

Abstract
DNA damage induces cell cycle arrest through both Chk1 and the p53 tumor suppressor protein, the latter arresting cells through induction of p21waf1 protein. Arrest permits cells to repair the damage and recover. The frequent loss of p53 in tumor cells makes them more dependent on Chk1 for arrest and survival. However, some p53 wild type tumor cell lines, such as HCT116 and U2OS, are also sensitive to inhibition of Chk1 due to attenuated p21waf1 induction upon DNA damage. The purpose of this study is to determine the cause of this attenuated p21waf1 protein induction. We find that neither the induction of p21waf1 mRNA nor protein half-life is sufficient to explain the low p21waf1 protein levels in HCT116 and U2OS cells. The induced mRNA associates with polysomes but little protein is made suggesting these two cell lines have a reduced rate of p21waf1 mRNA translation. This represents a novel mechanism for disruption of the p53-p21waf1 pathway as currently known mechanisms involve either mutation of p53 or reduction of p53 protein levels. As a consequence, this attenuated p21waf1 expression may render some p53 wild type tumors sensitive to a combination of DNA damage plus checkpoint inhibition.
Keywords: cell cycle checkpoints, Chk1, MK-8776, p21waf1, p53 response, UCN-01
Introduction
The DNA of a cell is constantly under attack by both external insults, such as the sun’s radiation, and internal insults, such as free radicals produced during normal metabolism. To ensure integrity of the DNA, the cell utilizes DNA damage checkpoints to arrest cell cycle progression and allow time for DNA repair. When DNA double-strand breaks are detected, ATM kinase is activated, which, in turn, activates Chk2 via phosphorylation of threonine 68.1 Double-strand breaks are also processed to single-stranded DNA that activates ATR, and, as a consequence, Chk1 is phosphorylated at serine 345.2,3 Chk1 is then autophosphorylated at serine 296 to become fully activated.4 Subsequently, activated Chk1 and Chk2 inhibit the CDC25 family of phosphatases that remove the inhibitory phosphorylation on the cyclin-dependent kinase (CDK)/cyclin complexes.5 Thus, Chk1 and Chk2 activation leads to rapid cell cycle arrest. In addition, ATM, ATR, Chk1 and Chk2 phosphorylate the p53 tumor suppressor at serines 15 and 20, which disrupts the interaction between p53 and its negative regulator, MDM2.6 Once activated, p53 induces transcription of the CDK inhibitor p21waf1 and thus provides a second mechanism to arrest cell cycle progression.7
As the p53-p21waf1 pathway requires the transcription and accumulation of newly synthesized p21waf1 protein, it is slower to induce arrest than the Chk1/2-CDC25 pathway.7 However, the p53-p21waf1 pathway is crucial for maintenance of arrest, as shown by our studies comparing isogenic cell lines.8 For example, the topoisomerase I inhibitor SN38 induces S-phase arrest in the p53 wild-type MCF10A cells as well as their p53- and p21waf1-suppressed derivatives.8,9 Chk1 inhibition by 7-hydroxystaurosporine (UCN-01) had no impact on the p53 wild-type cells but abrogated arrest in both the derivatives resulting in S and G2 phase progression. Based on these observations, it was expected that all p53 wild-type tumors would be resistant to inhibition of Chk1 by UCN-01, but we identified several that remained sensitive. In HCT116 and MCF7 cells, Chk1 inhibition abrogated SN38-induced arrest.9 We also demonstrated that this sensitivity to checkpoint abrogation correlated with an attenuated induction of p21waf1.9
In this study, we examined the cause of the attenuated p21waf1 induction in HCT116 cells and in another p53 wild-type cell line, U2OS. We find that this defect is not due to a failure to induce p21waf1 mRNA or to a shorter protein half-life. The induced mRNA associates with polysomes, but little protein is made, suggesting these two tumor cell lines have a reduced rate of p21waf1 mRNA translation.
Results
Abrogation of cell cycle arrest by MK-8776 in HCT116 and U2OS.
Our previous studies using MCF10A cells showed that p53 can prevent UCN-01-mediated abrogation of S-phase arrest induced by SN38.8,10 We extended these experiments to p53 wild-type tumors, and found that p53 could also prevent UCN-01-mediated abrogation of arrest in some, but not all, cell lines.9 Cell lines that remained sensitive to checkpoint abrogation included HCT116 and MCF7. Here, we report that U2OS cells are also sensitive to checkpoint abrogation.
As UCN-01 has been shown to have many off-target effects, we reconfirmed these findings with a more specific Chk1 inhibitor, MK-8776 (previously known as SCH900776).11,12 SN38 at 10 ng/ml induces S-phase arrest in MCF10A and U2OS cells, but primarily a G2 arrest in HCT116 cell (Fig. 1). The limited S‑phase arrest in HCT116 cells has been attributed to a defect in Mre11.13 On removal of SN38 after 24 h, MCF10A cells remained arrested in S phase for at least an additional 24 h, whereas U2OS slowly progressed to G2 and HCT116 remained in G2.

Figure 1. Comparison of the efficacy of MK-8776 to abrogate SN38-induced S and G2 arrest in p53 wild-type cell lines. Cell were incubated with 10 ng/ml SN38 for 24 h and then incubated in either media with or without 1 µM MK-8776. Cells were harvested and assayed for DNA content by flow cytometry.
Addition of MK-8776 to SN38-arrested cells did not abrogate arrest in MCF10A cells (Fig. 1), while a similar experiment in the p53 mutant MDA-MB-231 cells rapidly abrogated both S and G2 arrest.12 In the U2OS cells, MK-8776 accelerated the rate of progression through S phase and through mitosis. After 24 h in MK-8776, a large proportion of the U2OS cells are seen with G1 and sub‑G1 DNA content. In the HCT116 cells, the majority of the cells remained in G2/M upon incubation with MK-8776. However, flow cytometry cannot discriminate between the G2/M populations. We have previously shown that Chk1 inhibition abrogates G2 arrest and induces mitosis in HCT116 cells but they fail to undergo cytokinesis.9 The result is tetraploid cells with numerous micronuclei. This mitotic catastrophe was also observed with MK-8776. These results demonstrate that some p53 wild-type cancer cells have limited capacity to maintain cell cycle arrest when damaged, and that the ability to arrest is dependent on Chk1.
Attenuated p21waf1 induction by SN38 in HCT116 and U2OS.
We previously noted a delayed p21waf1 protein induction in HCT116 cells compared with MCF10A in SN38-treated cells. We thus hypothesized that an adequate p21waf1 protein level is crucial for maintenance of cell cycle arrest, and this was confirmed when we observed abrogation of S-phase arrest by Chk1 inhibition in MCF10A/Δp21waf1 cells.9 To extend this research, we compared the absolute p53 and p21waf1 protein levels between MCF10A, HCT116 and U2OS cells following incubation with SN38 (Fig. 2A). The levels of protein presented here and in subsequent figures were obtained from densitometry of multiple exposures of western blots to avoid analysis of over-exposed bands and from comparison to a standard curve generated for each antigen demonstrating that the values recorded were in the linear range of detection. In addition, the same number of cells was loaded in each lane, and this resulted in a constant amount of actin in each lane.
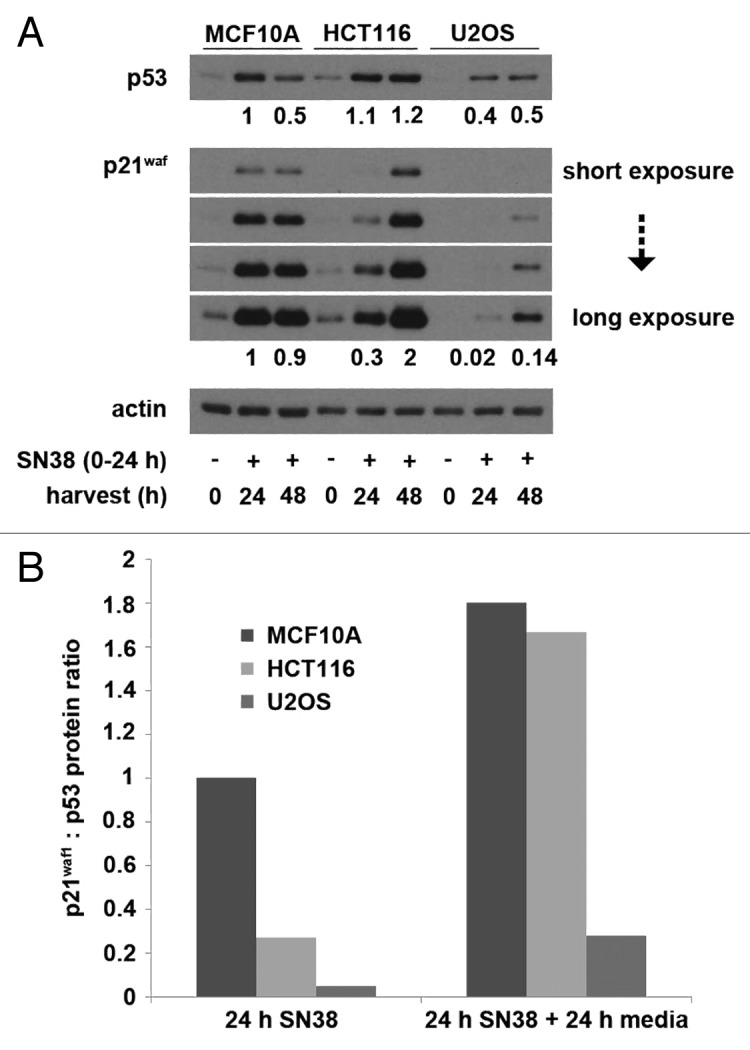
Figure 2. SN38-induced p53 and p21waf1 protein levels. (A) Cells were incubated with 10 ng/ml SN38 for 24 h and then released from SN38 for 24 h. The levels of p53 and p21waf1 protein were assessed by western blotting. Numerical values compare expression level to the 24 h-treated MCF10A cells. (B) The ratio of p21waf1 to p53 protein levels after 24 h of SN38 and after an additional 24 h in fresh medium were compared with that of MCF10A cells after 24 h of SN38.
Overall, MCF10A and HCT116 cells showed fairly similar levels of p53, while U2OS cells demonstrated only about 2-fold less by 24 h of SN38 treatment. In contrast, there were marked differences in the absolute levels of p21waf1. The p21waf1 expression in HCT116 and U2OS was about 30% and 2%, respectively, compared with MCF10A at 24 h of SN38. Interestingly, both the p53 and p21waf1 levels continued to increase in HCT116 and U2OS cells after removal of SN38, whereas both proteins decreased in MCF10A. To show that this attenuated p21waf1 induction is not due to low p53 induction, we compared the ratio of p21waf1 to p53 protein level. The results clearly show that despite strong activation of p53, the HCT116 and U2OS cells exhibit a very attenuated induction of p21waf1 at 24 h of SN38 (Fig. 2B). This p21waf1 to p53 ratio increased in all three cell lines within the next 24 h.
Comparison of p53 and p21waf1 protein kinetics induced by SN38.
To obtain further perspective on the relative induction of p53 and p21waf1 between 0 h and 24 h, we performed a detailed kinetic analysis during incubation with SN38. The p53 protein was markedly elevated by 4 h and remained elevated through 24 h in all three cell lines (Fig. 3A–C). The results were expressed in two different ways: (1) relative to the protein level at 0 h for each cell line or (2) relative to the protein level at 0 h for MCF10A cells (Fig. 3D and E). The latter expression provides a comparison of the absolute level of the protein. Although the fold induction of p53 was similar in all three cell lines, the lower basal levels of p53 in U2OS resulted in lower absolute p53 levels than MCF10A after 24 h.

Figure 3. Kinetics of p53 and p21waf1 protein expression following SN38 treatment. (A) MCF10A, (B) HCT116 and (C) U2OS cells were incubated with 10 ng/ml SN38 from 0–24 h. The drug was removed and cells incubated for an additional 24 h in fresh medium. Cells were harvested at the indicated times and proteins detected by western blotting. (D) p53 and (e) p21waf1 protein levels were quantified by densitometry of multiple exposures of western blots and from comparison to a standard curve generated for each antigen. The left panels show protein induction compared with the untreated control of each cell line. The right panels show protein induction compared with the level in untreated MCF10A cells.
Induction of p21waf1 protein was slightly slower than p53 in MCF10A cells but was clearly detectable by 8–10 h. At comparable exposures of the western blots, p21waf1 did not become detectable until 20–24 h in the other two cell lines. When expressed as fold induction, MCF10A and HCT116 cells were fairly similar, but the absolute level of protein present was significantly less in HCT116 cells. In U2OS cells, both the fold induction and the absolute level of protein expressed was significantly less than MCF10A cells. These results reiterate that the amount of p21waf1 protein does not reflect the level of p53 protein induced.
We concurrently assessed several DNA damage responses that occur when cells are damaged. In all the cells, Chk1 was phosphorylated at serines 345 and 296 within 2 h, suggesting that the delayed p21waf1 induction in HCT116 and U2OS is not due to lack of drug uptake or DNA damage. Phosphorylation of p53 at serine 15 occurred with the same kinetics in MCF10A and HCT116 cells, but only in the former did this correlate with the kinetics of p21waf1 induction. U2OS cells showed a lower level of p53 phosphorylation that may relate to the slightly lower level of p53 induced in these cells. Overall, these results show that SN38 is able to damage the DNA, activate Chk1, induce phosphorylation and accumulation of p53 in all the cell lines, but they differ markedly in their ability to express p21waf1.
We also analyzed the kinetics of protein expression following removal of SN38. Upon release from SN38, MCF10A cells appeared to partially recover, as reflected in the decrease in phosphorylation of Chk1 and p53, albeit p21waf1 remained high. In HCT116 and U2OS cells, Chk1 and p53 remained phosphorylated, and in fact, serine 345-Chk1 phosphorylation continued to rise in U2OS cells, suggesting that they were not recovering from the insult. Most notable is the continued and dramatic increase in p21waf1 in HCT116 and U2OS that occurs during the 24 h following drug removal.
p21waf1 mRNA is insufficient to explain the low p21waf1 protein levels in HCT116 and U2OS.
We next determined whether the p21waf1 mRNA levels could explain the difference in SN38-induced p21waf1 protein levels. The results were expressed either (1) relative to the mRNA level at 0 h for each cell line or (2) relative to the mRNA level at 0 h for MCF10A cells (Fig. 4). The basal level of mRNA was only 2-fold lower in the HCT116 and U2OS cells compared with MCF10A. The fold induction by SN38 was quite similar in all three cell lines. By 24 h, the absolute level in HCT116 was the same as in MCF10A, while the level in U2OS was about half. Hence, the difference is insufficient to explain the very low protein level observed. Upon removal of SN38, the p21waf1 mRNA began to decrease in MCF10A cells but continued to increase in both HCT116 and U2OS cells. The mRNA levels in these latter two cell lines seem to correlate with the increase in protein between 24 and 48 h.

Figure 4. Kinetics of p21waf1 mRNA induction by SN38 treatment. Cells were incubated with 10 ng/ml SN38 from 0–24 h. The drug was removed and cells were incubated for an additional 24 h in fresh medium. Cells were harvested at the indicated times and p21waf1 mRNA quantified by RT-qPCR. GAPDH was used as an internal control. The left panel shows mRNA induction compared with the untreated control of each cell line. The right panel shows mRNA induction compared with the untreated control of MCF10A. The error bars represent SE of at least three independent experiments.
p21waf1 protein half-life is insufficient to explain the low p21waf1 protein levels in HCT116 and U2OS.
We next assessed whether the difference in SN38-induced p21waf1 protein levels could be explained by differences in p21waf1 protein half-life. SN38-damaged cells were incubated with cycloheximide and the decay of p21waf1 assessed by western blotting (Fig. 5). The p21waf1 protein half-life was about 30 min for all three cells lines and therefore cannot explain the differences in p21waf1 protein levels. Additionally, the p21waf1 protein half-life remained about 30 min for all three cell lines 24 h after removal of drug, suggesting the increase of p21waf1 protein in HCT116 and U2OS is not due to changes in the protein half-life.

Figure 5. p21waf1 protein half-life following SN38 treatment. Cells were incubated with 10 ng/ml SN38 for 24 h or for an additional 24 h in fresh medium. Cells were then incubated with the protein synthesis inhibitor cyclohexmide (10 µg/ml) for the indicated times. The half-lives are presented as the means ± SE for three independent experiments.
Inhibition of p21waf1 mRNA translation.
Having been unable to attribute the level of p21waf1 protein to transcriptional differences or protein half-life, we next investigated whether there are differences in p21waf1 mRNA association with polysomes. Polysomes were purified from SN38-damaged cells, and polysome-associated p21waf1 mRNA was assessed (Fig. 6A). The majority of p21waf1 mRNA was associated with polysomes (> 4 ribosomes/transcript) in all three cell lines, suggesting that translation initiation had occurred. As the HCT116 and U2OS cells synthesize little p21waf1 protein, we assume that these polysomes are arrested on the transcript.

Figure 6. Inhibition of p21waf1 mRNA translation. (A) Cells were incubated with 10 ng/ml SN38 for 24 h and then collected for polysome profiling. Samples were divided into four fractions. Fraction one includes the free mRNAs and monosomes, while fractions two, three and four include polysome of increasing size. The bars represent the range of two independent experiments. (B) Cells were incubated with 10 ng/ml SN38 for 24 h and analyzed for levels of 44 miRNAs shown to affect p21waf1 protein levels. (C) Cells were left untreated or incubated with 10 ng/ml SN38 for 24 h. Changes in miR-106b, miR-93 and miR-130b levels were assessed by real-time PCR.
As miRNAs have been shown to affect p21waf1 mRNA translational efficiency, we hypothesized that high levels of miRNAs in HCT116 and U2OS are inhibiting p21waf1 mRNA translation.14 Recent studies identified a total of 44 miRNAs that inhibit p21waf1 expression and/or Ras-induced p21waf1-mediated growth arrest.15,16 We thus used microRNA microarray to analyze whether any of these 44 miRNAs were highly expressed in HCT116 and U2OS cells compared with MCF10A cells (Fig. 6B). The majority of these miRNA were expressed at negligible levels. Three miRNAs (miR-17-5p, miR-20a and miR-106a) were expressed at similar levels in all three cell lines. However, miR-106b, miR-93 and miR-130b were significantly elevated in the two tumor cell lines compared with MCF10A cells. We confirmed this with real-time PCR (Fig. 6C). These three miRNAs were not induced by SN38, rather, they were constitutively high in both HCT116 and U2OS compared with MCF10A cells. Additionally, we found miR125a to be highly expressed in U2OS cells compared with the other two cell lines (Fig. 6B). As miR-125a has also been shown to repress p53 expression and subsequently p21waf1 levels, this correlates with the low p53 induction observed in SN38-treated U2OS cells.17 These results suggest that expression of these miRNAs may be responsible for the arrested translation observed in HCT116 and U2OS cells.
Discussion
As the guardian of the genome, p53 suppresses cellular transformation by inducing arrest, apoptosis, DNA repair and differentiation in damaged cells.18 Thus, it is no surprise that the p53 function is almost always compromised in tumor cells.18 This allows these tumor cells to survive DNA damage and oncogene activation that would normally result in cell death.19 Approximately 50% of cancers have mutated p53, and some of the remaining p53 wild-type cancers have subdued p53 levels or activities.20 In this study, we show how dysregulation of the p53 pathway can still occur despite normal levels and activity of the p53 protein.
We found two well-characterized p53 wild-type cancer cell lines, HCT116 and U2OS, which exhibit an attenuated p21waf1 induction upon treatment with SN38 when compared with the non-tumorigenic MCF10A cells. This defect is not due to low p53 induction or lack of p53 activity, as all the cell lines produce similar levels of p21waf1 mRNA. As the p21waf1 protein half-life in both cell lines is also comparable to MCF10A cell lines, we looked for defects in p21waf1 mRNA translation. The majority of p21waf1 mRNA was associated with polysomes in all three cell lines, hence, we conclude that the differential expression of p21waf1 protein is due to differences in translation of the mRNA, a process that is regulated by microRNA.
MicroRNAs (miRNAs) regulate gene expression by affecting translational efficiency and/or stability of the target mRNA.14 Of the 44 miRNAs shown to either inhibit p21waf1 expression or Ras-induced p21waf1-mediated growth arrest,15,16 we identified three (miR-106b, miR-93 and miR-130b) that were constitutively high in HCT116 and U2OS compared with MCF10A cells. miR-106b and miR-93 are both part of the miR-106b family and have previously been shown to directly downregulate p21waf1 expression through binding to its 3' UTR.14,21-23 The other elevated miRNA, miR-130b, has been predicted to bind to an alternate sequence in the p21waf1 3' UTR. Overexpression of these miRNAs cause increased cell proliferation and accelerated G1/S transition.15,22,23 Consequently, it is likely that the presence of these miRNA in HCT116 and U2OS cells contributes to the attenuated induction of p21waf1 protein.
Upon DNA damage, the rapidly activated Chk1-Cdc25 pathway induces an immediate arrest, whereas the slow-operating p53-p21waf1 pathway sustains the arrest. In this study, we observed that Chk1 is downregulated once the p53-p21waf1 pathway becomes fully activated in MCF10A cells. This may be accomplished through p21waf1-mediated inhibition of Chk1 transcription. A previous study also observed Chk1 downregulation in daunorubicin-treated HCT116 cells,24 but we did not observe Chk1 suppression in SN38-treated HCT116 cells. Even when the levels of p21waf1 in HCT116 became twice that in MCF10A cells 24 h after the removal of SN38, the Chk1 levels remained unchanged. In U2OS cells, p21waf1 production is very low, and p21waf1 levels never reach more than 20% of the levels observed in the other two cell lines. As the impact of p53-activating agents on the cellular environment can be different, it will be interesting to see if this defective p21waf1 protein production in HCT116 and U2OS also occurs with other p53-activating agents that are either genotoxic, such as doxorubin, or non-genotoxic, such as Nutlin-3.
The tumor suppressive activity of p53 was first made clear with the identification of p21waf1 as its mediator of growth suppression.25 As p53 is often mutated in cancer, one might expect to find inactivating mutations of its downstream target, p21waf1, in wild-type p53 tumors.26 However, p21waf1 mutations are rare, yet reduced p21waf1 expression has been associated with colorectal, cervical, head and neck and small-cell lung cancers.27 Why would cancer cells repress p21waf1 expression rather than delete its function through mutation? This can perhaps be explained by evidence showing that low basal levels of p21waf1 promote active cyclin-CDK complex formation, and that cytoplasmic p21waf1 seems to exhibit antiapoptotic activites.14,27-29 But how would cancer cells maintain p21waf1 protein levels low enough to avoid arrest, especially when its mRNA is induced upon p53 activation? In this study, we show that this can be accomplished through suppression of p21waf1 mRNA translation. This helps explain why certain p53 wild-type tumors are attenuated in DNA damage-induced p21waf1 protein and thus sensitive to Chk1 inhibition.
Materials and Methods
Chemicals.
SN38 (7-ethyl-10-hydroxycamptothecin), the active metabolite of the topoisomerase I inhibitor irinotecan, was provided by Pfizer Global. MK-8776 (previously known as SCH900776) was provided by Merck. Both drugs were dissolved in DMSO at > 1,000 times the final concentration used in experiments. Cycloheximide was obtained from Sigma-Aldrich.
Cell culture.
MCF10A immortalized breast cell lines and U2OS osteosarcoma cell lines were obtained from American Type Culture Collection. HCT116 colorectal carcinoma cell lines were obtained from Dr. Vogelstein (Johns Hopkins University).30 U2OS and MCF10A cells were maintained in DMEM/F12 supplemented with 10% fetal bovine serum plus antibiotic/antimycotic. For MCF10A cells, the medium was also supplemented with 8 ∝g/ml insulin, 20 ng/ml epidermal growth factor and 500 ng/ml hydrocortisone. HCT116 cells were maintained in McCoy’s 5a medium supplemented with serum and antibiotics as above.
Cell cycle analysis.
Cell cycle analysis was performed as described previously, whereby cells were harvested, fixed in ethanol, incubated with ribonuclease and stained with propidium iodide.31 DNA content was determined on a FACScan flow cytometer (Becton Dickinson).
Western blot analysis.
Cells were rinsed with phosphate-buffered saline and then lysed by addition of urea sample buffer [4 M urea, 5% β-mercaptoethanol, 2% sodium dodecyl sulfate, 50 mM Tris (pH 6.8), 0.01% bromophenol blue and protease/phosphatase inhibitor cocktail]. Samples were immediately boiled for 5 min and stored at -80°C. Proteins were separated by SDS‑PAGE and transferred to polyvinylidene difluoride membranes. Membranes were blocked with 5% non-fat milk in Tris-buffered saline, 0.1% Tween 20, and then probed with the appropriate primary antibody in 5% non-fat milk or 5% BSA overnight at 4°C [p53 (DO-1) (Santa Cruz Biotechnology, sc-126), p21waf1 (C-19) (Santa Cruz Biotechnology, sc-397), Chk1 (G-4) (Santa Cruz Biotechnology, sc-8408), phosphoserine-15-p53 (Cell Signaling Technology, 9286), phosphoserine-345-Chk1 (Cell Signaling Technology, 2341), phosphoserine-296-Chk1 (Cell Signaling Technology, 2349), actin (Ab-1) (Calbiochem, CP01)]. Subsequently, membranes were washed in Tris-buffered saline, 0.1% Tween 20, and incubated with secondary antibody conjugated to horseradish peroxidase (Bio-Rad). Proteins were visualized by enhanced chemiluminescence (GE Healthcare).
Quantification of p21waf1 protein half-life.
Cells were incubated with the protein synthesis inhibitor cycloheximide (10 ∝g/ ml) for the indicated times. p21waf1 protein levels were detected by western blotting, and densitometry was performed within the quantitative range to measure the intensity of the bands. We assume that protein degradation follows first-order decay kinetics.32 The quantified protein intensity data was initially log-transformed, and then a linear best-fit curve was used to determine the decay rate constant (k). From the decay rate constant, the half-life (T1/2) was calculated.
Polysome fractionation.
Cells were incubated with 0.1 mg/ml cycloheximide for 3 min at 37°C and washed with ice-cold phosphate-buffered saline containing 0.1 mg/ml cycloheximide. Cells were lysed in 20 mM Tris, 2.5 mM MgCl2, 120 mM KCl (pH 7.5), 0.5 mM dithiothreitol, 0.1 mg/ml cycloheximide, 0.5 mg/ml heparin, 0.5% NP-40 and 100 U/ml SUPERase-In (Ambion, AM2696). The lysate was subsequently centrifuged at 3,000 rpm in a microcentrifuge for 5 min at 4°C. The supernatant was layered on a 10–50% w/v linear sucrose gradient and centrifuged for 3 h at 4°C at 29,000 rpm in a Surespin 630 Rotor (Sorvall). Gradients were fractionated using Gradient Station (Biocomp) while continuously monitoring absorbance at 254 nm. RNA was extracted from individual fractions using RNeasy Mini Kit (Qiagen).
Microarray.
Total RNA was extracted using Tri Reagent (Molecular Research Center Inc.,) according to the manufacturer’s instructions. 500 ng of total RNA was poly(A) tailed and then directly ligated to a biotin-labeled dendrimer using the FlashTag Biotin HSR kit (Genisphere). Labeled RNA was used for hybridization to the GeneChip miRNA Array (Affymetrix) for 16 h at 48°C and 60 rpm. Following hybridization, the miRNA arrays were washed and stained with streptavidin-PE (Affymetrix). Fluorescent images were obtained with 500GX scanner (Illumina) and processed with GeneChip Command Console software (Affymetrix).
Real-time PCR for mRNA.
Total RNA was isolated using RNeasy Mini Kit (Qiagen). cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad). Real-time PCR was performed with CFX96 real-time system (Bio-Rad) using iQSYBR Green PCR Supermix (Bio-Rad). Amplification cycles were: 95°C for 10 min, then 30 cycles of 94°C for 30 sec, 56.7°C for 30 sec and 72°C for 1 min. The fold change of p21waf1 mRNA was determined by the equation of Pfaffl using GAPDH as the endogenous control.33 The mRNA specific primer sequences are provided in Table 1.
Table 1. Primers used for Real-Time PCR.
| Name | Primer (5′-3′) | |
|---|---|---|
| p21waf1 (CDKN1A) |
Forward |
5′ - ATGTCAGAACCGGCTGGGGA - 3′ |
| |
Reverse |
5′ - GCCGTTTTCGACCCTGAGAG - 3′ |
| GAPDH |
Forward |
5′ - CTCAGACACCATGGGGAAGGTGA - 3′ |
| |
Reverse |
5′ - ATGATCTTGAGGCTGTTGTCATA - 3′ |
| miScript Universal Primer |
Reverse |
miScript SYBR Green PCR Kit (Qiagen) |
| miR-93 |
Forward |
5′ - CAAAGTGCTGTTCGTGCAGGTAG - 3′ |
| miR-106b |
Forward |
5′ - TAAAGTGCTGACAGTGCAGAT - 3′ |
| miR-130b |
Forward |
5′ - CAGTGCAATGATGAAAGGGCAT -3′ |
| Z30 | Forward | 5′ - TGGCTTTGACCAGGGTATGATC - 3′ |
Real-time PCR for miRNA.
Total RNA was isolated using Tri Reagent (Molecular Research Center Inc.,) as above. cDNA was synthesized from 400 ng of total RNA using miScript Reverse Transcription Kit (Qiagen). Real-time PCR was performed with CFX96 real-time system (Bio-Rad) using miScript SYBR Green PCR Kit (Qiagen) according to the manufacturer’s protocol. Amplification cycles were: 95°C for 15 min, then 45 cycles of 94°C for 15 sec, 55°C for 30 sec and 70°C for 30 sec. The fold change of miRNA was determined by the equation of Pfaffl using Z30 as the endogenous control.33 The miRNA specific primer sequences are provided in Table 1.
Glossary
Abbreviations:
- CDK
cyclin-dependent kinase
- UCN-01
7-hydroxystaurosporine
- miRNA
microRNA
- SN38
7-ethyl-10-hydroxycamptothecin
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant Support
This research was supported by NIH grant CA117874 (A. Eastman) and Cancer Center Support grant CA23108 to the Norris Cotton Cancer Center.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/20208
References
- 1.Melchionna R, Chen XB, Blasina A, McGowan CH. Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat Cell Biol. 2000;2:762–5. doi: 10.1038/35036406. [DOI] [PubMed] [Google Scholar]
- 2.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 3.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clarke CA, Clarke PR. DNA-dependent phosphorylation of Chk1 and Claspin in a human cell-free system. Biochem J. 2005;388:705–12. doi: 10.1042/BJ20041966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis. 2006;21:3–9. doi: 10.1093/mutage/gei063. [DOI] [PubMed] [Google Scholar]
- 6.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 8.Levesque AA, Kohn EA, Bresnick E, Eastman A. Distinct roles for p53 transactivation and repression in preventing UCN-01-mediated abrogation of DNA damage-induced arrest at S and G2 cell cycle checkpoints. Oncogene. 2005;24:3786–96. doi: 10.1038/sj.onc.1208451. [DOI] [PubMed] [Google Scholar]
- 9.Levesque AA, Fanous AA, Poh A, Eastman A. Defective p53 signaling in p53 wild-type tumors attenuates p21waf1 induction and cyclin B repression rendering them sensitive to Chk1 inhibitors that abrogate DNA damage-induced S and G2 arrest. Mol Cancer Ther. 2008;7:252–62. doi: 10.1158/1535-7163.MCT-07-2066. [DOI] [PubMed] [Google Scholar]
- 10.Kohn EA, Ruth ND, Brown MK, Livingstone M, Eastman A. Abrogation of the S phase DNA damage checkpoint results in S phase progression or premature mitosis depending on the concentration of 7-hydroxystaurosporine and the kinetics of Cdc25C activation. J Biol Chem. 2002;277:26553–64. doi: 10.1074/jbc.M202040200. [DOI] [PubMed] [Google Scholar]
- 11.Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol Cancer Ther. 2011;10:591–602. doi: 10.1158/1535-7163.MCT-10-0928. [DOI] [PubMed] [Google Scholar]
- 12.Montano R, Chung I, Garner KM, Parry D, Eastman A. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol Cancer Ther. 2012;11:427–38. doi: 10.1158/1535-7163.MCT-11-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garner KM, Eastman A. Variations in Mre11/Rad50/Nbs1 status and DNA damage-induced S-phase arrest in the cell lines of the NCI60 panel. BMC Cancer. 2011;11:206–, 1-13. doi: 10.1186/1471-2407-11-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung YS, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010;22:1003–12. doi: 10.1016/j.cellsig.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borgdorff V, Lleonart ME, Bishop CL, Fessart D, Bergin AH, Overhoff MG, et al. Multiple microRNAs rescue from Ras-induced senescence by inhibiting p21(Waf1/Cip1) Oncogene. 2010;29:2262–71. doi: 10.1038/onc.2009.497. [DOI] [PubMed] [Google Scholar]
- 16.Wu S, Huang S, Ding J, Zhao Y, Liang L, Liu T, et al. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene. 2010;29:2302–8. doi: 10.1038/onc.2010.34. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Gao JS, Tang X, Tucker LD, Quesenberry P, Rigoutsos I, et al. MicroRNA 125a and its regulation of the p53 tumor suppressor gene. FEBS Lett. 2009;583:3725–30. doi: 10.1016/j.febslet.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–13. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 19.Massagué J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 20.Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–42. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- 21.Hu S, Dong TS, Dalal SR, Wu F, Bissonnette M, Kwon JH, et al. The microbe-derived short chain fatty acid butyrate targets miRNA-dependent p21 gene expression in human colon cancer. PLoS One. 2011;6:e16221. doi: 10.1371/journal.pone.0016221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim YK, Yu J, Han TS, Park SY, Namkoong B, Kim DH, et al. Functional links between clustered microRNAs: suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res. 2009;37:1672–81. doi: 10.1093/nar/gkp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol. 2008;28:2167–74. doi: 10.1128/MCB.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gottifredi V, Karni-Schmidt O, Shieh SS, Prives C. p53 down-regulates CHK1 through p21 and the retinoblastoma protein. Mol Cell Biol. 2001;21:1066–76. doi: 10.1128/MCB.21.4.1066-1076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-P. [DOI] [PubMed] [Google Scholar]
- 26.McKenzie KE, Siva A, Maier S, Runnebaum IB, Seshadri R, Sukumar S. Altered WAF1 genes do not play a role in abnormal cell cycle regulation in breast cancers lacking p53 mutations. Clin Cancer Res. 1997;3:1669–73. [PubMed] [Google Scholar]
- 27.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–14. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erol A. Deciphering the intricate regulatory mechanisms for the cellular choice between cell repair, apoptosis or senescence in response to damaging signals. Cell Signal. 2011;23:1076–81. doi: 10.1016/j.cellsig.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 29.Gartel AL. p21(WAF1/CIP1) and cancer: a shifting paradigm? Biofactors. 2009;35:161–4. doi: 10.1002/biof.26. [DOI] [PubMed] [Google Scholar]
- 30.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–90. [PubMed] [Google Scholar]
- 31.Demarcq C, Bunch RT, Creswell D, Eastman A. The role of cell cycle progression in cisplatin-induced apoptosis in Chinese hamster ovary cells. Cell Growth Differ. 1994;5:983–93. [PubMed] [Google Scholar]
- 32.Belle A, Tanay A, Bitincka L, Shamir R, O’Shea EK. Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci U S A. 2006;103:13004–9. doi: 10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]