

Abstract
The recently published discrete mathematical method, extended consensus partition (ECP), identifies nucleotide types at each position that are strictly absent from a given sequence set, while occur in other sets. These are defined as discriminating elements (DEs). In this study using the ECP approach, we mapped potential hidden identity elements that discriminate the 20 different tRNA identities. We filtered the tDNA data set for the obligatory presence of well-established tRNA features, and then separately for each identity set, the presence of already experimentally identified strictly present identity elements. The analysis was performed on the three kingdoms of life. We determined the number of DE, e.g. the number of sets discriminated by the given position, for each tRNA position of each tRNA identity set. Then, from the positional DE numbers obtained from the 380 pairwise comparisons of the 20 identity sets, we calculated the average excluding value (AEV) for each tRNA position. The AEV provides a measure on the overall discriminating power of each position. Using a statistical analysis, we show that positional AEVs correlate with the number of already identified identity elements. Positions having high AEV but lacking published identity elements predict hitherto undiscovered tRNA identity elements.
Keywords: tRNA, identity element prediction, extended consensus partition
1. Introduction
In all organisms, the 20 aminoacyl-tRNA synthetase (AARS) enzymes have to recognize their amino acid substrates and the corresponding tRNA molecules with high precision to produce only legitimate aminoacyl-tRNA products. This exquisite specificity is of central importance as this enables the genetic information to be faithfully translated into protein sequences by following the rules defined in the genetic code. Although principles and many fine details of this selective recognition event have already been discovered,1–4 several questions remained still unanswered.5 tRNA positions that have utmost roles in the selective interaction with the cognate AARS and thus define the identity of the tRNA are denoted as identity elements. While only laboratory experiments can decisively define the identity elements, the large number of potential positions and the laborious nature of the experiments prompted a great variety of bioinformatics studies to predict such elements. These studies require large numbers of individual input tRNA sequences to locate statistically significant identity-related sequence properties. This magnitude of input data became available in the form of genomic DNA sequences, from which tRNA-detecting algorithms6,7 can identify functionally relevant tDNA sequences. Such analyses yielded numerous different tDNA databases.8–10 Several computational studies reported successful functional annotation and in silico identity element determination.11,12 Improved secondary structure-predicting algorithm-driven tRNA alignments13 yielded high-quality input data sets. These high-quality sets allowed for innovative sequence logo and inverse sequence logo-based analyses of tRNA features and identity element predictions.14 An information theory-based approach opened new frontiers in visualizing tRNA sequence features and predicting determinants and anti-determinants.15,16 Computational tRNA identity analysing methods were compared in a recent review of Ardell.17
In this paper, we introduce a new approach based on the recently published ‘extended consensus partition’ (ECP) algorithm.18 The ECP algorithm provides a discrete mathematical measure of pairwise distances of functionally related aligned sequence sets. It was first introduced to reveal characteristic sequence features that discriminate the two tRNA sets corresponding to Class I and Class II AARS enzymes.
In this study, we applied the ECP algorithm to assess the potential of each tRNA position to discriminate the 20 different tRNA identity sets from each other. The ECP method heavily relies on characteristic positional absence of nucleotide base types. Because of that, the method is sensitive even to the rare occurrence of atypical sequences. For removing such sequences, we filtered the tDNA data sets for the obligatory presence of well-established tRNA features. Moreover, as all bioinformatic studies, the ECP analysis also requires a large number of input sequences. In this case, it is needed to reliably identify nucleotide types that are strictly absent from a given position of the aligned sequence set, i.e. their absence is not due to stochastic sampling error.
In order to provide the necessary large input sets, we performed the ECP analyses on the three kingdoms of life instead of individual species. Nevertheless, we aimed to compare tRNA identity sets that contain isofunctional sequences in spite of being originated from different species. Therefore, separately for each identity set, we further filtered the sets for the presence of experimentally verified strictly present identity elements. As a control experiment, we also performed the analysis by omitting this second filtration step.
We argued that tRNA molecules sharing a large set of experimentally verified identity elements should interact with their corresponding AARS enzyme similarly and therefore should also share yet unidentified common identity elements.
By combining the ECP method with simple statistics, we generated average excluding values (AEVs) providing a measure on the overall discriminating power of each tRNA position. We show that both with and without the second filtering step, positional AEVs correlate with the number of already identified identity elements. We argue that positions having high AEV but lacking already published identity elements predict hitherto undiscovered tRNA identity elements.
The analysis located such potential identity elements on the anticodon arm (30:40 and 31:39) and suggests that the core region also contributes to defining tRNA identity.
2. Materials and methods
2.1. Data set building
The tDNA sequences of Bacteria and Eukaryotes were downloaded from the tRNAdb database.9 The Archaea set of this database has not yet contained the recently discovered and characterized19–22 split tRNA, which have been organized in the SPLITSdb database.20 Split tRNA data are already included in the tRNADB-CE database,10 which however (unlike Sprinzl and tRNAdb) does not contain aligned sequences. Therefore, we downloaded both normal and split Archaea tDNA from the tRNADB-CE database and aligned the sequences by the ClustalW software and manually as described by Fujishima et al.23,24 In the case of Archaea data set, we omitted the variable loop sequences from the analysis because of alignment complications.
The downloaded set was filtered for sequences that fulfil several criteria.
2.2. First filtering step for all data sets
In the first ECP-based study,18 we used the database from the tRNomics study of Marck and Grosjean.25 Although for the present study, we used a larger, updated database, in the case of Bacteria and Eukarya, we could still use the well-established kingdom-specific strictly present elements—as filtering rules—defined by that tRNomics study. For Archaea, we used the filtering rules of Fujishima et al.23 The obligatory presence of these elements established our first filtering criteria. The element sets for the three kingdoms were as follows. Bacteria: H14, G18, R19, Y33, G53:C61, T54, T55, Y56, D57, A58. Archaea: Y8, A14, G15, G18, G19, R21, T33, Y48, G53, T54, T55, C56, R57, A58. Eukarya: Y8, Y11, A14, -17a, G18, G19, R21, R24, H32, Y33, R37, H38, G53, H54, T55, C56, R57, A58, C61. (Note that nucleotides and their sets are denoted by IUPAC nucleotide codes.) Discarding sequences that lacked any of the strictly present kingdom-specific elements removed incorrectly sequenced or most likely non-functional tDNA data.
2.3. Second filtering step for the bacterial and eukaryotic data sets
We grouped each sequence based on identity and filtered for the presence of already identified and published major, strictly present identity elements characteristic to the given amino acid identity set.3 These sets for Escherichia coli and Saccharomyces cerevisiae are listed in Supplementary Table S1. By excluding sequences that lacked these elements, we aimed to generate tRNA identity sets that contain isofunctional sequences expected to function in E. coli or yeast, respectively. We argued that if hidden identity elements still exist, those could also be shared by these filtered sequences.
Out of the published identity elements, only the determinants were included, while the anti-determinants were not considered. One determinant, the G15:G48 Levitt pair of tRNACys, was omitted from the filtering as this is idiosyncratic to E. coli (more accurately, it could have emerged in the common ancestor of E. coli and Haemophilus influenzae).26
2.4. Third filtering step for all data sets
Finally, in order not to bias the statistical analysis, we removed any redundancy from the data set by keeping only unique sequences.
Supplementary Table S2 organized by the three kingdoms contains the species list corresponding to our raw data set and indicates the number of sequences contributed by each species in the raw set and after each filtering step.
Supplementary Data S1 shows the resulted sets after the final filtering step. It contains six databases in a multi-fasta format, two for each kingdom. For each kingdom, one database contains a set of non-redundant sequences, while the other set contains all sequences minus the non-redundant set, thus it contains all ‘siblings’.
2.5. Determination of discriminating elements by the extended consensus partition algorithm
Filtered data sets for each kingdom were analysed by the already published extended consensus partition (ECP) algorithm.18 Principles of the analysis and the algorithm remained the same. However, in this case, not the two AARS-based tRNA classes were compared, but all pairs of the 20 tRNA identity sets. The logic of the algorithm is illustrated in Fig. 1. Because the pairwise ECP analysis is non-symmetrical, from the filtered data set and separately for each kingdom, we produced all 380 (20 × 19) identity pairs. For each pair, we identified the discriminating elements (DEs) through the ECP algorithm as follows.
Figure 1.

Illustration of calculating the AEV using short artificial sequences. (A) Identifying DEs using an artificial set of short sequences belonging to three amino acid identity sets. Identities are labelled as 1aa, 2aa and 3aa and are highlighted as cyan, yellow and magenta, respectively. Each identity set contains four short, tetramer sequences. The calculated DE is either one base or a combination of bases and it is labelled by IUPAC codes of bases or base sets. Each step of the DE-generating algorithm is explained in Section 2. (B) The calculated DE for each identity pairs. Note that the DE-generating relationship is non-symmetrical, i.e. the 1aa vs. 2aa pair has a DE different from that of the 2aa vs. 1aa pair. (C) Positional summation of DE. Positional sum of the DE values (shown in the lowest row) provides the number of pairwise discriminations provided by the given position. The sums of the DE values are input data for calculating the AEVs). AEV is generated by dividing the positional sum of DE with the number of the identities (which in a real case is 20 while in this didactic case 3). Formalism and more detailed description are provided in Section 2. This figure can be viewed in colour online.
In each identity set and for each kingdom, we scanned the positions of the filtered and aligned9 data set. At each position, we documented the strictly absent elements, i.e. bases that at the given position are missing from each sequence of the given identity set. For each detected strictly absent element, we checked each other identity set in a pairwise manner whether any of the sequences of the other identity set contains that element. If yes, the detected strictly absent element is a DE that discriminates the two identity sets. For each kingdom and each position, these pairwise-interpreted DE elements were documented.
Mathematical description of the above procedure is described below.
We introduce the variable, Y. Elements of Y are nucleotide bases; therefore, elements are Y∈χ, where χ = {A,C,G,T}. The value of 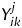 is the nucleotide base corresponding to position j (j = 1, …, L, L = 96; from normal position 0 to position 73, including the extra positions from e1 to e22) of the sequence k (k= 1, …, Mi, where the value of Mi varies for individual species) of amino acid identity i (i= 1, …, N, N = 20).
is the nucleotide base corresponding to position j (j = 1, …, L, L = 96; from normal position 0 to position 73, including the extra positions from e1 to e22) of the sequence k (k= 1, …, Mi, where the value of Mi varies for individual species) of amino acid identity i (i= 1, …, N, N = 20).
Then, we introduce the set of bases existing at the position j of identity i:
DE of identity set i against identity set l (where l= 1, …, N, N = 20) at position j is defined as follows:
2.6. The AEV
We introduced the AEV to determine the weighted average frequency of DE at each position as follows. At each identity set and at each position, we determined how many identity set pairs are discriminated by the given position. These numbers from each set were summed up and were divided by 20 (the number of identities), resulting in the AEVs that demonstrate the discriminating potential of each tRNA position.
In mathematical terms, the AEV is defined by the following functions:
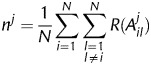 |
The n j value is denoted as the AEV.
2.7. Statistical analysis
We performed a simple statistical analysis to assess how the positional AEVs relate to the number of hitherto identified identity determinants. We assume that unidentified determinants still exist; therefore, the determinant set is only a subset of all existing elements. The test was done only on the bacterial set because only that sequence set contained enough input sequences. We correlated the AEVs with the number of published determinants (NPDs) with both the Pearson and Spearman analyses.
As both types of correlation were relatively weak, we also performed a bootstrap analysis as explained in Supplementary Data S2. In addition, the positional NPD and AEVs were compared with their respective weighted average values and ranked from very low to very high values based on standard deviation.
3. Results and discussion
The ECP analysis was performed both on the final filtered data sets and omitting the second filtration step (see later). At each position, the AEV was calculated and compared with the positional NPD value. Positional AEV/NPD values for bacterial and eukaryotic data and AEVs for Archaea are listed in Supplementary Table S3. In Fig. 2 at each position, AEV and NPD values were plotted and colour coded in the same diagram as explained in the figure legend.
Figure 2.

Results of the Bacterial (A and B); Eukaryote (C and D) and Archaea (E and F) data set. ECP-diagrams (A, C, E). In each ECP diagram, each column belongs to a position. The upper column set refers to positional AEVs. The colour codes for statistical analysis. For each kingdom, the weighted average of AEV calculated for all positions from 0 to 73 as a single set and the corresponding standard deviation is as follows: Bacteria 6.45 ± 3.54; Eukaryote 6.31 ± 3.49 and Archaea 6.72 ± 3.69. Segments of columns are colour coded based on their deviation from the weighted average as follows: below −1.5 SD blue; between −1.5 and −0.5 SD cyan; between −0.5 SD and +0.5 SD green; between +0.5 and +1.5 yellow; above +1.5 SD red. White illustrates positions where the analysis is not applicable. These are the 3′ CCA end for each kingdom, and the unpopulated e1 position in the Achaea set. The lower column set illustrates the positional NPDs. The logic of the colour-coding scheme is the same as for the AEVs. For each kingdom, the weighted average of NPD calculated for all positions from 0 to 73 as a single set and the corresponding standard deviation is as follows: Bacteria 1.63 ± 3.63; Eukaryote 0.49 ± 1.7. Archaea have too few experimentally verified determinants (listed in Table 2) to perform this analysis. ECP-cloverleaf (B, D, F). The cloverleaf structure illustrates spatial relationships of many base-pairing residues. Each position is illustrated as a circle. The upper half of the circle corresponds to the AEV and its colour coding is the same as for the corresponding ECP diagram. The lower half of the circle corresponds to the number of published identity elements. The colour coding is similar to that in the corresponding ECP diagrams except for positions where the NPD is zero. These positions are indicated as gray. White illustrates positions where the analysis is not applicable. These are the 3′ CCA end for each kingdom, and the single unpopulated position e1 in the Achaea set. This figure can be viewed in colour online.
The positional AEV/NPD patterns show characteristic similarities. In the following paragraphs, we organized the results from the highest to the lowest AEVs.
3.1. Bacterial (coli-like) data set
The density function of the AEVs in the bacterial set shows a normal distribution as the number of positions having values over the weighted average plus 0.5 SD (31 positions) practically equals those that are below the weighted average minus 0.5 SD values (32 positions). In order to facilitate the visual comparison of NPD and the AEVs at each position, these are illustrated in a composite plot as shown in Fig. 2A and B.
Importantly, two anticodon positions, 35 and 36, have the highest AEVs (located in the red zone) and these have the highest NPD values as well. On the other hand, position 34 (located in the yellow zone), which pairs with the third, wobbling codon position, has significantly (over one sigma) lower discriminating potential. Notably, out of the three anticodon positions, this contains the lowest NPD as well. Most positions with high AEVs (located in the yellow zone) give place to known identity elements and it is also clear that the AEVs of positions that base pair with each other correlate. In the acceptor arm, three position pairs contain determinant for many identities. These are 1:72 (Trp, Gly, Thr, Gln), 2:71 (Met, Trp, Asp, Gly, Ser, Cys, Ala, Gln) and 3:70 (Val, Met, Trp, Gly, Ser, Cys, Ala, Gln), which as a set carry known determinants for half of the identities.3 The discriminator base in position 73 has the third highest AEV right behind positions 35 and 36.
In the tRNA core region,27 the 12:23 pair having high AEVs overlaps with the know tRNAIle identity element T12:A23. Furthermore, high AEV positions 13, 22 and 46 have a published role as tRNAGlu identity element T13:G22:A4628–30 and deletion of position 47 with high AEV was also identified as tRNAGlu determinant. The 13:22 pair has been identified as determinant for tRNACys as well.31,32
Other positions having higher than average AEVs host several identity elements as follows. Position 38 in the anticodon loop contains determinant for tRNAIle, tRNAAsp and tRNAGln,33,34 while positions 10 (tRNAAsp, tRNAGln); 11:24 (tRNASer, tRNAGlu); 15:84 (tRNACys, tRNAPro); 20 (tRNAPhe, tRNAArg, tRNAAla); 29:41 (tRNAIle) and 45 (tRNAPhe) also have a few indicated determinants.3,35
At the more conserved T-loop position 60 having average AEV, there is another tRNAPhe determinant.3,35 The variable loop position having the highest, but otherwise average, AEV is e2, which is a known identity element of tRNASer.36
The position pair 31:39 at the anticodon loop has no known identity element associated. In 2338 out of the 2406 analysed non-redundant bacterial sequences, these form normal Watson–Crick base pair, but based on the high AEVs, the exact identity of the given base pair might serve an identity element function.
Based on the high AEV but low NPD values, position pairs 12:23 and 13:22 as well as Position 46 might contain hitherto unidentified determinants.
The facultative elements (17, 17a, 20a and 20b) in the D-loop and in position 47 might also have identity functions (that will be detailed in Section 3.5).
Several elements with the lowest AEVs indicated in blue in Fig. 2A and C do not show any discrimination potentials. These are functionally highly conserved elements shared by all tRNA sequences and most of these were used in the first filtering step. Positions with still lower than average AEVs highlighted with cyan or those with average AEV highlighted as green typically coincide with regions that do not contain known identity elements. For example, at the 6:67 pair, the NPD is zero. Nevertheless, there are some exceptions. For the 5:68 position pair, a single identity element has been published, the A5:T68 for tRNAMet.37 Moreover, the conserved A37 in the anticodon loop has been identified as Ile, Met, Glu and Gln determinant,27,37–40 although over half of the tRNA sequences harbour adenine at this position. Finally, in spite of being located at low AEV regions, the U8:A14 for tRNALeu41 and the G27:C43, G28:C42 and T59 for tRNAPhe have also been shown to be determinants.42
3.2. Eukaryotic (yeast-like) data set
While the bacterial data set represented coli-like identity rules, the eukaryotic set contained sequences that conformed to the already established yeast identity rules. The results are illustrated in Fig. 2 in the same way as for the bacterial set.
Just like in the case of the bacterial set, the majority of known determinants are located at positions having the highest AEVs, namely the three anticodon positions and the discriminator position 73. The three base pairs of the acceptor arm have the next highest NPD values and these also represent higher than average AEVs. Another high AEV position where a single known identity element for tRNAPhe exists is position 20.43
There are several positions where higher than average AEVs exist, but no determinants have been identified. At some of these positions such as 12:23; 13:22 and 45; 46; 47 that participate in establishing the core region, there are already published determinants for the bacterial set.
Interestingly, the high AEV positions 31:39 and 30:40 in the anticodon stem contain no identified yeast determinants, but harbour published determinants for human tRNAPhe.44
The lower than average AEVs indicated with blue and cyan colours in Fig. 2C and D are again all associated with conserved positions lacking any known identity elements. Very few published identity elements coincide with positions having average AEVs indicated with green in Fig. 2C and D. Position 3 harbours determinants for tRNAGly and tRNAAla, the anticodon loop position 37 for tRNALeu and positions 38 and 10:25 for tRNAAsp.45–49 Position 70, which pairs with position 3, has a higher than average AEV.
3.3. Testing potential biases introduced by the second filtering step
We have checked how the second filtering step alters the input sequence set from various species. For bacteria, the effect of filtering nicely mirrored evolutionary relations. Data in Supplementary Table S2 show that after the second filtering step, species containing the highest number of retained sequences are the closest relatives of E. coli. Namely, from the Gammaproteobacteria class (genera Escherichia, Haemophilus, Salmonella, Yersinia, Buchnera, Shigella), 75–90% of the input sequences are retained by the second filtering step. Moving away from E. coli on the phylogenetic tree, the proportion of retained sequences gradually decreases. Still high (around 70%) proportion of sequences are retained in the case of Proteobacteria (genera Desulfovibrio, Brucella, Campylobacter), but, for example, in the case of Firmicutes (genera Streptococcus, Bacillus, Lactobacillus, Lactococcus, Staphylococcus) only 50–70%, while in the case of Tenericutes (Mycoplasma, Ureaplasma) or Actinobacteria (Mycobacterium, Streptomyces), only 30–50% of the input sequences are retained.
In the case of Eukarya, no such trend was observed. This might be due to the—compared with bacteria—much lower number and possibly more general nature of determinants published for yeast.
The positional AEVs measure the average distance of functionally defined sets. One might think that the second filtering for the presence of identity elements could increase the separation of the identity sets and thus increase the AEVs at identity element positions. In order to assess this potential effect, we repeated the analysis for the bacterial and the eukaryotic sets by omitting the second filtration. The data are organized in Supplementary Table S3, Table 1 and Supplementary Figure S1.
Table 1.
Representative data for tDNA sequence set processing and analysis with and without filtering for the presence of known identity elements
| Bacteria |
Eukaryota |
Archaea | |||
|---|---|---|---|---|---|
| Withouta second filtration | With second filtration | Without second filtration | With second filtration | No second filtration | |
| No. of sequences | |||||
| Raw data | 6243 | 2222 | 1552 | ||
| First filtration | 6144 | 1930 | 1384 | ||
| Second filtration | — | 3901 | — | 1672 | — |
| Non-redundant data set | 3946 | 2406 | 1495 | 1264 | 1041 |
| AEV | |||||
| Average | 6.45 | 5.59 | 5.79 | 6.31 | 7.34 |
| SD | 3.54 | 3.51 | 3.43 | 3.49 | 3.97 |
| Pearsonb (R) | 0.53 | 0.55 | — | — | — |
| Spearmanb (ρ) | 0.39 | 0.54 | — | — | — |
| Bootstrapc | |||||
| Mean | 224 | 258 | — | — | — |
| SD | 16.9 | 17.1 | — | — | — |
| Cumulative AEV threshold | 358.95 | 344.55 | — | — | — |
| Significance (P) | 1.33e−15 | 3.54e−7 | — | — | — |
aThe optional second filtration was performed based on the presence of experimentally verified E. coli (for Bacteria) or yeast (for Eukaryota) identity elements. Archaea lack enough verified identity elements; therefore, the second filtration was not performed.
bCorrelation of the NPDs and the AEVs was done as described in Section 2.
cThe bootstrap analysis is described in Supplementary Data S2.
Briefly, in the case of the bacterial data set, where 40 positions carry known identity elements, second filtration removed 39% of the input sequences, e.g. those that lacked at least one required coli identity element. As a result, at positions, where known identity elements exist (positive NPD), the sum of the AEV increased with 22%, while at positions with no reported identity elements (zero NPD), the increase was only 8%. Out of the 40 positions, this AEV increase exceeded the standard deviation at 10 positions.
In the case of the eukaryotic data set, where only 15 positions carry known identity elements, second filtration caused a much smaller effect. It removed only 15% of the input sequences, e.g. those that lacked at least one required yeast identity element. As a result, at positions where known identity elements exist (positive NPD), the sum of the AEV decreased with 5%, while at positions with no reported identity elements (zero NPD), the decrease was 9%. However, this decrease was statistically significant only at position 23, where no identity element has been reported.
For the bacterial set, we performed several statistical analyses both in the case of the second filtered and non-filtered data sets (Table 1). The Pearson correlation gave an R-value of 0.55 for the filtered and 0.53 for the non-filtered case, while the Spearmen correlation yielded ρ-values 0.54 and 0.39, respectively.
We also applied a bootstrap analysis to test the statistical significance of high AEV positions overlapping with positions harbouring known identity elements (Table 1, Supplementary Data S2). Importantly, the overlap was statistically significant both in the non-filtered and in the filtered case, demonstrating that omitting the second filtration step did not change the overall distribution of the AEVs. The highest AEVs are at the anticodon positions, the discriminator base and the acceptor arm (1:72; 2:71). These are well-known positions of identity elements.
This suggests that this filtration does not introduce artefacts. On the other hand, as we emphasized, only the second filtration yields a data set, in which sequences belonging to the same identity set share known identity elements, suggesting that they might also share yet unidentified common identity elements.
3.4. Archaea data set
The second filtration step was not performed for the Archaea data set as only sparse experimental data are available for such elements (listed in Table 2). The only comprehensive analysis available for each identity set is an in silico study based on sequence alignments.50
Table 2.
Experimentally determined and published Archaea identity elements
| Identity | Element | Species | Refs. |
|---|---|---|---|
| Ala | G3:U70 | Archaeoglobus fulgidus; Pyrococcus horikoshii | 71,72 |
| Asp | C36 | Pyrococcus kodakaraensis | 73–75 |
| Gly | C35, C36, C2:G71, G3:C70 | Aeropyrum pernix K1 | 76 |
| His | C73 | Aeropyrum pernix K1 | 77 |
| Phe | G34, A35 A36, A73, G20 | Aeropyrum pernix K1 | 55 |
| Pro | G35, G36, A73, G1:C72 | Aeropyrum pernix K1 | 78 |
| Ser | G30:C40, G73, variable loop G1:C72, C3:G70 variable loop | Methanosarcina barkeri (Archaeal RS); Methanococcus maripaludis | 56,79 |
| Thr | U73, C2:G71 | Haloferax volcanii; Aeropyrum pernix K1 | 54,80,81 |
| Trp | C34, C35, A36, A73, G1:C72, G2:C71 | Aeropyrum pernix K1 | 82 |
| Tyr | C1:G72, A73 | Aeropyrum pernix K1 | 57,58 |
Positions of typical determinants, such as the discriminator base or members of the anticodon, have significantly higher than average AEVs (yellow and red zones in Fig. 2A and C). For some Archaea species and some identity sets, these have been experimentally verified as determinants (see in Table 2). However, it is noteworthy how differently the individual kingdoms (or sometimes groups within a kingdom) use identity elements on the acceptor stem. This can be illustrated through the example of tRNAThr.
In the case of E. coli,51 the discriminator base is not a tRNAThr determinant, while the first three base pairs (1:72; 2:71; 3:70) are identity elements. The most important one is the 2:71 pair. For yeast,52 the discriminator base and the first and third acceptor stem base pairs are determinants, and similar results were published for the Thermus thermophilus bacterium.53 Studies on the tRNAThr identity elements for two Archaea species revealed that their discriminator base and acceptor stem base pairs are used differently. In the case of Haloferax volcanii, these were used similarly as in yeast and T. thermophilus, while in Aeropyrum pernix, these elements were used as in E. coli.54 Based on the AEVs at this area in the archahea set, the majority of the Archaea might have an identity element distribution like that in A. pernix. Moreover, the role of the discriminator base relative to the anticodon set appears to be dampened in this kingdom, suggesting that it might have roles in fewer identity sets than in the other two kingdoms.
The 3:70 base pair positions have excelling AEVs and this pair was shown to be determinant for tRNAAla, tRNAGly and tRNASer in a few Archaea species (Table 2). Position 20 also has a higher than average AEV (yellow zone) and this position carries an identity element for tRNAPhe in Archaea.55
The position pairs 29:41 and 31:39 at the anticodon stem also have higher than average AEVs, yet no determinants have been identified at these positions. On the other hand, in the case of Methanosarcina barkeri, at the 30:40 pair having average or low AEV, a Ser identity element has been found.56 Just like in the case of the bacterial set, there are several positions in the core region and at the facultative base positions where the AEVs are higher than average, yet no identity element has been published.
Similar to the bacterial set, the AEV of position 34 is lower than those of positions 35 and 36. However, in this case, position 34 has a much lower, only average AEV (green zone), while in bacteria, it belonged to the highest category (red zone).
Positions having the lowest AEVs, just like in the case of the bacterial data set, are located at conserved tRNA architecture defining positions. A noteworthy difference compared with the bacterial data is that in Archaea, the 1:72 position pair has only average AEVs (Fig. 2A and C). This appears to be due to the fact that in ∼90% of the Archaea tRNA sequences, there is a G1:C72 pair at this position. Based on the already published sequence analysis,50 the only few exceptions are detected for the initiator tRNAMet and for tRNAGln and tRNATyr. The C1:G72 pair of tRNATyr has been experimentally verified to be a genuine identity element.57,58
3.5. Potential hidden identity elements
Positions having higher than average AEVs but containing no published determinants that would have been used in our second filtering step might harbour hitherto unidentified determinants. The most likely hidden identity elements are illustrated in Fig. 3. We searched the literature for any identified determinants at such positions reported for species other than coli-like bacteria or yeast. Such cases were already mentioned above for the 30:40 and 31:39 base pairs that are identified human determinants.44 Therefore, we analysed these two base pairs by checking our data set for each identity set. As a simplest scenario, we searched for base pairs that, in any one of the 20 identity sets, differ from all other 19 sets. In the bacterial set, there was a single such case, a normal Watson–Crick base pair, while for the eukaryotic set, all such pairs were non-Watson–Crick base pairs.
Figure 3.

Hitherto unidentified potential identity elements. Positions having high AEV but low NPDs could harbour hitherto unidentified identity elements. Grey or black highlights the most likely positions of such elements. For positions highlighted as black, we also propose the corresponding identity: bold corresponding to yeast, while italic to E. coli. Middle grey highlights ‘core region’ positions having high AEV and low NPD in all three kingdoms. Light grey highlights the highest AEV base pair in the Archaea kingdom.
3.5.1. Escherichia coli tRNATrp T31:A39
This base pair is present in the tRNATrp sequences in all but a very few species that provided tRNATrp sequences to our filtered coli-like data set. In four coli-like species, only a single tRNASer, while in two species, only a single tRNAGln, has the same base pair. We note that only the anticodon loop, the discriminator base and the acceptor stem were experimentally tested for coli tRNATrp determinants,59–61 thus the T31:A39 pair might be a hitherto undetected identity element.
3.5.2. Yeast Met T31:T39
This unconventional base pair is characteristic to eukaryotic elongator tRNAMet. Both eukaryotic as well as coli initiator tRNAMet and the coli elongator tRNAMet contain a normal G31:C39 pair at these positions. In the coli initiator tRNA, the normal G31:C39 base pair is required for binding to the P site on the ribosome and for protein synthesis initiation, while in the coli elongator tRNAMet, the same pair is required for proper acylation by the corresponding AARS enzyme. Eukaryotic initiator tRNAMet from a wide source were shown to be good substrates of the coli Met-RS enzyme, while the eukaryotic elongator tRNAMet from the same wide source were all poor substrates. When the original T31:T39 pair in the eukaryotic tRNAMet was replaced with a G31:C39 pair, it became a good substrate for the coli enzyme, and symmetrically, when the G31:C39 pair was replaced with a T31:T39 pair in the coli elongator tRNAMet, it became a good substrate for the eukaryotic enzyme. Thus, the kingdom-specific base pair is a determinant in both kingdoms. Moreover, it was also shown that in the elongator tRNA, it is not the identity of the bases that matter, but the fact whether these form a Watson–Crick base pair or not. If they do, the corresponding tRNAMet is a good substrate of the bacterial enzyme and a poor substrate of the eukaryotic enzyme. If they do not, the corresponding tRNAMet is a good substrate for the eukaryotic enzyme and a poor substrate for the bacterial enzyme. Thus, a Watson–Crick base pair at these positions or the lack of it affects the structure and/or malleability of the anticodon loop, which has an important role in the proper interaction with the AARS enzyme.
It has also been shown that replacing the G31:C39 base pair with a T31:T39 pair in the yeast initiatior tRNA renders it being able to participate in the elongation phase.
Note that this base pair has not been defined as a determinant per se, as the studies compared either isospecific elongator tRNAs from two different kingdoms or elongator vs. initiator tRNAs rather than two different elongator tRNA identities from the same species.62–64
3.5.3. Yeast Ile T30:G40
In the case of yeast tRNAIle, only the three anticodon positions were studied as identity elements,65 and the potential role of this nearby non-Watson–Crick base pair has not been tested. In our data set, this base pair is present in the majority of eukaryotic species from Arabidopsis through Drosophila to Human for several Ile isoacceptors, suggesting that this base pair might have an identity defining role.
3.5.4. Yeast Asp G30:T40
At the same positions as for tRNAIle, a different non-Watson-Crick base pair is present in yeast tRNAAsp sequences. This base pair is not conserved in eukaryotic species. In our data set, it is present only in yeast and Caenorhabditis elegans. As high AEVs suggest that the G30:T40 pair might be an identity element in yeast (and also in C. elegans), we checked the PDB for other yeast tRNA/AARS complex structures. Unfortunately, besides the Asp,66 only two other structures are available. One is for tRNATyr (PDB: 2DLC), which is incomplete, and another one for tRNAArg.67 Both have a normal G30:C40 base pair. Note that ArgRS and TyrRS belong to Class I, while AspRS belongs to Class II. As enzymes from the two classes approach the tRNA from opposite sides, we did not expect any similarities in the enzyme–tRNA interactions themselves.
As could be expected, the interactions are not homologous. In the case of tRNAAsp, the ribose-phosphate portion of G30 is in 2.9–3.3 Å (there are two complexes in the unit cell) distance from the Lys88 side chain of the enzyme, which suggests a salt bridge between the tRNA and the enzyme at this position. The equivalent residue of the AspRS Lys88 in the ArgRS is Lys78, which does not interact with the tRNA. On the other hand in the tRNAArg structure, the phosphate of C40 appears to form a H-bond with Ser440 of the enzyme, which is in 2.6 Å distance. The incomplete tRNATyr structure either does not have an interaction with the enzyme at this position or it is not resolved in the model. It is plausible that a G:T pair is disrupted more readily than a G:C pair. It appears that such local disruption of a G:T pair is required for forming a stable salt bridge with the enzyme in the case of tRNAAsp. On the other hand, tRNAArg has a different stabilizing interaction, which does not necessitate such a local perturbation.
We also checked whether a different base pair exists at these positions in bacterial tRNAAsp and if so, whether it affects the recognition mode by their respective AARS enzymes. In the coli tRNA homologue, there is a traditional G30:C40 pair and fortunately there is a tRNA:AARS complex structure for bacterial tRNAAsp in the PDB,68 so we could compare the two homologous interactions from the two different kingdoms. Differences are shown in Fig. 4. As already mentioned, in the yeast system, the ribose-phosphate portion of G30 of the tRNA forms a salt bridge with the Lys88 side chain of the enzyme. A Needleman–Wunsch alignment of the coli and yeast synthetase sequences identified Lys58 as the coli equivalent of Lys88. This side chain is in 10.3 Å distance from the G30 phosphate moiety therefore no salt bridge is formed. This comparison clearly shows that identical tRNA positions are differently recognized by the two iso-functional AARS enzymes.
Figure 4.

Comparison of tRNAAsp/AspRS structures from E. coli and yeast. (A) Structure of the E. coli tRNAAsp/AspRS complex68; PDB: 1IL2. (B) Structure of the yeast tRNAAsp/AspRS complex66; PDB: 1ASY. Colour coding: AARS, ‘wheat’; tRNA, pale cyan; Lys58 (E. coli, panel A) and Lys88 (yeast, panel B) and the G30:C40 (E. coli, panel A) and G30:U40 (yeast, panel B) base pairs are green. Other bases illustrated as sticks are already published identity elements. See Section 3 for details. This figure can be viewed in colour online.
Furthermore, the observed direct interaction of the G30:T40 pair with the AARS enzyme suggests that it might be an identity element in yeast.
3.5.5. Core region
In E. coli, it has been shown that the core region, formed by the 15:48 pair and by [13:20]:46, has identity functions. The G15:G48 was shown to be a Cys identity element.26 The core region was shown to be important for tRNAPro identity69 and for discriminating tRNAGlu from tRNAAsp.30 While in the bacterial set, the 15:48 pair has medium level (green zone) AEV, positions 22 and 46 have high (yellow zone) and position 13 very high (red zone) AEV. The eukaryote set also shows high AEVs at these positions (all in the yellow zone), suggesting that the core region might have similar identity roles in the eukaryotic kingdom as well.70
3.6. Conclusions
Deciphering identity elements of tRNAs has been one of the most interesting problems of molecular biology that has a long and successful history.2–5 It is unquestionable that only properly designed mutations combined with in vitro and/or in vivo experiments can identify such elements beyond any doubts. However, the large number of even the ‘reasonable’ mutations and the astronomical number of their combinations renders these laborious studies rather challenging. As a consequence, the existing collection of already determined identity element set cannot be considered complete, and several bioinformatics studies aimed to predict hitherto hidden such elements.6,13–17 Most of these studies applied classical sequence analysing tools and searched for conserved sequence features.
The ECP analysis, on the other hand, is based on strictly absent elements and provides a simple but meaningful measure of pairwise mathematical distances of functionally related sequence sets.18
The reliability of bioinformatic studies is strongly related to the number of input sequences that are compared. Individual species have only a few tRNA sequences for each identity set. Comparing tRNA sequences from a pool of related species could improve the signal/noise ratio of the analysis, but it can be justified only if the comparison is functionally relevant, i.e. tRNA from one species would be properly charged by the corresponding AARS from the other species.
We produced starting data sets, in which sequences grouped for the 20 identity from bacteria were filtered for the presence of major coli identity elements while eukaryotic sequences for the presence of yeast identity elements. We argue that common presence of such identity elements in the starting data set favours interspecies compatibility. If so, isofunctional tRNAs from different species sharing already published identity elements should also share their yet undiscovered determinants as well. It means that such an analysis should identify elements that have not been recognized yet and in the same time identify those too that had already been published, but not included in the filtering set. If hitherto unknown elements are detected, they should be considered potential identity elements only in bacterial or eukaryotic species that also use the coli or the yeast identity element sets, respectively, that were applied for filtering. Thus, this analysis could clearly miss elements that are not conserved across species.
We demonstrated that positional AEVs measure sequence feature distances of functional groups and correlate with the number of already identified determinants (Table 1). Based on the above arguments, we suggested that positions having high AEV but few or no identified determinants predict locations of hidden identity elements. We listed the most characteristic such positions and for one of these suggested a structural rationale for being identity element.
We also showed that omitting the second filtration in the case of both Bacteria and Eukarya still preserves the characteristic pattern of high AEV positions, which overlaps with the positions of the most important identity elements such as the anticodon bases and the discriminator base.
In this study, we have demonstrated that the ECP algorithm is capable of assessing the level of discriminating power of positions in separating functionally different sequence sets. As genomic sequencing continues at an increasing rate, our ECP analysis can be performed over and over on ever-increasing filtered databases.
We believe that when enough input data are available, we can analyse the discriminating power not only of individual positions but combinations of positions as well.
Nevertheless, only carefully designed mutations and laboratory experiments can assess the predicting potentials of the ECP algorithm to identify hidden determinants.
Supplementary Material
Acknowledgements
The authors are grateful to Dr Catherine Florentz for critical reading of the manuscript and for her highly valuable suggestions. We also thank Márk Szenes for his help in defining the mathematical formalism of the AEV calculating algorithm and for the useful discussions about the statistical analyses.
Footnotes
Edited by Hiroyuki Toh
Supplementary Data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Funding
The study was supported by the Hungarian Scientific Research Fund (OTKA) K68408, NK81950 and NK100769 as well as by the European Union and European Social Fund (TÁMOP) 4.2.1./B-09/KMR-2010-0003. G.P. is supported by the János Bolyai Research Fellowship.
References
- 1.Cavarelli J., Moras D. Recognition of tRNAs by aminoacyl-tRNA synthetases. FASEB J. 1993;7:79–86. doi: 10.1096/fasebj.7.1.8422978. [DOI] [PubMed] [Google Scholar]
- 2.McClain W.H. Rules that govern tRNA identity in protein synthesis. J. Mol. Biol. 1993;234:257–80. doi: 10.1006/jmbi.1993.1582. [DOI] [PubMed] [Google Scholar]
- 3.Giege R., Sissler M., Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–35. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ibba M., Soll D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000;69:617–50. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 5.Giege R. Toward a more complete view of tRNA biology. Nat. Struct. Mol. Biol. 2008;15:1007–14. doi: 10.1038/nsmb.1498. [DOI] [PubMed] [Google Scholar]
- 6.Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laslett D., Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–6. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan P.P., Lowe T.M. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–7. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Juhling F., Morl M., Hartmann R.K., Sprinzl M., Stadler P.F., Putz J. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–62. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abe T., Ikemura T., Sugahara J., et al. tRNADB-CE 2011: tRNA gene database curated manually by experts. Nucleic Acids Res. 2011;39:D210–3. doi: 10.1093/nar/gkq1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atilgan T., Nicholas H.B., Jr., McClain W.H. A statistical method for correlating tRNA sequence with amino acid specificity. Nucleic Acids Res. 1986;14:375–80. doi: 10.1093/nar/14.1.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholas H.B., Jr., McClain W.H. Searching tRNA sequences for relatedness to aminoacyl-tRNA synthetase families. J. Mol. Evol. 1995;40:482–6. doi: 10.1007/BF00166616. [DOI] [PubMed] [Google Scholar]
- 13.Eddy S.R., Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;22:2079–88. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ardell D.H., Andersson S.G. TFAM detects co-evolution of tRNA identity rules with lateral transfer of histidyl-tRNA synthetase. Nucleic Acids Res. 2006;34:893–904. doi: 10.1093/nar/gkj449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freyhult E., Moulton V., Ardell D.H. Visualizing bacterial tRNA identity determinants and antideterminants using function logos and inverse function logos. Nucleic Acids Res. 2006;34:905–16. doi: 10.1093/nar/gkj478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freyhult E., Cui Y., Nilsson O., Ardell D.H. New computational methods reveal tRNA identity element divergence between Proteobacteria and Cyanobacteria. Biochimie. 2007;89:1276–88. doi: 10.1016/j.biochi.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Ardell D.H. Computational analysis of tRNA identity. FEBS Lett. 2010;584:325–33. doi: 10.1016/j.febslet.2009.11.084. [DOI] [PubMed] [Google Scholar]
- 18.Jako E., Ittzes P., Szenes A., Kun A., Szathmary E., Pal G. In silico detection of tRNA sequence features characteristic to aminoacyl-tRNA synthetase class membership. Nucleic Acids Res. 2007;35:5593–609. doi: 10.1093/nar/gkm598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujishima K., Sugahara J., Kikuta K., et al. Tri-split tRNA is a transfer RNA made from 3 transcripts that provides insight into the evolution of fragmented tRNAs in archaea. Proc. Natl Acad. Sci. USA. 2009;106:2683–7. doi: 10.1073/pnas.0808246106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugahara J., Kikuta K., Fujishima K., Yachie N., Tomita M., Kanai A. Comprehensive analysis of archaeal tRNA genes reveals rapid increase of tRNA introns in the order thermoproteales. Mol. Biol. Evol. 2008;25:2709–16. doi: 10.1093/molbev/msn216. [DOI] [PubMed] [Google Scholar]
- 21.Maruyama S., Sugahara J., Kanai A., Nozaki H. Permuted tRNA genes in the nuclear and nucleomorph genomes of photosynthetic eukaryotes. Mol. Biol. Evol. 2010;27:1070–6. doi: 10.1093/molbev/msp313. [DOI] [PubMed] [Google Scholar]
- 22.Chan P.P., Cozen A.E., Lowe T.M. Discovery of permuted and recently split transfer RNAs in Archaea. Genome Biol. 2011;12:R38. doi: 10.1186/gb-2011-12-4-r38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujishima K., Sugahara J., Tomita M., Kanai A. Sequence evidence in the archaeal genomes that tRNAs emerged through the combination of ancestral genes as 5′ and 3′ tRNA halves. PLoS One. 2008;3:e1622. doi: 10.1371/journal.pone.0001622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilm A., Mainz I., Steger G. An enhanced RNA alignment benchmark for sequence alignment programs. Algorithms Mol. Biol. 2006;1:19. doi: 10.1186/1748-7188-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marck C., Grosjean H. tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA. 2002;8:1189–232. doi: 10.1017/s1355838202022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hou Y.M., Motegi H., Lipman R.S., Hamann C.S., Shiba K. Conservation of a tRNA core for aminoacylation. Nucleic Acids Res. 1999;27:4743–50. doi: 10.1093/nar/27.24.4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nureki O., Niimi T., Muramatsu T., et al. Molecular recognition of the identity-determinant set of isoleucine transfer RNA from Escherichia coli. J. Mol. Biol. 1994;236:710–24. doi: 10.1006/jmbi.1994.1184. [DOI] [PubMed] [Google Scholar]
- 28.Sylvers L.A., Rogers K.C., Shimizu M., Ohtsuka E., Soll D. A 2-thiouridine derivative in tRNAGlu is a positive determinant for aminoacylation by Escherichia coli glutamyl-tRNA synthetase. Biochemistry. 1993;32:3836–41. doi: 10.1021/bi00066a002. [DOI] [PubMed] [Google Scholar]
- 29.Sekine S., Nureki O., Sakamoto K., et al. Major identity determinants in the ‘augmented D helix’ of tRNA(Glu) from Escherichia coli. J. Mol. Biol. 1996;256:685–700. doi: 10.1006/jmbi.1996.0118. [DOI] [PubMed] [Google Scholar]
- 30.Sekine S., Nureki O., Tateno M., Yokoyama S. The identity determinants required for the discrimination between tRNAGlu and tRNAAsp by glutamyl-tRNA synthetase from Escherichia coli. Eur J Biochem. 1999;261:354–60. doi: 10.1046/j.1432-1327.1999.00293.x. [DOI] [PubMed] [Google Scholar]
- 31.Hou Y.M., Westhof E., Giege R. An unusual RNA tertiary interaction has a role for the specific aminoacylation of a transfer RNA. Proc. Natl Acad. Sci. USA. 1993;90:6776–80. doi: 10.1073/pnas.90.14.6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipman R.S., Hou Y.M. Aminoacylation of tRNA in the evolution of an aminoacyl-tRNA synthetase. Proc. Natl Acad. Sci. USA. 1998;95:13495–500. doi: 10.1073/pnas.95.23.13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nameki N., Tamura K., Himeno H., Asahara H., Hasegawa T., Shimizu M. Escherichia coli tRNA(Asp) recognition mechanism differing from that of the yeast system. Biochem. Biophys. Res. Commun. 1992;189:856–62. doi: 10.1016/0006-291x(92)92282-3. [DOI] [PubMed] [Google Scholar]
- 34.Giege R., Florentz C., Kern D., Gangloff J., Eriani G., Moras D. Aspartate identity of transfer RNAs. Biochimie. 1996;78:605–23. doi: 10.1016/s0300-9084(96)80007-1. [DOI] [PubMed] [Google Scholar]
- 35.Fender A., Sissler M., Florentz C., Giege R. Functional idiosyncrasies of tRNA isoacceptors in cognate and noncognate aminoacylation systems. Biochimie. 2004;86:21–9. doi: 10.1016/j.biochi.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 36.Asahara H., Himeno H., Tamura K., Nameki N., Hasegawa T., Shimizu M. Discrimination among E. coli tRNAs with a long variable arm. Nucleic Acids Symp. Ser. 1993:207–8. [PubMed] [Google Scholar]
- 37.Meinnel T., Mechulam Y., Lazennec C., Blanquet S., Fayat G. Critical role of the acceptor stem of tRNAs(Met) in their aminoacylation by Escherichia coli methionyl-tRNA synthetase. J. Mol. Biol. 1993;229:26–36. doi: 10.1006/jmbi.1993.1005. [DOI] [PubMed] [Google Scholar]
- 38.Rogers K.C., Soll D. Discrimination among tRNAs intermediate in glutamate and glutamine acceptor identity. Biochemistry. 1993;32:14210–9. doi: 10.1021/bi00214a021. [DOI] [PubMed] [Google Scholar]
- 39.Ibba M., Hong K.W., Sherman J.M., Sever S., Soll D. Interactions between tRNA identity nucleotides and their recognition sites in glutaminyl-tRNA synthetase determine the cognate amino acid affinity of the enzyme. Proc. Natl Acad. Sci. USA. 1996;93:6953–8. doi: 10.1073/pnas.93.14.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freist W., Gauss D.H., Ibba M., Soll D. Glutaminyl-tRNA synthetase. Biol. Chem. 1997;378:1103–17. [PubMed] [Google Scholar]
- 41.Normanly J., Ollick T., Abelson J. Eight base changes are sufficient to convert a leucine-inserting tRNA into a serine-inserting tRNA. Proc. Natl Acad. Sci. USA. 1992;89:5680–4. doi: 10.1073/pnas.89.12.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McClain W.H., Foss K. Nucleotides that contribute to the identity of Escherichia coli tRNA(Phe) J Mol Biol. 1988;202:697–709. doi: 10.1016/0022-2836(88)90551-7. [DOI] [PubMed] [Google Scholar]
- 43.Sampson J.R., DiRenzo A.B., Behlen L.S., Uhlenbeck O.C. Nucleotides in yeast tRNAPhe required for the specific recognition by its cognate synthetase. Science. 1989;243:1363–6. doi: 10.1126/science.2646717. [DOI] [PubMed] [Google Scholar]
- 44.Nazarenko I.A., Peterson E.T., Zakharova O.D., Lavrik O.I., Uhlenbeck O.C. Recognition nucleotides for human phenylalanyl-tRNA synthetase. Nucleic Acids Res. 1992;20:475–8. doi: 10.1093/nar/20.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nameki N., Tamura K., Asahara H., Hasegawa T. Recognition of tRNA(Gly) by three widely diverged glycyl-tRNA synthetases. J. Mol. Biol. 1997;268:640–7. doi: 10.1006/jmbi.1997.0993. [DOI] [PubMed] [Google Scholar]
- 46.Imura N., Weiss G.B., Chambers R.W. Reconstitution of alanine acceptor activity from fragments of yeast tRNA-Ala II. Nature. 1969;222:1147–8. doi: 10.1038/2221147a0. [DOI] [PubMed] [Google Scholar]
- 47.Soma A., Kumagai R., Nishikawa K., Himeno H. The anticodon loop is a major identity determinant of Saccharomyces cerevisiae tRNA(Leu) J. Mol. Biol. 1996;263:707–14. doi: 10.1006/jmbi.1996.0610. [DOI] [PubMed] [Google Scholar]
- 48.Putz J., Puglisi J.D., Florentz C., Giege R. Identity elements for specific aminoacylation of yeast tRNA(Asp) by cognate aspartyl-tRNA synthetase. Science. 1991;252:1696–9. doi: 10.1126/science.2047878. [DOI] [PubMed] [Google Scholar]
- 49.Frugier M., Soll D., Giege R., Florentz C. Identity switches between tRNAs aminoacylated by class I glutaminyl- and class II aspartyl-tRNA synthetases. Biochemistry. 1994;33:9912–21. doi: 10.1021/bi00199a013. [DOI] [PubMed] [Google Scholar]
- 50.Mallick B., Chakrabarti J., Sahoo S., Ghosh Z., Das S. Identity elements of archaeal tRNA. DNA Res. 2005;12:235–46. doi: 10.1093/dnares/dsi008. [DOI] [PubMed] [Google Scholar]
- 51.Hasegawa T., Miyano M., Himeno H., Sano Y., Kimura K., Shimizu M. Identity determinants of E. coli threonine tRNA. Biochem. Biophys. Res. Commun. 1992;184:478–84. doi: 10.1016/0006-291x(92)91219-g. [DOI] [PubMed] [Google Scholar]
- 52.Nameki N. Identity elements of tRNA(Thr) towards Saccharomyces cerevisiae threonyl-tRNA synthetase. Nucleic Acids Res. 1995;23:2831–6. doi: 10.1093/nar/23.15.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nameki N., Asahara H., Hasegawa T. Identity elements of Thermus thermophilus tRNA(Thr) FEBS Lett. 1996;396:201–7. doi: 10.1016/0014-5793(96)01094-0. [DOI] [PubMed] [Google Scholar]
- 54.Nagaoka Y., Yokozawa J., Umehara T., et al. Molecular recognition of threonine tRNA by threonyl-tRNA synthetase from an extreme thermophilic archaeon, Aeropyrum pernix K1. Nucleic Acids Res. 2002;(Suppl):81–2. doi: 10.1093/nass/2.1.81. [DOI] [PubMed] [Google Scholar]
- 55.Tsuchiya W., Kimura M., Hasegawa T. Determination of phenylalanine tRNA recognition sites by phenylalanyl-tRNA synthetase from hyperthermophilic archaeon, Aeropyrum pernix K1. Nucleic Acids Symp. Ser. (Oxf) 2007:367–8. doi: 10.1093/nass/nrm184. [DOI] [PubMed] [Google Scholar]
- 56.Korencic D., Polycarpo C., Weygand-Durasevic I., Soll D. Differential modes of transfer RNASer recognition in Methanosarcina barkeri. J. Biol. Chem. 2004;279:48780–6. doi: 10.1074/jbc.M408753200. [DOI] [PubMed] [Google Scholar]
- 57.Iwaki J., Asahara H., Nagaoka Y., et al. Differences in tyrosine tRNA identity between Escherichia coli and archaeon, Aeropyrum pernix K1. Nucleic Acids Res. 2002;(Suppl):225–6. doi: 10.1093/nass/2.1.225. [DOI] [PubMed] [Google Scholar]
- 58.Iwaki J., Suzuki R., Fujimoto Z., Momma M., Kuno A., Hasegawa T. Overexpression, purification and crystallization of tyrosyl-tRNA synthetase from the hyperthermophilic archaeon Aeropyrum pernix K1. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2005;61:1003–5. doi: 10.1107/S1744309105033245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pak M., Pallanck L., Schulman L.H. Conversion of a methionine initiator tRNA into a tryptophan-inserting elongator tRNA in vivo. Biochemistry. 1992;31:3303–9. doi: 10.1021/bi00128a001. [DOI] [PubMed] [Google Scholar]
- 60.Pak M., Willis I.M., Schulman L.H. Analysis of acceptor stem base pairing on tRNA(Trp) aminoacylation and function in vivo. J. Biol. Chem. 1994;269:2277–82. [PubMed] [Google Scholar]
- 61.Rogers M.J., Adachi T., Inokuchi H., Soll D. Switching tRNA(Gln) identity from glutamine to tryptophan. Proc. Natl Acad. Sci. USA. 1992;89:3463–7. doi: 10.1073/pnas.89.8.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.von Pawel-Rammingen U., Astrom S., Bystrom A. S. Mutational analysis of conserved positions potentially important for initiator tRNA function in Saccharomyces cerevisiae. Mol. Cell. Biol. 1992;12:1432–42. doi: 10.1128/mcb.12.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drabkin H.J., Helk B., RajBhandary U.L. The role of nucleotides conserved in eukaryotic initiator methionine tRNAs in initiation of protein synthesis. J. Biol. Chem. 1993;268:25221–8. [PubMed] [Google Scholar]
- 64.Meinnel T., Mechulam Y., Fayat G., Blanquet S. Involvement of the size and sequence of the anticodon loop in tRNA recognition by mammalian and E. coli methionyl-tRNA synthetases. Nucleic Acids Res. 1992;20:4741–6. doi: 10.1093/nar/20.18.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Senger B., Auxilien S., Englisch U., Cramer F., Fasiolo F. The modified wobble base inosine in yeast tRNAIle is a positive determinant for aminoacylation by isoleucyl-tRNA synthetase. Biochemistry. 1997;36:8269–75. doi: 10.1021/bi970206l. [DOI] [PubMed] [Google Scholar]
- 66.Moulinier L., Eiler S., Eriani G., et al. The structure of an AspRS-tRNA(Asp) complex reveals a tRNA-dependent control mechanism. EMBO J. 2001;20:5290–301. doi: 10.1093/emboj/20.18.5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delagoutte B., Moras D., Cavarelli J. tRNA aminoacylation by arginyl-tRNA synthetase: induced conformations during substrates binding. EMBO J. 2000;19:5599–610. doi: 10.1093/emboj/19.21.5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruff M., Krishnaswamy S., Boeglin M., et al. Class II aminoacyl transfer RNA synthetases: crystal structure of yeast aspartyl-tRNA synthetase complexed with tRNA(Asp) Science. 1991;252:1682–9. doi: 10.1126/science.2047877. [DOI] [PubMed] [Google Scholar]
- 69.Liu H., Musier-Forsyth K. Escherichia coli proline tRNA synthetase is sensitive to changes in the core region of tRNA(Pro) Biochemistry. 1994;33:12708–14. doi: 10.1021/bi00208a023. [DOI] [PubMed] [Google Scholar]
- 70.Ryckelynck M., Giege R., Frugier M. Yeast tRNA(Asp) charging accuracy is threatened by the N-terminal extension of aspartyl-tRNA synthetase. J. Biol. Chem. 2003;278:9683–90. doi: 10.1074/jbc.M211035200. [DOI] [PubMed] [Google Scholar]
- 71.Naganuma M., Sekine S., Fukunaga R., Yokoyama S. Unique protein architecture of alanyl-tRNA synthetase for aminoacylation, editing, and dimerization. Proc. Natl Acad. Sci. USA. 2009;106:8489–94. doi: 10.1073/pnas.0901572106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sokabe M., Okada A., Yao M., Nakashima T., Tanaka I. Molecular basis of alanine discrimination in editing site. Proc. Natl Acad. Sci. USA. 2005;102:11669–74. doi: 10.1073/pnas.0502119102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tumbula-Hansen D., Feng L., Toogood H., Stetter K.O., Soll D. Evolutionary divergence of the archaeal aspartyl-tRNA synthetases into discriminating and nondiscriminating forms. J. Biol. Chem. 2002;277:37184–90. doi: 10.1074/jbc.M204767200. [DOI] [PubMed] [Google Scholar]
- 74.Schmitt E., Moulinier L., Fujiwara S., Imanaka T., Thierry J.C., Moras D. Crystal structure of aspartyl-tRNA synthetase from Pyrococcus kodakaraensis KOD: archaeon specificity and catalytic mechanism of adenylate formation. EMBO J. 1998;17:5227–37. doi: 10.1093/emboj/17.17.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feng L., Tumbula-Hansen D., Toogood H., Soll D. Expanding tRNA recognition of a tRNA synthetase by a single amino acid change. Proc. Natl Acad. Sci. USA. 2003;100:5676–81. doi: 10.1073/pnas.0631525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Okamoto K., Kuno A., Hasegawa T. Recognition sites of glycine tRNA for glycyl-tRNA synthetase from hyperthermophilic archaeon, Aeropyrum pernix K1. Nucleic Acids Symp. Ser. (Oxf) 2005:299–300. doi: 10.1093/nass/49.1.299. [DOI] [PubMed] [Google Scholar]
- 77.Nagatoyo Y., Iwaki J., Suzuki S., Kuno A., Hasegawa T. Molecular recognition of histidine tRNA by histidyl-tRNA synthetase from hyperthermophilic archaeon, Aeropyrum pernix K1. Nucleic Acids Symp. Ser. (Oxf) 2005:307–8. doi: 10.1093/nass/49.1.307. [DOI] [PubMed] [Google Scholar]
- 78.Yokozawa J., Okamoto K., Kawarabayasi Y., Kuno A., Hasegawa T. Molecular recognition of proline tRNA by prolyl-tRNA synthetase from hyperthermophilic archaeon, Aeropyrum pernix K1. Nucleic Acids Res. 2003;(Suppl):247–8. doi: 10.1093/nass/3.1.247. [DOI] [PubMed] [Google Scholar]
- 79.Gruic-Sovulj I., Jaric J., Dulic M., Cindric M., Weygand-Durasevic I. Shuffling of discrete tRNASer regions reveals differently utilized identity elements in yeast and methanogenic archaea. J. Mol. Biol. 2006;361:128–39. doi: 10.1016/j.jmb.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 80.Yokozawa J., Nagaoka Y., Umehara T., et al. Recognition of tRNA by aminoacyl-tRNA synthetase from hyperthermophilic archaea, Aeropyrum pernix K1. Nucleic Acids Res. 2001;(Suppl):117–8. doi: 10.1093/nass/1.1.117. [DOI] [PubMed] [Google Scholar]
- 81.Ishikura H., Nagaoka Y., Yokozawa J., Umehara T., Kuno A., Hasegawa T. Threonyl-tRNA synthetase of archaea: importance of the discriminator base in the aminoacylation of threonine tRNA. Nucleic Acids Symp. Ser. 2000:83–4. doi: 10.1093/nass/44.1.83. [DOI] [PubMed] [Google Scholar]
- 82.Tsuchiya W., Umehara T., Kuno A., Hasegawa T. Determination of tryptophan tRNA recognition sites for tryptophanyl-tRNA synthetase from hyperthermophilic archaeon, Aeropyrum pernix K1. Nucleic Acids Symp. Ser. (Oxf) 2004:185–6. doi: 10.1093/nass/48.1.185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}