

Abstract
A hyperglutamatergic state has been hypothesized to drive escalation of alcohol intake. This hypothesis predicts that an impairment of glutamate clearance through inactivation of the astrocytic glutamate transporter, GLAST (EAAT1), will result in escalation of alcohol consumption. Here, we used mice with a deletion of GLAST to test this prediction. WT and GLAST KO mice were tested for alcohol consumption using two-bottle free-choice drinking. Alcohol reward was evaluated using conditioned place preference (CPP). Sensitivity to depressant alcohol effects was tested using the accelerating rotarod, alcohol-induced hypothermia, and loss of righting reflex. Extracellular glutamate was measured using microdialysis, and striatal slice electrophysiology was carried out to examine plasticity of the cortico-striatal pathway as a model system in which adaptations to the constitutive GLAST deletion can be studied. Contrary to our hypothesis, GLAST KO mice showed markedly decreased alcohol consumption, and lacked CPP for alcohol, despite a higher locomotor response to this drug. Alcohol-induced ataxia, hypothermia, and sedation were unaffected. In striatal slices from GLAST KO mice, long-term depression (LTD) induced by high frequency stimulation, or by post-synaptic depolarization combined with the L-type calcium channel activator FPL 64176 was absent. In contrast, normal synaptic depression was observed after application of the cannabinoid 1 (CB1) receptor agonist WIN55,212-2. Constitutive deletion of GLAST unexpectedly results in markedly reduced alcohol consumption and preference, associated with markedly reduced alcohol reward. Endocannabinoid signaling appears to be down-regulated upstream of the CB1 receptor as a result of the GLAST deletion, and is a candidate mechanism behind the reduction of alcohol reward observed.
Keywords: glutamate transporter, alcohol, reward, endocannabinoid
1. Introduction
Glutamatergic dysregulation has been hypothesized as a key pathophysiological factor in alcoholism, and a mechanism that could be targeted by pharmacotherapies for this disorder (De Witte et al., 2003; Spanagel, 2009; Spanagel and Kiefer, 2008; Tsai and Coyle, 1998). Brain microdialysis in rats has directly shown an increase in extracellular glutamate levels during withdrawal from alcohol (Dahchour et al., 1998; Rossetti and Carboni, 1995), and has shown that this increase is progressive with consecutive cycles of intoxication and withdrawal (Dahchour and De Witte, 2003). Conversely, glutamate reuptake inhibition using dl-threo-β-benzyloxyaspartic acid (TBOA) has been shown to increase voluntary alcohol consumption (Kapasova and Szumlinski, 2008). Accordingly, acamprosate, a putative functional glutamate antagonist, is an approved alcoholism medication (Bouza et al., 2004; Spanagel and Kiefer, 2008).
Clearance of extracellular glutamate is critical for glutamate homeostasis, and occurs in large part through the activity of the glutamate transporters GLAST (EAAT1) and GLT-1 (EAAT2), primarily expressed by astrocytes (Danbolt, 2001; Rothstein et al., 1996). Up-regulation of GLAST gene expression accompanies escalation of alcohol consumption in rats following prolonged intermittent brain alcohol exposure (Rimondini et al., 2002), and has also been reported in post-mortem brain tissue from alcoholics (Flatscher-Bader et al., 2006; Flatscher-Bader and Wilce, 2006). In these cases, up-regulation of GLAST can be viewed as a homeostatic adaptation to elevated extracellular glutamate levels caused by repeated cycles of intoxication and withdrawal. Once this mechanism becomes insufficient to maintain glutamate homeostasis, a persistent hyperglutamatergic state can emerge, and continue to drive escalated alcohol consumption. In other cases, deficient GLAST function may be pre-existing due to e.g. genetic factors, and directly drive a hyperglutamatergic state accompanied by escalation of alcohol consumption.
Both these scenarios predict that disruption of GLAST function should lead to escalation of alcohol consumption. Support for this prediction has come from work with mutant mice in which deletion of Per2 (periodic clock gene 2) was associated with deficient GLAST expression and function (Spanagel et al., 2005). This was accompanied by an increase in extracellular glutamate and escalation of voluntary alcohol consumption, both of which were rescued by acamprosate (Spanagel et al., 2005). In a parallel to these findings, acamprosate, an approved alcoholism treatment, suppressed central glutamate levels in recently detoxified alcoholics as measured by magnetic resonance spectroscopy (MRS) (Umhau et al., 2010).
The Per2 study found that alcohol consumption was inversely correlated with GLAST function, but did not establish a causal relationship between the two. Strong evidence for causality, and support for the hyperglutamatergic theory of alcoholism would be obtained if disruption of GLAST function were found to result in escalation of alcohol consumption. This would also offer an attractive in vivo model to evaluate novel candidate medications that target the glutamatergic system. Here, we examined this possibility. Pharmacological tools to modulate GLAST function with a high degree of specificity are lacking; for instance, the reuptake inhibitor TBOA does not distinguish between the EEATs. Here, we therefore used mice with a genetic deletion of GLAST (Watase et al., 1998), and evaluated them for alcohol consumption and reward, as well as sensitivity to depressant effects of alcohol. In search of the mechanistic underpinnings of our behavioral findings, we then performed microdialysis for glutamate in the Nc. Accumbens, a key structure for drug reward. Synaptic plasticity in the cortico-striatal pathway was finally assessed because it offers a well characterized model of glutamate dependent plasticity, in which potential adaptations to the constitutive GLAST deletion can be studied. Because endocannabinoid transmission is an established modulator of both glutamatergic transmission and alcohol consumption, its integrity was examined in this model system.
2. Material and Methods
2.1 Animals
Generation of the GLAST knock-out (KO) mice has been described (Watase et al., 1998). The GLAST gene was disrupted in embryonic stem (ES) cells from 129/Sv mice, and replaced by a neomycin resistance cassette in exon 6. The targeted ES clone was injected into C57BL/6J blastocysts to create chimeric mice, and mice were back-crossed onto a C57 background for >10 generations. In order to avoid a potential confound from genotypic differences in maternal behavior and early life environment (Millstein and Holmes, 2007), GLAST KO, GLAST HET and WT mice were all generated from HET × HET matings.
Mice were housed in groups of 1-4 per cage, in a temperature and humidity controlled vivarium, under 12-h light/dark cycle, and with ad libitum access to food and water in the home cage. Males and females at least 8 weeks of age were tested, and the experimenter remained blind to the genotype during testing. All experimental procedures were approved by the National Institute of Alcohol Abuse and Alcoholism Animal Care and Use Committee and followed the NIH guidelines ‘Using Animals in Intramural Research’; or were approved by the Committee on Animal Care and Use (Regierungspräsidium Karlsruhe), and carried out in accordance with the local Animal Welfare Act and the European Communities Council Directives.
2.2 Two-bottle free-choice alcohol consumption
Mice were single caged, habituated to a new animal room for two weeks, and then given continuous free access to increasing concentrations of an aqueous alcohol solution or water. For the first 7 days, access was given to two bottles containing tap water, and intake was examined for possible side preference. Alcohol was then provided in one of the bottles and faded in (2%, vol/vol; 4 - 5 days, 4%, 4 – 5 days), after which consumption was measured at 8%, 12%, and 16%, obtaining 6 data points at each concentration over the course of 7-8 days. The amount of alcohol ingested was expressed as g/24h/kg body weight.
2.3 Conditioned place preference for alcohol
CPP for alcohol was examined as previously described (Boyce-Rustay and Cunningham, 2004). Time spent and locomotor activity within each compartment were measured by photobeams placed 1.2 cm apart across the full length of the apparatus. The grid and mesh floors were covered with solid Plexiglas to prevent conditioning to the floor texture. Mice were first given a habituation session in which they received an i.p. saline injection and were allowed to freely explore the apparatus for 5 min. Twenty-four hours later, conditioning started, using an unbiased design in which half of the subjects received 2 g/kg alcohol (CS+) or saline (CS-), and were placed into the appropriate compartment for 5 min. CS+ and CS- trials were alternated daily, and their order was counterbalanced within genotype. One complete trial consisted of a CS+ trial and a CS- trial. After 6 conditioning trials, preference was tested by giving the animal a saline injection, and placing it into the center compartment to freely explore the whole apparatus for 10 min.
2.4 Fear conditioning
Fear conditioning was assessed as previously described (Karlsson et al., 2005; Kim and Fanselow, 1992). Briefly, the Freeze Monitor system (San Diego Instruments, San Diego, CA, USA) was used, and automatically delivered a 30 sec 80 dB auditory conditioned stimulus (CS) and a 0.6 mA 2 sec footshock unconditioned stimulus (US). Twenty-four hours after conditioning, mice were placed in a novel context, allowed to habituate for 180 s, and then presented with the auditory CS and evaluated for freezing for 180 s. Additional twenty-four hours later, freezing induced by re-exposure to the training context was tested for 300 s. The presence of freezing behavior was scored every 10 sec, and was defined as the absence of any movement except respiration. Data were calculated as the proportion of observations in which the subject was scored as freezing.
2.5 Sensitivity to depressant effects of alcohol
Depressant effects of alcohol could interfere with place conditioning, and could also be aversive. To examine possible genotype differences in the sensitivity to depresant alcohol actions, we assessed alcohol-induced ataxia, hypothermia and sedation/hypnosis. A single cohort of mice was tested in all three assays. Because the respective behaviors are sensitive to different doses of alcohol, testing started with the assay involving the lowest dose (i.e., ataxia), followed by hypothermia, and last sedation/hypnosis. At least 1 week was allowed between tests. This regimen has been employed in previous studies (Boyce Rustay et al., 2006; Boyce-Rustay and Holmes, 2006) and is not thought to produce lasting tolerance to alcohol (Crabbe et al., 2008).
To assess alcohol-induced ataxia, mice were placed onto the rotarod dowel, which was accelerated at a constant rate of 8 rpm/min up to 40 rpm. The latency to fall off was recorded by photocell beams, with a maximum cutoff latency of 5 min. Mice received 10 training trials separated by a 30-sec inter-trial interval. Twenty-four hours later, there was a baseline habituation trial, followed by 2 baseline trials that were averaged to obtain a single measure of pre-alcohol treatment performance. Mice were then injected with 2 or 2.25 g/kg alcohol. Thirty min thereafter, they were given an additional habituation trial, followed by 2 test trials that were averaged to obtain a single measure of post-alcohol treatment performance. The dependent measure was the difference between pre- and post-alcohol treatment performance (=delta latency).
Basal core body temperature was measured by inserting a Thermalert TH-5 thermometer (Physitemp, Clifton, NJ, USA) 2 cm into the rectum until a stable reading was obtained. Mice were then injected with 3.0 or 4.0 g/kg alcohol, and temperature was measured 30, 60, 90, and 120 min later to provide an average post-alcohol measure. The difference between pre-alcohol versus post-alcohol treatment temperature was the dependent measure (=delta temperature). Ambient room temperature was 23°C.
To assess alcohol-induced sedation, loss of righting reflex (LORR) was studied. Mice were injected with 3.0 or 4.0 g/kg alcohol and placed into the supine position in a V-shaped chamber. The time from injection to recovery of the righting reflex (turning onto all 4 paws twice in 30 sec after initial self-righting) was measured.
Finally, to exclude the possibility that any genotypic difference in alcohol effects might be related to differences in the pharmacokinetics of alcohol, separate groups of alcohol-naïve mice were injected with 3.5 g/kg alcohol, placed into an empty cage, and sacrificed via cervical dislocation and decapitation at different time points. Trunk blood was taken for analysis of blood alcohol concentration (BAC) using the Analox AM1 Alcohol Analyzer (Analox Instruments USA Inc, Lunenburg, MA).
2.6 Microdialysis
Alcohol naive WT and KO mice were used. Surgeries were performed under inhalation anaesthesia using isoflurane. Mice were mounted in a Cunningham stereotactic mouse device (Stoelting Co., Wood Dale, IL, USA), and unilaterally implanted with CMA7 guide cannulas (CMA Microdialysis, Solna, Sweeden) aimed at the nucleus accumbens (+1.5 mm rostral to bregma, 0.7 mm lateral to midline, -3.4 mm below dura). Guides were fixed to the skull using two anchor-screws and dental cement (GC FujiCEM Automix, GC Corporation, Tokyo, Japan). After surgery, mice were placed back in their home cage. After five to seven days of recovery, microdialysis probes (CMA7/2, 2 mm membrane length; CMA Microdialysis AB, Solna, Sweden) were slowly inserted in the Nc. Accumbens through the guide cannulas. Microdialysis probes were perfused with sterile artificial cerebrospinal fluid (CMA Microdialysis AB, Solna, Sweden) at a flow rate of 1 μl/min using a PHD2000 microinfusion pump (Harvard Apparatus, USA) while mice were connected to counterbalanced dual-channel liquid swivels (Instech Laboratories, Plymouth Meeting, PA, USA), allowing them to move freely. The sampling period started after overnight stabilisation. Microdialysis samples were collected every 30 min over a period of 6h in 300 μl plastic tubes using a CMA/470 fraction-cooling collector (CMA Microdialysis AB, Solna, Sweden) and stored at −80°C until HPLC analysis.
Four baseline samples were first collected, after which mice were injected with saline (i.p.), and two additional samples were obtained. Alcohol (2g/kg; i.p.) was then given, and 6 additional samples were obtained. At the end of the experiments, brains were collected, frozen in isopentane, and stored at -80°C until histological verification of the probe placement. Glutamate content was measured as described previously (Umhau et al., 2010). Briefly, a 10-μL aliquot of sample was derivatized with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate, and analyzed using ultraperformance liquid chromatography (Aquity UPLC; Waters Corp, Milford, Massachusetts) with fluorescent detection using an amino acid kit (MassTrak; Waters Corp).
2.7 Electrophysiology
Coronal brain slices (350 μm) containing both striatum and cortex were prepared from postnatal day 16–19 (for patch-clamp experiments), or 2 month old (for field potential recordings) wild type and GLAST KO mice, as described previously (Adermark and Lovinger, 2007b). Slices were allowed to equilibrate for at least one hour in artificial cerebrospinal fluid (aCSF) containing (in mM); 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4 and 10 D-glucose, continuously bubbled with a mixture of 95% O2 / 5% CO2 gas, before transfer to a recording chamber perfused with aCSF at approximately 2 mL/min. For all experiments, the temperature of the bath was maintained at 28–31°C and was stable within ±1°C during any given experiment.
Whole cell patch clamp recordings were performed as previously described (Adermark and Lovinger, 2007b). Baseline excitatory postsynaptic currents (EPSCs) were evoked in medium spiny neurons (MSNs) voltage clamped at -70 mV. The internal solution consisted of (in mM); 120 CsMeSO3, 5 NaCl, 10 TEA-Cl, 10 HEPES, 5 QX-314, 1.1 EGTA, 4 Mg-ATP, 0.3 Na-GTP. AMPA/NMDA ratio was determined by measuring the peak EPSC amplitude at -70 mV divided by amplitude at 50 ms post-stimulus when clamped at +40 mV. EPSCs were allowed to completely stabilize before amplitudes were measured.
To assess baseline synaptic responses and paired-pulse ratios, paired pulse stimulation (50 ms interpulse interval) was delivered every 20 second through a twisted tungsten wire placed at the border of the overlaying white matter and the dorsolateral part of the striatum, and adjusted to elicit baseline EPSC amplitudes between 200 and 400 pA. For induction of endocannabinoid release and LTD-formation we used a high frequency stimulation (HFS) protocol, consisting of four 1 sec 100 Hz trains delivered every 10 sec through the wire stimulating electrode, paired with depolarization of the postsynaptic cell to 0 mV. Synaptic depression was also induced with the L-type calcium channel activator 2,5-Dimethyl-4-[2-(phenylmethyl)benzoyl]-1H-pyrrole-3-carboxlic acid methyl ester (FPL64176) (Sigma-Aldrich) (Adermark and Lovinger, 2007a). In these experiments the postsynaptic cell was clamped at -50 mV throughout the recording, and FPL 64176 was washed on for 10 min. FPL 64176 was dissolved in alcohol to 50 mM, and further diluted to 0.5 μM in aCSF (final EtOH concentration of 0.001%). Synaptic depression was also induced by the CB1 receptor agonist WIN55,212-2, dissolved in DMSO to 10 mM and diluted to 1 μM in aCSF containing bovine serum albumin (0.5 g/l). Throughout all recordings, GABA-ergic transmission was blocked by the GABAA antagonist picrotoxin (50 μM, Sigma-Aldrich). The NMDA receptor antagonist d-(−)-2-amino-5-phosphonopentanoic acid (APV; 50 μM, Sigma-Aldrich) was included in all recordings except those shown in Figure 4A. Picrotoxin and APV were dissolved in aCSF shortly before each experiment. Recordings were discontinued if the series resistance varied more than 20% or increased over 30 MΩ.
Figure 4.
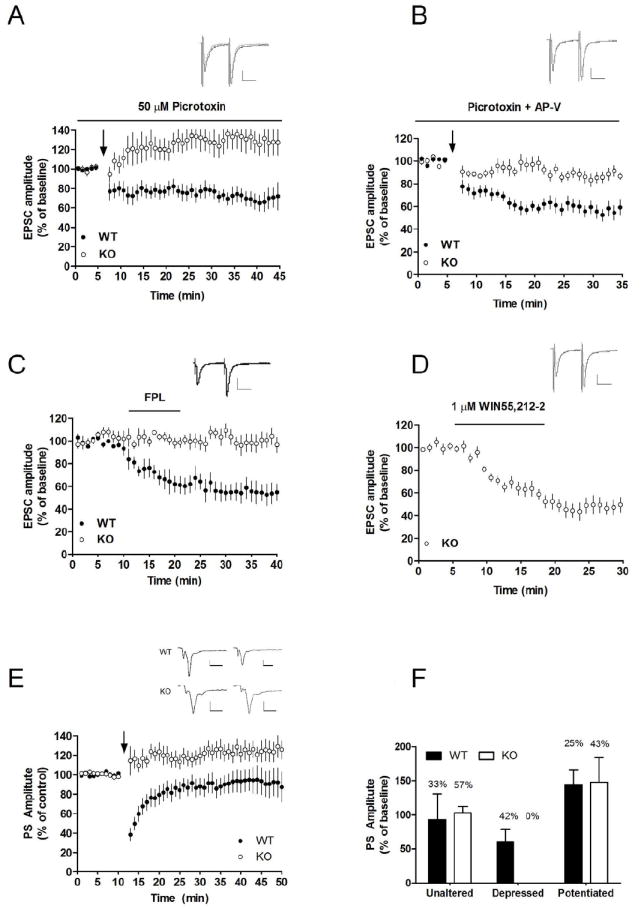
LTD-induction but not CB1R activation is impaired in slices from GLAST KO mice. HFS was insufficient to induce LTD in slices from GLAST KO mice (A), even during blocked NMDA receptors (B). Application of the L-type calcium channel activator FPL 64176 (500 nM) paired with slight depolarization of the postsynaptic cell (-50 mV) induced LTD in control slices, but not in slices from GLAST KO mice (C). WIN55,212-2 induced a robust depression, showing that activation of CB1R and downstream mechanism from this receptor is functional in slices from GLAST KO mice (D). EPSC values from MSNs located in the dorsolateral striatum are mean±SEM. Example traces show evoked EPSCs at baseline (black) and after HFS stimulation (gray) in KO (A), and WT (B), and in KO following 20 min of FPL or WIN 55,212-2 treatment (C, D). Arrows mark time point for HFS. n=5-11. Field potential recordings reviled that LTP could be induced by HFS in slices from GLAST KO mice, while both LTP and LTD was observed in WT (E, F). n=12-14. Scale bar is 0.025 ms and 100 pA.
Extracellular field recordings were performed as previously described (Yin et al., 2007). Test stimuli were delivered via a S48 stimulator (Grass Instruments, West Warwick, RI) at a frequency of 0.05 Hz through a bipolar twisted tungsten wire placed in the dorsomedial striatum, and set to evoke a population spike (PS) with an amplitude half the maximum. The high frequency stimulation (HFS) protocol used to induce plastic changes in striatal output consisted of four 1 s, 100 Hz trains delivered every 10 s, paired with an increase in stimulation amplitude to that which evoked a maximal baseline PS.
2.8 Statistics
Data were analyzed using Statistica (Statsoft, Tulsa, OK, USA). Behavioral data were analyzed with analysis of variance, using models specified in conjunction with the results of the respective analysis, and followed by Tukey post hoc tests where appropriate. Electrophysiological data in text are presented as mean EPSC amplitude at t=25-30 min compared to baseline with 95% confidence interval (CI). EPSC data in time course figures are plotted as mean EPSC amplitude compared to baseline, with standard error of the mean (SEM). Paired t-test was used for the majority of statistical analyses on the electrophysiological data.
3. Results
3.1 Alcohol consumption is markedly reduced in GLAST KO mice
Alcohol consumption was analyzed with genotype as between subjects factor, and two hierarchical within subjects factors, alcohol concentration (8, 12 and 16%), and measurement day (1-6) within the respective concentration. There was a significant main effect of genotype (F2,126=6.9, P<0.01), concentration (F2,252=88.9, P<0.001), as well as a genotype × concentration interaction (F4,252=10.9, P<0.001). Post hoc analysis showed significantly reduced alcohol consumption in GLAST KO compared to WT mice at 16% alcohol (Figure 1A). To examine a possible contribution of sex, data were also evaluated following introduction of this factor into the model above, but there was no genotype × sex, or genotype × concentration × sex interaction. To rule out the possibility that a contribution of sex went undetected because of the inherently lower power of an interaction test, we also carried out follow up analyses within each sex. These followed the same pattern as the global model presented, making a confound of sex highly unlikely. The decrease in consumption associated with GLAST deletion was similar in both sexes, although it was most pronounced in females (genotype: F2,57=9.8, P<0.001; concentration: F2,114=72.3, P<0.001; genotype × concentration: F4,114=6.7, P<0.001; post-hoc analysis: significantly lower consumption at 12 and 16% alcohol). In males, the main effect of genotype did not reach significance (F2,66=0.7, P>0.05), but in addition to a significant main effect of concentration (F2,132=31.0, P<0.001) there was a genotype × concentration interaction (F4,132=5.5, P<0.001); post-hoc analysis did not reach significance at any individual alcohol concentration.
Figure 1.
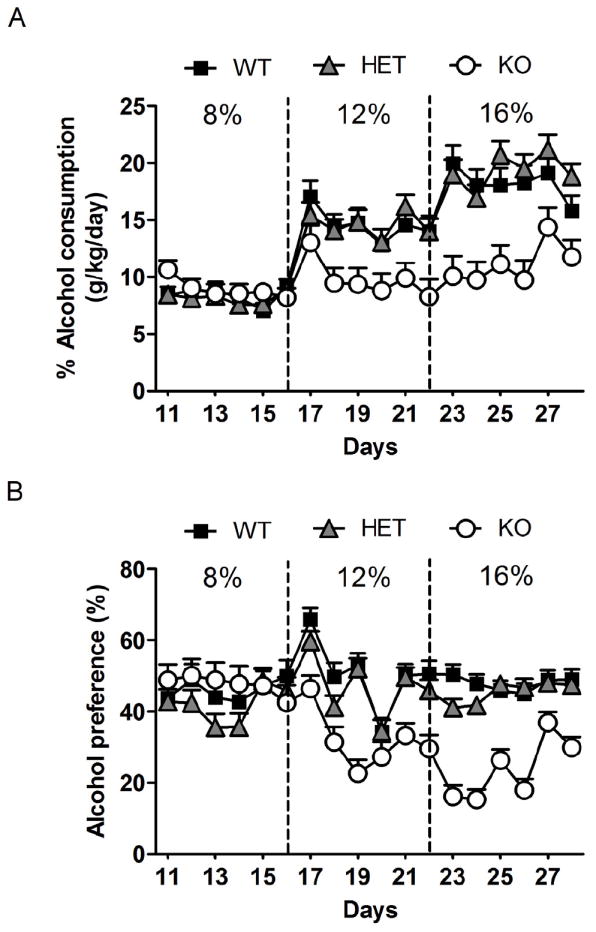
Decreased 2-bottle free choice consumption of alcohol in GLAST KO mice. GLAST KO mice showed decreased alcohol consumption at the highest alcohol concentration tested, 16% (A), and showed a decreased preference for alcohol vs water both at 12 and 16% (B). Data are mean±SEM. n=28-46/genotype for consumption. For detailed statistics, see Results.
Similarly, alcohol preference was significantly affected by genotype (F2,104=10.4, P<0.01) as well as concentration (F2,208=5.5, P<0.001), and there was a significant genotype × concentration interaction (F4,208=11.1, P<0.001). Post hoc analysis showed a significantly reduced alcohol preference for GLAST KO mice at 16% (Figure 1B). Once again, introduction of sex as factor did not produce any significant interactions. Analysis by sex showed the same pattern as the consumption data, although the reduction of alcohol preference reached significance in both male and female GLAST KO mice (males: genotype: F2,51=2.7, P=0.07, concentration: F2,102=13.1, P<0.001, genotype × concentration: F4,102=6.4, P<0.001; post-hoc analysis: significantly lower preference at 16%; females: genotype: F2,50=9.0, P<0.001, concentration: F2,100=0.2, P=0.03, genotype × concentration: F4,100=5.0, P<0.01; post-hoc analysis: significantly lower preference at 12 and 16%).
3.2 GLAST KO mice lack conditioned place preference for alcohol
During habituation to the CPP apparatus, no side preference was present in any of the genotypes, and there was no different between genotypes (F1,50=1. 9, P>0.05; Figure 2A). Similarly, no genotype difference in locomotor activity was found (WT=1439.9±92.9, HET=1498.9±78.6, KO=1551.2±81.0 beam breaks; mean±SEM; F2,52=0.4, P>0.05).
Figure 2.
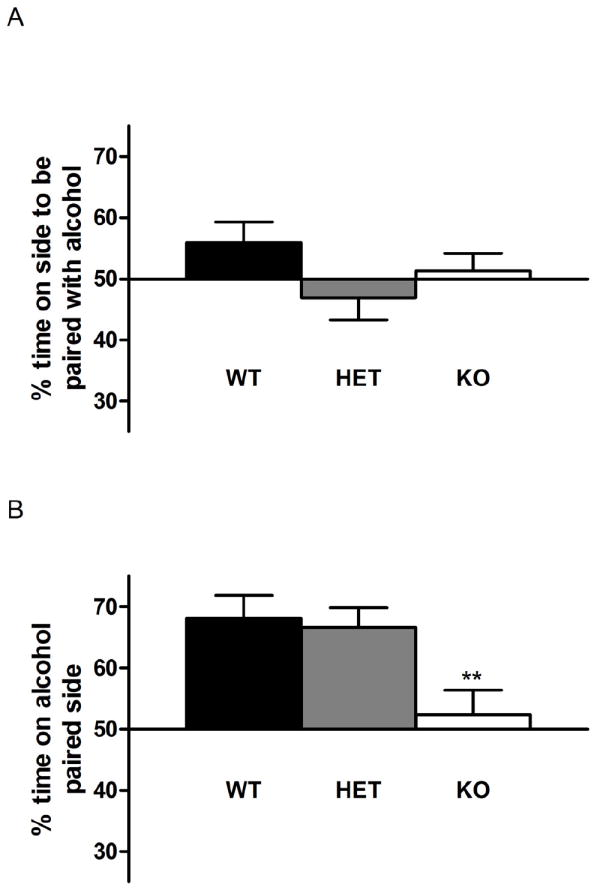
Loss of alcohol reward in GLAST KO mice. There was no significant difference between genotypes in preference for side going to become alcohol-paired in the initial habituation session (A). WT and GLAST HET mice showed robust place preference for the alcohol-paired side, while GLAST KO mice did not show any side preference, and consequently differed from WT mice. (B). Data are mean±SEM, n=17-18 for condition place preference, **P<0.01 compared to WT.
During preference testing, WT and HET mice displayed robust CPP for alcohol, and spent about 80% time in the alcohol-associated compartment. In contrast, this measure was at chance level for GLAST KO:s. Accordingly, there was a significant effect of genotype for %time spent on the alcohol associated side (F2,50=5.6, P<0.01), and post hoc analysis showed that GLAST KO:s spent significantly less time on this side compared to both HET and WT mice (Figure 2B). There was no significant effect of sex when this factor was included in the analysis.
Locomotor activity was higher in the GLAST KO:s during alcohol-conditioning trials, but not during saline trials (data not shown), and also during preference testing, both in the saline and the alcohol associated compartment (alcohol associated compartment: WT=369.2±59.5, HET=770.5±77.8, KO=800.9±62.0; saline associated compartment: WT=469.5±55.0, HET=407.9±45.2, KO=731.5±85.5 beam breaks; mean±SEM; n=17-18; main effect of genotype F2,100=5.95, P<0.001; main effect of compartment: F1,100=14.2, P<0.001; no genotype × compartment interaction). However, in the GLAST KO:s, activity was not correlated with preference (r=-0.26, n=17, P=0.32).
3.3 GLAST KO mice do not have a general impairment of associative learning
Classical fear conditioning was unaffected by the GLAST deletion. First, freezing did not differ between genotypes during acquisition of conditioned fear (WT=59.1±10.5, HET=47.2±6.7, KO=43.6±10.4 % of observations; mean±SEM; n=10-15,). Second, no genotype differences were observed when conditioned fear was recalled by re-exposure to the shock-associated auditory cue (WT=77.8±6.3, HET=70.2±6.0, KO=55.8±7.1) or the shock-associated context (WT=35.3±6.1, HET=25.2±3.9, KO=33.0±8.3).
3.4 Sensitivity to depressant effects of alcohol is not altered by deletion of GLAST
There was no effect of sex in any of the analysis performed for the sensitivity to depressant effects of alcohol. For rotarod training, there were no difference between genotype at the final level, nor during baseline performance on the test day (data not shown). There was a robust effect of alcohol dose (F1,86=11.6, P<0.001), but no effect of genotype, or genotype × dose interaction with regard to the alcohol-induced impairment on the rotarod, measured as delta in the latency to fall off the dowel (2g/kg: WT=-51.7±9.2, HET=-63.5±8.8, KO=-61.3±12.2; 2.25 g/kg: WT=-90.1±12.2, HET=-87.5±8.8, KO=-93.7±10.2 sec; mean±SEM; n=13-17).
Baseline body temperature did not differ between genotypes (not shown). There was a significant effect of alcohol dose (F1,86=12.0, P<0.001), but no genotype effect, or genotype × dose interaction (delta temperature at 3g/kg alcohol: WT=-1.5±0.2, HET=-1.1±0.2, KO=-1.2±0.1; 4g/kg alcohol: WT=-1.7±0.2, HET=-1.8±0.2, KO=2.0±0.3°C; mean±SEM).
There was a robust effect of alcohol dose (F1,79=93.9, P<0.001), but no effect of genotype or genotype × dose interaction effect on the time to regain the righting reflex (3g/kg alcohol: WT=28.6±4.0, HET=34.2±4.0, KO=46.1±10.7; 4g/kg alcohol: WT=101.7±9.0, HET=96.5±8.8, KO=99.3±8.6 min; mean±SEM).
As expected, BACs declined significantly over time after the bolus injection (F3,78=45.8, P<0.001), but there was no effect of genotype or genotype × time interaction (5 min: WT=392.9±24.1 mg/dL, HET=390.3±42.0, KO=416.9±38.3; 30 min: WT=337.6±16.3, HET=331.6±45.0, KO=328.4±22.8; 360 min: WT=102.53±32.7, HET=89.3 ±29.0, KO=126.2 ±27.3 mg/dl; mean±SEM).
3.5 Extracellular glutamate levels are not altered in GLAST KO mice
Extracellular concentrations of glutamate in the nucleus accumbens were analyzed with genotype as between subjects, and sample number, a measure of time, as within subjects factor. There was a significant effect of time (F10,160=2.5, P<0.01) but no difference between genotypes (F1,16=0.003, P>0.05), or genotype × time interaction (F10,160=1.0, P>0.05; Figure 3).
Figure 3.
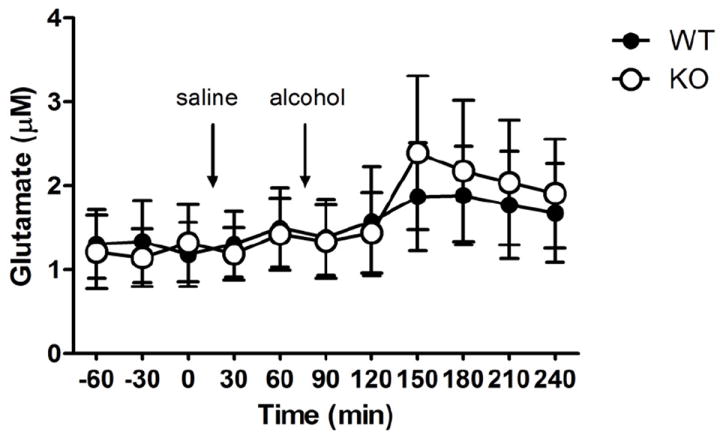
Extracellular glutamate in the Nc. Accumbens was not affected by the GLAST deletion, neither at baseline, nor following systemic administration of alcohol (2g/kg) n=7-8 / genotype / time point. Data are mean±SEM μM.
3.6 Endocannabinoid signaling is impaired in striatal slices from GLAST KO mice
The basal properties of striatal synaptic transmission were not significantly altered in slices from GLAST KO mice relative to WT controls. Using paired pulses (50 ms interpulse interval) delivered every 20 sec, we observed that decay time of evoked EPSCs was 19±2.3 ms (EPSC 1), and 20±2.6 ms (EPSC 2) in slices from GLAST KO mice (n=16), and 17±1.6 ms (EPSC 1), and 18±1.7 (EPSC 2) in slices from WT mice (n=14). The paired pulse ratio (PPR) of EPSC 2 and EPSC 1 amplitudes (EPSC 2/EPSC1) did not differ in MSNs from GLAST KO (1.2±0.1, n=16), compared to WT (1.2±0.1, n=13; KO vs. WT, P>0.05), nor was the AMPA/NMDA ratio altered (WT: 2.3±0.9 (n=10), KO: 3.2±0.9 (n=11), F = 2.84, p > 0.05).
Repetitive activation of afferent fibers by high frequency stimulation (HFS) was insufficient to induce LTD in slices from KO mice (EPSC amplitude at t=20-25 min: 126±22% of control, n=7, P<0.05), while the same stimulation protocol induced a robust depression of EPSC amplitude in slices from WT mice (EPSC amplitude at t=20-25min: 76±15% of control, n=8, P<0.05; KO vs. WT, 15-20 min after HFS, P<0.01; Figure 4A). To determine if a possible depression at these synapses could be unmasked by blockade of NMDA receptors, we perfused slices with picrotoxin (50 μM) and APV (50 μM). Blockade of NMDA receptors with APV prevented the small but significant (p<0.05) potentiation detected after HFS in slices from KO mice. More importantly, it did not unmask a depression, suggesting that striatal LTD is significantly impaired in slices from GLAST KO mice (Figure 4B). EPSC amplitude 20-25 min after HFS was 93±8.8% of control, n=9, P>0.05 in GLAST KO slices, and 60±8.7% of control in slices from WT mice, n=8, P<0.001; KO vs. WT, P<0.001. PPR was increased compared to baseline after HFS in slices from WT (106±1.4% of control, n=13, P<0.001), while no change was detected in slices from GLAST KO mice (99±3.1% of control, n=16, P>0.05).
Treatment with the L-type calcium channel activator FPL64176 has previously been shown to induce mGluR and D2R independent LTD (FPL-LTD) at glutamatergic synapses (Adermark and Lovinger, 2007a). Similar to HFS, treatment with FPL64176 induced a robust LTD in slices from WT mice (EPSC amplitude=59±15% of control, n=5, P<0.05), while no depression was detected in slices from KO mice (EPSC amplitude=104±11.9% of control, n=5, P>0.05; Figure 4C). In contrast, activation of presynaptic CB1 receptors by WIN55,212-2 (1 μM) induced a robust depression of EPSC amplitude in MSNs from GLAST KO mice (EPSC amplitude at t=20-25 min: 46±9.3% of control, n=5, P<0.001; Figure 4D), and significantly enhanced PPR (109±4.2% of control, n=6, P<0.05).
3.7 LTP-induction is normal in striatal slices from GLAST KO mice
Field potential recordings dorsomedial striatum were carried out to examine possible changes in LTP-formation in GLAST KO mice. HFS-induced LTP was not significantly altered in GLAST KO mice (Fisher’s exact test, two tailed, p=0.43). Similar to whole-cell recordings from the dorsolateral striatum, LTD was only detected in slices from WT mice (Figure 4E-F). Striatal output (0-1 mV), evaluated by varying the stimulation intensity (0-150μA), was not altered in slices from GLAST KO mice (data not shown).
4. Discussion
We found an unexpected and pronounced reduction of voluntary alcohol consumption in mice with homozygous GLAST deletion. With increasing alcohol concentrations over time, alcohol intake increased in WT controls and HET mice, and daily consumption ultimately approached 20 g/kg at the highest concentration of alcohol offered. In contrast, GLAST KO mice only slightly escalated their consumption with increasing alcohol concentrations, and their daily consumption remained around 10 g/kg throughout the experiment. Relative preference for alcohol over water closely followed the consumption data, making it unlikely that non-specific effects on fluid consumption caused the reduction of alcohol intake. The suppression of alcohol consumption in GLAST KO:s was selective for the highest levels of alcohol concentration and intake. In contrast, there was no genotype effect on the lower levels of consumption observed at low alcohol concentrations, which can occur for non-pharmacological properties such as taste or caloric content (Leeman et al., 2010). The data from the GLAST KO:s are therefore suggestive of a specifically reduced motivation to consume alcohol for its pharmacodynamic properties.
A marked reduction of motivation to consume alcohol for its rewarding properties in the GLAST KO:s is further suggested by the CPP data. In close agreement with our prior experience in C57BL/6 mice [see e.g. (Thorsell et al., 2010)], WT and HET mice showed robust place preference for a compartment associated with a 2 g/kg dose of alcohol. In contrast, GLAST KO mice failed to show preference for the alcohol associated compartment, and performed at chance level during recall testing. Because we only evaluated a 2 g/kg alcohol dose, we can not exclude the possibility that residual alcohol CPP might be detected in the GLAST mutants at a different alcohol dose. Alcohol CPP does not lend itself well to detailed dose-response studies, and similar levels of place preference have previously been found with 1.5, 2.0, and 4.0 g/kg doses of alcohol, while no CPP was observed after 0.5 g/kg (Groblewski et al., 2008). The lack of detectable place preference for alcohol in the GLAST mutants at 2.0 g/kg suggests a loss, or, at a minimum, a marked reduction of this alcohol action. Before drawing this conclusion, however, we several alternative interpretations should be considered.
First, differential effects of alcohol on aversive properties of the apparatus, rather than altered reward, could conceivably contribute to a genotype difference in CPP (Tzschentke, 1998; Tzschentke, 2007). We believe that this is unlikely, because our results were obtained in an unbiased CPP paradigm, and none of the genotypes showed a side preference prior to conditioning. Second, increased activity observed during preference testing in the GLAST mutants could counfound preference measures, as hyperactivity has previously been shown to disrupt the expression of place preference (Gremel and Cunningham, 2007). However, our control analysis did not find an inverse correlation between preference and activity in the GLAST KO:s, making a confound from increased activity less likely. Third, alcohol is a mixed rewarding and aversive stimulus, and decreased preference for an alcohol associated context could therefore represent either decreased sensitivity to alcohol reward, or increased sensitivity the aversive properties of this drug. We believe that the latter possibility is less likely, because alcohol has been shown to produce place aversion when administered after conditioning sessions, while rewarding properties dominate across as wide dose range when alcohol administration, as here, precedes conditioning (Cunningham and Henderson, 2000). Fourth, differences in sensitivity to depressant alcohol effects could influence place conditioning, but this also appears unlikely, because neither alcohol-induced ataxia, hypothermia or the latency to regain the righting reflex were affected by the GLAST deletion. Finally, a global disruption of associative learning could impair the ability of GLAST mutants to acquire place preference, but is made unlikely by the demonstration that the KO:s acquired and expressed classical fear conditioning, both to cue and to context, as effectively as WT controls or HET mice. Collectively, the data suggest that reduction of alcohol consumption observed in the GLAST KO:s is associated with, and potentially caused by, a loss or a marked reduction of alcohol reward.
We have previously reported that the GLAST KO:s respond with a more pronounced locomotor response both to novelty, and to NMDA antagonism (Karlsson et al., 2008). In the course of the present experiments, we observed that this is also the case in response to alcohol, despite the marked reduction in alcohol reward as measured by CPP. These data provide further evidence that psychomotor stimulant effect of alcohol and alcohol reward can be dissociated. An influential theory postulated a close link between psychomotor stimulant effects of addictive drugs and their addictive properties (Wise and Bozarth, 1987), and this link is also implied by more recent theoretical frameworks (Robinson and Berridge, 2003). It has, however, been shown that psychomotor stimulant and rewarding drug actions can be dissociated for stimulants, opioids and glutamatergic drugs (Carr et al., 1988; Gong and Justice, 1997; Sellings and Clarke, 2003; Tiffany Cunningham and Kelley, 1992). We have previously found an enhanced locomotor response to the NMDA antagonist MK-801 in GLAST KO mice (Karlsson et al., 2008), and it is possible that their similarly increased response to an acute alcohol challenge is related to a hyperexcitability within the prefrontal cortex (Homayoun and Moghaddam, 2007; Kargieman et al., 2007).
Our hypothesis that alcohol consumption would be elevated in GLAST KO:s was based on an expected impairment of glutamate clearance and concomitantly elevated extracellular levels of glutamate as a result of GLAST inactivation (Danbolt, 2001; Rothstein et al., 1996). Impaired glutamate uptake was originally demonstrated ex vivo in brain tissue from the GLAST KO:s, and was accompanied by increased epileptogenesis (Watase et al., 1998). Microdialysis subsequently showed elevated glutamate levels in the perilymph of the otic bulla in GLAST KO:s following acoustic stimulation, and this was associated with an excitotoxic hearing loss (Hakuba et al., 2000). However, extracellular glutamate levels have to our knowledge not been directly evaluated in vivo in the forebrain of GLAST KO mice. Given the unexpected finding of reduced rather than increased alcohol consumption in the GLAST KO:s, we examined whether their extracellular glutamate levels are in fact elevated within the ventral striatum, a structure of key importance for drug reward (Koob and Volkow, 2010). Unexpectedly, extracellular glutamate levels were remarkably similar between genotypes, both at baseline and following administration of a 2 g/kg alcohol dose. Effects of alcohol on extracellular glutamate are biphasic, with stimulation at lower, and suppression at higher doses (Moghaddam and Bolinao, 1994). We can therefore not exclude that other alcohol doses could have influenced glutamate levels, or revealed a genotype difference. The significance of the data obtained with the 2 g/kg dose is that they parallel the dose used in the place conditioning model. Together, these observations make it unlikely that the difference in alcohol reward observed in that paradigm can be accounted for by differences in alcohol-induced glutamate release. The dissociation between extracellular glutamate levels in the ventral striatum on one hand, and alcohol intake and CPP on the other is of also interest, because it indicates that glutamatergic transmission in this region does not always play a critical role in regulation of alcohol related behaviors.
A major role for GLAST in glutamate clearance was originally established using antisense oligonucleotide treatment to knock down GLAST expression (Rothstein et al., 1996). In contrast, our study used a constitutive KO model, where developmental compensations can occur. Widespread compensatory changes in glutamatergic gene expression have indeed been found in the forebrain of the GLAST KO:s (Ueda et al., 2002). Although expression within the striatum was not analyzed in this study, analysis of frontal cortical tissue showed that both GLT-1 and EAAC-1 transporters were markedly up-regulated in the mutants, an adaptation clearly likely to compensate for impaired glutamate clearance due to loss of GLAST. Instead, the authors concluded that the increased susceptibility to seizures in GLAST KO mice is related to their increased expression of Glu-R1 in the hippocampus, coupled with decreased cortical expression of Glu-R2 and generally increased NMDA-R1 and -2A, -2B expression. The absence of elevated extracellular glutamate levels in the ventral striatum of the GLAST KO:s supports the notion that compensatory mechanisms have in fact been activated as a result of the GLAST deletion.
Endocannabinoid signaling is crucial for alcohol consumption and reward [e.g. (Economidou et al., 2006; Hansson et al., 2007; Hungund et al., 2003; Lallemand et al., 2001; Lallemand and De Witte, 2006; Wang et al., 2003)]. Developmental neuroadaptations encompassing the endocannabinoid system could therefore contribute to the decreased alcohol consumption and reward observed in the GLAST KO:s. Corticostriatal LTD in slice preparations of dorsal striatum is endocannabinoid dependent, and has been extensively characterized as a model system in which endocannabinoid function can be studied [e.g. (Adermark and Lovinger, 2007a; Adermark and Lovinger, 2007b; Yin et al., 2007)]. Using this model system, we found a loss of LTD induced by HFS in slices from GLAST KO:s. We considered the possibility that impaired mGluR1 or mGluR5 function could account for this observation, because post-synaptic metabotropic glutamate receptors are required for LTD induction by HFS at striatal synapses (Sung et al., 2001). However, depolarization of postsynaptic neurons induced by activation of L-type calcium channels was also unable to induce LTD in slices from GLAST KO, making it unlikely that effects on postsynaptic mGluRs can explain the observed loss of LTD (Adermark and Lovinger, 2007a). In contrast, when presynaptic CB1 receptors were directly stimulated by administration of the exogenous agonist WIN55,212-2, robust synaptic depression was observed, indicating normal levels and function of these receptors. Collectively, these data suggest that developmental neuroadaptations in GLAST KO:s involve a postsynaptic impairment of retrograde endocannabinoid signaling under conditions when this is normally triggered. A loss of endocannabinoid function is a plausible candidate mechanism to account for the reduced alcohol consumption and reward found following a constitutive deletion of GLAST.
In summary, we report that deletion of GLAST results in an unexpected decrease in alcohol consumption and preference, suggestive of decreased alcohol reward. This behavioral phenotype is associated with impaired endocannabinoid function, potentially reflecting impaired postsynaptic endocannabinoid synthesis or release. Observations of compensatory mechanisms in the GLAST mutants cannot be directly generalized to normal physiology. They may, however, point to mechanism that could be exploited for medications development. Thus, the CB1 receptor antagonist rimonabant was recently evaluated clinically as a candidate treatment for alcoholism (George et al., 2010; Soyka et al., 2008). Despite the extensive preclinical literature supporting a role of endocannabinoids in alcohol reward, the results of these studies were negative, presumably because rimonabant doses that are safe and well tolerated are insufficient to achieve therapeutic effects, while higher doses are associated with unacceptable side effects. Based on our present findings, it might be speculated that more subtle modulation of endocannabinoid signaling, such as e.g. inhibition of their on-demand synthesis, may offer alternative therapeutic strategies in alcoholism that would not be limited by adverse effects.
Highlights.
A hyperglutamatergic state might contribute to alcohol addiction
We studied alcohol related behaviors in mice deficient in the glutamate transporter GLAST
Alcohol consumption and reward were markedly reduced in the GLAST KO:s
Several measures of sedative – ataxic alcohol effects were unaffected by the deletion
Microdialysis and electrophysiology indicated endocannabinoid adaptations in the KO:s
Acknowledgments
We thank Jessica Mensch for assistance with the breeding at NIAAA and Dr Yi-Chyan Chen for technical assistance. Authors MH, DML and and AH are supported by the National Institute on Alcohol Abuse and Alcoholism Intramural Research Program; RS by the German Bundesministerium für Bildung und Forschung (NGFN Plus; FKZ: 01GS08152). KT is supported by The Novartis Foundation (Japan) for the promotion of Science, Takeda Science Foundation, The Tokyo Biochemical Research Foundation, Research Foundation for Opto-Science and Technology and by Grants-in-Aids for Scientific Research on Priority Area (20022013 and 18053006) provided by the Ministry of Education, Culture, Sports, Science, and Technology of Japan. AM is supported by Svenska Stiftelsen för Medicinsk Forskning and Svenska Läkarsällskapet.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Adermark L, Lovinger DM. Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. The Journal of neuroscience. 2007a;27:6781. doi: 10.1523/JNEUROSCI.0280-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adermark L, Lovinger DM. Retrograde endocannabinoid signaling at striatal synapses requires a regulated postsynaptic release step. Proceedings of the National Academy of Sciences. 2007b;104:20564. doi: 10.1073/pnas.0706873104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouza C, Angeles M, Munoz A, Amate JM. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction. 2004;99:811–828. doi: 10.1111/j.1360-0443.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- Boyce Rustay JM, Wiedholz LM, Millstein RA, Carroll J, Murphy DL, Daws LC, Holmes A. Ethanol Related Behaviors in Serotonin Transporter Knockout Mice. Alcoholism: Clinical and Experimental Research. 2006;30:1957–1965. doi: 10.1111/j.1530-0277.2006.00241.x. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Cunningham CL. The Role of NMDA Receptor Binding Sites in Ethanol Place Conditioning. Behavioral Neuroscience. 2004;118:822. doi: 10.1037/0735-7044.118.4.822. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Holmes A. Ethanol-related behaviors in mice lacking the NMDA receptor NR2A subunit. Psychopharmacology. 2006;187:455–466. doi: 10.1007/s00213-006-0448-6. [DOI] [PubMed] [Google Scholar]
- Carr GD, Phillips AG, Fibiger HC. Independence of amphetamine reward from locomotor stimulation demonstrated by conditioned place preference. Psychopharmacology. 1988;94:221–226. doi: 10.1007/BF00176849. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Cameron AJ, Munn E, Bunning M, Wahlsten D. Overview of mouse assays of ethanol intoxication. Current Protocols in Neuroscience. 2008 doi: 10.1002/0471142301.ns0926s42. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Henderson CM. Ethanol-induced conditioned place aversion in mice. Behavioural Pharmacology. 2000;11:591–602. doi: 10.1097/00008877-200011000-00006. [DOI] [PubMed] [Google Scholar]
- Dahchour A, De Witte P. Effects of acamprosate on excitatory amino acids during multiple ethanol withdrawal periods. Alcohol Clin Exp Res. 2003;27:465–470. doi: 10.1097/01.ALC.0000056617.68874.18. [DOI] [PubMed] [Google Scholar]
- Dahchour A, De Witte P, Bolo N, Nedelec JF, Muzet M, Durbin P, Macher JP. Central effects of acamprosate: part 1. Acamprosate blocks the glutamate increase in the nucleus accumbens microdialysate in ethanol withdrawn rats. Psychiatry Research. 1998;82:107–114. doi: 10.1016/s0925-4927(98)00016-x. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Progress in Neurobiology. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- De Witte P, Pinto E, Ansseau M, Verbanck P. Alcohol and withdrawal: from animal research to clinical issues. Neuroscience and Biobehavioral Reviews. 2003;27:189–197. doi: 10.1016/s0149-7634(03)00030-7. [DOI] [PubMed] [Google Scholar]
- Economidou D, Mattioli L, Cifani C, Perfumi M, Massi M, Cuomo V, Trabace L, Ciccocioppo R. Effect of the cannabinoid CB1 receptor antagonist SR-141716A on ethanol self-administration and ethanol-seeking behaviour in rats. Psychopharmacology (Berl) 2006;183:394–403. doi: 10.1007/s00213-005-0199-9. [DOI] [PubMed] [Google Scholar]
- Flatscher-Bader T, van der Brug MP, Landis N, Hwang JW, Harrison E, Wilce PA. Comparative gene expression in brain regions of human alcoholics. Genes Brain Behav. 2006;5(Suppl 1):78–84. doi: 10.1111/j.1601-183X.2006.00197.x. [DOI] [PubMed] [Google Scholar]
- Flatscher-Bader T, Wilce PA. Chronic smoking and alcoholism change expression of selective genes in the human prefrontal cortex. Alcohol Clin Exp Res. 2006;30:908–915. doi: 10.1111/j.1530-0277.2006.00106.x. [DOI] [PubMed] [Google Scholar]
- George D, Herion D, Jones C, Phillips M, Hersh J, Hill D, Heilig M, Ramchandani V, Geyer C, Spero D, Singley E, O’Malley S, Bishai R, Rawlings R, Kunos G. Rimonabant (SR141716) has no effect on alcohol self-administration or endocrine measures in nontreatment-seeking heavy alcohol drinkers. Psychopharmacology. 2010;208:37–44. doi: 10.1007/s00213-009-1704-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W, Justice JB. Dissociation of locomotor and conditioned place preference responses following manipulation of GABA-A and AMPA receptors in ventral pallidum. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 1997;21:839–852. doi: 10.1016/s0278-5846(97)00084-5. [DOI] [PubMed] [Google Scholar]
- Gremel CM, Cunningham CL. Role of test activity in ethanol-induced disruption of place preference expression in mice. Psychopharmacology. 2007;191:195–202. doi: 10.1007/s00213-006-0651-5. [DOI] [PubMed] [Google Scholar]
- Groblewski PA, Bax LS, Cunningham CL. Reference-dose place conditioning with ethanol in mice: empirical and theoretical analysis. Psychopharmacology. 2008;201:97–106. doi: 10.1007/s00213-008-1251-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakuba N, Koga K, Gyo K, Usami S, Tanaka K. Exacerbation of noise-induced hearing loss in mice lacking the glutamate transporter GLAST. The Journal of neuroscience. 2000;20:8750. doi: 10.1523/JNEUROSCI.20-23-08750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson AC, Bermudez-Silva FJ, Malinen H, Hyytia P, Sanchez-Vera I, Rimondini R, Rodriguez de FF, Kunos G, Sommer WH, Heilig M. Genetic impairment of frontocortical endocannabinoid degradation and high alcohol preference. Neuropsychopharmacology. 2007;32:117–126. doi: 10.1038/sj.npp.1301034. [DOI] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. The Journal of neuroscience. 2007;27:11496. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hungund BL, Szakall I, Adam A, Basavarajappa BS, Vadasz C. Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol-induced dopamine release in the nucleus accumbens. J Neurochem. 2003;84:698–704. doi: 10.1046/j.1471-4159.2003.01576.x. [DOI] [PubMed] [Google Scholar]
- Kapasova Z, Szumlinski KK. Strain Differences in Alcohol Induced Neurochemical Plasticity: A Role for Accumbens Glutamate in Alcohol Intake. Alcoholism: Clinical and Experimental Research. 2008;32:617–631. doi: 10.1111/j.1530-0277.2008.00620.x. [DOI] [PubMed] [Google Scholar]
- Kargieman L, Santana N, Mengod G, Celada P, Artigas F. Antipsychotic drugs reverse the disruption in prefrontal cortex function produced by NMDA receptor blockade with phencyclidine. Proceedings of the National Academy of Sciences. 2007;104:14843. doi: 10.1073/pnas.0704848104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson RM, Heilig M, Holmes A. Loss of Glial Glutamate and Aspartate Transporter (Excitatory Amino Acid Transporter 1) Causes Locomotor Hyperactivity and Exaggerated Responses to Psychotomimetics: Rescue by Haloperidol and Metabotropic Glutamate 2/3 Agonist. Biological Psychiatry. 2008;64:810–814. doi: 10.1016/j.biopsych.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson RM, Holmes A, Heilig M, Crawley JN. Anxiolytic-like actions of centrally-administered neuropeptide Y, but not galanin, in C57BL/6J mice. Pharmacol Biochem Behav. 2005;80:427–436. doi: 10.1016/j.pbb.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of Addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand F, De Witte P. SR147778, a CB1 cannabinoid receptor antagonist, suppresses ethanol preference in chronically alcoholized Wistar rats. Alcohol. 2006;39:125–134. doi: 10.1016/j.alcohol.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Lallemand F, Soubrie PH, Witte PH. Effects of CB1 cannabinoid receptor blockade on ethanol preference after chronic ethanol administration. Alcoholism: Clinical and Experimental Research. 2001;25:1317–1323. [PubMed] [Google Scholar]
- Leeman RF, Heilig M, Cunningham CL, Stephens DN, Duka T, O’Malley SS. Ethanol consumption: how should we measure it? Achieving consilience between human and animal phenotypes. Addict Biol. 2010;15:109–124. doi: 10.1111/j.1369-1600.2009.00192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millstein RA, Holmes A. Effects of repeated maternal separation on anxiety-and depression-related phenotypes in different mouse strains. Neuroscience & Biobehavioral Reviews. 2007;31:3–17. doi: 10.1016/j.neubiorev.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Bolinao ML. Biphasic effect of ethanol on extracellular accumulation of glutamate in the hippocampus and the nucleus accumbens. Neuroscience Letters. 1994;178:99–102. doi: 10.1016/0304-3940(94)90299-2. [DOI] [PubMed] [Google Scholar]
- Rimondini R, Arlinde C, Sommer W, Heilig M. Long-lasting increase in voluntary ethanol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. FASEB Journal. 2002;16:27–35. doi: 10.1096/fj.01-0593com. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Addiction. Annual Review of Psychology. 2003;54:25–53. doi: 10.1146/annurev.psych.54.101601.145237. [DOI] [PubMed] [Google Scholar]
- Rossetti ZL, Carboni S. Ethanol withdrawal is associated with increased extracellular glutamate in the rat striatum. European Journal of Pharmacology. 1995;283:177–183. doi: 10.1016/0014-2999(95)00344-k. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Sellings LHL, Clarke P. Segregation of amphetamine reward and locomotor stimulation between nucleus accumbens medial shell and core. The Journal of neuroscience. 2003;23:6295. doi: 10.1523/JNEUROSCI.23-15-06295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyka M, Koller G, Schmidt P, Lesch OM, Leweke M, Fehr C, Gann H, Mann KF. Cannabinoid receptor 1 blocker rimonabant (SR 141716) for treatment of alcohol dependence: results from a placebo-controlled, double-blind trial. J Clin Psychopharmacol. 2008;28:317–324. doi: 10.1097/JCP.0b013e318172b8bc. [DOI] [PubMed] [Google Scholar]
- Spanagel R. Alcoholism: A Systems Approach From Molecular Physiology to Addictive Behavior. Physiological Reviews. 2009;89:649–705. doi: 10.1152/physrev.00013.2008. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Kiefer F. Drugs for relapse prevention of alcoholism: ten years of progress. Trends in Pharmacological Sciences. 2008;29:109–115. doi: 10.1016/j.tips.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Pendyala G, Abarca C, Zghoul T, Sanchis-Segura C, Magnone MC, Lascorz J, Depner M, Holzberg D, Soyka M, Schreiber S, Matsuda F, Lathrop M, Schumann G, Albrecht U. The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nature Medicine. 2005;11:35–42. doi: 10.1038/nm1163. [DOI] [PubMed] [Google Scholar]
- Sung KW, Choi S, Lovinger DM. Activation of group I mGluRs is necessary for induction of long-term depression at striatal synapses. Journal of Neurophysiology. 2001;86:2405. doi: 10.1152/jn.2001.86.5.2405. [DOI] [PubMed] [Google Scholar]
- Thorsell A, Schank JR, Singley E, Hunt SP, Heilig M. Neurokinin-1 receptors (NK1R:s), alcohol consumption, and alcohol reward in mice. Psychopharmacology. 2010;209:103–111. doi: 10.1007/s00213-010-1775-1. [DOI] [PubMed] [Google Scholar]
- Tiffany Cunningham S, Kelley AE. Opiate infusion into nucleus accumbens: contrasting effects on motor activity and responding for conditioned reward. Brain Research. 1992;588:104–114. doi: 10.1016/0006-8993(92)91349-j. [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. The role of glutamatergic neurotransmission in the pathophysiology of alcoholism. Annual Review of Medicine. 1998;49:173–184. doi: 10.1146/annurev.med.49.1.173. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Progress in Neurobiology. 1998;56:613–672. doi: 10.1016/s0301-0082(98)00060-4. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addiction Biology. 2007;12:227. doi: 10.1111/j.1369-1600.2007.00070.x. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Doi T, Tsuru N, Tokumaru J, Mitsuyama Y. Expression of glutamate transporters and ionotropic glutamate receptors in GLAST knockout mice. Molecular Brain Research. 2002;104:120–126. doi: 10.1016/s0169-328x(02)00325-x. [DOI] [PubMed] [Google Scholar]
- Umhau JC, Momenan R, Schwandt ML, Singley E, Lifshitz M, Doty L, Adams LJ, Vengeliene V, Spanagel R, Zhang Y, Shen J, George DT, Hummer D, Heilig M. Effect of Acamprosate on Magnetic Resonance Spectroscopy Measures of Central Glutamate in Detoxified Alcohol-Dependent Individuals A Randomized Controlled Experimental Medicine Study. Archives of General Psychiatry. 2010;67:1069–1077. doi: 10.1001/archgenpsychiatry.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu J, Harvey-White J, Zimmer A, Kunos G. Endocannabinoid signaling via cannabinoid receptor 1 is involved in ethanol preference and its age-dependent decline in mice. Proc Natl Acad Sci U S A. 2003;100:1393–1398. doi: 10.1073/pnas.0336351100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. European Journal of Neuroscience. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychological Review. 1987;94:469. [PubMed] [Google Scholar]
- Yin HH, Park BS, Adermark L, Lovinger DM. Ethanol reverses the direction of long term synaptic plasticity in the dorsomedial striatum. European Journal of Neuroscience. 2007;25:3226–3232. doi: 10.1111/j.1460-9568.2007.05606.x. [DOI] [PubMed] [Google Scholar]