

Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) have been widely reported to display strong efficacy for cancer chemoprevention, although their mechanism of action is poorly understood. The most well documented effects of NSAIDs include inhibition of tumor cell proliferation and induction of apoptosis, but their effect on tumor cell invasion has not been well studied. Here we show that the NSAID, sulindac sulfide (SS) can potently inhibit the invasion of human MDA-MB-231 breast and HCT116 colon tumor cells in vitro at concentrations less than those required to inhibit tumor cell growth. To study the molecular basis for this activity, we investigated the involvement of microRNA (miRNA). A total of 132 miRNAs were found to be altered in response to SS treatment including miR-10b, miR-17, miR-21, and miR-9, which have been previously implicated in tumor invasion and metastasis. We confirmed that these miRNA can stimulate tumor cell invasion and show that SS can attenuate their invasive effects by down-regulating their expression. Employing luciferase and chromatin immunoprecipitation assays, NF-κB was found to bind the promoters of all four miRNAs to suppress their expression at the transcriptional level. We show that SS can inhibit the translocation of NF-κB to the nucleus by decreasing the phosphorylation of IKKβ and IκB. Analysis of the promoter sequences of the miRNAs suppressed by SS revealed that 81 of 115 sequences contained NF-κB binding sites. These results show that SS can inhibit tumor cell invasion by suppressing NF-κB mediated transcription of miRNAs.
Keywords: sulindac, invasion, microRNA, NF-κB
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are a chemically diverse family of drugs commonly used to treat a variety of inflammatory conditions and pain associated with arthritis. The long-term use of NSAIDs has been reported to significantly reduce the incidence and risk of death from colorectal and other types of cancer 1. In addition, the NSAID sulindac displays strong efficacy in patients with familial adenomatous polyposis to suppress adenoma size and number by as much as 60–70% 2. These observations are consistent with preclinical studies that have shown pronounced inhibitory effects of sulindac and other NSAIDs on tumorigenesis in experimental rodent models 3–6. The pharmacological basis for the anti-inflammatory activity of NSAIDs involves the inhibition of two distinct cyclooxygenases (COX-1 and -2) that share similar catalytic activity but have different patterns of expression and sensitivity to inhibitors. The antineoplastic activity of NSAIDs is primarily believed to involve both anti-proliferative and pro-apoptotic effects by the inhibition of COX-2, which is elevated in tumor cells 7. However, other studies support the involvement of a COX-independent mechanism 8–14.
Although indomethacin (a sulindac analog) has been shown to significantly increase survival of patients with metastastic disease 15, there have been only a few studies describing the effects of NSAIDs on tumor invasion and metastasis. For example, a recent report demonstrated that sulindac can inhibit metastasis by disrupting β-catenin signaling 16. Tumor invasion and metastasis involves multiple steps that induce neoplastic cells to spread and migrate to surrounding tissue, beyond the borders of the original tumor, and which are hallmarks of malignant tumors that lead to failure of chemotherapy 17. Numerous studies have focused on the identification and characterization of the markers associated with tumor cell invasion and metastasis, but the precise molecular mechanisms that regulate these complex biological processes are largely unknown.
MicroRNAs (miRNAs) are naturally occurring, single-stranded, non-coding sequences of small RNAs that regulate gene expression at the post-transcriptional and translational levels 18, 19. In contrast with messenger RNAs, miRNAs are a small set of approximately 1,500 RNA molecules. Each miRNA can control the expression of several hundred cognate messenger RNA targets simultaneously, and more than 30% of human genes are known to be regulated by miRNAs 20. MiRNAs have been implicated in many biological events such as cell differentiation, proliferation, apoptosis, tumorigenesis, as well as tumor cell invasion and metastasis 21–24.
In this study, we found that the NSAID, sulindac sulfide (SS) can potently inhibit the invasion of human breast and colon tumor cells at concentrations less than those required to inhibit tumor cell growth in vitro. SS treatment altered the expression of 132 miRNAs, in which several are known to be associated with tumor invasion and metastasis, including miR-10b, miR-17, miR-21, and miR-9 25–31. Bioinformatic analysis revealed that more than 70% of the down-regulated miRNAs contain NF-κB binding sites in their promoter regions, which suggest that NF-κB can mediate the effects of SS on miRNA expression. This study functionally demonstrates that SS can inhibit tumor cell invasion by a novel mechanism involving the suppression of NF-κB signaling to inhibit the transcription of miRNAs involved in tumor cell invasion and metastasis.
Results
SS inhibits tumor cell invasion without affecting tumor cell growth
To determine the effects of SS on tumor cell invasion in vitro, the human breast MDA-MB-231 and colorectal HCT116 tumor cell lines were plated on matrigel-coated inserts and treated with different concentrations of SS for 36 h. Counting of viable cell number was performed in parallel to simultaneously measure the tumor cell growth inhibitory activity of SS. Figures 1A and 1B show that SS can inhibit the invasion of both tumor cell lines in a dose-dependent manner, whereby a concentration of 50 µM was found to have a significant inhibitory effect when compared to vehicle-treated control cells (>2-fold; p<0.05). In contrast, viable cell number was not significantly affected at this concentration level (p>0.05), although longer treatments of 48–72 h, resulted in reduced numbers of viable cells as expected (Figure 1C and 1D). These results show that SS can inhibit tumor cell invasion at concentrations lower than those required to inhibit tumor cell growth, which suggest that a distinct molecular mechanisms may be responsible for these effects.
Figure 1. SS inhibits tumor cell invasion without affecting tumor cell growth.
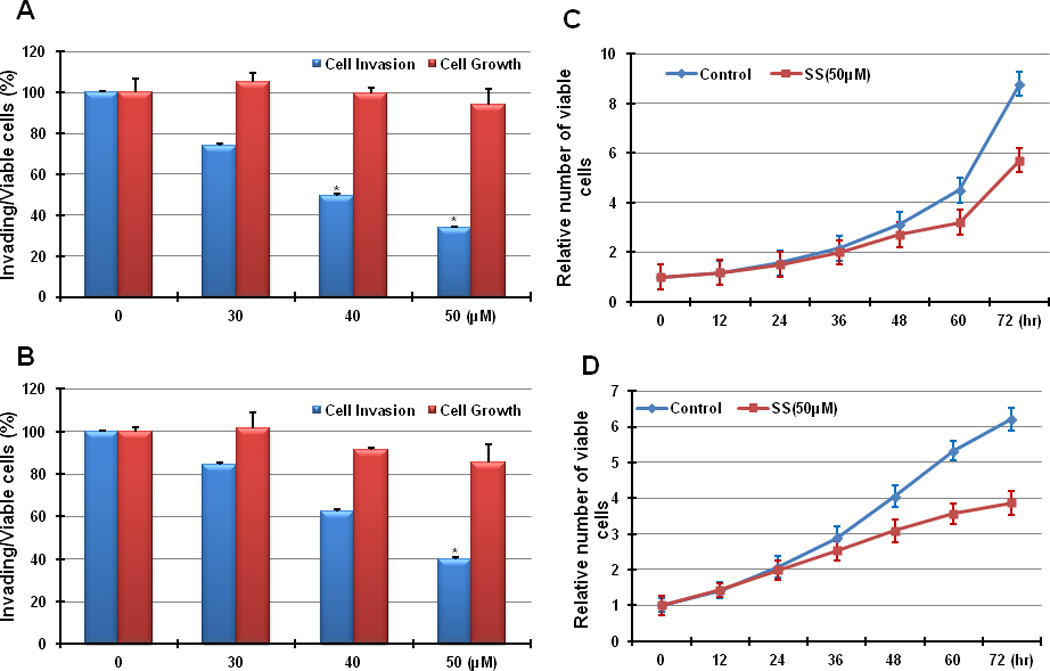
(A) MDA-MB-231 and (B) HCT116 cells were treated with SS at 0, 30 µM, 40 µM, and 50 µM for 36 h. The inhibition of cell invasion by SS was dose dependent, and 50 µM had a significant effect on both MDA-MB-231 and HCT116 cells (p<0.05). The same condition did not significantly affect cell growth (p>0.05). (C) MDA-MB-231 and (D) HCT116 cells were treated with SS at 50 µM, and cell growth was determined after various treatment times. The viability of both tumor cell lines was not significantly influenced until 48 h of treatment (p<0.05). (T-test was used for determining statistical significance; * indicates p<0.05)
MiRNAs are altered in response to treatment with SS in HCT 116 cells
Employing Taqman Low Density Array (TLDA), we determined miRNA expression profiles in HCT116 cells treated with vehicle (0.1% DMSO) or SS (50 µM) for 36 h. The results showed that SS treatment induced 17 miRNAs, while 115 miRNAs were suppressed by 2-fold or greater (Supplementary Figure 1). To confirm this finding, we measured the expression of miR-9, miR-10b, miR-17, miR-21, and miR-125, which have been previously reported to be involved in tumor cell invasion or metastasis 25–32 by employing the SYBR green-based qRT-PCR assay and using the same sample sets tested by the array. The results showed high consistency between the two data sets (Supplementary Figure 2).
MiRNAs mediate the SS-induced inhibition of tumor cell invasion
MiR-10b, miR-17, miR-21, and miR-9 are four of the most well documented miRNAs that are elevated during tumor cell invasion and metastasis 25–31. To determine if these miRNAs could mediate the inhibition of tumor cell invasion by SS, we transfected synthetic miRNA oligonucleotides into MDA-MB-231 and HCT116 cells, as well as two additional tumor cell lines, SUM1315 (breast) and HT29 (colon). The results showed that the elevation of miR-10b, miR-17, miR-21, and miR-9 not only stimulated the invasion of all four tumor cell lines (Figure 2A), but that SS can attenuate their effect on tumor cell invasion (Figure 2B). Interestingly, the miR-pool consisting of miR-10b, miR-17, miR-21, and miR-9 at an equal percent (25%) displayed greater effects than each single miRNA on promoting tumor cell invasion and rescuing the inhibition of SS.
Figure 2. MiRNAs mediate the SS-mediated inhibition of cancer cell invasion.
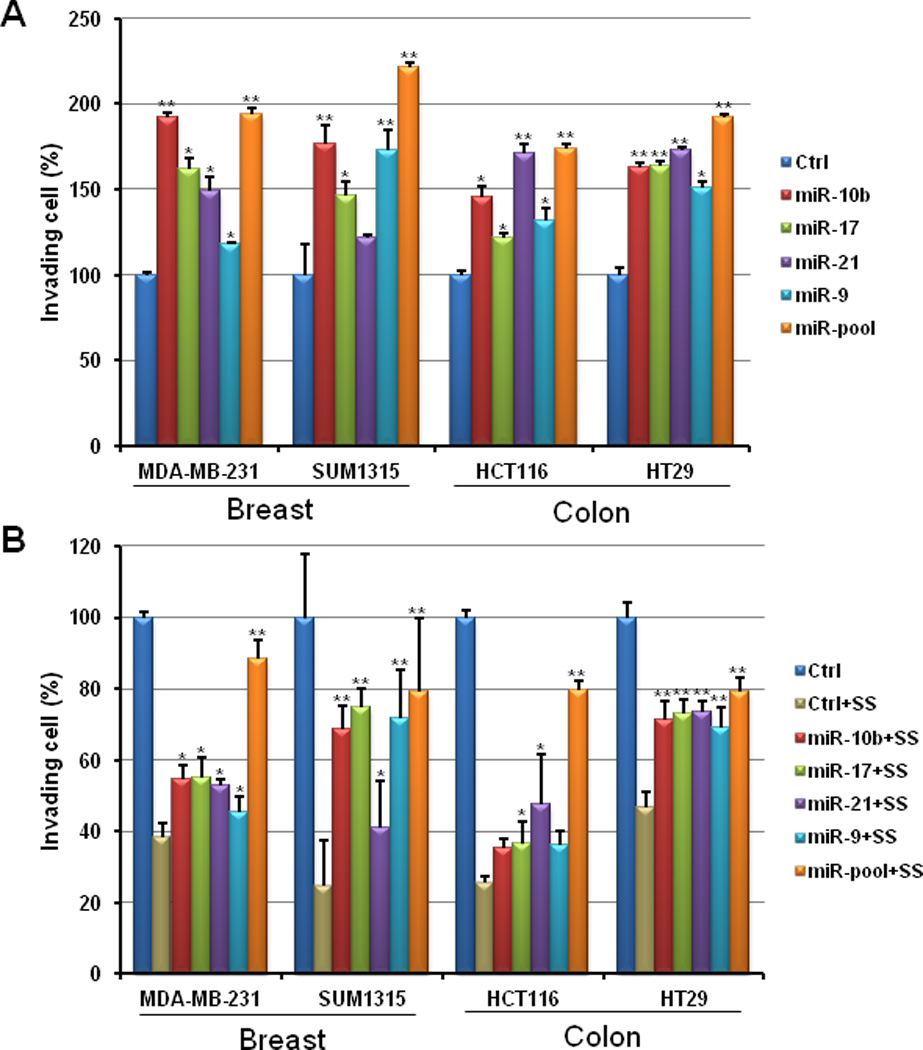
Over-expression of miR-10b, miR-17, miR-21, and miR-9 by transfection of their mimics not only can promote cell invasion in human breast cancer MDA-MB-231 and SUM1315 cells and human colon cancer HCT116 and HT29 cells (A), but also attenuate the inhibitory effect of SS on invasion of tumor cells (B). (T-test was used for determining statistical significance; * indicates p<0.05; ** indicates p<0.01; the error bars represent the standard deviation)
NF-κB regulates the selected miRNAs at the transcriptional level
To study the mechanism by which SS regulates miRNA expression, we screened the promoter regions (−2,000 to +500 bp) of 132 miRNAs from the array data using the interface provided by SITECON (http://wwwmgs.bionet.nsc.ru/mgs/programs/sitecon/) and found that 81 of the 115 miRNAs that were suppressed by SS contained NF-κB binding sites (>70%; Supplementary Figure 1; the miRNA list is shown in Supplementary Table 2), which implies that NF-κB may play an important role in regulation of miRNA expression by SS. As known, the biogenesis of miRNAs is similar to gene transcription and the primary miRNA (pri-miRNA) is the transcript of the miRNA gene. Given that NF-κB can regulate miRNA expression at transcriptional level, pri-miRNA can be up-regulated after NF-κB is induced.
TNFα is commonly used to induce NF-κB by activating the IKK complex 33–35. The activation of IKK complex results in phosphorylation of IκB family members (IκBα, IκBβ and IκBε) that accounts for ubiquitination and proteasomal degradation of IκB 36, 37. Before moving to the nucleus and activating target genes regulated by κB sites, NF-κB is retained in the cytoplasm by unphosphorylated IκB, while the phosphorylation of IκB can promote the translocation of NF-κB. Deoxycholic acid (DCA) is another NF-κB inducer involving direct degradation of IκB, which causes release of NF-κB from the cytoplasm 38. A p65 NF-κB plasmid construct, which can result in the over-expression of NF-κB in MDA-MB-231 and HCT116 cells (Supplementary Figure 3) was employed as a positive control to determine if the induction of pri-miRNAs by TNFα and DCA involve NF-κB signaling. As shown in Figure 3, all four pri-miRNAs, miR-10b, miR-17, miR-21, and miR-9, could be up-regulated by TNFα, DCA, and the p65 construct in both MDA-MB-231 and HCT116 cells, which implies the involvement of NF-κB in regulating miRNA expression through transcriptional modulation.
Figure 3. Induced NF-κB regulates the expression of the selected miRNAs at the transcriptional level.
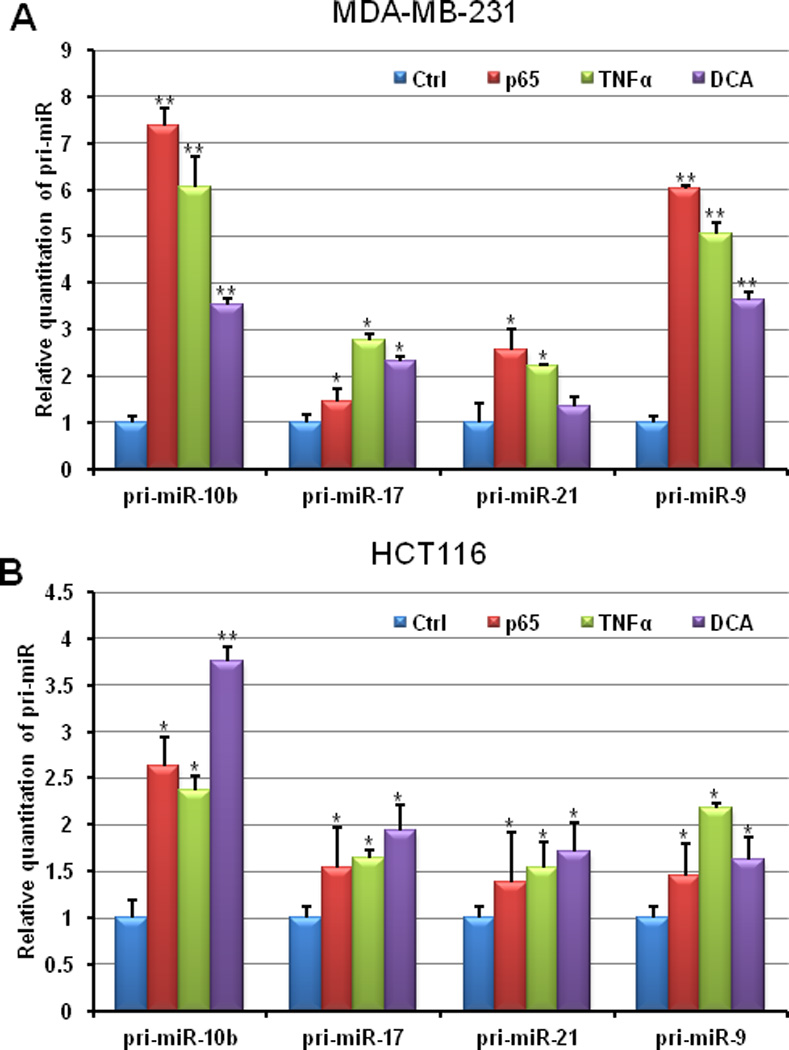
After being exposed to 25ng/ml TNFα for 5h or 250µM DCA for 2h, non-treated control and treated MDA-MB-231 and HCT116 cells were harvested for RNA isolation. QRT-PCR was employed for examine the relative expression of pri-miR-10b, pri-miR-17, pri-miR-21, and pri-miR-9. A p65 construct was transiently transfected into MDA-MB-231 and HCT116 cells as a positive control for NF-κB over-expression. (T-test was used for determining statistical significance; * indicates p<0.05; ** indicates p<0.01; the error bars represent the standard deviation)
To further study the mechanistic role of NF-κB in mediating expression of miRNAs inhibited by SS, we measured the relative expression of miR-10b, miR-17, miR-21, and miR-9 using qRT-PCR in response to treatment with the NF-κB inducer, TNFα and the inhibitor, Bay11-7082 39. The results, as summarized in Table 1, show that the expression of miR-10b, miR-17, miR-21, and miR-9 can either be significantly induced by TNFα or reduced by Bay11-7082 in both MDA-MB-231 and HCT116 tumor cells. After adding Bay11-7082 to cells that were pre-treated by TNFα, the inductive effect of TNFα was attenuated. These results together indicate that NF-κB is capable of regulating the expression of miR-10b, miR-17, miR-21, and miR-9 at the transcriptional level. When treating MDA-MB-231 and HCT116 cells with SS, we found that SS not only can reduce the expression of miR-10b, miR-17, miR-21, and miR-9 as did Bay11-7082, but also can prevent the inductive effect of TNFα on the expression of these miRNAs. These results demonstrate that NF-κB signaling can mediate the inhibition of miR-10b, miR-17, miR-21, and miR-9 by SS to account for its inhibitory effect on tumor cell invasion.
Table 1.
The miRNA expression in response to treatments of TNF- α, Bay11-7082, and SS
| Cell | MDA-MB-231 | HCT116 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Treatment | pri-miR-10b | pri-miR-17 | pri-miR-21 | pri-miR-9 | pri-miR-10b | pri-miR-17 | pri-miR-21 | pri-miR-9 | |
| Non-treated | 1.00±0.431 | 1.00±0.41 | 1.00±0.15 | 1.00±0.15 | 1.00±0.211 | 1.00±0.22 | 1.00±0.21 | 1.00±0.22 | |
| TNFα2 | 6.05±0.69 | 2.76±0.14 | 2.21±0.05 | 5.05±0.24 | 2.37±0.16 | 1.64±0.09 | 1.54±0.27 | 2.18±0.25 | |
| Bay11-70823 | 0.12±0.04 | 0.40±0.20 | 0.55±0.03 | 0.61±0.12 | 0.25±0.06 | 0.23±0.08 | 0.27±0.07 | 0.33±0.08 | |
| SS4 | 0.65±0.15 | 0.38±0.02 | 0.53±0.07 | 0.47±0.02 | 0.57±0.07 | 0.24±0.02 | 0.55±0.10 | 0.42±0.18 | |
| TNFα+Bay5 | 1.14±0.36 | 0.82±0.19 | 1.19±0.64 | 1.38±0.66 | 0.54±0.14 | 0.65±0.14 | 0.48±0.10 | 0.52±0.16 | |
| TNFα+SS6 | 1.30±0.20 | 1.00±0.23 | 1.26±0.01 | 1.25±0.11 | 0.87±0.20 | 0.57±0.21 | 0.82±0.09 | 0.48±0.17 | |
Note:
mean+ standard deviation;
25ng/ml for 5hr;
10µM for 2hr;
50uM for 24hr;
25ng/ml TNFα for 3hr, then 10µM Bay11-7082 for 2hr;
50µM SS for 24h, then 25ng/ml TNFα for 5h.
We next determined if NF-κB can directly bind to the promoter of the selected miRNAs. First, we used a luciferase assay that measures the interaction of NF-κB and the promoter of miR-10b. Bioinformatic analysis showed that the DNA upstream of the miR-10b gene contained two putative binding sites of p65 (W1: −1078 to −1065; W2: −379 to −365). Both binding sites were amplified and cloned into luciferase reporter vectors. The corresponding mutated constructs with deletion of the binding sequence (M1 and M2) were also amplified (Figure 4A). All constructs were transfected into HCT116 cells. After using TNFα to induce NF-κB, W2 significantly increased luciferase activity when compared to M2, but W1 displayed a similar response as M1 to TNFα (Figure 4B). Secondly, we co-transfected the p65 construct and luciferase reporters with miR-10b promoter fragments into HCT116 cells and obtained the same result as with TNFα stimulation (Figure 4C). To confirm the direct binding of NF-κB to the miR-10b, miR-17, miR-21, and miR-9 genes, we performed a chromatin immunoprecipitation (ChIP) assay using HCT116 cells. The chromatin was immunoprecipitated by p65 NF-κB antibody, and the immunoprecipitated DNA fragments were amplified using the primers designated for the each miRNA’s promoter. MiR-17 and miR-21 were documented to be regulated by NF-κB at the transcription level 40, and the same primer sets were used again in this study. The PCR results showed that the DNA fragments immunoprecipitated by the p65 NF-κB antibody contain the promoter sequences of miR-10b, miR-17, miR-21, and miR-9 (Figure 4D), which demonstrates the direct binding of NF-κB to these miRNA genes. Moreover, the ChIP results verified the luciferase assay results that W2 (−379 to −365) is the true binding sites of NF-κB in the miR-10b promoter.
Figure 4. NF-κB directly binds to the selected miRNAs promoters.
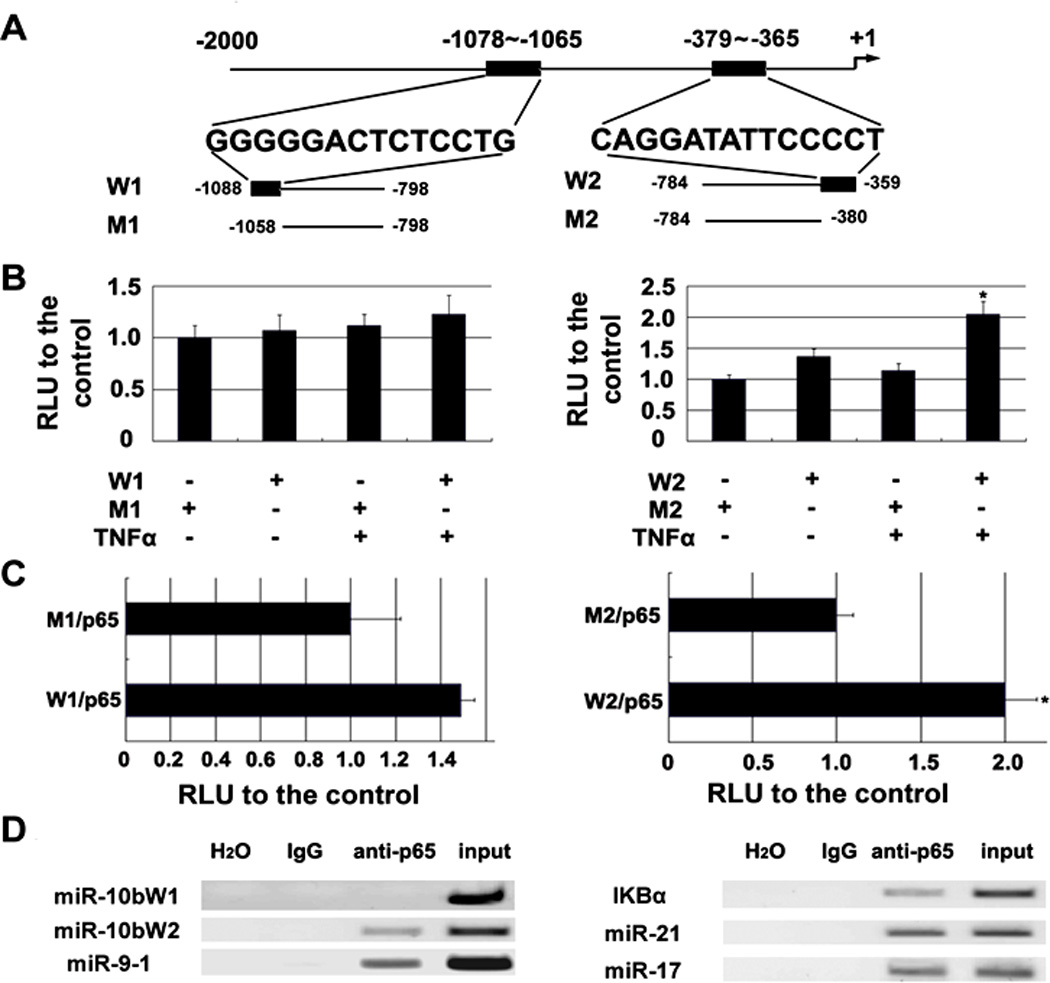
(A) Schematic of miR-10b promoter fragments containing p65 NF-κB binding sites. DNA fragments including two putative binding sequences of p65 (W1: −1078 to −1065; W2: −379 to −365) and the corresponding mutated sequences (M1 and M2) were cloned. (B) TNFα can induce the relative luciferase activity through W2 (p<0.05) but not W1. (C) Transfection of a p65 NF-κB construct increases the relative luciferase activity via W2 (p<0.05) but not W1 (RLU means relative luminescence units; T-test was used to determine statistical significance; * indicates p<0.05; the error bars represent the standard deviation). (D) ChIP assay of chromatin isolated from HCT116 cells treated with 25 ng/ml TNFα for 20 min and immunoprecipitated by anti-p65 or control IgG, followed by PCR analysis with primers targeted to the upstream sequence (299bp) of the IκBα promoter (the positive control from the kit), or to the sequences including W2 (233bp) and W1 (290bp) at the 5′-end of miR-10b, or to the binding sequence (202bp) in the miR-9-1 promoter. MiR-17 and miR-21 were tested using the previously published primer sequences 40.
SS prevents the translocation of NF-κB through inhibiting the phosphorylation of IKKβ
The activation of IKK complex can regulate the transcriptional activity of NF-κB through IκB phosphorylation as discussed above. Previous studies reported that SS can induce apoptotic cell death through inhibition of IKKβ, which implies that NF-κB regulation is an important pathway for mediating the antineoplastic properties of sulindac 41, 42. However, this effect has not been explored with regard to the inhibitory effect of SS on tumor cell invasion. We therefore examined the expression levels of IKKβ and phosphorylated IKKβ (p-IKKβ) in MDA-MB-231 and HCT116 cells following the treatment with SS. Figure 5A showed that p-IKKβ is significantly decreased in response to SS treatment at a concentration of 50 µM for 36 h. After induction by TNFα, the expression of phosphorylated IκBα (p-IκBα) was elevated in both MDA-MB-231 and HCT116 cells. However, SS treatment significantly decreased p-IκBα (Figure 5B), which might account for the attenuation of the inductive effect of TNFα by SS. The accumulation of unphosphorylated IκBα by SS retains NF-κB in the cytoplasm resulting in the suppression of NF-κB transcriptional activity. Moreover, using an NF-κB immunofluorescence assay, we showed that SS can prevent NF-κB nuclear localization. In both MDA-MB-231 and HCT116 cells, SS treatment was found to dramatically reduce the expression of NF-κB in the nucleus even after TNFα induction (Figure 5C). Altogether, these results show that SS is capable of suppressing the phosphorylation of IKKβ and IκBα, which leads to the retention of NF-κB in the cytoplasm to reduce the transcription of miRNA genes, such as miR-10b, miR-17, miR-21, and miR-9 that are involved in tumor cell invasion and metastasis.
Figure 5. SS prevents the translocation of NF-κB through inhibiting the phosphorylation of IKKβ and IκB.
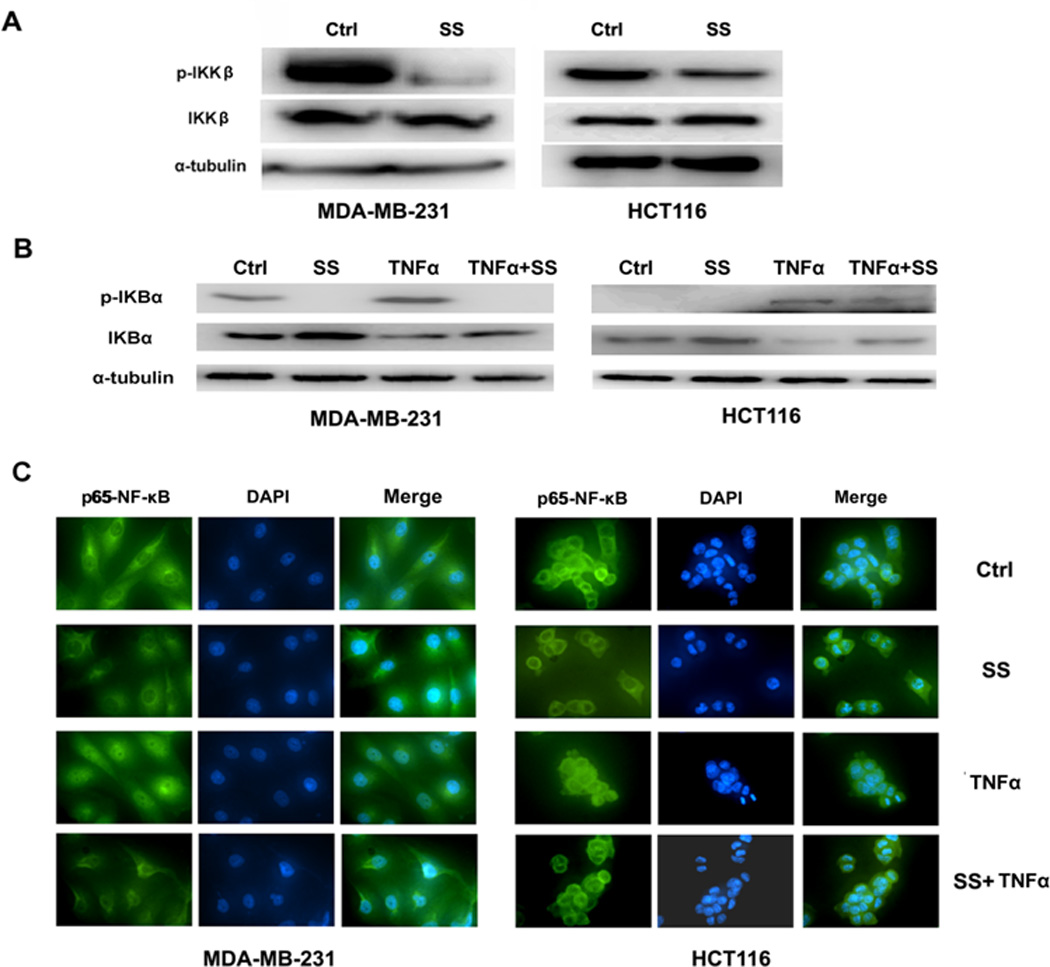
(A)The Western blot assay showed the phosphorylation of IKKβ is decreased in both MDA-MB-231 and HCT116 cells in response to SS treatment. (B) The Western blot assay showed that the decline of phosphorylated IκBα versus the accumulation of IκBα when MDA-MB-231 and HCT116 cells were treated by SS. TNFα (25 ng/ml for 20 min) was used to stimulated the expression of nuclear NF-κB. (C) NF-κB immunofluorescence of MDA-MB-231 and HCT116 cells. The conditions were as follows: (1) control; (2) treatment with 50 µM SS for 12 h; (3) treatment with 25 ng/ml TNFα for 20 min; and (4) both TNFα and SS treatments. The green (anti-NF-κB) indicates NF-κB distribution, and blue indicates the location of the nucleus.
Discussion
A large body of evidence indicates that sulindac has strong cancer chemopreventive efficacy, although its use for patients with malignant disease have not been well studied. Several publications have reported that sulindac can inhibit the invasion of tumor cells from glioblastoma 21, hepatoma 43, and colorectal cancer 16, but the mechanism of action has not been well defined. Here we demonstrate that SS can potently inhibit tumor cell invasion at concentrations less than those required to inhibit tumor cell growth by a novel mechanism that involves the inhibition of NF-κB signaling to suppress specific miRNAs and their target genes.
MiRNAs have been recognized as important regulators of gene expression based on the repression on their cognate genes, which affects many essential biological processes, including proliferation, differentiation, apoptosis, tumorigenesis, tumor cell invasion and metastasis 21–24. The majority of human miRNAs are transcribed from miRNA genes 44, 45 in which their biogenesis is spatially and temporally regulated in response to extracellular stimuli 18, 19. Compared to a large number of studies on the regulatory functions of miRNAs, only a few reports have described the transcriptional regulation of miRNAs. Similar to the mechanism of messenger RNA transcription, miRNA expression is regulated by a number of transcription factors. In addition to the report by our group first describing the transcriptional regulation of miRNA expression by p53 46, other groups have reported that different transcription factors such as c-Myc, NF-κB, STAT3 and C/EBPα are also involved in the regulation of miRNAs 47–51. Because the nature of miRNA is non-coding RNA molecules, the transcriptional process is of importance for regulating expression of miRNA. Our findings suggest that NF-κB regulation of miRNA transcription mediates the inhibitory effect of SS on tumor cell invasion.
Although sulindac has been previously reported to inhibit NF-κB activation42, there have been no reports describing an association between miRNAs and the antineoplastic properties of sulindac. In this study we report that SS treatment leads to the suppression of 115 miRNAs and up-regulation of 17 miRNAs. Interestingly, more than 70% of the down-regulated miRNAs contain NF-κB binding sequences within their promoter regions (−2,000 to +500bp; Supplementary Figure 1). This is consistent with previous studies reporting that a number of miRNAs, such as miR-132, miR-146, miR-155, miR-9, the miR-17-92 cluster, miR-125b-1, miR-23b-27b-24-1, miR-21, miR-30b, and miR-130a, can be regulated by NF-κB in human cancer cell lines 40, 47, 52, 53. Although miRNAs are regulated by certain transcription factors such as p53, c-Myc, STAT3, and NF-κB, the mechanism of transcriptional regulation of miRNAs is different from that of messenger RNA. First, miRNAs that are in the same genomic loci are organized in a single gene and share the same promoter to generate a polycistronic primary transcript, which ultimately produces multiple mature miRNAs 54. For example, the miR-17-92 gene encodes six miRNAs: miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92-1 55. In this study, we found that SS can suppress all members of the miR-17-92 cluster in HCT116 cells (Supplementary Table 2), and confirmed the NF-κB binding site in the promoter of the miR-17 gene. Second, many miRNA genes are located in the introns of host genes and are referred to as intronic miRNAs that share the promoters with the host genes for transcription 56. Third, approximately 40% of human miRNA loci are located less than 3 kb from an adjacent miRNA locus 7, 57, which implies that multiple miRNA genes can be transcribed together using the same promoter. In this study, we found that miR-125b, let-7c and miR-99a are suppressed in response to SS treatment (Supplementary Table 2). MiR-125b, but not let-7c or miR-99a, has a putative binding site for NF- κB. These three miRNA genes have been found to be clustered and co-transfected in a coordinated manner 58.
All NF-κB family members including RelA (p65), RelB, c-Rel, NF-κB1(p50), and NF-κB2(p52) are able to recognize κB binding sites within their target gene’s promoter regions 59. The activation domain (TAD) is the most important component that is responsible for the transcriptional activity of NF-κB 60. In this study, we demonstrate that SS can decrease phosphorylated IKKβ and IκB. The phosphorylation of IκB can release NF-κB to enter the nucleus but the accumulation of unphosphorylated IκB by SS retains NF-κB in the cytoplasm. Therefore, SS can inhibit the nuclear translocation of NF-κB. Moreover, we found that miR-10b, miR-17, miR-21, and miR-9, are directly regulated by p65 NF-κB using the luciferase reporter assay and/or ChIP assay. Altogether, these data suggest SS can inhibit tumor cell invasion by a molecular pathway that is mediated by NF-κB and miRNAs.
We demonstrated that miR-10b, miR-17, miR-21, and miR-9 are involved in tumor cell invasion in response to SS treatment, which is consistent with previous studies showing the involvement of these miRNA in breast tumor cell invasion and metastasis 25–31. MiR-10b is well documented to be associated with metastasis via activation of the pro-metastatic gene Ras homolog gene family member C by inhibiting the translation of gene Homeobox D10 (HOXD10) 28, 61. Recently, it is also reported that the silencing of miR-10b can directly inhibit metastasis of breast cancer cells 27. MiR-17, a member of the miR-17-92 cluster, was shown to promote breast cancer metastasis by targeting the metastasis suppressor type II transforming growth factor-β receptor (TβR2) 62 and by suppressing HMG box-containing protein 1 (HBP1) 63. MiR-21 is another well studied miRNA that is expressed in cells with oncogenic characteristics 64. The high level of miR-21 expression in breast cancer represents a significant lymph node metastasis 26, 30, and recently it was identified to promote breast cancer invasion and metastasis by down-regulating multiple tumor suppressor genes tropomyosin 1 (TPM1), programmed cell death 4 (PDCD4) and maspin 31, modulating the tissue inhibitor of the metalloproteinase-3 gene, whose encoding product is involved in extracellular matrix (ECM) degradation 26, 30. MiR-21 also was demonstrated to be over-expressed in colorectal cancer compared with normal tissue, and the high expression of miR-21 was associated with lymph node metastasis and distant metastasis 65. MiR-9 is a well-studied metastasis activator. Its expression was found to be higher in the primary tumors from breast cancer patients with metastasis than ones from metastasis-free patients 29. Also, miR-9 was identified to promote breast cancer metastasis by not only inducing epithelial-mesenchymal transition (EMT) and invasion of cancer cells, but also promoting angiogenesis through down-regulating E-cadherin expression 66. A clinical study showed that the expression of miR-9 was associated with colorectal cancer lymph node metastasis (Bandres, 2009a). In this study, we demonstrated that miR-10b, miR-17, miR-21, and miR-9 are regulated by NF-κB at the transcriptional levels. Inducing or repressing NF-κB activities by different stimulations can alter the expression of these miRNAs accordingly. Because SS can prevent the transcriptional activity of NF-κB, we believe that the down-regulation of miR-10b, miR-17, miR-21, and miR-9, in response to SS is mediated by NF-κB to suppress tumor cell invasion.
Increasing numbers of publications have reported that the antineoplastic activity of sulindac and other NSAIDs is COX-independent 8–14, 67. In support of this possibility, the tumor cell lines used in this study, MDA-MB-231 and HCT116 cells, are known to express low levels of COX-2 68, 69. We also examined COX-2 expression using qRT-PCR but did not find significant changes in either tumor cell line in response to treatment (data not shown). This implies that SS inhibition of tumor cell invasion is a COX-independent process, although further studies are required to identify alternative targets, for example, using non-COX inhibitory derivatives of sulindac as previously reported67.
In summary, these data show that SS can inhibit tumor cell invasion by suppressing NF-κB-mediated transcription of specific miRNAs that play an important regulatory role in tumor cell invasion and metastasis. These results support further studies to determine the potential use of sulindac for the prevention of metastasis in patients with advanced malignant disease.
Materials and methods
Cell culture and reagents
The human colon cancer cell line HCT116 was kindly provided by Dr. Bert Vogelstein (Johns Hopkins University, USA) and cultured using McCoy’s 5A medium (Invitrogen, Carlsbad, CA, USA). The human breast cancer cell line MDA-MB-231 and colon cancer line HT29 were purchased from ATCC (Manassas, VA, USA) and cultured using MEM-α medium (Invitrogen) and McCoy’s 5A medium, respectively. Mediums contained 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA). The human breast cancer cell line SUM1315 was purchased from Asterand, Inc (Detroit, MI, USA) and cultured in the DMEM/F12 medium (Invitrogen) containing 5% FBS, 5 µg/ml insulin (Sigma-Aldrich, St Louis, MO, USA), and 1µg/ml epidermal growth factor (EGF; Sigma-Aldrich) in accordance to the vendor’s instruction. The cells were maintained in humidified atmosphere of 5% CO2-95% air. Sulindac sulfide, TNFα, DCA, and Bay11-7082, were purchased from Sigma-Aldrich.
RNA isolation
Total RNA was extracted using Trizol reagent (Invitrogen) as previously reported 70. Briefly, cells were harvested and dissolved in 1 ml of Trizol reagent, and then 100 µl of 1-bromo-3-chloropropane (BCP) solution was added (Molecular Research Center, Inc. Cincinnati, OH, USA) and vigorously vortexed. After 10 min of centrifugation at 14,000 rpm at 4°C, the upper aqueous phase was transferred to a new tube, and an equal volume of isopropanol (Sigma-Aldrich) was added for precipitation. After washing pellets using 75% ethanol, RNA was dried in air and dissolved in nuclease free water for quantitation using a Nanodrop (Thermo, Worcester, MA, USA).
Quantitative Real-Time PCR
TaqMan Low Density Array (TLDA) Human MicroRNA Panel v2.0 (Applied Biosystems, Foster City, CA, USA) was employed for miRNA global profiling using our previously published protocol 71. Total RNA was employed for cDNA synthesis using a high capacity cDNA reverse transcriptase kit (Applied Biosystems). The specific stem-loop RT primers were designed for miRNAs following the published guideline 72. RT was performed at 37°C for 2 h by incubating the 20 µl mixture including 2 µg of total RNA, 2 µM RT primer, 2 µl 10× reverse transcription buffer, 0.8 µl 100 mM dNTP, and nuclease-free water. The quantitative real-time PCR reaction mixtures, consisting of 10 µl 2× SYBR master mix (Roche, Indianapolis, IN, USA), 2 µl synthesized forward primer (7 µM) and reverse primer (7 µM) mixture, 1 µl cDNA, and 7 µl nuclease-free water, were incubated for 30 cycles on a Bio-Rad IQ-5 real-time PCR System (Bio-Rad, Hercules, CA, USA). Each cycle includes denaturing for 10 sec at 94°C, annealing, and extension for 30 sec at 58°C. The comparative Ct method was used to compute relative levels of target miRNAs by subtracting the Ct values of the endogenous control (U6) and comparing to a designated calibrator in a batch of samples 72. Given that the relative value of the calibrator is 1.0, the other samples were n-fold relative to the calibrator.
Cell growth assay
HCT116 and MDA-MB-231 cells were seeded in 24-well plates at a density of 5×104 cells and 2.5×104 cells per well, respectively, and treated with SS for a designed time. Cell number was determined by trypan blue staining and manual counting. Growth curves were plotted as the relative cell number compared with vehicle (0.1% DMSO) treated controls.
Invasion assay
Cell invasion was measured using the Biocoat matrigel invasion chamber kit (BD Bioscience, Sparks, MD, USA). First, cells were transfected with 100 nmol/L of the mimic oligonucleotides of miR-10b, miR-17, miR-21, miR-9, and nonspecific control miR (Applied Biosystems) using Oligofectamine (Invitrogen). Then, the Matrigel coated plates were rehydrated by warm bicarbonate based culture medium for 2 h. After removing the medium, 2.5×104 cells were suspended in 500 µl blank medium on the insert, and then 750 µl chemoattractant (medium with 10% FBS) was added to the 24-well chamber. Cells were then incubated in 5% CO2 atmosphere at 37°C for 36 h. For the measurement of invading cells, non-invading cells were removed from the upper surface of the membrane by scraping, and invading cells were fixed with formaldehyde and then stained with crystal violet for counting.
Luciferase reporter constructs and luciferase assay
Primers were designed to amplify fragments of the miR-10b promoter (Supplementary Table 1), and PCR reactions were performed for 30 cycles consisting of denaturing for 10 sec at 94°C, annealing for 30 sec at 58°C and extension for 1 min at 72°C. The amplified DNA fragments were separated by agarose gel electrophoresis and purified by a gel extraction kit (Qiagen, Valencia, CA, USA). After digesting by MluI and XhoI (Promega, Madison, WI, USA), these fragments were ligated into the pGL3 basic vector using T4 DNA ligase (Promega). Reporter plasmid (150 ng) was co-transfected into HCT116 cells with 5 ng of pRL-TK control plasmid (Promega) or 350 ng of p65 expression vector (gift from Dr. Xianming Chen) using 1.5 µl of Lipofectamine 2000 (Invitrogen). Additionally, after co-transfection for 24 h, HCT116 cells were treated with TNFα for 4 h before assessing luciferase activity. Luciferase activity was measured by using a dual luciferase reporter assay (Promega). The results were computed for relative luciferase activity using the ratio of firefly Luc/Renilla Luc.
Chromatin immunoprecipitation assay (ChIP)
ChIP analysis was performed with the ChIPAb-NFκB p65 (Cat.#17-10060) and EZ-Magna ChIP™ kit (Cat.# 17-409) from Millipore (Billerica, MA, USA). The procedure strictly followed the manufacturer’s instructions. Briefly, 1 × 107 HCT116 cells were cultured in a 15-cm culture dish and stimulated with 20 ng/ml TNFα for 20 min before crosslinking using 1% formaldehyde (Sigma-Aldrich). The fixed cells were lysed, and the chromatin was sheared by sonication using an optimized condition. The chromatin fraction was immunoprecipitated overnight at 4°C with the anti-p65 antibody and goat-anti-mouse IgG. The DNA was extracted and purified after the immunoprecipitation. PCR amplification was performed in a total volume of 20 µl with pre-designed primers, and the sequences of primers are listed in Supplementary Table 1. PCR reactions were performed for 30 cycles consisting of denaturing for 10 sec at 94°C, annealing for 30 sec at 58°C and extension for 1 min at 72°C.
Western blot assay
Total proteins were extracted from cells using RIPA lysis buffer (Sigma-Aldrich) and quantified with a BCA protein assay kit (Thermo). Total proteins were separated on a 10% SDS-PAGE gel and then transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad). The membrane was blocked with 5% nonfat milk and then incubated with mouse anti-human α-tubulin monoclonal antibody (Santa Cruz, Santa Cruz, CA, USA), rabbit anti-human IKKβ polyclonal antibody (Millipore, Billerica, MA, USA), rabbit anti-human phosphorylated IKKβ polyclonal antibody (Abcam, Cambridge, MA, USA), and rabbit anti-human IκBα and phosphorylated IκBα polyclonal antibodies (Santa Cruz) at 4°C overnight. After washing with TBST (Tris-buffered saline containing 0.1% Tween 20, both from Bio-Rad), peroxidase-linked secondary goat anti-mouse IgG or goat anti-rabbit IgG antibodies (Santa Cruz) were incubated with membranes for 1 h at room temperature. After washing using TBST, the enhanced chemiluminescent substrate for detection of HRP (Thermo) was applied, and images were captured by a Fujifilm Las-3000 imager (Fujifilm, Inc. Stamford, CT, USA).
NF-κB immunofluorescence assay
MDA-MB-231 and HCT116 cells (1×105) were seeded in a 6-well plate overnight at 37°C and then treated with 50 µM SS or same volume of 0.1% DMSO for 12 h. Before fixation using 4% formaldehyde (Sigma-Aldrich), TNFα at a concentration of 25 ng/ml was added to the cells for 20 min. The cells were then permeabilized with 1% Triton X-100 (Sigma-Aldrich) and blocked with 1% BSA (Sigma-Aldrich) before incubating with p65 antibody at 4°C overnight. After washing with phosphate buffered solution, the cells were incubated with the FITC-conjugated secondary antibody (Invitrogen) for 1 hour at 37°C. After washing and staining with 5 ng/ml DAPI (Invitrogen), pictures were taken using the Nikon TE2000-U fluorescence microscope system (Nikon, Inc., Melville, NY, USA).
Supplementary Material
Acknowledgement
This study was supported by the institutional start-up funding (Dr. Yaguang Xi) and the National Cancer Institute grant R01CA148817 (Dr. Gary A. Piazza). We sincerely appreciate Mrs. Margaret Sullivan for proof-reading the manuscript.
Footnotes
Conflict of Interest: Authors declare there are no competing financial interests in relation to the work described.
Supplementary information is available at ONCOGNE’s website.
Reference
- 1.Smalley W, Ray WA, Daugherty J. Griffin MR Use of nonsteroidal anti-inflammatory drugs and incidence of colorectal cancer: a population-based study. Arch Intern Med. 1999;159:161–166. doi: 10.1001/archinte.159.2.161. [DOI] [PubMed] [Google Scholar]
- 2.Giardiello FM, Hamilton SR, Krush AJ, Piantadosi S, Hylind LM, Celano P, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328:1313–1316. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 3.Beazer-Barclay Y, Levy DB, Moser AR, Dove WF, Hamilton SR, Vogelstein B, et al. Sulindac suppresses tumorigenesis in the Min mouse. Carcinogenesis. 1996;17:1757–1760. doi: 10.1093/carcin/17.8.1757. [DOI] [PubMed] [Google Scholar]
- 4.Mahmoud NN, Boolbol SK, Dannenberg AJ, Mestre JR, Bilinski RT, Martucci C, et al. The sulfide metabolite of sulindac prevents tumors and restores enterocyte apoptosis in a murine model of familial adenomatous polyposis. Carcinogenesis. 1998;19:87–91. doi: 10.1093/carcin/19.1.87. [DOI] [PubMed] [Google Scholar]
- 5.Piazza GA, Alberts DS, Hixson LJ, Paranka NS, Li H, Finn T, et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997;57:2909–2915. [PubMed] [Google Scholar]
- 6.Thompson HJ, Jiang C, Lu J, Mehta RG, Piazza GA, Paranka NS, et al. Sulfone metabolite of sulindac inhibits mammary carcinogenesis. Cancer Res. 1997;57:267–271. [PubMed] [Google Scholar]
- 7.Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, et al. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005;33:2697–2706. doi: 10.1093/nar/gki567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piazza GA, Rahm AL, Krutzsch M, Sperl G, Paranka NS, Gross PH, et al. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Res. 1995;55:3110–3116. [PubMed] [Google Scholar]
- 9.Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, et al. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol. 1996;52:237–245. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 10.Elder DJ, Halton DE, Hague A, Paraskeva C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin Cancer Res. 1997;3:1679–1683. [PubMed] [Google Scholar]
- 11.Piazza GA, Rahm AK, Finn TS, Fryer BH, Li H, Stoumen AL, et al. Apoptosis primarily accounts for the growth-inhibitory properties of sulindac metabolites and involves a mechanism that is independent of cyclooxygenase inhibition, cell cycle arrest, and p53 induction. Cancer Res. 1997;57:2452–2459. [PubMed] [Google Scholar]
- 12.Rigas B, Shiff SJ. Is inhibition of cyclooxygenase required for the chemopreventive effect of NSAIDs in colon cancer? A model reconciling the current contradiction. Med Hypotheses. 2000;54:210–215. doi: 10.1054/mehy.1999.0023. [DOI] [PubMed] [Google Scholar]
- 13.Kashfi K, Rigas B. Non-COX-2 targets and cancer: expanding the molecular target repertoire of chemoprevention. Biochem Pharmacol. 2005;70:969–986. doi: 10.1016/j.bcp.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Alberts DS, Hixson L, Ahnen D, Bogert C, Einspahr J, Paranka N, et al. Do NSAIDs exert their colon cancer chemoprevention activities through the inhibition of mucosal prostaglandin synthetase? J Cell Biochem Suppl. 1995;22:18–23. doi: 10.1002/jcb.240590804. [DOI] [PubMed] [Google Scholar]
- 15.Lundholm K, Gelin J, Hyltander A, Lonnroth C, Sandstrom R, Svaninger G, et al. Anti-inflammatory treatment may prolong survival in undernourished patients with metastatic solid tumors. Cancer Res. 1994;54:5602–5606. [PubMed] [Google Scholar]
- 16.Stein U, Arlt F, Smith J, Sack U, Herrmann P, Walther W, et al. Intervening in beta-Catenin Signaling by Sulindac Inhibits S100A4-Dependent Colon Cancer Metastasis. Neoplasia. 2011;13:131–144. doi: 10.1593/neo.101172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–1757. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 19.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 20.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 21.Lee HC, Park IC, Park MJ, An S, Woo SH, Jin HO, et al. Sulindac and its metabolites inhibit invasion of glioblastoma cells via down-regulation of Akt/PKB and MMP-2. J Cell Biochem. 2005;94:597–610. doi: 10.1002/jcb.20312. [DOI] [PubMed] [Google Scholar]
- 22.Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 23.Carmell MA, Xuan Z, Zhang MQ, Hannon GJ. The Argonaute family: tentacles that reach into RNAi, developmental control, stem cell maintenance, and tumorigenesis. Genes Dev. 2002;16:2733–2742. doi: 10.1101/gad.1026102. [DOI] [PubMed] [Google Scholar]
- 24.Karube Y, Tanaka H, Osada H, Tomida S, Tatematsu Y, Yanagisawa K, et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005;96:111–115. doi: 10.1111/j.1349-7006.2005.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu Z, Willmarth NE, Zhou J, Katiyar S, Wang M, Liu Y, et al. microRNA 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc Natl Acad Sci U S A. 2010;107:8231–8236. doi: 10.1073/pnas.1002080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang GL, Zhang XH, Guo GL, Huang KT, Yang KY, Shen X, et al. Clinical significance of miR-21 expression in breast cancer: SYBR-Green I-based real-time RT-PCR study of invasive ductal carcinoma. Oncol Rep. 2009;21:673–679. [PubMed] [Google Scholar]
- 27.Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, et al. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28:341–347. doi: 10.1038/nbt.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 29.Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song B, Wang C, Liu J, Wang X, Lv L, Wei L, et al. MicroRNA-21 regulates breast cancer invasion partly by targeting tissue inhibitor of metalloproteinase 3 expression. J Exp Clin Cancer Res. 2010;29:29. doi: 10.1186/1756-9966-29-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo YY. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18:350–359. doi: 10.1038/cr.2008.24. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Yan LX, Wu QN, Du ZM, Chen J, Liao DZ, et al. miR-125b is methylated and functions as a tumor suppressor by regulating the ETS1 proto-oncogene in human invasive breast cancer. Cancer Res. 2011;71:3552–3562. doi: 10.1158/0008-5472.CAN-10-2435. [DOI] [PubMed] [Google Scholar]
- 33.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 34.Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-kappaB in p53-mediated programmed cell death. Nature. 2000;404:892–897. doi: 10.1038/35009130. [DOI] [PubMed] [Google Scholar]
- 35.Thanos D, Maniatis T. NF-kappa B: a lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 36.Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, Baldwin AS., Jr I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: a mechanism for cytoplasmic retention. Genes Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 37.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 38.Shah SA, Volkov Y, Arfin Q, Abdel-Latif MM, Kelleher D. Ursodeoxycholic acid inhibits interleukin 1 beta [corrected] and deoxycholic acid-induced activation of NF-kappaB and AP-1 in human colon cancer cells. Int J Cancer. 2006;118:532–539. doi: 10.1002/ijc.21365. [DOI] [PubMed] [Google Scholar]
- 39.Mori N, Yamada Y, Ikeda S, Yamasaki Y, Tsukasaki K, Tanaka Y, et al. Bay 11-7082 inhibits transcription factor NF-kappaB and induces apoptosis of HTLV-I-infected T-cell lines and primary adult T-cell leukemia cells. Blood. 2002;100:1828–1834. doi: 10.1182/blood-2002-01-0151. [DOI] [PubMed] [Google Scholar]
- 40.Zhou R, Hu G, Gong AY, Chen XM. Binding of NF-kappaB p65 subunit to the promoter elements is involved in LPS-induced transactivation of miRNA genes in human biliary epithelial cells. Nucleic Acids Res. 2010;38:3222–3232. doi: 10.1093/nar/gkq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamamoto Y, Yin MJ, Lin KM, Gaynor RB. Sulindac inhibits activation of the NF-kappaB pathway. J Biol Chem. 1999;274:27307–27314. doi: 10.1074/jbc.274.38.27307. [DOI] [PubMed] [Google Scholar]
- 42.Seo AM, Hong SW, Shin JS, Park IC, Hong NJ, Kim DJ, et al. Sulindac induces apoptotic cell death in susceptible human breast cancer cells through, at least in part, inhibition of IKKbeta. Apoptosis. 2009;14:913–922. doi: 10.1007/s10495-009-0367-1. [DOI] [PubMed] [Google Scholar]
- 43.Jiang MC, Liao CF, Lee PH. Aspirin inhibits matrix metalloproteinase-2 activity, increases E-cadherin production, and inhibits in vitro invasion of tumor cells. Biochem Biophys Res Commun. 2001;282:671–677. doi: 10.1006/bbrc.2001.4637. [DOI] [PubMed] [Google Scholar]
- 44.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. Embo J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. Rna. 2004;10:1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xi Y, Shalgi R, Fodstad O, Pilpel Y, Ju J. Differentially regulated micro-RNAs and actively translated messenger RNA transcripts by tumor suppressor p53 in colon cancer. Clin Cancer Res. 2006;12:2014–2024. doi: 10.1158/1078-0432.CCR-05-1853. [DOI] [PubMed] [Google Scholar]
- 47.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermuller J, Kretzschmar AK, et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007;110:1330–1333. doi: 10.1182/blood-2007-03-081133. [DOI] [PubMed] [Google Scholar]
- 49.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005;123:819–831. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 51.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, et al. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci U S A. 2009;106:5282–5287. doi: 10.1073/pnas.0810909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown JW, Marshall DF, Echeverria M. Intronic noncoding RNAs and splicing. Trends Plant Sci. 2008;13:335–342. doi: 10.1016/j.tplants.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 57.Saini HK, Griffiths-Jones S, Enright AJ. Genomic analysis of human microRNA transcripts. Proc Natl Acad Sci U S A. 2007;104:17719–17724. doi: 10.1073/pnas.0703890104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gefen N, Binder V, Zaliova M, Linka Y, Morrow M, Novosel A, et al. Hsa-mir-125b-2 is highly expressed in childhood ETV6/RUNX1 (TEL/AML1) leukemias and confers survival advantage to growth inhibitory signals independent of p53. Leukemia. 2010;24:89–96. doi: 10.1038/leu.2009.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nabel GJ, Verma IM. Proposed NF-kappa B/I kappa B family nomenclature. Genes Dev. 1993;7:2063. doi: 10.1101/gad.7.11.2063. [DOI] [PubMed] [Google Scholar]
- 60.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 61.Hurst DR, Edmonds MD, Welch DR. Metastamir: the field of metastasis-regulatory microRNA is spreading. Cancer Res. 2009;69:7495–7498. doi: 10.1158/0008-5472.CAN-09-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu S, Goldstein RH, Scepansky EM, Rosenblatt M. Inhibition of rho-associated kinase signaling prevents breast cancer metastasis to human bone. Cancer Res. 2009;69:8742–8751. doi: 10.1158/0008-5472.CAN-09-1541. [DOI] [PubMed] [Google Scholar]
- 63.Li H, Bian C, Liao L, Li J, Zhao RC. miR-17-5p promotes human breast cancer cell migration and invasion through suppression of HBP1. Breast Cancer Res Treat. 2010 doi: 10.1007/s10549-010-0954-4. [DOI] [PubMed] [Google Scholar]
- 64.Selcuklu SD, Donoghue MT, Spillane C. miR-21 as a key regulator of oncogenic processes. Biochem Soc Trans. 2009;37:918–925. doi: 10.1042/BST0370918. [DOI] [PubMed] [Google Scholar]
- 65.Slaby O, Svoboda M, Fabian P, Smerdova T, Knoflickova D, Bednarikova M, et al. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology. 2007;72:397–402. doi: 10.1159/000113489. [DOI] [PubMed] [Google Scholar]
- 66.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 67.Piazza GA, Keeton AB, Tinsley HN, Gary BD, Whitt JD, Mathew B, et al. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prev Res (Phila) 2009;2:572–580. doi: 10.1158/1940-6207.CAPR-09-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Agarwal B, Swaroop P, Protiva P, Raj SV, Shirin H, Holt PR. Cox-2 is needed but not sufficient for apoptosis induced by Cox-2 selective inhibitors in colon cancer cells. Apoptosis. 2003;8:649–654. doi: 10.1023/A:1026199929747. [DOI] [PubMed] [Google Scholar]
- 69.Singh B, Berry JA, Shoher A, Lucci A. COX-2 induces IL-11 production in human breast cancer cells. J Surg Res. 2006;131:267–275. doi: 10.1016/j.jss.2005.11.582. [DOI] [PubMed] [Google Scholar]
- 70.Xi Y, Nakajima G, Gavin E, Morris CG, Kudo K, Hayashi K, et al. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA. 2007;13:1668–1674. doi: 10.1261/rna.642907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang B, Howel P, Bruheim S, Ju J, Owen LB, Fodstad O, et al. Systematic Evaluation of Three microRNA Profiling Platforms: Microarray, Beads Array, and Quantitative Real-Time PCR Array. PLoS One. 2011;6:e17167. doi: 10.1371/journal.pone.0017167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP. Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods. 2007;3:12. doi: 10.1186/1746-4811-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.