

Abstract
Neurodegenerative diseases, such as Parkinson's disease (PD) and Alzheimer's disease(AD), are a group of pathologies characterized by a progressive and specific loss of certain brain cell populations. Oxidative stress, mitochondrial dysfunction, and apoptosis play interrelated roles in these disorders. It is well documented that free radical oxidative damage, particularly on neuronal lipids, proteins, DNA, and RNA, is extensive in PD and AD brains. Moreover, alterations of glutathione (GSH) metabolism in brain have been implicated in oxidative stress and neurodegenerative diseases. As a consequence, the reduced GSH levels observed in these pathologies have stimulated a number of researchers to find new potential approaches for maintaining or restoring GSH levels. Unfortunately, GSH delivery to the central nervous system (CNS) is limited due to a poor stability and low bioavailability. Medicinal-chemistry- and technology-based approaches are commonly used to improve physicochemical, biopharmaceutical, and drug delivery properties of therapeutic agents. This paper will focus primarily on these approaches used in order to replenish intracellular GSH levels, which are reduced in neurodegenerative diseases. Here, we discuss the beneficial properties of these approaches and their potential implications for the future treatment of patients suffering from neurodegenerative diseases, and more specifically from PD and AD.
1. Introduction
Neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's diseases and amyotrophic lateral sclerosis make up a group of pathologies characterized by a separated etiology with distinct morphological and pathophysiological features. These disorders are defined by a multifactorial nature and have common neuropathological hallmarks such as (a) abnormal protein dynamics with defective protein degradation and aggregation; (b) oxidative stress and free radical formation; (c) impaired bioenergetics and mitochondrial dysfunctions; (d) neuroinflammatory processes [1, 2]. It is difficult to establish the correct sequence of these events, but it has been shown that the oxidative damage to the brains of affected individuals is one of the earliest pathological markers. Oxidative and nitrosative stresses arise from the imbalance between the increased production of both the reactive oxygen species (ROS) and the reactive nitrogen species (RNS) and the cellular antioxidant defense systems [3]. At low levels, ROS function as signaling intermediates for the modulation of cellular activities but, at higher concentrations, they contribute to neuronal membrane damage. The ROS mainly involved in neurodegeneration are the superoxide anion (O2 −), the hydrogen peroxide (H2O2), and the hydroxyl radical (HO●). RNS, such as nitric oxide (NO), can react with O2 − to produce peroxynitrite (ONOO−), a powerful oxidant that may decompose itself to form HO● [4]. Cells normally employ a number of defense mechanisms against free radical such as enzymes (Cu/Zn- and Mn-superoxide dismutase, GSH peroxidase, GSH reductase, catalase, and methionine sulfoxide reductase) and low-molecular-weight antioxidants (vitamin E, ascorbate, and GSH) [5]. Macromolecules such as lipids, proteins, and DNA undergo damage and subsequently cell death mainly by apoptosis when the antioxidant defense network is not sufficient [6].
2. GSH Depletion in PD and AD
The brain is especially vulnerable to free radical damage because of its high oxygen consumption rate, high content of lipids, and relative paucity of antioxidant enzymes compared with other organs [7]. Significant biological changes, related to a condition of oxidative stress, have been found in brain tissue of individuals affected by PD, AD, and other diseases [5, 8–11]. In particular, data from postmortem studies of brains from patients with PD suggest that oxidative stress plays a role in neural degeneration of the pigmented dopaminergic neurons in the substantia nigra pars compacta (SNpc) [12]. The normal metabolism of dopamine can generate free radicals and other ROS. Furthermore, in the human SNpc the autooxidation of dopamine leads to neuromelanin and can generate quinone and semiquinone species and ROS [13]. Finally, enzymatic oxidation of dopamine catalyzed by monoamine oxidase leads to formation of H2O2, which can react with Fe2+ and form the highly reactive radical HO● via the Fenton reaction [14]. All these unfavorable events contribute to alter the antioxidant defenses suggesting that the oxidative stress plays an important role in PD. The strongest alteration in the antioxidant defense is a decrease in GSH concentration [15–17]. According to postmortem studies, GSH levels in the SNpc of PD patients are remarkably lower than those of healthy subjects (60% compared to control subjects) while oxidized glutathione (GSSG) levels are slightly increased [18, 19]. Although GSH is not the only antioxidant molecule reported to be altered in PD, it is hypothesized that the magnitude of its depletion is the earliest indicator of nigrostriatal degeneration [20]. Moreover, striatal DA content and GSH levels are not altered in areas of the brain other than SNpc, or in other diseases affecting dopaminergic neurons [21–24]. GSH loss in PD is also accompanied by a reduction in mitochondrial complex I activity, which is regionally selective for the SNpc in PD and does not occur in related basal ganglia degenerative disorders [25]. These findings suggest that decreased nigrostriatal GSH levels can initiate or facilitate a cascade of further oxidative stress with consequent degeneration of dopaminergic neurons in idiopathic PD [26, 27].
AD is characterized by the loss of pyramidal neurons in the hippocampus and cortex, as well as cholinergic neurons in the basal forebrain. The etiology of AD is not completely known yet, although there are different hallmarks that seem to play significant roles in the disease, such as β-amyloid (Aβ) deposits, τ-protein aggregation, oxidative damage in cellular structures, and low levels of acetylcholine (ACh) [28, 29]. In AD patients there is a strong evidence that Aβ-associated free radicals and the resultant oxidative stress are a part of the mechanism that is involved in the pathogenic cascade that leads to neurodegeneration in AD brain [30]. Furthermore, alterations to GSH metabolism have been found in these pathological conditions [16]. In this context, Gu et al. [31] reported that GSH levels are depressed in AD cingulated cortex and AD substantia innominata, while Liu et al. [32] found these reduced levels only in red blood cells of male AD patients. Nevertheless, increased GSH levels have been observed by Adams et al. [33] in the midbrain and in the caudate nucleus, while normal GSH contents have been determined by Perry et al. [34]. Presumably, dissenting results are due to differences in techniques or difficulty in sample collection after death of AD patients. In any case, it has been observed that GSH protects cultured neurons against oxidative damage resulting from β-peptide and 4-hydroxynonenal (HNE), a lipid peroxidation product that is increased in AD [35]. A significant decrease in Cu and significant increases in Zn and Fe were found in AD hippocampus and amygdale, while Cu, Fe, and Zn are elevated in senile plaques of AD. These metal ions can catalyze free radical reactions and contribute to oxidative damage observed in AD brain [36]. GSH protects these areas through formation of metal complexes via nonenzymatic reactions and may also be beneficial for normalizing the adverse effects of iron accumulation in the aging brain [37].
3. Antioxidant Neuroprotection in PD and AD
Neuroprotective antioxidants are considered a promising approach to slow down the progression and limit the extent of neuronal cell loss in neurodegenerative disorders [38–41]. These agents were classified by Behl and Moosmann according to their mode of action in (a) compounds that prevent the formation of free radicals; (b) compounds that chemically interfere with formed free radicals; (c) compounds which limit the damage extent to the cell by alleviating the secondary metabolic burden of increased levels of free radicals [42]. N-acetylcysteine, lipoic acid, GSH, and its thiol derivatives belong to the last class of neuroprotective antioxidants. In this context, the GSH system is especially important for cellular defense against ROS in brain cells, acting directly in detoxification of radicals in non-enzymatic reactions and working as a substrate for various peroxidases [43]. Astrocytes appear to play a key role in the GSH metabolism in the brain since astroglial GSH export is essential for providing GSH precursor to neurons. Normally astrocytes release GSH and protect it against oxidation by releasing a protecting factor into the medium. Astroglial release of GSH is the first step in the supply of the GSH precursor cysteine to neurons. The extracellular GSH is processed by γ-glutamyl transpeptidase (γ-GT) and aminopeptidase N (ApN) to generate the cysteine, which limits the synthesis of GSH in neurons. Alterations of the release rate of GSH from astrocytes and reduced activities of the ectoenzymes may contribute to a lowered antioxidant defense in neurons and to an increased susceptibility to oxidative stress, both involved in the progression of neurodegenerative diseases [44]. Alterations of GSH metabolism in brain have been found in neurodegenerative disorders as PD and AD [5, 45–47]. The causes of GSH depletion are not well understood yet, but their consequences are quite serious. GSH depletion can inhibit complex I, E1 ubiquitin ligase, and proteasome activity; it can also exacerbate oxidative stress and activate the JNK pathway, leading to an inflammatory response. All these effects cause dopaminergic neuronal death and accumulation of proteins into Lewy bodies in patients affected by PD [48]. Furthermore, an emerging evidence indicates that the total antioxidant capacity (including GSH, ascorbic acid, uric acid, and bilirubin) has shown to be reduced by 24% in plasma samples from AD patients [49]. An increased number of mutations in mitochondrial DNA have been found in AD, such as increased concentrations of 8-hydroxy-2-deoxyguanosine, a marker of oxidative damage to DNA. These deletions or point mutations, which may result from oxidative stress, can cause mitochondrial dysfunction and trigger apoptotic cell death. In addition to DNA damage, several mitochondrial key enzymes involved in ROS detoxification are also affected. In vivo studies on animal models of AD have also shown the implication of mitochondria in the disease pathogenesis [50]. In this regard, several groups have focused their efforts on developing neuroprotective strategies targeting mitochondria. Some of the major mitochondrial targets used as therapeutics against ROS-mediated damage are members of the quinone family. An ubiquinone derivative, mitoquinone mesylate or MitoQ, has been used to prevent oxidative damage in AD [51]. MitoQ consists of CoQ10 linked to a triphenylphosphonium ion, which has a positive charge; therefore, it accumulates in mitochondria, which have a strongly negative membrane potential (about −120 mV). More precisely, MitoQ is adsorbed in the inner mitochondrial membrane facing the matrix. This ROS-enriched region provides a real potency to MitoQ. In addition, MitoQ prevents AD-like pathology in mouse cortical neurons in cell culture, attenuates β-amyloid-induced neurotoxicity, and prevents increased production of ROS.
Mitochondrial dysfunction, oxidative stress, glutamate excitotoxicity, and formation of high-molecular-weight aggregates also define the most common adult-onset motoneuron disease: amyotrophic lateral sclerosis (ALS) caused by the progressive degeneration of moto-neurons in the spinal cord, brain stem, and motor cortex [52]. Dominant mutation in Cu/Zn-superoxide dismutase (SOD1) causes familial forms of ALS. In order to investigate the role of GSH in this pathology, knockout mice for the glutamate-cysteine ligase modifier (GCLM) subunit were used. Results suggested that the lack of GCLM significantly accelerates disease and mitochondrial pathology in hSOD1 mice [53].
A promising therapeutic intervention in the above reported diseases could be the antioxidant neuroprotection [54]. In this context, the increase of GSH availability in neurons is a logical therapeutical target in neural impairment related to oxidative stress. Due to the difficulty in elevating GSH directly as described by Zeevalk et al. [55], other strategies to raise brain levels of this antioxidant have been investigated [56]. In this paper we will focus on the medicinal chemistry and technological approaches aimed at maintaining or restoring GSH levels in PD and AD patients. Particular attention will be paid to different strategies for increasing GSH levels by supplying GSH codrugs and GSH nanocarrier systems able to cross the cellular membrane more easily than GSH.
4. Medicinal-Chemistry-Based Strategies to Increase GSH Levels
Medicinal-chemistry-based strategies include analogues [57, 58], as well as prodrugs and codrugs approaches [59]. While each of these strategies may be equally promising to increase GSH levels, this paper will mainly focus on codrugs approach since the other medicinal-chemistry-based strategies have been previously discussed [56, 60–62]. The codrug approach consists in linking, via a covalent chemical linkage, two different pharmacophores with similar or different pharmacological activities in order to improve physiochemical, biopharmaceutical, and drug delivery properties of therapeutic agents. The resulting codrug has to be stable at gastrointestinal level and transported to the target site of action where it provides the two parent drugs following hydrolysis [63].
The codrug approach has been used for the treatment of PD and AD joining antioxidant or chelating molecules with a therapeutic compound (antiparkinson or anti-alzheimer's drugs) [64–66]. In particular, codrugs containing antioxidant molecules such as GSH, N-acetyl-cysteine, methionine, and cysteinyl derivatives have been synthesized in order to permit a targeted delivery of antioxidant directly to specific groups of neurons where cellular stress is associated with PD and AD. The dual advantage of these antioxidant molecules lies in the fact that the antioxidant portion, in addition to acting as a scavenger directly or indirectly of free radicals, can be used as a carrier. In fact, GSH and cysteinyl derivatives can be used as BBB shuttles for delivery of antiparkinson or ant-Ialzheimer's drugs since the presence of GSH transporters at the BBB is well documented [67, 68]. In this context, the research of new codrugs for the treatment of PD and AD has gained our attention. L-Dopa-GSH codrugs (LD-GSH, 1-2), obtained via an amide bond between LD and the C- and N-terminal GSH, respectively, have been synthesized and evaluated as potential anti-Parkinson agents with antioxidant properties (Figure 1) [69]. These codrugs permit a targeted delivery of GSH directly to SNpc neurons of PD patients and contribute in attenuating the damage caused by the prooxidant effects of traditional LD therapies. Codrugs 1-2 showed good stability toward gastrointestinal simulated fluids and released LD in rat and human plasma after enzymatic hydrolysis. Furthermore, they prolonged the plasma LD levels and were able to induce sustained delivery of dopamine (DA) in rat striatum with respect to equimolar dose of LD. Taken together, these results demonstrated the possible therapeutic application of codrugs 1-2 in PD, being able to protect against the oxidative stress deriving from autoxidation and the MAO-mediated metabolism of DA [69].
Figure 1.
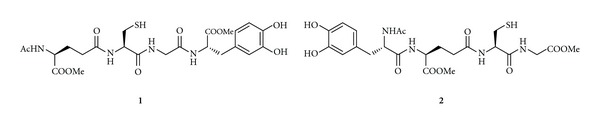
Chemical structures of GSH codrugs 1-2.
Later, More and Vince [70] reported two GSH bioconjugates (3-4) containing a metabolically stable urea analogue of GSH resistant to the enzyme γ-GT (Figure 2). The antioxidant portion has been covalently joined to the therapeutic drugs, as DA and adamantine, via a heterodisulfide linkage. This suitable junction is stable in plasma and able to release DA or adamantine, and the antioxidant portion due to the abundance of the enzyme disulfide reductases in the brain [71]. More importantly, these bioconjugates cross the BBB through recognition by GSH transporters on the luminal side of BBB [68]. Studies successfully confirmed the carrier-mediated transport of conjugates 3-4 in an in vitro BBB model and their ability to release the active drug at the target site, thus representing an innovative approach for the targeted delivery of anti-Parkinson drugs into the CNS using the GSH transport system [70]. In particular, the MDCK cell monolayer has been used to study the bioconjugates 3-4 transport. At concentration of 100 μM, the transport of bioconjugates 3-4 from the apical to the basal side was greater than the transport in the reverse direction. Moreover, to ensure that the codrugs 3-4 were not being metabolized as they crossed the MDCK cell monolayer, the integrity of codrugs 3-4 was confirmed by HPLC studies [70]. These experiments successfully demonstrated the GSH-carrier-mediated transport of the bioconjugates 3-4 in an in vitro BBB model.
Figure 2.
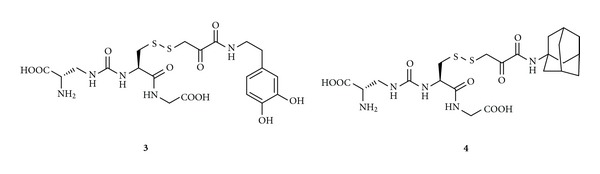
Chemical structures of GSH codrugs 3-4.
Another bioconjugate that could use the GSH transporters on the luminal side of BBB might be the hybrid 5 (Figure 3). This molecule is characterized by the replacement of cysteine with methionine in order to obtain stable GSH analogue at γ-GT [72]. Furthermore, the GSH analogue has been linked to LD to obtain CNS drug delivery. This compound was demonstrated to cross unaltered the acidic environment of the stomach, to be stable enough to be absorbed from the intestine, to have radical scavenging activity, and to release LD in human plasma after enzymatic hydrolysis. Taken together, these data suggest a therapeutic potential of 5 in pathological events associated with free radical damage and decreasing DA concentration in the brain [72].
Figure 3.
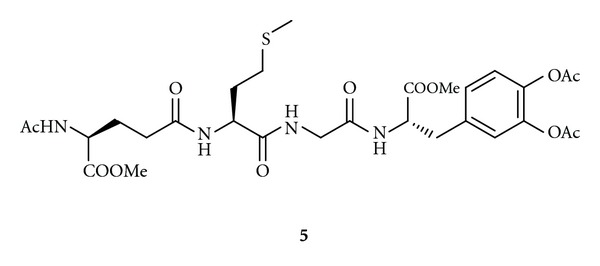
Chemical structure of GSH hybrid 5.
Recently, Ehrlich et al. [73] designed and synthesized a library of new GSH codrugs (called UPF peptides) with powerful hydroxyl radical scavenging activities. They have been obtained via an amide bond between GSH and tyrosine derivatives as shown in Figure 4. In particular, the enzyme free hydroxyl radical scavenging assay showed that substitution of γ-glutamyl moiety (UPF1, 4-methoxy-L-tyrosinyl-γ-L-glutamyl-L-cysteinyl-glycine, 6) with α-glutamyl moiety (UPF17, 4-methoxy-L-tyrosinyl-α-L-glutamyl-L-cysteinyl-glycine, 7) improved hydroxyl radical scavenging activity of about 500-fold [74]. UPF1 (6) is an effective and potential agent that diminishes neuronal injury in global cerebral ischemia [75]; it acts as a free radical scavenger or a modulator of G-protein in frontocortical membrane preparations. Although the exact mechanisms of the protective action of UPF1 still remain unclear, it can possibly act as a scavenger or a signal molecule increasing GSH levels or the GSH redox ratio; UPF1 could be a promising lead for the design of powerful antioxidants for the treatment of conditions associated with reduced GSH levels [76]. Unfortunately, the role of UPF peptides for the treatment of PD has not been studied yet. It could be interesting to investigate the activity of UPF peptides in patients affected by PD since these peptides contain the tyrosine moiety, the metabolic precursor of DA.
Figure 4.
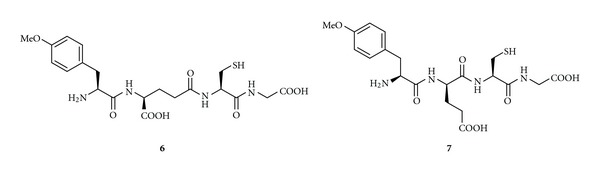
Chemical structures of GSH codrugs 6-7.
Few data are available in the literature about GSH codrugs for the treatment of AD. We recently synthesized Ibuprofen-GSH (IBU-GSH, 8) obtained via amide bond between GSH and IBU, a nonsteroidal anti-inflammatory drug (NSAID) (Figure 5) [77]. NSAIDs treatment reduces AD risk, delays disease progression, and reduces microglia activation [78]. In particular, Lim et al. [79] reported that six months of treatment of a transgenic animal model of AD with IBU resulted in a significant reduction of amyloid plaque burden and total Aβ peptide levels. Furthermore, IBU treatment led to a reduction of plaque-associated microglia and a corresponding attenuation in proinflammatory cytokine levels in brain [80]. Codrug 8 possessed good stability toward human plasma enzymatic activity and displayed in vitro free radical scavenging activity in time- and concentration-dependent manner. More importantly, it antagonizes the deleterious and cognitive effects of β-amyloid(1–40) in a rat model for AD, as also confirmed by behavioral tests of long-term spatial memory. In conclusion, IBU-GSH might permit targeted delivery of IBU and GSH directly to neurons, where oxidative stress and inflammatory processes are associated with AD [77].
Figure 5.
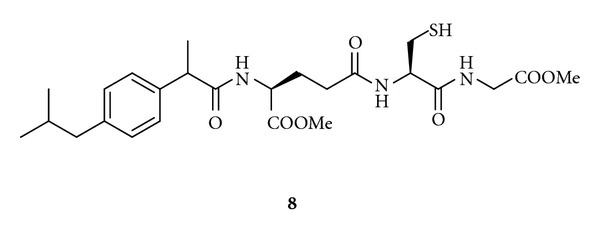
Chemical structure of GSH codrug 8.
Almost all the codrugs (1-2, 5, 9–14) have been tested for their chemical and enzymatic stabilities in order to check both their stability in aqueous medium and their sensitivity towards enzymatic cleavage in rat and human plasma (Tables 1 and 2) [69, 72, 81]. Stability studies were performed at 37°C in isotonic sodium phosphate buffer (pH 7.4), in simulated gastric fluid (SGF, pH 1.3), and in rat and human plasma diluted to 80% with isotonic sodium phosphate buffer (pH 7.4). All codrugs showed good stability toward gastrointestinal hydrolysis (t 1/2 > 20 h) (Table 1). On the contrary, in rat and human plasma the codrugs (1-2, 5, 9–14) underwent rapid bioconversion of the codrugs into their constituents (Table 2).
Table 1.
Kinetic data for chemical hydrolysis of codrugs 1-2, 5, and 9–14 at 37°C.
| pH 1.3a | pH 7.4a | |||
|---|---|---|---|---|
| Compd | t 1/2 (h) | K obs (h−1) | t 1/2 (h) | K obs (h−1) |
| 1 | 20.14 (±0.73) | 0.034 (±1.2 × 10−3) | 7.22 (±0.31) | 0.096 (±4.1 × 10−3) |
| 2 | 28.12 (±1.21) | 0.025 (±1.1 × 10−3) | 12.23 (±0.49) | 0.057 (±2.3 × 10−3) |
| 5 | 20.67 (±0.83) | 0.094 (±0.3 × 10−3) | 10.80 (±0.40) | 0.018 (±0.8 × 10−3) |
| 9 | 301.0 (±10.5) | 0.002 (±0.07 × 10−3) | 46.2 (±0.90) | 0.015 (±0.3 × 10−3) |
| 10 | 290.6 (±5.8) | 0.002 (±0.04 × 10−3) | 48.0 (±1.70) | 0.015 (±0.45 × 10−3) |
| 11 | 296.3 (±11.8) | 0.002 (±0.08 × 10−3) | 30.2 (±1.40) | 0.023 (±1.04 × 10−3) |
| 12 | 292.1 (±4.4) | 0.002 (±0.03 × 10−3) | 26.9 (±0.70) | 0.026 (±0.65 × 10−3) |
| 13 | 292.8 (±8.8) | 0.002 (±0.06 × 10−3) | 48.50 (±0.70) | 0.005 (±0.25 × 10−3) |
| 14 | 293.4 (±14.7) | 0.002 (±0.1 × 10−3) | 21.3 (±0.60) | 0.033 (±0.99 × 10−3) |
aValues are means of three experiments, and standard deviation is given in parentheses.
Table 2.
Rate constants for the hydrolysis of codrugs 1-2, 5, and 9–14 in 80% rat plasma and 80% human plasma at 37°C.
| Rat plasmaa | Human plasmaa | |||
|---|---|---|---|---|
| Compd | t 1/2 (min) | K obs (min−1) | t 1/2 (min) | K obs (min−1) |
| 1 | immediate hydrolysis | — | 3.2 (±0.1) | 0.217 (±6 × 10−3) |
| 2 | 2.7 (±0.1) | 0.257 (±8 × 10−3) | 15.1 (±0.4) | 0.046 (±1 × 10−3) |
| 5 | 4.7 (±0.1) | 0.150 (±0.01) | 7.3 (±0.3) | 0.100 (±0.01) |
| 9 | 46.8 (±1.4) | 0.010 (±0.20 × 10−3) | 69.6 (±1.4) | 0.015 (±0.44 × 10−3) |
| 10 | 36.6 (±1.6) | 0.019 (±0.85 × 10−3) | 65.4 (±1.6) | 0.011 (±0.26 × 10−3) |
| 11 | 115.2 (±11.0) | 0.002 (±0.07 × 10−3) | 315.0 (±4.6) | 0.006 (±0.24 × 10−3) |
| 12 | 93.0 (±10.2) | 0.003 (±0.17 × 10−3) | 263.4 (±1.9) | 0.007 (±0.14 × 10−3) |
| 13 | 55.8 (±10.5) | 0.003 (±0.11 × 10−3) | 203.4 (±0.6) | 0.030 (±0.75x10 − 3) |
| 14 | 69.6 (±3.5) | 0.010 (±0.50 × 10−3) | 90.0 (±2.7) | 0.008 (±0.24 × 10−3) |
aValues are means of three experiments, standard deviation is given in parentheses.
Sulfur-containing amino acids have gained great attention as source of thiols for GSH synthesis [82]. A series of multifunctional thiol codrugs (9–14) were synthesized to overcome the prooxidant effect associated with LD therapy in parkinsonian models (Figure 6) [81]. In this regard, thiol antioxidants (N-acetyl-L-cysteine, methionine, dithiothreitol) prevent DA autoxidation, production of dopamine-melanin, and inhibition of dopamine-induced apoptosis [83]. Moreover, they increase levels of intracellular cysteine, the limiting amino acid in GSH biosynthesis, thus potentiating the natural cellular defense mechanisms against oxidative damage. The multifunctional codrugs 9-14 proved to be good radical scavengers. The LD and DA striatal level profiles indicate that codrugs 11 and 12 were able to induce sustained delivery of both LD and DA in rat striatum with respect to equimolar doses of LD [82].
Figure 6.
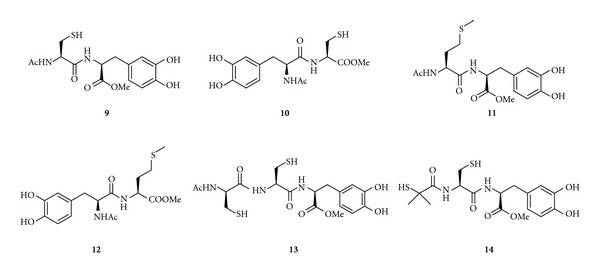
Chemical structures of cysteinyl codrugs 9–14.
In addition, Minelli et al. [84] showed that administration of codrug 11 to mice treated with Z-ILeu-Glu(OtBu)-Ala-Leu-CHO (PSI), used as a PD model, resulted in a reduction in dopaminergic neuronal death and a significant raise in GSH levels. In particular, codrug 11 could control the LD-induced oxidative stress in primary mesencephalic cultures and in newborn mice pups since in both cases GSH content results increased. Using newborn mice pups, characterized by incomplete formation of BBB, Minelli et al. [84] found that buthionine-[S,R]-sulfoximine-(BSO-) mediated GSH depletion prevented the increase of GSH levels promoted by codrug 11, supporting the role of GSH for codrug-11-induced protection. To investigate whether heme oxygenase (HO) activity was related to GSH levels, ZnPPIX was used as HO inhibitor. Compared to untreated control, and LD-treated newborn mice, brain GSH levels were increased by ZnPPIX indicating that HO activity was not essential to GSH synthesis. An injection of codrug 11 induced a significant increase in GSH levels that was markedly reduced by BSO indicating the essential role of γ-glutamylcysteine synthetase in increasing GSH brain levels. Codrug 11 exhibited in vivo protective effect against LD-induced stress through a mechanism via Nrf2 activation leading to a decrease in ROS generation and an increase in GSH. Therefore, this codrug might offer benefits in the treatment of PD and provide a potential alternative to LD therapy by avoiding nigrostriatal oxidative degeneration [84].
Maher et al. [85, 86] demonstrated that the conjugation of catechins with cysteine generates antioxidant compounds (15–20) with enhanced neuroprotective activity (Figure 7). The thiol conjugates 15–20 were active in protecting HT-22 nerve cells (EC50 between 36 and 65 μM) from oxidative stress-induced death. Although all the conjugates were able to scavenge mitochondrial generated ROS inside the cells, the majority of their neuroprotective activity seems to be dependent on their ability to maintain GSH levels. These compounds were able to maintain cellular GSH levels by enhancing the uptake of cystine/cysteine into cells by a mechanism that uncouples the uptake from system x c −, a Na+-independent cystine/glutamate antiporter [87]. System x c − transports cysteine into cells in a 1 : 1 exchange with glutamate. The importance of this system for the maintenance of the GSH levels in cells is demonstrated by the loss of GSH and subsequent cell death in nerve and other types of cells following exposure to millimolar concentrations of extracellular glutamate. Treatments able to maintain GSH levels, in presence of an induced stress by GSH loss, have a significant potential for the treatment of neurodegenerative diseases.
Figure 7.
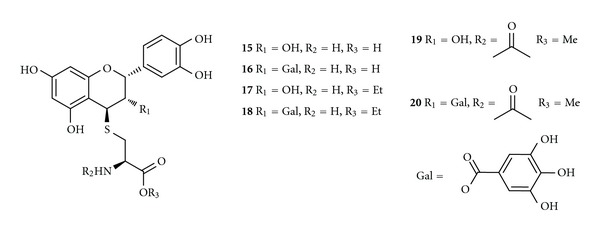
Chemical structures of thiol conjugates 15–20.
5. Technology-Based Strategies to Increase GSH Levels
The effectiveness of exogenous antioxidants to protect tissues from oxidative stress in vivo depends on the antioxidant used, its physicochemical and biopharmaceutical properties, and its bioavailability at the site of action [88, 89]. With the aim of improving the physicochemical, biopharmaceutical and drug delivery properties of neuroprotective antioxidants, the technology-based strategy could be useful for the treatment of several diseases in which oxidative stress plays an important role [59, 90]. Particularly, this approach could be adopted in order to selectively deliver antioxidants to tissues in sufficient concentrations to reduce the oxidative damage. In order to afford neuroprotection and to facilitate the delivery of GSH across the BBB, several GSH delivery systems, such as liposomes, nanoparticles, and dendrimers, were developed.
Liposomes are considered as carrier systems for therapeutically active compounds due to their unique characteristics such as capability of incorporating hydrophilic and hydrophobic drugs, good compatibility, low toxicity, lack of immune system activation, and targeted delivery of bioactive compounds to the site of action [91]. Liposome technology has been recently used in the treatment of neurodegenerative diseases. In this context, GSH has been encapsulated in liposomes in order to replenish intracellular GSH and provide neuroprotection in an in vitro model of PD [92]. The formulation of GSH has been encapsulated in lipid vesicles made of lecithin and glycerol and then tested on mixed mesencephalic cultures treated with paraquat plus maneb. Zeevalk et al. [92] observed that liposomal GSH was taken up into neurons and astrocytes via an endosomal process, and subsequently the endosomes containing liposomal-GSH were fused with lysosomes (Figure 8).
Figure 8.
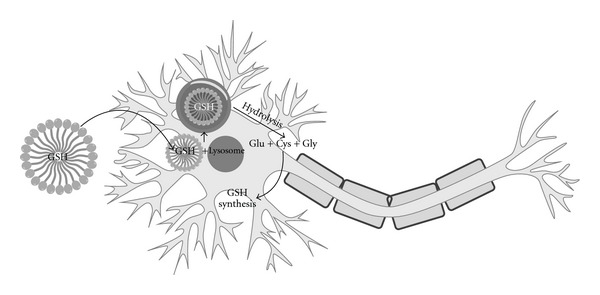
Liposomal GSH delivery to neurons and relative hydrolysis following fusion with lysosome.
In these conditions, GSH was hydrolyzed and its constituent amino acids (glutamate, cysteine, and glycine) released from lysosomes could be used for the GSH biosynthesis. The results obtained by Zeevalk et al. [92] suggested that this formulation was 100-fold more potent than nonliposomal-GSH in providing substrates for the maintenance of intracellular GSH in neuronal cells. Moreover, liposomal-GSH dose-dependently provided complete neuroprotection of dopaminergic neurons treated with paraquat plus maneb with an EC50 of 10.5 μM ± 1.08. These findings suggest that liposomal-GSH represents a promising therapeutic strategy for neuronal maintenance in pathologies characterized by GSH depletion.
Nanoparticles (NPs) are solid colloidal particles made of polymeric materials ranging in size from 1–1000 nm. NPs are used as carrier systems in which the drug is dissolved, entrapped, encapsulated, adsorbed, or chemically linked to the surface [93]. The advantages of NPs are high drug-loading capacity and resistance against chemical and enzymatic degradation. Coating NPs with hydrophilic polymer is a promising strategy in order to prolong their presence in plasma and the therapeutic effect. The surface modification of NPs can be achieved using polyethylene glycol (PEG) or polysaccharides such as chitosan, dextran, pectin, and hyaluronic acid [94, 95]. NPs have been employed for delivering GSH to CNS. In this regard, a series of NPs containing GSH (GS-PEG-GS, 21) were prepared with PEGs of various molecular weights (Figure 9) [96]. PEG was used because of its well-established biocompatibility, low immunogenicity, low antigenicity, and low toxicity [97].
Figure 9.
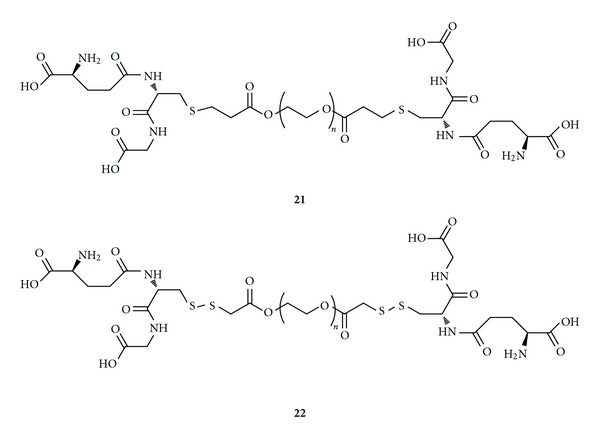
Chemical structures of GS-PEG-SG (21) and GS-SPEGS-SG (22).
Unfortunately, GS-PEG-GS nanoparticles were not able to exert their antioxidant activity because the thiol groups were consumed during Michael addition. Thus, a disulfide bridge was proposed for antioxidant delivery in order to release GSH when the pH was low enough to enable thiol formation. Disulfide-linked GSH NPs (GS-SPEGS-SG, 22) were synthesized and tested on SH-SY5Y cells challenged with 100 μM H2O2, a compound that induces oxidative stress. The GS-SPEGS-SG NPs were 100% at protecting SH-SY5Y cells at 250 μM from oxidative stress, while the GS-PEG-GS did not offer protection [96]. According to these data, this approach could be employed in treating diseases typically associated with increased ROS levels.
Chitosan-GSH nanoparticles (CS-GSH NPs) have been developed by Koo et al. [98] as delivery system for enhancing stability and bioavailability of GSH. Chitosan (CS) nanodelivery system offers many advantages: (a) it is not toxic; (b) it is biodegradable; (c) it is biocompatible; d) it has good bioadhesibility and water dispersibility [99, 100]. Thus, CS-GSH was synthesized using a radical polymerization method, and CS-GSH NPs were prepared by ionic gelation of CS-GSH with sodium tripolyphosphate (TPP) (Figure 10). The resulting NPs showed a good entrapment and loading efficiency. Furthermore, to investigate the CS-GSH NPs stability under oxidative stress, the effect of the presence of H2O2 on their thiol groups was evaluated. The reduction of thiol groups of the CS-GSH NPs under oxidative stress resulted in being 1.5-fold lower than that of free GSH [98]. These results suggest that CS-GSH NPs could be used as effective delivery carriers of GSH under oxidative insults, but further studies on animal models of PD and AD are necessary in order to evaluate their true efficacy.
Figure 10.
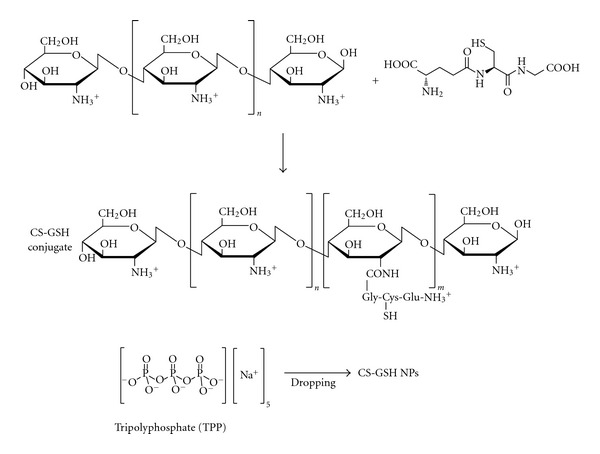
Scheme for GS-GSH NPs preparation as reported by Koo et al. [98].
The use of appropriate nanocarrier systems for GSH may be useful because they are noninvasive systems and protect the molecule to be delivered against inactivation mechanism and clearance. However, they are characterized by limiting factors as safety and toxicity. At present, few data are available about the utilization and human application of nanocarrier systems for transport across the BBB and CNS delivery. Thus, the future clinical study of GSH delivery systems for neurodegenerative diseases is strongly recommended.
6. Conclusions
The drug delivery to CNS is a complex and challenging task requiring close collaboration of several scientific areas including pharmaceutical and technological sciences, biological chemistry, and pharmacology. In this context, this paper has investigated multidisciplinary approaches such as the codrug and nanocarriers strategies that could be used to treat neurodegenerative disorders associated with GSH deficiency. GSH and cysteinyl codrugs have been designed on the basis of combining suitable groups of the antioxidant portion with available drugs without altering their inherent pharmacodynamic properties with improved physiochemical properties of drugs. This promising approach has also been used to resolve the issues like permeation, solubility of drug, stability, drug resistance, oral absorption, and brain delivery but still there are very few well-established candidates that have been approved for clinical applications.
Novel experimental neuroprotective strategies include formulations containing GSH, such as nanocarrier systems. This approach could be adopted in order to selectively deliver GSH to tissues in sufficient concentrations to reduce the oxidative damage, but few data are available about clinical studies.
Although the potential use of these strategies needs further exhaustive studies, they may offer a promising therapeutic alternative for reducing the GSH functional loss related to human diseases such as PD and AD.
References
- 1.Jellinker KA. General aspects of neurodegeneration. Journal of Neural Transmission . 2003;65:101–144. doi: 10.1007/978-3-7091-0643-3_7. [DOI] [PubMed] [Google Scholar]
- 2.Jellinger KA. Basic mechanisms of neurodegeneration: a critical update. Journal of Cellular and Molecular Medicine. 2010;14(3):457–487. doi: 10.1111/j.1582-4934.2010.01010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. International Journal of Biochemistry and Cell Biology. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Melo A, Monteiro L, Lima RMF, de Oliveira DM, de Cerqueira MD, El-Bacha RS. Oxidative stress in neurodegenerative diseases: mechanism and therapeutic perspectives. Oxidative Medicine and Cellular Longevity. 2011;2011:14 pages. doi: 10.1155/2011/467180. Article ID 467180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Markesbery WR. The role of oxidative stress in Alzheimer disease. Archives of Neurology. 1999;56(12):1449–1452. doi: 10.1001/archneur.56.12.1449. [DOI] [PubMed] [Google Scholar]
- 6.Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Current Medicinal Chemistry. 2001;8(7):721–738. doi: 10.2174/0929867013372922. [DOI] [PubMed] [Google Scholar]
- 7.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262(5134):689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 8.Jenner P. Oxidative stress in Parkinson’s disease. Annals of Neurology. 2003;53(supplement 3):S26–S38. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- 9.Polidori MC, Griffiths HR, Mariani E, Mecocci P. Hallmarks of protein oxidative damage in neurodegenerative diseases: focus on Alzheimer’s disease. Amino Acids. 2007;32(4):553–559. doi: 10.1007/s00726-006-0431-x. [DOI] [PubMed] [Google Scholar]
- 10.Moreira PI, Honda K, Quan L, et al. Alzheimer’s disease and oxidative stress: the old problem remains unsolved. Current Medicinal Chemistry. 2005;5(1):51–62. [Google Scholar]
- 11.Farooqui T, Farooqui AA. Aging: an important factor for the pathogenesis of neurodegenerative diseases. Mechanisms of Ageing and Development. 2009;130(4):203–215. doi: 10.1016/j.mad.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 12.Fahn S, Cohen G. The oxidant stress hypothesis in Parkinson’s disease: evidence supporting it. Annals of Neurology. 1992;32(6):804–812. doi: 10.1002/ana.410320616. [DOI] [PubMed] [Google Scholar]
- 13.Offen D, Gorodin S, Melamed E, Hanania J, Malik Z. Dopamine-melanin is actively phagocytized by PC12 cells and cerebellar granular cells: possible implications for the etiology of Parkinson’s disease. Neuroscience Letters. 1999;260(2):101–104. doi: 10.1016/s0304-3940(98)00950-1. [DOI] [PubMed] [Google Scholar]
- 14.Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. European Journal of Biochemistry. 2000;267(16):4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 15.Bains JS, Shaw CA. Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Research Reviews. 1997;25(3):335–358. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- 16.Lee M, Cho T, Jantaratnotai N, Wang YT, McGeer E, McGeer PL. Depletion of GSH in glial cells induces neurotoxicity: relevance to aging and degenerative neurological diseases. The FASEB Journal. 2010;24(7):2533–2545. doi: 10.1096/fj.09-149997. [DOI] [PubMed] [Google Scholar]
- 17.Pearce RKB, Owen A, Daniel S, Jenner P, Marsden CD. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. Journal of Neural Transmission. 1997;104(6-7):661–677. doi: 10.1007/BF01291884. [DOI] [PubMed] [Google Scholar]
- 18.Sian J, Dexter DT, Lees AJ, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Annals of Neurology. 1994;36(3):348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 19.Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience. 1993;52(1):1–6. doi: 10.1016/0306-4522(93)90175-f. [DOI] [PubMed] [Google Scholar]
- 20.Garrido M, Tereshchenko Y, Zhevtsova Z, Taschenberger G, Bähr M, Kügler S. Glutathione depletion and overproduction both initiate degeneration of nigral dopaminergic neurons. Acta Neuropathologica. 2011;121(4):475–485. doi: 10.1007/s00401-010-0791-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albers DS, Beal MF. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. Journal of Neural Transmission. 2000;(59):133–154. doi: 10.1007/978-3-7091-6781-6_16. [DOI] [PubMed] [Google Scholar]
- 22.Jenner P. Oxidative mechanisms in nigral cell death in Parkinson’s disease. Movement Disorders. 1998;13(1):24–34. [PubMed] [Google Scholar]
- 23.Spencer JPE, Jenner P, Daniel SE, Lees AJ, Marsden DC, Halliwell B. Conjugates of catecholamines with cysteine and GSH in Parkinson’s disease: possible mechanisms of formation involving reactive oxygen species. Journal of Neurochemistry. 1998;71(5):2112–2122. doi: 10.1046/j.1471-4159.1998.71052112.x. [DOI] [PubMed] [Google Scholar]
- 24.Andersen JK, Mo JQ, Horn DG, et al. Effect of buthionine sulfoximine, a synthesis inhibitor of the antioxidant glutathione, on the murine nigrostriatal neurons. Journal of Neurochemistry. 1996;67(5):2164–2171. doi: 10.1046/j.1471-4159.1996.67052164.x. [DOI] [PubMed] [Google Scholar]
- 25.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 26.Chinta SJ, Andersen JK. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson’s disease. Free Radical Biology and Medicine. 2006;41(9):1442–1448. doi: 10.1016/j.freeradbiomed.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 27.Chinta SJ, Kumar MJ, Hsu M, et al. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. Journal of Neuroscience. 2007;27(51):13997–14006. doi: 10.1523/JNEUROSCI.3885-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lau LF, Brodney MA. Therapeutic approaches for the treatment of Alzheimer’s disease: an overview. Topics in Medicinal Chemistry. 2008;2:1–24. [Google Scholar]
- 29.Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Review: Alzheimer’s amyloid β-peptide-associated free radical oxidative stress and neurotoxicity. Journal of Structural Biology. 2000;130(2-3):184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- 30.Maltsev AV, Bystryak S, Galzitskaya OV. The role of β-amyloid peptide in neurodegenerative diseases. Ageing Research Reviews. 2011;10:440–452. doi: 10.1016/j.arr.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Gu M, Owen AD, Toffa SEK, et al. Mitochondrial function, GSH and iron in neurodegeneration and Lewy body diseases. Journal of the Neurological Sciences. 1998;158(1):24–29. doi: 10.1016/s0022-510x(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Harrell LE, Shenvi S, Hagen T, Liu RM. Gender differences in glutathione metabolism in Alzheimer’s disease. Journal of Neuroscience Research. 2005;79(6):861–867. doi: 10.1002/jnr.20424. [DOI] [PubMed] [Google Scholar]
- 33.Adams JD, Klaidman LK, Odunze IN, Shen HC, Miller CA. Alzheimer’s and Parkinson’s disease: brain levels of glutathione, glutathione disulfide, and vitamin E. Molecular and Chemical Neuropathology. 1991;14(3):213–226. doi: 10.1007/BF03159937. [DOI] [PubMed] [Google Scholar]
- 34.Perry TL, Yong VW, Bergeron C, Hansen S, Jones K. Amino acids, glutathione, and glutathione transferase activity in the brains of patients with Alzheimer’s disease. Annals of Neurology. 2005;21(4):331–336. doi: 10.1002/ana.410210403. [DOI] [PubMed] [Google Scholar]
- 35.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid β- peptide. Journal of Neurochemistry. 1997;68(1):255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 36.Butterfield DA, Pocernich CB, Drake J. Elevated glutathione as a therapeutic strategy in Alzheimer’s disease. Drug Development Research. 2002;56(3):428–437. [Google Scholar]
- 37.Hammond CL, Lee TK, Ballatori N. Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. Journal of Hepatology. 2001;34(6):946–954. doi: 10.1016/s0168-8278(01)00037-x. [DOI] [PubMed] [Google Scholar]
- 38.Ghosh N, Ghosh R, Mandal SC. Antioxidant protection: a promising therapeutic intervention in neurodegenerative disease. Free Radical Research. 2011;45(8):888–905. doi: 10.3109/10715762.2011.574290. [DOI] [PubMed] [Google Scholar]
- 39.di Matteo V, Esposito E. Biochemical and therapeutic effects of antioxidants in the treatment of Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Current Drug Targets, CNS & Neurological Disorders. 2003;2(2):95–107. doi: 10.2174/1568007033482959. [DOI] [PubMed] [Google Scholar]
- 40.Cacciatore I, Cornacchia C, Baldassarre L, et al. GPE and GPE analogues as promising neuroprotective agents. Mini Reviews in Medicinal Chemistry. 2012;12(1):13–23. doi: 10.2174/138955712798868995. [DOI] [PubMed] [Google Scholar]
- 41.Cornacchia C, Cacciatore I, Baldassarre L, Mollica A, Feliciani F, Pinnen F. 2,5-Diketopiperazines as neuroprotective agents. Mini Reviews in Medicinal Chemistry. 2012;12:2–12. doi: 10.2174/138955712798868959. [DOI] [PubMed] [Google Scholar]
- 42.Moosmann B, Behl C. Antioxidants as treatment for neurodegenerative disorders. Expert Opinion on Investigational Drugs. 2002;11(10):1407–1435. doi: 10.1517/13543784.11.10.1407. [DOI] [PubMed] [Google Scholar]
- 43.Dringen R. Metabolism and functions of glutathione in brain. Progress in Neurobiology. 2000;62(6):649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 44.Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biological Chemistry. 2003;384(4):505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 45.Bains JS, Shaw CA. Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Research Reviews. 1997;25(3):335–358. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- 46.Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomedicine and Pharmacotherapy. 2003;57(3):145–155. doi: 10.1016/s0753-3322(03)00043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biological Chemistry. 2009;390(3):191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin HL, Teismann P. Glutathione—a review on its role and significance in Parkinson’s dysregulation and the etiology and progression of human diseases. The FASEB Journal. 2009;23:3263–3272. doi: 10.1096/fj.08-125443. [DOI] [PubMed] [Google Scholar]
- 49.Guidi I, Galimberti D, Lonati S, et al. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiology of Aging. 2006;27(2):262–269. doi: 10.1016/j.neurobiolaging.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Dumont M, Beal MF. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radical Biology and Medicine. 2011;51:1014–1026. doi: 10.1016/j.freeradbiomed.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mcmanus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant mitoq prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. The Journal of Neuroscience. 2011;31:15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beckman JS, Estévez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends in Neurosciences. 2001;24(supplement 11):S15–S20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- 53.Vargas MR, Johnson DA, Johnson JA. Decreased glutathione accelerates neurologiCal deficit and mitochondrial pathology in familial ALS-linked hSOD1G93A mice model. Neurobiology of Disease. 2011;43(3):543–551. doi: 10.1016/j.nbd.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Behl C, Moosmann B. Antioxidant neuroprotection in Alzheimer’s disease as preventive and therapeutic approach. Free Radical Biology and Medicine. 2002;33(2):182–191. doi: 10.1016/s0891-5849(02)00883-3. [DOI] [PubMed] [Google Scholar]
- 55.Zeevalk GD, Manzino L, Sonsalla PK, Bernard LP. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson’s disease. Experimental Neurology. 2007;203(2):512–520. doi: 10.1016/j.expneurol.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cacciatore I, Cornacchia C, Pinnen F, Mollica A, di Stefano A. Prodrug approach for increasing cellular glutathione levels. Molecules. 2010;15(3):1242–1264. doi: 10.3390/molecules15031242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cacciatore I, Cocco A, Costa M, et al. Biochemical properties of new synthetic carnosine analogues containing the residue of 2,3-diaminopropionic acid: the effect of N-acetylation. Amino Acids. 2005;28(1):77–83. doi: 10.1007/s00726-004-0142-0. [DOI] [PubMed] [Google Scholar]
- 58.Brunetti L, Cacciatore I, di Stefano A, et al. Synthesis and biological evaluation of a novel pyroglutamyl-modified TRH analogue. Farmaco. 2002;57(6):479–486. doi: 10.1016/s0014-827x(02)01232-6. [DOI] [PubMed] [Google Scholar]
- 59.Denora N, Trapani A, Laquintana V, Lopedota A, Trapani G. Recent advances in medicinal chemistry and pharmaceutical technology-strategies for drug delivery to the brain. Current Topics in Medicinal Chemistry. 2009;9(2):182–196. doi: 10.2174/156802609787521571. [DOI] [PubMed] [Google Scholar]
- 60.Cacciatore I, di Stefano A, Luisi G, Pinnen F, Sozio P. Transition state isosteres of the γ-glutamyl peptide bond hydrolysis: synthesis and characterization of the ψ (CH2NH) pseudopeptide analogue of glutathione. Journal of Peptide Science. 2004;10(2):109–114. doi: 10.1002/psc.501. [DOI] [PubMed] [Google Scholar]
- 61.Cacciatore I, Caccuri AM, di Stefano A, et al. Synthesis and activity of novel glutathione analogues containing an urethane backbone linkage. Farmaco. 2003;58(9):787–793. doi: 10.1016/S0014-827X(03)00135-6. [DOI] [PubMed] [Google Scholar]
- 62.Cacciatore I, Caccuri AM, Cocco A, et al. Potent isozyme-selective inhibition of human glutathione S-transferase A1-1 by a novel glutathione S-conjugate. Amino Acids. 2005;29(3):255–261. doi: 10.1007/s00726-005-0232-7. [DOI] [PubMed] [Google Scholar]
- 63.Das N, Dhanawat M, Dash B, Nagarwal RC, Shrivastava SK. Codrug: an efficient approach for drug optimization. European Journal of Pharmaceutical Sciences. 2010;41(5):571–588. doi: 10.1016/j.ejps.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 64.Decker M. Hybrid molecules incorporating natural products: applications in cancer therapy, neurodegenerative disorders and beyond. Current Medicinal Chemistry. 2011;18(10):1464–1475. doi: 10.2174/092986711795328355. [DOI] [PubMed] [Google Scholar]
- 65.Minelli A, Conte C, Cacciatore I, Cornacchia C, Pinnen F. Molecular mechanism underlying the cerebral effect of Gly-Pro-Glu tripeptide bound to L-dopa in a Parkinson’s animal model. doi: 10.1007/s00726-011-1210-x. Amino Acids. In press. [DOI] [PubMed] [Google Scholar]
- 66.Sozio P, Iannitelli A, Cerasa LS, et al. New L-dopa codrugs as potential antiparkinson agents. Archiv der Pharmazie. 2008;341(7):412–417. doi: 10.1002/ardp.200700228. [DOI] [PubMed] [Google Scholar]
- 67.Malakoutikhah M, Teixidó M, Giralt E. Toward an optimal blood-brain barrier shuttle by synthesis and evaluation of peptide libraries. Journal of Medicinal Chemistry. 2008;51(16):4881–4889. doi: 10.1021/jm800156z. [DOI] [PubMed] [Google Scholar]
- 68.Kannan R, Kuhlenkamp JF, Jeandidier E, Trinh H, Ookhtens M, Kaplowitz N. Evidence for carrier-mediated transport of glutathione across the blood-brain barrier in the rat. Journal of Clinical Investigation. 1990;85(6):2009–2013. doi: 10.1172/JCI114666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pinnen F, Cacciatore I, Cornacchia C, et al. Synthesis and study of L-dopa-glutathione codrugs as new anti-Parkinson agents with free radical scavenging properties. Journal of Medicinal Chemistry. 2007;50(10):2506–2515. doi: 10.1021/jm070037v. [DOI] [PubMed] [Google Scholar]
- 70.More SS, Vince R. Design, synthesis and biological evaluation of glutathione peptidomimetics as components of anti-Parkinson prodrugs. Journal of Medicinal Chemistry. 2008;51(15):4581–4588. doi: 10.1021/jm800239v. [DOI] [PubMed] [Google Scholar]
- 71.Bickel U, Kang YS, Pardridge WM. In vivo cleavability of a disulfide-based chimeric opioid peptide in rat brain. Bioconjugate Chemistry. 1995;6(2):211–218. doi: 10.1021/bc00032a009. [DOI] [PubMed] [Google Scholar]
- 72.Pinnen F, Cacciatore I, Cornacchia C, et al. CNS delivery of l-dopa by a new hybrid glutathione-methionine peptidomimetic prodrug. Amino Acids. 2012;42(1):261–269. doi: 10.1007/s00726-010-0804-z. [DOI] [PubMed] [Google Scholar]
- 73.Ehrlich K, Viirlaid S, Mahlapuu R, et al. Design, synthesis and properties of novel powerful antioxidants, glutathione analogues. Free Radical Research. 2007;41(7):779–787. doi: 10.1080/10715760701348611. [DOI] [PubMed] [Google Scholar]
- 74.Ehrlich K, Ida K, Mahlapuu R, et al. Characterization of UPF peptides, members of the glutathione analogues library, on the basis of their effects on oxidative stress-related enzymes. Free Radical Research. 2009;43(6):572–580. doi: 10.1080/10715760902918691. [DOI] [PubMed] [Google Scholar]
- 75.Põder P, Zilmer M, Starkopf J, et al. An antioxidant tetrapeptide UPF1 in rats has a neuroprotective effect in transient global brain ischemia. Neuroscience Letters. 2004;370(1):45–50. doi: 10.1016/j.neulet.2004.07.063. [DOI] [PubMed] [Google Scholar]
- 76.Karelson E, Mahlapuu R, Zilmer M, Soomets U, Bogdanovic N, Langel Ü. Possible signaling by glutathione and its novel analogue through potent stimulation of frontocortical G proteins in normal aging and in Alzheimer’s disease. Annals of the New York Academy of Sciences. 2002;973:537–540. doi: 10.1111/j.1749-6632.2002.tb04696.x. [DOI] [PubMed] [Google Scholar]
- 77.Pinnen F, Sozio P, Cacciatore I, et al. Ibuprofen and glutathione conjugate as a potential therapeutic agent for treating Alzheimer’s disease. Archiv der Pharmazie. 2011;344(3):139–148. doi: 10.1002/ardp.201000209. [DOI] [PubMed] [Google Scholar]
- 78.Prasad KN, Cole WC, Prasad KC. Risk factors for Alzheimer’s disease: role of multiple antioxidants, non-steroidal anti-inflammatory and cholinergic agents alone or in combination in prevention and treatment. Journal of the American College of Nutrition. 2002;21(6):506–522. doi: 10.1080/07315724.2002.10719249. [DOI] [PubMed] [Google Scholar]
- 79.Lim GP, Yang F, Chu T, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. Journal of Neuroscience. 2000;20(15):5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yan Q, Zhang J, Liu H, et al. Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer’s disease. Journal of Neuroscience. 2003;23(20):7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Atmaca G. Antioxidant effects of sulfur-containing amino acids. Yonsei Medical Journal. 2004;45(5):776–788. doi: 10.3349/ymj.2004.45.5.776. [DOI] [PubMed] [Google Scholar]
- 82.Pinnen F, Cacciatore I, Cornacchia C, et al. Codrugs linking L-dopa and sulfur-containing antioxidants: new pharmacological tools against Parkinson’s disease. Journal of Medicinal Chemistry. 2009;52(2):559–563. doi: 10.1021/jm801266x. [DOI] [PubMed] [Google Scholar]
- 83.Offen D, Ziv I, Sternin H, Melamed E, Hochman A. Prevention of dopamine-induced cell death by thiol antioxidants: possible implications for treatment of Parkinson’s disease. Experimental Neurology. 1996;141(1):32–39. doi: 10.1006/exnr.1996.0136. [DOI] [PubMed] [Google Scholar]
- 84.Minelli A, Conte C, Prudenzi E, et al. N-Acetyl-L-Methionyl-L-Dopa-Methyl Ester as a dual acting drug that relieves L-Dopa-induced oxidative toxicity. Free Radical Biology and Medicine. 2010;49(1):31–39. doi: 10.1016/j.freeradbiomed.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 85.Torres JL, Lozano C, Maher P. Conjugation of catechins with cysteine generates antioxidant compounds with enhanced neuroprotective activity. Phytochemistry. 2005;66(17):2032–2037. doi: 10.1016/j.phytochem.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 86.Maher P, Lewerenz J, Lozano C, Torres JL. A novel approach to enhancing cellular glutathione levels. Journal of Neurochemistry. 2008;107(3):690–700. doi: 10.1111/j.1471-4159.2008.05620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. Journal of Biological Chemistry. 1999;274(17):11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- 88.Steinhubl SR. Why have antioxidants failed in clinical trials? American Journal of Cardiology. 2008;101(supplement 10):S14–S19. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 89.Ratnam DV, Ankola DD, Bhardwaj V, Sahana DK, Kumar MNVR. Role of antioxidants in prophylaxis and therapy: a pharmaceutical perspective. Journal of Controlled Release. 2006;113(3):189–207. doi: 10.1016/j.jconrel.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 90.Gilmore JL, Yi X, Quan L, Kabanov AV. Novel nanomaterials for clinical neuroscience. Journal of NeuroImmune Pharmacology. 2008;3(2):83–94. doi: 10.1007/s11481-007-9099-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen C, Han D, Cai C, Tang X. An overview of liposome lyophilization and its future potential. Journal of Controlled Release. 2010;142(3):299–311. doi: 10.1016/j.jconrel.2009.10.024. [DOI] [PubMed] [Google Scholar]
- 92.Zeevalk GD, Bernard LP, Guilford FT. Liposomal-glutathione provides maintenance of intracellular glutathione and neuroprotection in mesencephalic neuronal cells. Neurochemical Research. 2010;35(10):1575–1587. doi: 10.1007/s11064-010-0217-0. [DOI] [PubMed] [Google Scholar]
- 93.de Jong WH, Borm PJA. Drug delivery and nanoparticles: applications and hazards. International Journal of Nanomedicine. 2008;3(2):133–149. doi: 10.2147/ijn.s596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aumelas A, Serrero A, Durand A, Dellacherie E, Leonard M. Nanoparticles of hydrophobically modified dextrans as potential drug carrier systems. Colloids and Surfaces B. 2007;59(1):74–80. doi: 10.1016/j.colsurfb.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 95.Esmaeili F, Ghahremani MH, Esmaeili B, Khoshayand MR, Atyabi F, Dinarvand R. PLGA nanoparticles of different surface properties: preparation and evaluation of their body distribution. International Journal of Pharmaceutics. 2008;349(1-2):249–255. doi: 10.1016/j.ijpharm.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 96.Williams SR, Lepene BS, Thatcher CD, Long TE. Synthesis and characterization of poly(ethylene glycol)-glutathione conjugate self-assembled nanoparticles for antioxidant delivery. Biomacromolecules. 2009;10(1):155–161. doi: 10.1021/bm801058j. [DOI] [PubMed] [Google Scholar]
- 97.Alcantar NA, Aydil ES, Israelachvili JN. Polyethylene glycol-coated biocompatible surfaces. Journal of Biomedical Materials Research. 2000;51(3):343–351. doi: 10.1002/1097-4636(20000905)51:3<343::aid-jbm7>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 98.Koo SH, Lee JS, Kim GH, Lee HG. Preparation, characteristics, and stability of glutathione-loaded nanoparticles. Journal of Agricultural and Food Chemistry. 2011;9:11264–11269. doi: 10.1021/jf2024648. [DOI] [PubMed] [Google Scholar]
- 99.Agnihotri SA, Mallikarjuna NN, Aminabhavi TM. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. Journal of Controlled Release. 2004;100(1):5–28. doi: 10.1016/j.jconrel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 100.Takeuchi H, Yamamoto H, Kawashima Y. Mucoadhesive nanoparticulate systems for peptide drug delivery. Advanced Drug Delivery Reviews. 2001;47(1):39–54. doi: 10.1016/s0169-409x(00)00120-4. [DOI] [PubMed] [Google Scholar]