

Abstract
It is usually assumed that to ensure proper function, membrane proteins must be inserted in a unique topology. However, a number of dimeric small multidrug transporters can function in the membrane in various topologies. Thus, the dimers can be a random mixture of NiCi (N- and C-termini facing the cell cytoplasm) and NoCo (N- and C-termini facing the outside) orientation. In addition, the dimer functions whether the two protomers are parallel (N- and C-termini of both protomers on the same side of the membrane) or anti-parallel (N- and C-termini of each protomer on opposite sides of the membrane). This unique phenomenon provides strong support for a simple mechanism of transport where the directionality is determined solely by the driving force
Much has been said about the topology of EmrE, a small (110 residues) homodimeric multidrug transporter from Escherichia coli that extrudes positively charged aromatic drugs in exchange for two protons, making bacteria resistant to a variety of toxic compounds.
EmrE is a dimer. Any single dimeric membrane protein may adopt three different topologies (denoted here as ‘topoforms’) schematically shown in figure 1 (top). Relative to each other, the two protomers can theoretically adopt either a parallel (P, N and C-termini of both protomers on the same side of the membrane) or anti-parallel orientation (AP, N and C-termini of each protomer on opposite sides of the membrane). Additionally, relative to the lipid bilayer, the ensemble of parallel dimers can theoretically adopt a single topology or a dual topology where dimers are either all in a NiCi (or NoCo) orientation or a mixture of both. However, while all the potential topologies could occur what topology makes biological sense? The answer to that question is open since there are constraints such as the way the protein is inserted into the membrane during synthesis and the requirement for the dimer to be catalytically active. So, determining the topology of EmrE is essential to learning how it functions and how the protein has evolved. As more complex membrane proteins could have evolved from smaller ones, this also has implications for the evolution of membrane proteins in general.
Figure 1. Native and artificially generated topoforms of EmrE.
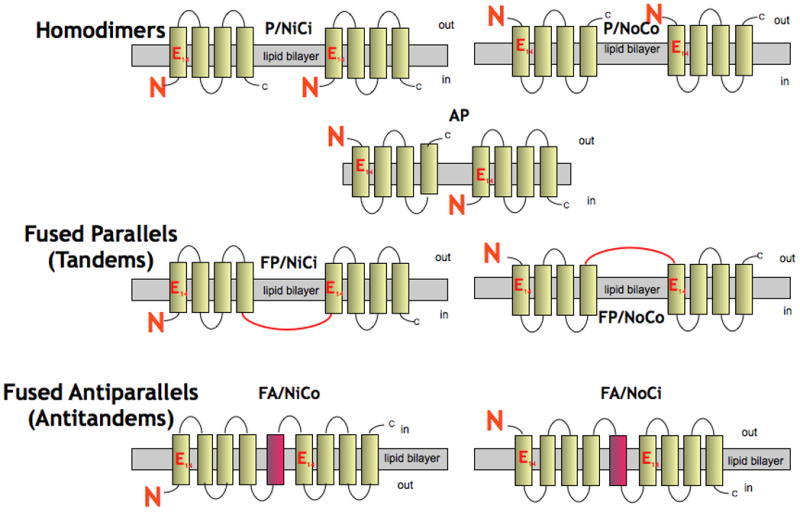
Four of them (P/NiCi, P/NoCo, FP/NiCi and FA/NiCo) have been experimentally demonstrated while a fifth one (AP) was suggested from models based on low-resolution 2D and 3D crystals and from FRET and crosslinking experiments in bicelles (see text for references and explanations).
One of the reasons for the controversy about the topology of EmrE was the assumption that to ensure proper function membrane proteins must be inserted in a unique topology. Indeed, it is obvious that proteins such as receptors must face the environment they are probing, channels that sense the electrical field across membranes depend on the polarity of the field and cannot be inserted randomly, pumps that derive their energy from molecules such as ATP, available only on one side of the membrane, need to have the ATP utilization machinery in the right location. However, is a unique topology a necessary prerequisite for uniporters or ion-coupled transporters? Work with EmrE demonstrates that this is not an essential prerequisite for function as will become clear in this Opinion paper.
Looking for the “right” unique topology
During almost a decade our group and others have published what looks like conflicting results regarding the topology of EmrE. I will briefly describe below the evidence supporting the opposing views. I must however start by clarifying from the very beginning that even though I critically evaluate the evidence that supports the antiparallel topology, I do believe that the antiparallel topology is also possible under some conditions and my group has presented experimental evidence that supports this view. However, I also present the evidence that supports the parallel topology (critically evaluated or disregarded by others) and I conclude that SMR proteins can function whether parallel or antiparallel and when parallel they can function whether facing one side of the membrane or the other. This is to my knowledge a unique phenomenon and its significance is discussed below.
A. Evidence supporting antiparallel topology
The possibility of an antiparallel topology of the protomers in the EmrE homodimer was first suggested by Ubarretxena-Belandia et al. to explain the quasi-symmetry observed in the 3-D structure of EmrE acquired by electron cryo-microscopy (cryo-EM) at 7.5 Å resolution in the membrane plane [1]., a model derived from a low-resolution 3D-crystal structure supported this suggestion [2]. Even though the structure was withdrawn [3], it stimulated investigators to pursue this unique avenue. Fleishman et al. used the symmetry relationship mentioned above, combined with sequence conservation data, to assign the transmembrane segments in EmrE to the densities seen in the cryo-EM structure. The C-α model of the transmembrane region was constructed so that the helices of one protomer have the topology opposite to the ones of the other protomer (antiparallel) [4]. A recalculated model based on the 3D-crystals is remarkably similar to the model suggested from the electron microscopy data [5]. The similarity is notable since the resolution of the structures used to build the models is very low in both cases. Moreover, the crystals used to derive the C-α model x-ray structure were obtained with protein solubilized with detergents that inhibit activity by dissociating monomers, as shown by us and in the crystallization paper [5, 6]. Protomers arrange in the crystal in a conformation that minimizes the energy needed for crystal formation and do not necessarily reflect the topology in the membrane.
Genetic experiments were designed to support the claim for an antiparallel topology. EmrE was fused to the “topology-reporter proteins” alkaline phosphatase or green fluorescent protein, and the results showed that the topology of the EmrE fusion proteins in the membrane is sensitive to the distribution of positive charges in the protein [7]. Manipulation of the positive charges generated a set of mutants, some with NoCo, and others with NiCi apparent topology [7, 8]. Since neither mutant conferred resistance to ethidium, the authors concluded that this was due to the modified topology. Co-expression of the inactive mutants restored the ethidium resistance to the same level as seen with wild type EmrE [8]. The suggested interpretation of this finding was that co-expression results in the generation of a functional, antiparallel heterodimer. However, this assumption is based solely on the contention that the NiCi and NoCo mutants are inactive. On closer investigation, we and others found that they are both functional as judged from the phenotype assayed following continuous growth in liquid medium [9] or in cells with the proper genetic background where the chromosomal gene of EmrE has been inactivated and expression of the mutants induced [10]. The lower activity of the single mutants compared with the coexpressed ones could be due to impaired dimerization and not necessarily due to a different topology [10]. The coexpressed mutants display high activity because they can interact with high affinity and form a functional heterodimer where each monomer differs from the other by six amino acids [8].
A comprehensive study of the orientation in the membrane of several SMR proteins was performed using reporter genes [11]. The results suggest a fraction of the protein faces the cytoplasmic side while the other faces the periplasm. The authors conclude that the findings support an antiparallel topology but the results cannot distinguish between this and a mixed population of parallel dimers (Fig. 1, top).
Amadi et al. performed a systematic spin labeling and EPR analysis to study the structure and dynamics of EmrE in liposomes [12]. The study revealed at least two spin label populations arising from different packing interfaces of the EmrE dimer. One population is consistent with antiparallel arrangement of the monomers, although the EPR parameters suggest deviations from the crystal structure of substrate-bound EmrE. It is not yet clear whether the parallel dimer accounts for the rest of the population [12].
A recent study provides support for the presence of antiparallel dimers with protein reconstituted in bicelles [13]. The experimental evidence does not rule out the existence of parallel dimers as well. Crosslinking experiments were performed with two classes of crosslinkers: one that crosslinks only parallel dimers and another one only antiparallel ones. Despite the fact that both crosslink the dimers it is claimed that the “parallel” crosslinking reflects an artifact due to crowding and proximity between different dimers. However even at high dilutions the crosslinking is observed and no such control is reported for the other crosslinker. The elegant FRET experiments that were carried out cannot rule out the existence of populations where the fluorophores are very close. Energy transfer efficiency depends not only on the distance but also on the dipole orientation and this parameter may be specially relevant in membrane proteins where the dipoles cannot always freely randomize [14].
It is possible that in bicelles, the very specific lipid environment and the manipulation of the protein pushed the equilibrium towards the antiparallel conformation [13]. This finding can provide interesting information about the determinants that drive the protein to an antiparallel conformation and it will be interesting to see whether further experimentation demonstrates functionality of the protein under these conditions.
B. Evidence supporting parallel topology
To evaluate the antiparallel structural model, we tested one of its most basic predictions, the existence of antiparallel dimers, using a cross-linking approach. All our experiments were consistent only with parallel dimers. When unique cysteines were engineered in the hydrophilic loops, or in the termini, crosslinkers that could react with residues 9–11 Å apart quantitatively crosslinked the protomers [15]. This finding is inconsistent with an antiparallel topology, which predicts the two cysteine residues to be at least 35–40 Å apart. Since crosslinking could be amenable to artifacts we also purified two proteins crosslinked in loops 1 and 3 and showed that they are fully active supporting the contention that the crosslinking experiments reflect the protomer symmetry in functional dimers [6].
Generation of genetic fusions (tandems) where the C-terminus of one protomer was fused to the N-terminus of the second protomer provided a paradigm to study the topology both in vivo and in vitro. The linkers were designed such that they are very short or very hydrophilic. This ensured that both termini are in the same side of the membrane and force the dimer into a parallel topology (Fig. 1, middle panel) [16]. All the tandems built as described were functional in vivo and after purification. To ensure that the functional unit is the dimer and not a result of interaction between dimers we showed that, unlike the wild type protein, the fused dimer does not interact with inactive mutants (negative dominance experiments). Furthermore, we did not detect larger oligomeric species as judged from pull-down experiments [6, 16].
A solution to the controversy
How can we reconcile the controversy? Our biochemical work very strongly supports the existence of a parallel dimer that, in our view, is the native form of EmrE. However, we wanted to know if the antiparallel association also exists. We were not able to identify any sizable fraction of the “native” population in antiparallel configuration [6, 17, 18]. However, evolution has provided a partial answer to this question. In the SMR family closely related SMR parallel homodimers, such as EmrE, and heterodimers, putatively antiparallel, such as EbrAB from B. subtilis [19], perform identical functions (ethidium and acriflavine efflux) even though the relative topology of the protomers is different (Fig. 2 top). Moreover EbrB is capable of forming homodimers, most likely in a parallel mode because it has strong topogenic signals (Fig 2 and [10]). In addition, individual EbrA and EbrB mutants with altered (K+R) bias also function as homodimers (Fig 2 and [20]). In other words, the interaction between protomers is very promiscuous: the protomers can interact either with each other as an antiparallel heterodimer or as a parallel homodimer in the absence of the other protomer or if the topogenic signals are modified.
Figure 2. The many faces of a heterodimer.

EbrAB is a heterodimer from Bacillus subtilis [19]. EbrB can function also as a homodimer when expressed without EbrA in a ΔemrE host [10]. Both EbrA and EbrB can function as homodimers when their topogenic signals are properly manipulated [20]. None of the topologies was experimentally determined.
If very closely related heterodimers are so promiscuous and can interact parallel and antiparallel, the answer to the EmrE controversy may be quite simple: EmrE is also promiscuous in the interaction of the protomers and is functional in either of the proposed topologies (Fig 1, top). The topogenic determinants in the wild type EmrE are non-existent and therefore parallel dimers insert in random topologies: about half NiCi and half NoCo [10]. After insertion, the interaction of the protomers will result in a parallel or antiparallel mode depending only on their relative affinities. Since the interaction in a parallel mode is the only one identified in the “native” protein [6, 15–17], we propose that affinity for parallel association is higher, at least under the conditions tested thus far. However, the protomers can be forced to interact productively also in an antiparallel mode by fusing them head to tail with an additional membrane helix (Figure 1, bottom and ref. [10]). This fusion results in higher “local” concentrations of the topologically antiparallel protomer overcoming its lower affinity. This fused antiparallel dimer was also shown to be functional in vivo and in vitro [10]. As the case with the parallel tandem, negative dominance and pull down experiments supported the claim that the dimer is the functional unit [10].
Topology and function
As far as we know, the diversity of functional topoforms has not been shown yet in other systems. Two important questions are posed by such a behavior. First, an implicit conclusion from these findings is that the interaction between protomers is very promiscuous: homodimers can form functional heterodimers when pushed to do so, for example by modification of the topogenic signals [8] or by in vitro mixing with other SMRs [21]. On the other hand, heterodimers can also form homodimers as described above for EbrAB [10, 20]. Promiscuity of interaction between proteins is not unheard of. It is a well-documented necessity in the major signaling systems where one master protein can interact with many different partners (see for example [22]). Second and to account for the observation that both parallel and antiparallel dimers are functional, we propose that it is because the mechanism of coupling ion and substrate transport is so simple. This simplicity provides the robustness necessary to tolerate such a unique and unprecedented ambiguity in the interaction of the subunits and in the dimer topology relative to the membrane [17]. EmrE has a binding site that is occupied either by H+ or by substrate, providing a basis for the coupling reaction (Fig. 3). Its occupancy is mutually exclusive thanks to a carefully tuned pKa of two Glu residues located in a central location in the binding domain [17]. This essential property of the Glu residues is conserved whether the protomers interact in a parallel or antiparallel mode because both configurations provide a suitably similar environment in the hydrophobic cavity.
Figure 3. Transport mechanism in EmrE.
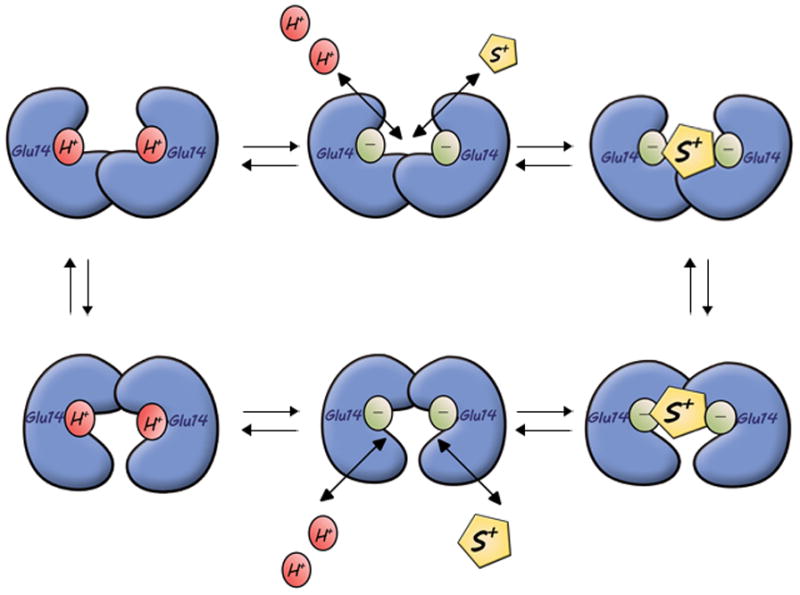
The pKa of the carboxyl at position 14 is around 8.0 [27]. Substrate binds only to the negatively charged EmrE and shifts the equilibrium so that 2H+ are released to the same side of the membrane. Substrate binding induces a conformational change that opens the binding chamber to the opposite side. Proton binding displaces the substrate and reorients the binding site. The substrate is represented as a yellow diamond and protons as red balls. Cartoon based on mechanism depicted in reference [27].
How are the binding determinants conserved in all the topoforms? In polyspecific proteins, binding determinants have been shown to be overlapping and even redundant so that substrates are capable of interacting with one of a number of them (see e.g. [23]). We conclude that such a redundancy allows for specific binding in both, the parallel and antiparallel dimers.
The findings described here support the concept that ion-coupled proteins are by nature symmetric in their function and the direction of the transport is determined solely by the direction of the driving force (see e.g. [24]). As suggested by the alternate access model, the coupled translocation of a substrate molecule and one or more ions across the membrane involves a global conformational change that allows alternating access of substrate- and H+-binding sites to either side of the membrane. Therefore, unless dictated otherwise by regulation, only the driving force determines the directionality of transport.
The behavior observed with EmrE and other SMRs may represent a stage in the evolution of the topology of membrane proteins [17, 25]. The evolutionary challenge of recognition and transport of a wide spectrum of substrates may have selected for SMR heterodimers that originated from gene duplication of the more ancient homodimers. After gene duplication, a relatively small number of mutations would allow either parallel or antiparallel orientation of the protomers within the heterodimer. In this manner, one protein with only a slightly modified sequence may extend the range of the substrate specificity [25]. Inverted repeats have been identified in many large modern transporters and have been suggested to play important roles in the transport mechanism [26]. The phenomenon described here suggests a way by which these inverted repeats may have evolved.
Acknowledgments
Work in our laboratory is supported by National Institutes of Health Grant NS16708 and Grant 119/04 from the Israel Science Foundation. I thank all the colleagues and friends that critically read this manuscript and suggested improvements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ubarretxena-Belandia I, et al. Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. EMBO J. 2003;22:6175–6181. doi: 10.1093/emboj/cdg611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pornillos O, et al. X-ray structure of the EmrE multidrug transporter in complex with a substrate. Science. 2005;310:1950–1953. doi: 10.1126/science.1119776. [DOI] [PubMed] [Google Scholar]
- 3.Chang G, et al. Retraction. Science. 2006;314:1875. doi: 10.1126/science.314.5807.1875b. [DOI] [PubMed] [Google Scholar]
- 4.Fleishman SJ, et al. Quasi-symmetry in the cryo-EM structure of EmrE provides the key to modeling its transmembrane domain. J Mol Biol. 2006;364:54–67. doi: 10.1016/j.jmb.2006.08.072. [DOI] [PubMed] [Google Scholar]
- 5.Chen YJ, et al. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci U S A. 2007;104:18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soskine M, et al. On parallel and antiparallel topology of an homodimeric multidrug transporter. J Biol Chem. 2006;281:36205–36212. doi: 10.1074/jbc.M607186200. [DOI] [PubMed] [Google Scholar]
- 7.Rapp M, et al. Identification and evolution of dual-topology membrane proteins. Nat Struct Mol Biol. 2006;13:112–116. doi: 10.1038/nsmb1057. [DOI] [PubMed] [Google Scholar]
- 8.Rapp M, et al. Emulating membrane protein evolution by rational design. Science. 2007;315:1282–1284. doi: 10.1126/science.1135406. [DOI] [PubMed] [Google Scholar]
- 9.McHaourab HS, et al. Role of sequence bias in the topology of the multidrug transporter EmrE. Biochemistry. 2008;47:7980–7982. doi: 10.1021/bi800628d. [DOI] [PubMed] [Google Scholar]
- 10.Nasie I, et al. Topologically random insertion of EmrE supports a pathway for evolution of inverted repeats in ion-coupled transporters. J Biol Chem. 2010;285:15234–15244. doi: 10.1074/jbc.M110.108746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolbusz MA, et al. Orientation of Small Multidrug Resistance Transporter Subunits in the Membrane: Correlation with the Positive-Inside Rule. J Mol Biol. 2010;402:127–138. doi: 10.1016/j.jmb.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 12.Amadi ST, et al. Structure, dynamics, and substrate-induced conformational changes of the multidrug transporter EmrE in liposomes. J Biol Chem. 2010;285:26710–26718. doi: 10.1074/jbc.M110.132621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison EA, et al. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature. 2011;481:45–50. doi: 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lakowicz JR. Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum Publishers; 1999. [Google Scholar]
- 15.Soskine M, et al. Crosslinking of membrane-embedded cysteines reveals contact points in the EmrE oligomer. Proc Natl Acad Sci U S A. 2002;99:12043–12048. doi: 10.1073/pnas.192392899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steiner-Mordoch S, et al. Parallel topology of genetically fused EmrE homodimers. Embo J. 2008;27:17–26. doi: 10.1038/sj.emboj.7601951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuldiner S. EmrE, a model for studying evolution and mechanism of ion-coupled transporters. Biochim Biophys Acta. 2009;1794:748–762. doi: 10.1016/j.bbapap.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 18.Ninio S, et al. The membrane topology of EmrE - a small multidrug transporter from Escherichia coli. FEBS Lett. 2004;562:193–196. doi: 10.1016/S0014-5793(04)00240-6. [DOI] [PubMed] [Google Scholar]
- 19.Masaoka Y, et al. A two-component multidrug efflux pump, EbrAB, in Bacillus subtilis. J Bacteriol. 2000;182:2307–2310. doi: 10.1128/jb.182.8.2307-2310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kikukawa T, et al. Two-component bacterial multidrug transporter, EbrAB: Mutations making each component solely functional. Biochim Biophys Acta. 2006;1758:673–679. doi: 10.1016/j.bbamem.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 21.Ninio S, et al. Functional analysis of novel multidrug transporters from human pathogens. J Biol Chem. 2001;276:48250–48256. doi: 10.1074/jbc.M108231200. [DOI] [PubMed] [Google Scholar]
- 22.Pawson T. Dynamic control of signaling by modular adaptor proteins. Curr Opin Cell Biol. 2007;19:112–116. doi: 10.1016/j.ceb.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Peters KM, et al. A single acidic residue can guide binding site selection but does not govern QacR cationic-drug affinity. PLoS One. 2011;6:e15974. doi: 10.1371/journal.pone.0015974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smirnova I, et al. Lactose permease and the alternating access mechanism. Biochemistry. 2011;50:9684–9693. doi: 10.1021/bi2014294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuldiner S. When biochemistry meets structural biology: the cautionary tale of EmrE. Trends Biochem Sci. 2007;32:252–258. doi: 10.1016/j.tibs.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Forrest LR, Rudnick G. The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology (Bethesda) 2009;24:377–386. doi: 10.1152/physiol.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adam Y, et al. The fast release of sticky protons: Kinetics of substrate binding and proton release in a multidrug transporter. Proc Natl Acad Sci U S A. 2007;104:17989–17994. doi: 10.1073/pnas.0704425104. [DOI] [PMC free article] [PubMed] [Google Scholar]