

Abstract
Nylon-3 polymers have a polyamide backbone reminiscent of that found in proteins (β- vs. α-amino acid residues, respectively), which makes these materials interesting for biological applications. Because of the versatility of the ring-opening polymerization process and the variety of β-lactam starting materials available, the structure of nylon-3 copolymers is highly amenable to alteration. A previous study showed that relatively subtle changes in the structure or ratio of hydrophobic and cationic subunits that comprise these polymers can result in significant changes in the ability of nylon-3-bearing surfaces to support cell adhesion and spreading. In the present study we have exploited the highly tailorable nature of these polymers to synthesize new versions possessing a wide range of chain lengths, with the intent of optimizing these materials for use as cell-supportive substrates. We find that longer nylon-3 chains lead to better fibroblast growth on modified surfaces, and that at the optimal chain lengths, less hydrophobic subunits are superior. The best polymers we identified are comparable to an RGD-containing peptide in supporting fibroblast attachment. The results described here will help to focus future efforts aimed at refining nylon-3 copolymer substrates for specific tissue engineering applications.
INTRODUCTION
Biomaterials that can control cell fate by regulating cell adhesion, proliferation, differentiation, and phenotype have attracted considerable attention because of their potential biomedical uses, especially as substrates for tissue regeneration.1–3 Ideal biomaterials should mimic the natural microenvironment encountered by cells by providing appropriate structural support, molecular signals, topography, nutrient and oxygen transport and waste removal.4, 5 Cell-adhesive biomaterials can be obtained from natural sources or generated via synthesis; each approach has advantages and limitations. Naturally-derived materials, especially those composed of extracellular matrix (ECM) components such as fibronectin (FN), vitronectin (VN), and collagen (Coll), can closely mimic natural microenvironments.6 However, it can be difficult to modify the structures of these materials, and their biological origins lead to concerns regarding pathogenicity and immunogenicity.7 To avoid these disadvantages, synthetic methods have been developed to create tailorable materials using structurally-defined molecules such as cell-adhesive segments of natural proteins8 and self-assembled peptides.9–12 The large-scale synthesis of sequence-specific peptides, however, is labor-intensive and expensive. Synthetic polymers, intrinsically heterogeneous at the molecular level, have been explored as potential replacements for sequence-specific peptides; polyethylene glycol (PEG), for example, has been widely used to construct cell-responsive scaffolds via incorporation of specific functional groups13 or cell-binding motifs.8, 14, 15 There is an ongoing need to identify new types of synthetic polymers that are biocompatible and readily modified.
Nylon-3 polymers have attracted our attention as potential biomaterials because the poly-β-amino acid backbone resembles the poly-α-amino acid backbone of proteins, and this similarity seems likely to result in biocompatibility. This polymer class has received relatively little attention, but recent studies have shown that nylon-3 materials can mimic the biological activities of host-defense peptides16–18 or the biophysical properties of lung-surfactant proteins.19 In addition, preliminary surveys of cationic-hydrophobic nylon-3 copolymers in our laboratories have identified subunit compositions that provide a highly favorable environment for 3T3 fibroblast adhesion and growth.20 The studies described here build upon these initial observations regarding the use of nylon-3 materials as cell-adhesive biomaterials.
The new work assesses two aspects of nylon-3 copolymer structure – subunit identity and chain length – in terms of the extent to which 3T3 fibroblasts interact with a surface that bears one of these covalently attached polymers. Because of the influential role of hydrophobicity in mediating protein adsorption and, hence, cell adhesion, we have expanded the set of building blocks used to construct the nylon-3 materials to encompass a greater range of subunit hydrophobicity, relative to our initial survey.20 Nylon-3 materials are prepared from β-lactam precursors via anionic ring-opening polymerization (ROP; Scheme 1). We focus on copolymers that contain both a hydrophobic and a cationic subunit, the latter derived either from β-lactam MM (monomethyl) or β-lactam DM (dimethyl; Figure 1). Previously we examined binary copolymers in which the hydrophobic subunit was derived from either CH (cyclohexyl) or CO (cyclooctyl; Figure 1). In the present study we have expanded the β-lactam set to include CP (cyclopentyl) and CD (cyclododecyl). By altering the size of the cycloalkane ring, we alter intrinsic hydrophobicity; the nylon-3 subunit derived from CP is less hydrophobic than that derived from either CH or CO, and the subunit derived from CD is more hydrophobic. Thus, in addition to examining the effect of altering net hydrophobicity of the polymer chains on cell adhesion by changing the proportion of hydrophobic subunits, we can evaluate the effect of altering subunit hydrophobicity over a wide range.
Scheme 1.
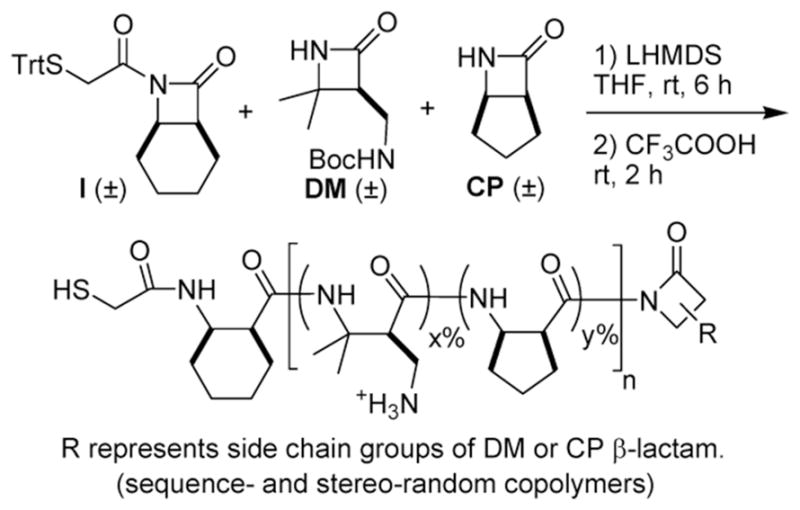
Synthesis of a Representative Nylon-3 Copolymers (DM:CP series)a
a All polymers in this study were synthesized in a similar way, but with varying pairs of β-lactams. After side chain deprotection, the cationic polymers were obtained in TFA salt forms. DM = dimethyl, CP = cyclopentyl
Figure 1.
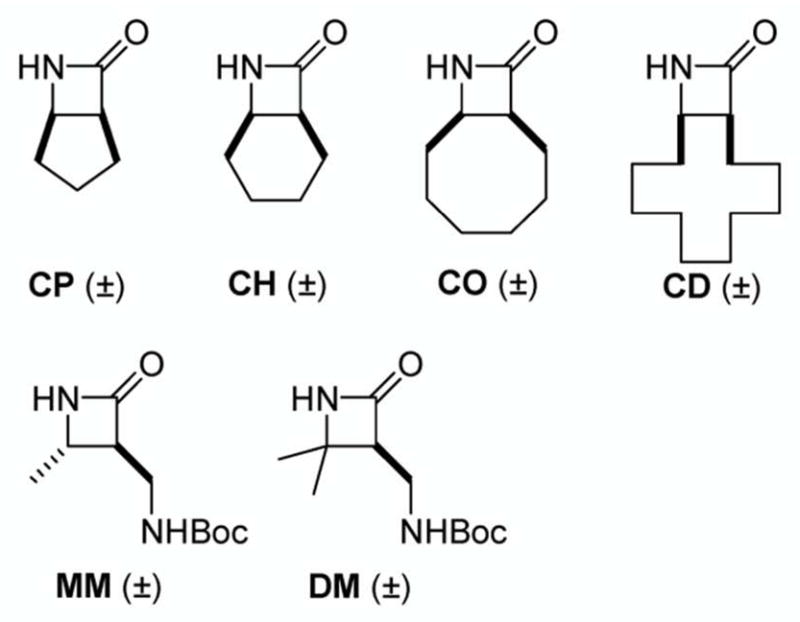
β-Lactams used to construct nylon-3 homo- and copolymers. CP = cyclopentyl; CH = cyclohexyl; CO = cyclooctyl; CD = cyclododecyl; MM = monomethyl; DM = dimethyl
The second important parameter explored in this work is polymer length. Initial studies were limited to relatively short chains (20–30 subunits on average; molecular weights in the range 4200–6400 g/mol),20 but in the present work we have considered nylon-3 materials with average lengths up to and beyond 100 subunits (MW up to 30,000 g/mol). Length variation could prove to be important in altering the amount of surface coverage, or for reasons that are not readily predicted.
EXPERIMENTAL SECTION
Polymer Synthesis
Sequence- and stereo-random nylon-3 polymers were generated via anionic ring-opening polymerization (ROP) from racemic β-lactams. The β-lactams (Figure 1) were prepared via reaction of the appropriate alkene and chlorosulfonyl isocyanate (CIS), as previously described.21, 22, 18, 23 The polymerization was conducted with lithium bis(trimethylsilyl)amide (LiHMDS); I was employed as the co-initiator (Scheme 1). Polymerization was allowed to continue for 6 h to consume all β-lactam; methanol was then added to terminate the reaction. The crude product, a polymeric material that retains Boc protecting groups on the side chain amines, was dissolved in THF and analyzed via gel permeation chromatography (GPC). The crude polymer was purified by precipitation, which was induced by adding pentane to the THF solution. The precipitate was collected after centrifugation. The protected MM and DM homopolymers were much less soluble in THF relative to copolymers containing as little as 10% of a hydrophobic subunit. We speculate that the hydrophobic subunits, all of which contain rings, somehow disrupt intermolecular associations among the polymer chains that are prevalent for the side chain-protected MM and DM homopolymers. Removal of Boc protecting groups with TFA afforded the final polymers as amine salts, which were purified by addition of diethyl ether (Et2O) to induce precipitation. The precipitates were collected after centrifugation. Nylon-3 polymer samples used in this study had an average molecular weight ranging from 4,400 to 30,000 g/mol, and polydispersity indices (PDI) ranging from 1.03 to 1.20.
Preparation of Polymer-Functionalized Substrates
CodeLink activated microarray slides from SurModics (Eden Prairie, MN, U.S.A.) were used as purchased. Each slide was covered with a 50-well silicone coverslip, and the wells were filled with solutions containing various polymers (1 mM); or aminoethanol (50 mM), as a negative control; or the peptide GRGDS (1 mM), as a positive control in 100 mM NaHCO3 aqueous buffer containing 15% glycerol. The glass slides were incubated in a water-loaded humidifying chamber for 12 h at room temperature, rinsed thoroughly with Milli-Q water, and dried with N2.
CodeLink slides were stored in a dry environment at room temperature until use. We did not detect any reduction of surface modification efficiency within 10 months of purchase. Control studies of surface modification efficiency with a fluorescently labeled amine (Supplementary Figure 11) indicated that a solution containing 0.125 mM amine was sufficient to achieve maximum surface functionalization, which is consistent with guidance provided in the CodeLink slide user guide.
Cell Attachment Analysis
NIH 3T3 fibroblast cells were cultured in DMEM containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin and 2 mM L-glutamine at 37 °C in a 5% CO2 environment. Cells were cultured in a petri dish until they reached 90% confluence, detached from the dish with a solution containing 0.05% trypsin and 0.02% EDTA, isolated via centrifugation, and resuspended in culture medium to a final concentration of 2.5 × 105 cell/mL. An 8 μL aliquot of this cell suspension was added to each well of the polymer-functionalized glass slide, which was placed on the bottom of a petri dish and incubated at 37 °C for 2 h. The whole slide was then immersed by addition of 30 mL DMEM containing 10% FBS to the petri dish. The slide was incubated for 2 days. After excess medium was removed, the slide was stained with 850 μL of a LIVE/DEAD staining solution containing 2 μM calcein AM and 4 μM ethidium homodimer-1. Cell adhesion was quantified by scanning the whole slide on a GeneTAC UC 4 × 4 scanner (Genomic Solution) at 512 nm after 20 minutes of staining. Data were analyzed using NIH MacBiophotonics ImageJ (version 1.43m) software.
Protein Adsorption Analysis
An 8 μl aliquot of DMEM containing 10% FBS was added to each individual well of a polymer-functionalized glass slide, which was then incubated in a humidifying chamber at room temperature for 2 h. Each well was washed with 10 μL PBS (× 3), 10 μL Milli-Q water (× 1), and dried with N2. Then, 8 μl of NanoOrange working solution was added to each well, and the slide was incubated in a humidifying chamber at 37 °C for 1 h, cooled to room temperature, and scanned on a GeneTAC UC 4 × 4 scanner at 595 nm. Data were analyzed using NIH MacBiophotonics ImageJ (version 1.43m) software.
Statistical Analysis
Data are presented as mean ± standard deviation and were analyzed statistically using ANOVA with Tukey’s HSD post-hoc test. Differences were taken as significant at p < 0.05.
RESULTS AND DISCUSSION
New polymer compositions
Previous work, involving binary nylon-3 copolymers containing cationic subunits derived from either MM or DM and hydrophobic subunits derived from either CH or CO, identified two compositions as particularly promising in terms of 3T3 cell adhesion: 60:40 MM:CO and 60:40 DM:CH. These two copolymers were comparable to conventional tissue culture polystyrene in supporting cell adhesion.20 We included these compositions as benchmarks in a new survey that explored hydrophobic subunits derived from β-lactam CP or CD as alternatives to subunits derived from CH or CO (Figure 2). CodeLink activated microarray slides bearing N-hydroxysuccinimide (NHS) ester groups on the surface were used for immobilization of each polymer. This initial survey involved polymer samples with average length in the 27mer-37mer range. Cells were seeded on the modified surfaces, incubated for two days in the presence of serum-containing medium, and then qualitatively assessed via microscopy and quantitatively assessed via measurement of fluorescence intensity following viability staining; these results are summarized in Figure 3. Among the new nylon-3 copolymers, 90:10 MM:CD appeared to be superior to all others, including the benchmarks, in terms of 3T3 cell attachment. In addition, 60:40 MM:CD and 90:10 DM:CP displayed moderate improvement in supporting 3T3 fibroblast attachment relative to the benchmark polymers 60:40 MM:CO and 60:40 DM:CH.
Figure 2.

Nylon-3 polymers used in this study. (a) The chemical structure of a representative copolymer family containing of subunits derived from β-lactams DM and CP. (b) The abbreviations of all homo- and copolymers examined in this study.
Figure 3.

Relative density of 3T3 fibroblasts on surfaces modified with nylon-3 polymers at two days post-seeding. The polymers used for these studies were all in the ~30mer length range. 60:40 MM:CO and 60:40 DM:CH were benchmark polymers that were identified in previous work to be highly supportive of cell attachment.
Effects of varying polymer length
For each of the six new nylon-3 copolymer compositions, as well as the two benchmark compositions and the cationic homopolymers poly-MM and poly-DM, a series of samples was prepared in which average chain length was increased incrementally from the ~30mer range to the >90mer range. Each polymer sample was covalently attached to a CodeLink activated microarray slide, and the resulting surface was evaluated for 3T3 cell attachment. As summarized in Figure 4, the number of living cells after a two-day incubation (as judged by calcein AM fluorescence) rose with increasing chain length for each polymer composition. In some cases this rise leveled off over the range chain length that was evaluated (60:40 DM:CH, 40:60 DM:CP, 60:40 MM:CD, 90:10 MM:CD and the DM homopolymer), but in the other cases, such a plateau was not evident. The most effective surfaces in terms of supporting 3T3 fibroblast culture were those bearing long-chain forms of 60:40 DM:CH and 40:60 DM:CP, which supported ~10-fold greater cell attachment than surfaces bearing the best materials identified in our previous report (short-chain forms of 60:40 DM:CH and 60:40 MM:CO).20
Figure 4.

Attachment of 3T3 fibroblasts on surfaces modified with nylon-3 polymers of varying chain length after two-day incubation. Within each graph, polymers are identical in subunit composition but varied in length (average number of subunits). The vertical axes indicate fluorescence intensity based on live-cell staining.
Two of the best copolymers identified in Figure 4, 60:40 DM:CH (102mer average length) and 40:60 DM:CP (142mer average length), were compared with a short peptide, Gly-Arg-Gly-Asp-Ser, which contains the RGD motif that is well-known to promote cell adhesion via direct interactions with cell surface integrin receptors.24, 25 In contrast, random nylon-3 copolymers presumably lack cell surface receptor-binding domains and likely attract cells via surface-adsorbed serum proteins. This difference in cell adhesion mechanism between an RGD-containing peptide and nylon-3 copolymers makes it challenging to choose a basis for peptide vs. polymer comparison. We chose to saturate glass slide surfaces with either the RGD-containing peptide or the nylon-3 copolymers by using functionalization solutions containing 1 mM of the appropriate reagent. Control studies showed that the slide surface is saturated when 0.125 mM of a fluorescent amine is used (Supplementary Figure 11).
Comparison of surfaces bearing the RGD peptide (attached via the N-terminal amino group) with surfaces bearing the nylon-3 copolymers in terms of 3T3 fibroblast attachment were conducted in standard serum-containing medium. Under these conditions, the two nylon-3 copolymers were similar to the RGD-containing peptide in terms of both cell density and cell morphology after two days (Figure 5).
Figure 5.

Quantitative and qualitative evaluation of 3T3 fibroblasts cultured on surfaces modified with the best nylon-3 long polymers or with an RGD-containing peptide after a two-day incubation. (a) The vertical axis indicates fluorescence intensity based on live-cell staining; (*) indicates p < 0.0001 compared to RGD. (b) Bright field micrographs (100x total magnification) of 3T3 cells on the surfaces modified with the nylon-3 copolymers or the RGD-containing peptide.
Relationship between protein adsorption and fibroblast attachment on polymer-functionalized surfaces
Fibroblast adhesion to a surface is presumably mediated by proteins adsorbed to that surface.26 Such adsorption is well-known to occur in the presence of serum, which is rich in proteins that are prone to adsorption. Our recent study on the mechanism of cell-nylon-3 polymer interaction indicated that cell adhesion is correlated to the adsorption of total serum proteins rather than a single protein (collagen, fibronectin, or vitronectin) in the serum-containing medium.27 Based on this result, surfaces bearing each of the polymers discussed in this work were incubated for two hours with Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS), and then rinsed. The amount of protein adsorbed to each surface was determined via NanoOrange staining. Figure 6 shows the results of these measurements; also shown are the cell attachment results from Figure 4, to facilitate comparison of cell attachment and protein adsorption for each polymer-bearing surface.
Figure 6.

Relationship between 3T3 fibroblast attachment and protein adsorption as a function of polymer composition and chain length. For each graph, the vertical axis on the left indicates fluorescence intensity that reports live cell density (gray bars), while the vertical axis on the right indicates fluorescence intensity that reports protein adsorption (NanoOrange; black squares). The cell attachment data represent a recasting of data shown in Figure 4.
Two important conclusions may be drawn regarding the relationship between polymer structure and protein adsorption. First, the extent of protein adsorption generally appears to be independent of nylon-3 chain length, although 60:40 MM:CD shows a strong and sharply defined length effect (higher adsorption to surfaces bearing longer polymer chains). Second, there is significant variation among the materials in terms of the extent of protein adsorption, which indicates that protein adsorption is affected by nylon-3 copolymer subunit composition; however, no simple trend is evident in terms of cationic:hydrophobic proportion. Across the MM:CP series, for example, the trend is non-monotonic. The cationic MM homopolymer displays moderate protein adsorption, as does the 40:60 MM:CP copolymer, but the 90:10 MM:CP copolymer, with an intermediate composition, displays much higher protein adsorption than the other two. A different trend is observed across the MM:CD series, with slightly higher adsorption for 90:10 MM:CD relative to MM homopolymer, and then much higher protein adsorption for 60:40 MM:CD, although only for the longer chains.
The data in Figure 6 suggest that there is no simple relationship between the extent of protein adsorption to surfaces bearing nylon-3 polymers and the ability of those surfaces to promote 3T3 fibroblast attachment. This lack of correlation can be seen by considering the longer-chain versions of three copolymers, 40:60 MM:CP, 60:40 DM:CH and 60:40 MM:CD. The former two are among the best materials we have found in terms of promoting fibroblast attachment on polymer-functionalized surfaces, as noted above. In both cases, the level of protein adsorption is moderate relative to the range of variations observed among all of the polymer-functionalized surfaces we evaluated. The longer-chain versions of 60:40 MM:CD are nearly as effective as the other two aforementioned copolymers in promoting fibroblast attachment, although this composition is much more effective at promoting protein adsorption. The lack of correlation between extent of protein adsorption and promotion of fibroblast attachment can be seen also in several copolymers that display significant length-dependent trends in terms of fibroblast attachment but no length-dependence in terms of protein adsorption (e.g., 40:60 MM:CP, 40:60 DM:CP, 90:10 MM:CP).
CONCLUSIONS
Our most significant finding is that nylon-3 copolymer chain length can exert a substantial impact on the attachment of 3T3 fibroblasts to a surface bearing the copolymer. The origin of this chain length effect is not yet clear, although our results indicate that trends in total protein adsorption from the culture medium cannot explain the length-dependence. Previously we screened copolymer compositions for the ability to promote 3T3 fibroblast adhesion by focusing on relatively short polymers (average lengths in the 20mer-30mer range),20 but our current results indicate that this approach is imperfect because the ranking of surface-immobilized nylon-3 copolymers, as a function of composition, can change with increasing chain length. This effect is seen by comparing Figures 3 and 4, the former comparing 3T3 fibroblast attachment on surfaces bearing short nylon-3 polymers containing different subunit combinations, and the latter showing how 3T3 fibroblast attachment is affected by increasing length for different nylon-3 compositions. Among the short polymers 90:10 MM:CD shows the best performance, but as chain length increases 60:40 DM:CH and 40:60 DM:CP surpass 90:10 MM:CD.
The results reported here expand the previously reported structure-activity series in that we have included new subunits that are either more hydrophobic (CD) or less hydrophobic (CP) than those previously considered (CH and CO). The most promising nylon-3 copolymers we have identified contain either a cyclohexyl or cyclopentyl ring (CH or CP) as the hydrophobic subunit, which suggests that the biological effects we seek are best served by avoiding very large and hydrophobic cycloalkyl components, such as a cyclooctyl or cyclododecyl ring (CO or CD). This information can be applied to the design of biomaterial environments whose chemical structures are optimized to elicit predictable and desired interactions with both proteins and cells.
Supplementary Material
Acknowledgments
This research was supported in part by the Nanoscale Science and Engineering Center at UW–Madison (DMR-0832760) and the NIH (R21EB013259, to K.S.M and S.H.G.).
Footnotes
Supporting Information Available: Additional details describing the preparation and characterization of polymers, fabrication of polymer-functionalized glass slides, protein adsorption, and cell attachment upon polymer-functionalized substrates. This information is available free of charge via the Internet at http://pubs.acs.org/.
Contributor Information
Kristyn S. Masters, Email: kmasters@wisc.edu.
Samuel H. Gellman, Email: gellman@chem.wisc.edu.
References
- 1.Shin H, Jo S, Mikos AG. Biomaterials. 2003;24(24):4353–4364. doi: 10.1016/s0142-9612(03)00339-9. [DOI] [PubMed] [Google Scholar]
- 2.Stevens MM, George JH. Science. 2005;310(5751):1135–1138. doi: 10.1126/science.1106587. [DOI] [PubMed] [Google Scholar]
- 3.Ma PX. Adv Drug Deliv Rev. 2008;60(2):184–198. doi: 10.1016/j.addr.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hersel U, Dahmen C, Kessler H. Biomaterials. 2003;24(24):4385–4415. doi: 10.1016/s0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- 5.Ratner BD, Bryant SJ. Annu Rev Biomed Eng. 2004;6:41–75. doi: 10.1146/annurev.bioeng.6.040803.140027. [DOI] [PubMed] [Google Scholar]
- 6.Ode A, Duda GN, Glaeser JD, Matziolis G, Frauenschuh S, Perka C, Wilson CJ, Kasper G. J Biomed Mater Res. 2010;95(4):1114–1124. doi: 10.1002/jbm.a.32909. [DOI] [PubMed] [Google Scholar]
- 7.Rosso F, Giordano A, Barbarisi M, Barbarisi A. J Cell Physiol. 2004;199(2):174–180. doi: 10.1002/jcp.10471. [DOI] [PubMed] [Google Scholar]
- 8.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23(1):47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 9.Zhang SG. Nat Biotechnol. 2003;21(10):1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 10.Davis ME, Motion JPM, Narmoneva DA, Takahashi T, Hakuno D, Kamm RD, Zhang SG, Lee RT. Circulation. 2005;111(4):442–450. doi: 10.1161/01.CIR.0000153847.47301.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kretsinger JK, Haines LA, Schneider JP. Biopolymers. 2005;80(4):493–493. [Google Scholar]
- 12.Palmer LC, Newcomb CJ, Kaltz SR, Spoerke ED, Stupp SI. Chem Rev. 2008;108(11):4754–4783. doi: 10.1021/cr8004422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benoit DSW, Schwartz MP, Durney AR, Anseth KS. Nature Materials. 2008;7(10):816–823. doi: 10.1038/nmat2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benton JA, Fairbanks BD, Anseth KS. Biomaterials. 2009;30(34):6593–6603. doi: 10.1016/j.biomaterials.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin CC, Anseth KS. Adv Funct Mater. 2009;19(14):2325–2331. doi: 10.1002/adfm.200900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mowery BP, Lee SE, Kissounko DA, Epand RF, Epand RM, Weisblum B, Stahl SS, Gellman SH. J Am Chem Soc. 2007;129(50):15474–15476. doi: 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]
- 17.Epand RM, Epand RF, Mowery BP, Lee SE, Stahl SS, Lehrer RI, Gellman SH. J Mol Biol. 2008;379(1):38–50. doi: 10.1016/j.jmb.2008.03.047. [DOI] [PubMed] [Google Scholar]
- 18.Mowery BP, Lindner AH, Weisblum B, Stahl SS, Gellman SH. J Am Chem Soc. 2009;131(28):9735–9745. doi: 10.1021/ja901613g. [DOI] [PubMed] [Google Scholar]
- 19.Dohm MT, Mowery BP, Czyzewski AM, Stahl SS, Gellman SH, Barron AE. J Am Chem Soc. 2010;132(23):7957–7967. doi: 10.1021/ja909734n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee MR, Stahl SS, Gellman SH, Masters KS. J Am Chem Soc. 2009;131(46):16779–16789. doi: 10.1021/ja9050636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodgame DML, Hill SPW, Lincoln R, Quiros M, Williams DJ. Polyhedron. 1993;12(23):2753–2762. [Google Scholar]
- 22.Dener JM, Fantauzzi PP, Kshirsagar TA, Kelly DE, Wolfe AB. Organic Process Research & Development. 2001;5(4):445–449. [Google Scholar]
- 23.Zhang JH, Kissounko DA, Lee SE, Gellman SH, Stahl SS. J Am Chem Soc. 2009;131(4):1589–1597. doi: 10.1021/ja8069192. [DOI] [PubMed] [Google Scholar]
- 24.Ruoslahti E, Pierschbacher MD. Science. 1987;238(4826):491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 25.Massia SP, Hubbell JA. J Cell Biol. 1991;114(5):1089–1100. doi: 10.1083/jcb.114.5.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steele JG, Johnson G, Underwood PA. J Biomed Mater Res. 1992;26(7):861–884. doi: 10.1002/jbm.820260704. [DOI] [PubMed] [Google Scholar]
- 27.Liu R, Vang KZ, Kreeger PK, Gellman SH, Masters KS. J Biomed Mater Res A. 2012 doi: 10.1002/jbm.a.34211. manuscript submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.