2002;40:1436–40. doi: 10.1128/JCM.40.4.1436-1440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.