

Abstract
Environmental enteropathy (also called tropical enteropathy) is a subclinical condition caused by constant fecal-oral contamination and resulting in blunting of intestinal villi and intestinal inflammation. Although these histological changes were discovered decades ago, the clinical impact of environmental enteropathy is just starting to be recognized. The failure of nutritional interventions and oral vaccines in the developing world may be attributed to environmental enteropathy, as the intestinal absorptive and immunologic functions are significantly deranged. Here we review the existing literature and examine potential mechanisms of pathogenesis for this poorly understood condition.
Keywords: Environmental enteropathy, tropical enteropathy, malnutrition, stunting, oral vaccine failure, mucosal immunity, fecal-oral contamination
The deleterious effects of unsanitary living conditions
On July 28, 2010, Bolivian Ambassador Pablo Solón pleaded before the United Nations General Assembly that access to potable water and sanitation should be considered a basic human right. “The vast majority of illnesses around the world are caused by fecal matter,” he proclaimed, “3.5 million people die of waterborne illness each year.” [1] While the Ambassador’s appeal was met with great support, in actuality he had grossly understated the impact of hygiene and sanitation on the health of impoverished people worldwide; he failed to recognize the effect of an insidious condition called environmental enteropathy (EE).
EE is a subclinical disorder that occurs among inhabitants of environments with poor sanitation and hygiene, such as those often found in developing countries. Chronic exposure to fecal pathogens is hypothesized to cause inflammation and structural changes in the small bowel, which ultimately result in functional changes. EE is marked by increased intestinal permeability, impaired gut immune function, malabsorption, growth faltering, and, potentially, oral vaccine failure, all in a seemingly asymptomatic individual without overt diarrhea [2,3].
The most significant impact of EE may be on malnutrition, a well-recognized problem in the developing world where 26% of children under age five are underweight. Even more alarming is the fact that 21% of deaths in children less than five are attributable to sequelae of malnutrition [http://www.un.org/millenniumgoals/pdf/MDG%20Report%202010%20En%20r15%20-low%20res%2020100615%20-.pdf]. EE is also thought to contribute to the poor response to nutritional therapy. Various trials of complementary supervised feeding interventions have failed to show significant improvement in growth [4,5]. Even when breastfeeding is adequate, growth stunting and associated pathologic changes in the intestinal mucosa have been found in infants as young as three months. Models depicting the use of all known interventions – including vitamin A and zinc supplementation, balanced energy protein supplementation, complementary feeding, breastfeeding promotion, and micronutrient supplementation in pregnancy – showed that the use of these interventions in 99% of children worldwide would only decrease stunting by 33% [6]. Clearly, there is a large knowledge gap in our understanding of growth stunting and its associated mortality that cannot be explained by food insecurity. We must consider the impact of subclinical yet pathologic changes that are occurring in the absorptive and immune function of the gastrointestinal tract of infants and children in the developing world.
In addition to the failure of nutritional interventions, there has been the observation that oral vaccines, including those for polio and rotavirus, are less immunogenic in children of developing countries, suggesting that EE may cause altered mucosal immunity in these children [7,8,9,10]. EE causes fundamental changes in both small intestinal structure and function, as evidenced by biopsies and abnormal sugar absorption tests. The immunologic function of the small bowel may also be significantly altered. Because of constant exposure to enteric pathogens, in EE there is breach of the nonspecific host defenses and engagement of the innate and adaptive immune responses (Table 1). If the mucosal immune system is chronically activated in children with EE, an oral vaccine might not elicit the same response in this population [6].
Table 1.
Differential for non-infectious Tropical malabsorption
| Environmental Enteropathy | Tropical Sprue | Celiac Sprue | Crohn’s Disease | |
|---|---|---|---|---|
| Characteristic signs and symptoms | Subclinical; can have mild malabsorption and be associated with growth faltering | Chronic diarrhea, weight loss, manifestations of nutritional deficiencies | A minority of patients have the classic diarrhea- predominant form; more patients have “silent” disease, characterized by lack of symptoms or extraintestinal symptoms, such as arthritis, infertility, anemia, and osteoporosis | Perianal abscesses, intestinal fistulas, and strictures; individuals are at increased risk for PSC, ankylosing spondylsis, and psoriasis |
| Histopathology | Blunting of intestinal villi, increased crypt lengthening, intraepithelial lymphocytes and lymphocytic infiltration of the lamina propria | Blunting of intestinal villi, increased crypt lengthening, increased mononuclear inflammatory cells and intraepithelial lymphocytes. | *indistinguishable from EE | Active and chronic inflammation, variable distortion to villous architecture, metaplastic epithelial changes, possible granulomas |
| Serologic diagnosis | No established biochemical parameters for diagnosis | No established biochemical parameters for diagnosis | Anti-tissue transglutaminse antibody; anti- endomysial antibody; anti- gliadin Ab (nonspecific) | No established biochemical parameters for diagnosis |
| Etiology | Recurrent environmental exposure to fecal contamination | Infectious | Genetic predisposition with presence of HLA-DQ2 or HLA- DQ8 initiated by exposure to gluten proteins. Pathogenesis due to both t-cell and antibody- mediated response. | Unknown; genetic predisposition to certain forms of the disease, involving NOD2 and autophagy gene (ATG161L). |
| Available treatment | None | May respond to tetracycline therapy | Avoidance of gluten in diet | Corticosteroids and immunomodulating drugs |
| Refs | 2 | 2,63 | 32, 80 | 64 |
Here, we propose that the failure of health interventions in developing nations, specifically those targeting malnutrition and employing oral vaccines, can be directly attributed to EE, a subclinical, pathologic process occurring in the gastrointestinal tract of millions of people worldwide living in impoverished and unsanitary conditions (Figure 1).
Figure 1. Model for the mechanism of Environmental Enteropathy development.

(Adapted with permission from [78].
Epidemiology
EE was originally dubbed “Tropical Enteropathy” in the 1960s when there were several reports of abnormalities on jejunal biopsy of asymptomatic individuals from tropical countries [11,12,13,14,15]. Publications from Asia, Africa, and Latin America confirmed these findings in all age groups studied, from infants to adults. Changes seen on biopsy included decreased villous height, increased crypt depth, lymphocytic infiltration of the lamina propria, and increased intra-epithelial lymphocytes (Figure 2). In addition, most of these individuals had abnormalities in the absorptive function of the small bowel.
Figure 2. Jejunal biopsies of normal (a) and diseased (b) intestines.
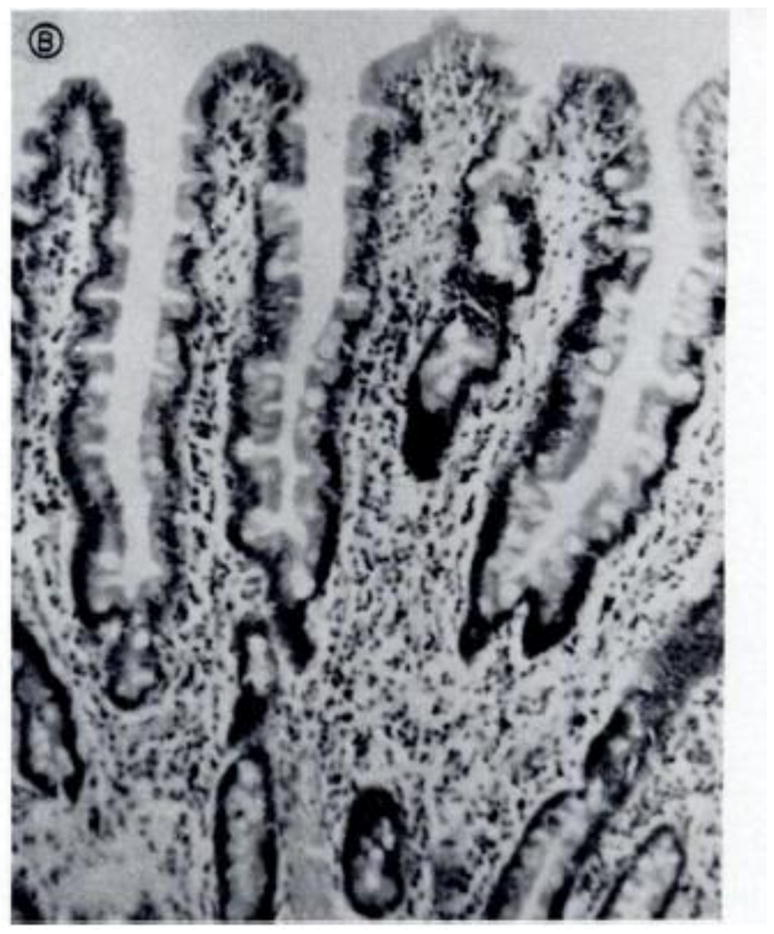

(a) Note the finger-like villi and short and narrow crypts of the normal intestine (a) compared with the severely flattened villi and inflammatory infiltration in the lamina propria and epithelium from the diseased tissue (b). This biopsy was taken from a Mexican adult with malnutrition, who may have had concurrent diarrhea. Reproduced with permission from [79].
Lindenbaum and colleagues set out to investigate whether this condition of subclinical malabsorption was acquired, and if so, whether it could be reversed. They studied Peace Corps volunteers living in Pakistan: many had mild diarrhea and a few had weight loss. On jejunal biopsy, the same abnormalities that were seen in native Pakistanis were observed, as well as abnormal carbohydrate absorption [16,17]. Two to three years after migrating back to the United States, the volunteers were found to have regained normal carbohydrate absorptive function. Five of these individuals also had repeat biopsies, all of which showed a return to normal jejunal histology. A second study, this time of a group of asymptomatic Indians and Pakistani immigrants living in New York City, demonstrated that almost half had malabsorption when studied within the first two years of migrating to the U.S., but absorption and changes on jejunal biopsies improved with increasing periods of residence in the US [18]. These observed changes in histopathology in the same individuals in both the developed and developing country settings suggested there may be an environmental etiology to this condition.
A study from Vellore, India found that stillborn fetuses had normal finger-like intestinal villi, but abnormalities in architecture were seen in infants as early as 8-weeks of life, again making an environmental rather than genetic etiology plausible [19]. In Brazil, EE was documented in slum-dwelling infants living in poor sanitary conditions [20]. A study of adults in Zambia found that none of the two hundred subjects had ‘normal’ jejunal biopsies. The subjects were noted to have more severe abnormalities in intestinal permeability than controls of higher socioeconomic status. As these subjects were studied serially over three years, it was also observed that there was seasonal variation in villous height and carbohydrate absorption [21].
Constant exposure to fecal-oral contamination, and sustained episodes of self-limited infectious gastroenteritis, may lead to a perpetual state of small bowel injury. This injury could cause hyperstimulation of the mucosal immune system, and result in the small bowel pathology that has been described. These structural and immune changes may be the body’s adaptive response to a particularly hostile environment. If there is constant pathogenic infiltration into the mucosa, it could be advantageous to remain in an inflammatory, hyperimmune state.
Although EE was widely reported in the 1970s and 1980s as an interesting phenomenon, there were few attempts to elucidate the mechanism of disease, and the condition appeared to be almost forgotten over the next 20 years. More recently, there has been increasing interest in the condition as the scientific community struggles to understand disparate responses to health interventions between the developing and developed worlds [22].
EE as a distinct entity
Even though EE is marked by increased intestinal permeability, impaired gut immune function, malabsorption, and growth faltering, individuals with this condition are seemingly asymptomatic, rarely having overt gastrointestinal symptoms [2,3,23]. The differential for conditions causing malabsorption in developing countries is quite long, and includes tropical sprue, a distinct entity from EE. (Table 2). Tropical sprue is a condition that affects both residents and travelers to countries in Asia, Africa, South and Central America, and the Caribbean [24,25]. Patients with tropical sprue have chronic diarrhea, steatorrhea, weight loss, and manifestations of nutritional deficiencies. While a specific pathogen has not been identified, the etiology of tropical sprue is thought to be infectious as these patients do respond to treatment with broad-spectrum antibiotics [26].
Table 2.
Biomarkers which may be informative in the study of Environmental Enteropathya
| Biomarker | Description | Refs |
|---|---|---|
| Zonulin | Human homolog of zonnula occudens toxin from V. cholerae that modulates tight junctions. In celiac sprue, gliadin induces zonulin release by binding to the CXCR3 receptor causing cytoskeletal rearrangement and ZO-1 reorganization, leading to increased intestinal permeability. | 65,66, 70 |
| Fecal Calprotectin | A complex of S100A8/S100A9, this protein accounts for 60% of neutrophilic cytosol. It has been used to predict relapse in IBD patients. | 54,55, 71,72 |
| Fecal S100A12 | Marker of neutrophil activation, elevated in IBD and bacterial enteritis, but not viral enteritis. | 72,73, 74 |
| Endocab a | Antibody to bacterial cell wall LPS core oligosaccharide. Endocab has is the only biomarker that has been studied in EE | 52,70 |
| Granulocyte macrophage Colony Stimulating Factor Autoantibody | Reported to be associated with increased intestinal permeability as measured by lactulose:mannitol test in patients with Crohn Disease. | 75 |
| Neopterin | Protein produced by monocytes and macrophages after stimulation with IFN-gamma. Marker of TH1 cell activation. | 76 |
| Myeloperoxidase | A lysosomal protein in neutrophilic granulocytes. Found to be elevated in mouse model of IBD. | 77 |
| Citrulline | Amino acid produced by enterocytes. Proposed as measure of gross enterocyte mass. | 57,58 |
| Reg 1A, Reg 1B | Gene family involved in tissue regeneration. Protein found in colonic crypt epithelial cells with anti-apoptotic and pro-proliferative properties. Up-regulated during amebiasis, and may play a protective role against parasite-induced apoptosis. Also being studied for diagnosis and monitoring of Celiac disease. | 78,79 |
| Lactoferrin | Iron-binding glycoprotein found in secondary granules of leukocytes. Used as a surrogate for fecal leukocytes, and a measure of intestinal inflammation. | 80,81, 82 |
Endocab is the only biomarker that has been evaluated in EE, the rest have been studied in other inflammatory bowel conditions and may be potentially useful in understanding EE.
There are reports that small bowel bacterial overgrowth (SBO) may play a role in EE. Khin-Maung et al found an association between abnormal breath hydrogen test (a measure of small bowel bacterial overgrowth) and growth faltering in Burmese children [27]. Another study from Brazil reported that slum-dwelling children were more likely to have an abnormal breath hydrogen test when compared to a control group with access to running water and adequate sewage [28]. This study, however, did not find an increased rate of growth faltering among slum-dwelling children with small bowel overgrowth. While both studies may suggest that children with SBO have malabsorption, they did not investigate measures of intestinal permeability. Trehan and colleagues treated Malawian children with rifaximin for seven days and found no improvement in markers of intestinal permeability, suggesting that while SBO may play a role in malabsorption, it may not contribute to leaky intestinal barrier [29].
T-cell mediated enteropathy
The majority of studies that have investigated EE have reported the same findings: blunting of the villi, crypt hyperplasia, and lymphocytic infiltration of the lamina propria [2,3]. Neonates born in developing countries had normal intestinal villi at birth, but then developed these pathologic changes within 2–12 weeks of age. Slum-dwelling infants in Brazil had abnormal villous/crypt ratio and inflammatory cell infiltration in the lamina propria. These infants were also found to have abnormal D-xylose absorption when compared to infants from middle-class families with access to potable water and sewage facilities [30].
Jejunal biopsies performed on children in the Gambia revealed a range of pathology, from normal architecture to flattened villi, with most specimens having crypt hypertrophy and elevated concentrations of intraepithelial CD8+ lymphocytes [31]. Further studies on children from the same region found histologic evidence of cell-mediated enteropathy, independent of nutritional status, with an increased ratio of inflammatory cytokines (IFN-gamma) over regulatory cytokines (TGF-B) [32]. The authors hypothesized that the mucosal changes seen in these children were histologically and immunologically suggestive of a pro-inflammatory state and indicated a T-cell-mediated response to an environmental antigen, in many ways analogous to celiac disease.
Celiac sprue is a T-cell mediated gluten insensitivity that occurs in genetically predisposed individuals. Histologically, celiac sprue and EE are similar, with villous atrophy and increased lymphocytic infiltration. Clinically, both can result in malabsorption, malnutrition, and their sequalae. Etiologies of both EE and celiac sprue are believed to be due to continued exposure to an environmental insult, therefore it may be helpful to draw upon the celiac sprue literature to gain better understanding into the pathology of EE.
Celiac sprue is triggered by gluten-exposure in genetically susceptible individuals. Almost all patients with the disease have HLA class II genes HLA-DQ2 or HLA-DQ8, which encode molecules that present gluten peptides and are expressed on antigen presenting cells (APCs). The gluten-presenting APCs activate CD4+ T-cells, which subsequently release IFN-gamma. This stimulates myofibroblast secretion of matrix metalloproteinases (MMPs) which induce matrix remodeling and villus atrophy. Additionally, mucosal interleukin-15 release in the small bowel activates intraepithelial lymphocytes to act as natural killer cells and results in epithelial damage and increased mucosal permeability. The increased permeability allows gluten peptides to be picked up by mucosal antigen-presenting cells and causes further T-cell activation [33].
Adults in Zambia with EE have evidence of increased T-cell activation as measured by CD69 and HLA-DR expression [34]. It is plausible that in EE there is constant T-cell stimulation due to luminal exposure of enteric pathogens, ultimately resulting in pathogenic changes in the small bowel (Figure 2). Thus, EE may simply be the body’s response to an over-stimulating environment. A state of chronic inflammation could be a protective mechanism against constant infiltration by foreign pathogens.
Chronic Immune Stimulation
In theory, persistent damage to the mucosal barrier could permit translocation of foreign macromolecules into the circulation that may cause activation of a systemic immune response [35]. Lipopolysaccharide (LPS) is a component of the gram negative bacterial cell wall and the TLR 4 receptor for endotoxin is present on monocytes, macrophages, and dendritic cells. Measurement of serum LPS is used to assess bacterial translocation across the gut wall. Presence of bacterial ribosomal 16S RNA is also indicative of microbial translocation, and has been shown to correlate with elevated LPS [36]. Naturally occurring antibodies to the LPS core oligosaccharide (EndoCab) are produced by B cells in a T-cell dependent manner after LPS exposure. In conditions involving acute bacterial translocation, such as with gastrointestinal surgery or sepsis, peripheral EndoCab levels are decreased because these antibodies are actively involved in clearing LPS from the circulation [37].
Bacterial translocation may play an important role in chronic immune activation and disease progression in patients with HIV/AIDS [38,39,40]. North American patients infected with HIV have higher levels of LPS in serum than HIV negative controls, indicating gut barrier breakdown and immune dysfunction. Acutely infected HIV patients have low levels of LPS and low levels of EndoCab, whereas chronically infected HIV patients have higher levels of LPS but low levels of EndoCab, suggesting that in chronic disease circulating EndoCab levels are inadequate to clear serum LPS, and thus unable to prevent systemic immune activation that occurs in response to LPS [35].
What are the implications of bacterial translocation? In HIV patients, Brenchly et al studied monocytes that were chronically stimulated by LPS in vivo and found these cells were hyporesponsive to further stimulation by LPS in vitro. Other studies have shown that elevated serum LPS levels correlate with decreased expression of IL-12 and IFN-alpha after stimulation with TLR agonists, suggesting chronic immune stimulation by LPS results in a blunted innate immune response [39].
Among HIV negative patients, although serum LPS levels do not differ significantly between inhabitants of North America versus those of East Africa, EndoCab levels are significantly higher among Africans, implicating chronic bacterial translocation in this population [41]. Presumably, sustained exposure to intestinal pathogens via ingestion leads to subsequent blunting of intestinal villi, increased intestinal permeability, microbial translocation, and chronic systemic exposure to LPS. Thus among residents of the developing world, this exposure causes chronic immune stimulation and, ultimately, a dampened mucosal immune response. This down-regulated mucosal immune response could be responsible for the poor immunogenicity of oral vaccines in the developing world.
Under normal conditions, the mucosal immune system can discriminate between commensal and pathogenic bacteria in the gut. Exposure to commensal bacteria causes down-regulation of TLR expression and induces regulatory T-cell differentiation. By contrast, dendritic cells in the lamina propria (on the basolateral side) recognize pathogenic bacteria and induce NF-kappa B activation. Recently, it has been found in mice that segmented filamentous bacteria induce the appearance of Th17 cells in the lamina propria. Thus the presence of commensal organisms may enhance mucosal immunity to pathogens [42].Conversely, there is recent data that commensal organisms may facilitate infection and reproduction of enteric viruses. Kane et al demonstrated that mouse mammary tumor virus, a retrovirus, binds to bacterial LPS in the gut [43]. Once bound to LPS, the virus can interact with TLR4 (a natural LPS receptor) on leukocytes, causing release of IL-10, an anti-inflammatory cytokine that suppresses the anti-viral immune response. Kuss et al reported that antibiotic depletion of gut microbiota in mice resulted in reduced susceptibility to enteric viruses, including polio virus [44]. They found that polio virus binds LPS and that exposure to commensal bacteria enhances infection of host cells, suggesting that viruses may exploit commensal flora. The interplay between commensal bacteria and enteric viruses could have important implications for the efficacy of oral vaccines against polio and rotavirus, both enteric viruses. We do not know how the microbiome varies in children with EE; presumably constant exposure to fecal contamination could cause marked alterations in commensal flora with resulting variation in mucosal immunity and response to oral vaccines.
The presence of helminth infection may also have a large influence on the modulation of mucosal immunity. Colonization by intestinal worms has been shown to induce certain populations of T-cells, influence production of regulatory cytokines, and suppress intestinal inflammation in animal models [45, 46]. The eradication of helminth infection from higher income countries is thought to contribute to the increased incidence of atopic and inflammatory disorders to the extent that there are ongoing clinical trials investigating the benefits of “helminth therapy” on a variety of inflammatory conditions, including crohn’s disease and ulcerative colitis [47] The impact of helminth colonization on EE is has not been well studied, but considering that prevalence of intestinal worm infection is as high as 50% in children in certain parts of the world, this is a crucial issue that needs further attention [48].
Noninvasive Measures of EE
Because this condition is incompletely understood, a clear gold standard for diagnosis of EE is not yet defined. The most widely accepted surrogate marker for epithelial integrity is the lactulose:mannitol test, in which the subject is given an oral solution containing lactulose and mannitol. Mannitol is a monosaccharide and is completely absorbed by the small intestine; lactulose is a disaccharide and is only partially absorbed. Two hours after ingestion, urine is collected from the subject and chromatography is used to assess the levels of the sugars. Urine collected at this time point will reflect permeability and absorption of both the small intestine and colon, whereas urine collected later than two hours may exclusively reflect colonic function. In patients with normal gut barrier, there should be a low level of lactulose compared with mannitol in the urine. In patients with leaky gut mucosa, the amount of absorbed lactulose, and thus lactulose in the urine, will be higher and result in a higher lactulose to mannitol ratio. This test has been validated as a screening test for celiac disease [49,50].
Studies from the Gambia have studied the association between increased intestinal permeability and growth faltering [51,52]. Lunn et al reported that 43% of growth faltering in the first 15 months of life was associated with mucosal enteropathy by biopsy [53]. These same children had an abnormal lactulose:mannitol test when compared to British children of the same age. Interestingly, the abnormal L:M ratio was marked by both elevated lactulose and decreased mannitol, suggesting increased gut permeability as well as malabsorption.
Several serum and fecal biomarkers have been studied as indicators of intestinal inflammation (Table 2). Most of these markers have been developed for evaluation in patients with inflammatory bowel disease (IBD), and a few have been studied in populations at risk for EE. Fecal calprotectin, a cytoplasmic protein within neutrophils, has been correlated with active intestinal inflammation in patients with inflammatory bowel disease [54,55]. As mentioned previously, Endocab is a measure of exposure to LPS core antigen. Endocab correlates with endotoxemia and increased intestinal permeability in patients with acute pancreatitis [56]. Among Gambian infants, elevated serum Endocab is associated with poor height and weight growth and increased intestinal permeability, as measured by the lactulose:mannitol test [52].
Other studies have focused on metabolic byproducts of enterocytes or regulators of tight junctions (Text Box 1, Table 2). Citrulline is an amino acid produced by intestinal epithelial cells, and levels of citrulline were lower in patients with villous atrophy from IBD when compared with controls. Additionally, citrulline levels correlated with length of remnant small bowel in patients with short gut syndrome. Thus, citrulline is being proposed as a marker of gross enterocyte mass and may be useful for following patients with villous atrophy [57]. Citrulline levels did not correlate with d-xylose absorption test in patients with HIV-related enterpathy [58].
Text Box 1. Zonulin.
Zonula occludens toxin (Zot), produced by Vibrio cholera, is a recently identified molecule that opens tight junctions in the intestine. Fasana et al identified a human analogue of Zot which they named Zonulin. Zonulin and Zot act on the same intestinal epithelial receptor. Fasana et al propose that just as Zot can reversibly open intestinal tight junctions, zonulin (activated by intestinal flora imbalance or other immunogenic stimulation) causes an intracellular signaling cascade that results actin filament rearrangement and opening of tight junctions. Increased intestinal permeability and disruption of intestinal tight junctions is an important aspect of the pathogenesis of EE. As zonulin is responsible for paracellular leakage, it may play a significant role in development of EE. Once tight junctions are opened, antigens can pass into the lamina propria and stimulate an immune response. Intestinal expression of zonulin and serum zonulin IgA are elevated in celiac patients with active disease, further supporting the hypothesis that zonulin modulates tight junction permeability. It has been proposed that by mediating immune exposure to luminal antigens, zonulin may be involved in the development of autoimmune diseases, such as diabetes and celiac sprue [65,66].
Concluding Remarks and Future Perspectives
Children and adults who inhabit a fecally contaminated environment are at risk for the development of EE, which is marked by alterations in intestinal architecture, increased intestinal inflammation and permeability, deranged gut immune response, and growth faltering. These changes have important implications for dealing with maladies that affect the developing world, including malnutrition and infectious diseases control.
Some studies have investigated potential treatments to ameliorate the detrimental effects of EE on nutritional status. Lima et al studied dietary supplementation with glutamine in underweight children aged two to sixty months [59]While there was no difference in weight gain among the placebo and treatment groups, the treatment group did show a significant improvement in lactulose:mannitol ratio, indicating improved intestinal barrier function and suggesting that glutamine supplementation may promote enterocyte proliferation and intestinal repair. Sullivan et al found significant histologic improvement in jejunal biopsies of children who underwent a four week feeding program, suggesting that improved nutrition can foster mucosal repair independent of effect on nutritional status [60]. Perhaps both of these studies demonstrate that while enterocyte repair may occur acutely with specific nutritional interventions, the “catch up” in growth and increase in overall body mass may take longer to appreciate, and therefore, long term follow-up of these children is necessary.
A few studies have shown a correlation between enteropathy and exposure to specific pathogens, including hookworm and Citrobacter rodentium [12,21,61]. In Malawi, treatment of children with the antibiotic rifaximin failed to improve gut barrier function [29]. However, if a clear relationship between specific pathogens and EE can be established, then antimicrobials or vaccines could play a role in preventing EE development.
Promotion of basic sanitation as a means to reduce fecal-oral contamination is necessary to reduce the impact of EE in the developing world [62,63][http://www.unicef.org/wash/index_43084.html]. Ensuring universal access to potable water and plumbing would render the sequelae of EE obsolete. However, until the political will, stability, and resources come to fruition to make this feasible, it is imperative that we better elucidate the complexities of EE.
This subclinical condition may have a larger impact on worldwide Disability Adjusted Life years (DALYs) than does any specific pathogen, particularly because EE increases susceptibility to other morbidities. Before treatments can be devised, better methods of diagnosing EE are needed. Noninvasive diagnostic parameters are especially important as endoscopy is impractical at the public health scale of intervention that is likely to be required for an ailment affecting up to 40% of the world’s children. Finally, treatment and prevention will require filling the “knowledge gap” of pathogenesis, for which in vivo animal models will play an important role [64]. It is this type of progressive approach that will be required to help us better understand and treat EE, and ultimately address undernutrition, growth faltering, and vaccine efficacy to promote a more healthy and productive global population.
Figure 3. Intestinal function in normal and diseased individuals.

(a) In normal intestines, goblet cells secrete a layer of mucus that protects epithelial cells from exposure to bacteria. Phagocytosis by macrophages protects against bacteria entering the lamina propria while minimizing tissue injury. Tight junctions are undisrupted. Villi are long and finger-like and crypts are shallow in comparison, with a villus:crypt ratio of 3:1. Adapted with permission from [60]. (b) Constant exposure to fecal-oral contamination and recurrent episodes of gastroenteritis lead to a perpetual state of small bowel injury and hyperstimulation of the mucosal immune system. Luminal bacteria bind to Toll-like receptors (not shown) on dendritic cells and intraepithelial lymphocytes (IELs) on the basolateral surface of epithelial cells. Dendritic cells release chemokines and cytokines and recruit polymorphonuclear neutrophils (PMNs), and act as antigen presenting cells that cause activation and differentiation of CD4+ T-cells. CD4+ cells then release TH1 cytokines stimulating myofibroblasts to release metalloproteases that cause remodeling and blunting of villi. CD4+ T-cells stimulate B-cell production of endotoxin-core antibodies. Release of IL-15 stimulates differentiation of IELs into natural killer cells, resulting in intestinal inflammation and increased permeability. The result is increased intraepithelial lymphocytes and lymphocytic infiltration in the lamina propria, as well as villus atrophy and crypt hyperplasia with a decreased villus:crypt ratio. Adapted with permission [32].
Outstanding Questions Box.
How prevalent is EE?
What causes EE? Does it have an infectious or toxic etiology?
What indicators (biomarkers) can we use to identify children at risk for development of EE?
Are there nutritional interventions that can facilitate healing of the gut?
How is the immune system different in children with EE?
How can we design vaccines that will elicit a response in children with EE?
What is the impact of EE in adults?
Is there a genetic predisposition?
Does EE in a mother result in malnutrition in her child?
How do helminth infections influence the development of EE?
What is the role of commensal versus pathogenic microbes in development of EE?
Can EE be transferred from one person to the next by fecal contamination?
Can one develop immunity to EE?
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are p roviding this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Solón Pablo. The Human Right to Water and Sanitation. 64th General Assembly Plenary, United Nations; New York, New York. 28 July 2010. [Google Scholar]
- 2.Ramakrishna BS, et al. Tropical malabsorption. Postgraduate Medical Journal. 2006;82:779–87. doi: 10.1136/pgmj.2006.048579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sullivan PB, et al. Chronic diarrhea and malnutrition--histology of the small intestinal lesion. Journal of Pediatric Gastroenterology and Nutrition. 1991;12:195–203. doi: 10.1097/00005176-199102000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Dewey KG, Adu-Afarwuah S. Systematic review of the efficacy and effectiveness of complementary feeding interventions in developing countries. Mater Child Nutr. 2008;4(Suppl 1):24–85. doi: 10.1111/j.1740-8709.2007.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prentice A. Nutrient requirements for growth, pregnancy, and lactation: the Keneba experience. SAfr J Clin Nutr. 1993;6:33–38. [Google Scholar]
- 6.Bhutta ZA, et al. What works? Interventions for maternal and child undernutrition and survival. Lancet. 2008;371:417–440. doi: 10.1016/S0140-6736(07)61693-6. [DOI] [PubMed] [Google Scholar]
- 7.Hasan AS, et al. Antibody levels against polioviruses in children following pulse polio immunization program. Indian Pediatr. 2004;41:1040–1044. [PubMed] [Google Scholar]
- 8.Naik TN, Krishnan T. Rotavirus vaccine: current status & future prospects. Indian J Med Res. 1996;104:76–85. [PubMed] [Google Scholar]
- 9.Ratnaswamy L, et al. Paralytic poliomyelitis: clinical and virological studies. Indian Pediatr. 1973;10:443–447. [PubMed] [Google Scholar]
- 10.Levine MM. Immunogenicity and efficacy of oral vaccines in developing countries: Lessons from a live cholera vaccine. BMC Biology. 2010;8:129. doi: 10.1186/1741-7007-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sprinz H, et al. Biopsy of small bowel of Thai people. With special reference to recovery from Asiatic cholera and to an intestinal malabsorption syndrome. Am J Clin Pathol. 1962;38:43–51. doi: 10.1093/ajcp/38.1.43. [DOI] [PubMed] [Google Scholar]
- 12.Cook GC, et al. Jejunal morphology of the African in Uganda. J Pathol. 1969;98:157–69. doi: 10.1002/path.1710980302. [DOI] [PubMed] [Google Scholar]
- 13.Lindenbaum J, et al. Subclinical small-intestinal disease in east pakistan. Br Med J. 1966;2:1616–1619. doi: 10.1136/bmj.2.5530.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cowell EJ, et al. Jejunal morphological characteristics in South Vietnamese residents. JAMA. 1968;206:2273. [PubMed] [Google Scholar]
- 15.Garcia S. Malabsorption and Malnutrition in Mexico. Am J Clin Nutr. 1968;21:1066–1076. doi: 10.1093/ajcn/21.9.1066. [DOI] [PubMed] [Google Scholar]
- 16.Lindenbaum J, et al. Recovery of small-intestinal structure and function after residence in the tropics. I. studies in peace corps volunteers. Ann Intern Med. 1971;74:218–222. doi: 10.7326/0003-4819-74-2-218. [DOI] [PubMed] [Google Scholar]
- 17.Lindenbaum J. Small intestine dysfunction in Pakistanis and Americans resident in Pakistan. Am J Clin Nutr. 1968;21:1023–1029. doi: 10.1093/ajcn/21.9.1023. [DOI] [PubMed] [Google Scholar]
- 18.Lindenbaum J, et al. Subclinical malabsorption in developing countries. Am J Clin Nutr. 1972;25:1056–1061. doi: 10.1093/ajcn/25.10.1056. [DOI] [PubMed] [Google Scholar]
- 19.Stanfield JP, et al. Intestinal biopsy in kwashiorkor. Lancet. 1965;2:519–523. doi: 10.1016/s0140-6736(65)91474-1. [DOI] [PubMed] [Google Scholar]
- 20.Fagundes-Neto U. Tropical enteropathy (environmental enteropathy) in early childhood: A syndrome caused by contaminated environment. Journal of Tropical Pediatrics. 1984;30(4):204–209. doi: 10.1093/tropej/30.4.204. [DOI] [PubMed] [Google Scholar]
- 21.Kelly P, et al. Responses of small intestinal architecture and function over time to environmental factors in a tropical population. Am J Trop Med Hyg. 2004;70:412–419. [PubMed] [Google Scholar]
- 22.Patriarca PA, et al. Factors affecting the immunogenicity of oral poliovirus vaccine in developing countries: Review. Rev Infect Dis. 1991;13:926–939. doi: 10.1093/clinids/13.5.926. [DOI] [PubMed] [Google Scholar]
- 23.Lunn PG. The impact of infection and nutrition on gut function and growth in childhood. The Proceedings of the Nutrition Society. 2000;59:147–54. doi: 10.1017/s0029665100000173. [DOI] [PubMed] [Google Scholar]
- 24.Mathan VI, Baker SJ. An epidemic of tropical sprue in southern India. I Clinical features. Ann Trop Med Parasitol. 1970;64:439–451. doi: 10.1080/00034983.1970.11686715. [DOI] [PubMed] [Google Scholar]
- 25.The epidemiology of tropical sprue; Case records of the massachusetts general hospital. weekly clinicopathological exercises. case 15–. A 78-year-old woman from the dominican republic with chronic diarrhea. N Engl J Med. 1990;322:1067–1075. doi: 10.1056/NEJM199004123221509. [DOI] [PubMed] [Google Scholar]
- 26.Batheja MJ, et al. The face of tropical sprue in 2010. Case Reports in Gastroenterology. 2010;4:168–172. doi: 10.1159/000314231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khin-Maung-U, et al. Epidemiology of small bowel bacterial overgrowth and rice carbohydrate malabsorption in burmese (myanmar) village children. Am J Trop Med Hyg. 1992;47:298–304. doi: 10.4269/ajtmh.1992.47.298. [DOI] [PubMed] [Google Scholar]
- 28.dos Reis JC, et al. Breath hydrogen test in the diagnosis of environmental enteropathy in children living in an urban slum. Digestive Diseases and Sciences. 2007;52:1253–1258. doi: 10.1007/s10620-006-9288-9. [DOI] [PubMed] [Google Scholar]
- 29.Trehan I, et al. A randomized, double-blind, placebo-controlled trial of rifaximin, a nonabsorbable antibiotic, in the treatment of tropical enteropathy. Am J Gastroenterol. 2009;104:2326–33. doi: 10.1038/ajg.2009.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fagundes Neto U, et al. Asymptomatic environmental enteropathy among slum-dwelling infants. J Am Coll Nutr. 1994;13:51–56. doi: 10.1080/07315724.1994.10718371. [DOI] [PubMed] [Google Scholar]
- 31.Sullivan PB, et al. Persistent diarrhea and malnutrition--the impact of treatment on small bowel structure and permeability. J Pediatr Gastroenterol Nutr. 1992;14:208–15. doi: 10.1097/00005176-199202000-00016. [DOI] [PubMed] [Google Scholar]
- 32.Campbell DI, et al. Chronic T Cell-Mediated Enteropathy in Rural West African Children: Relationship with Nutritional Status and Small Bowel Function. Pediatr Res. 2003;54:306–311. doi: 10.1203/01.PDR.0000076666.16021.5E. [DOI] [PubMed] [Google Scholar]
- 33.Schuppan D, et al. Celiac Disease: From Pathogenesis to Novel Therapies. Gastroenterology. 2009;137:1912–1933. doi: 10.1053/j.gastro.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 34.Veitch AM, et al. Tropical enteropathy: a T-cell-mediated crypt hyperplasia enteropathy. Eur J Gastroenterol Hepatol. 2001;13:1175–81. doi: 10.1097/00042737-200110000-00009. [DOI] [PubMed] [Google Scholar]
- 35.Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 36.Jiang W, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199:1177–1185. doi: 10.1086/597476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strutz F, et al. Relationship of antibodies to endotoxin core to mortality in medical patients with sepsis syndrome. Intensive Care Medicine. 1999;25:435–444. doi: 10.1007/s001340050877. [DOI] [PubMed] [Google Scholar]
- 38.Cassol E, et al. Persistent microbial translocation and immune activation in HIV-1-infected South Africans receiving combination antiretroviral therapy. J Infect Dis. 2010;202:723–733. doi: 10.1086/655229. [DOI] [PubMed] [Google Scholar]
- 39.Nowroozalizadeh S, et al. Microbial translocation correlates with the severity of both HIV-1 and HIV-2 infections. J Infect Dis. 2010;201:1150–1154. doi: 10.1086/651430. [DOI] [PubMed] [Google Scholar]
- 40.Redd AD, et al. Is microbial translocation a cause or consequence of HIV disease progression? J Infect Dis. 2011;203:744–745. doi: 10.1093/infdis/jiq107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Redd AD, et al. Microbial translocation, the innate cytokine response, and HIV-1 disease progression in africa. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6718–6723. doi: 10.1073/pnas.0901983106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ivanov II, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kane M, et al. Successful transmission of a retrovirus depends on the commensal microbiota. Science. 2011;334:245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuss S, et al. Intestinal Microbiota Promote Enteric Virus Replication and Systemic Pathogenesis. Science. 2011;334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ince MN, et al. Heligomosomoides polygyrus induces TLR4 on murine mucosal T cells that produce TGF beta after lipopolysaccharide stimulation. J Immunol. 2006;176:726–729. doi: 10.4049/jimmunol.176.2.726. [DOI] [PubMed] [Google Scholar]
- 46.Metwali A, et al. Induction of CD8+ regulatory T cells in the intestine by Heligmosomoides polygyrus infection. Am J Physiol Gastrointest Liver Physiol. 291:G253–G259. doi: 10.1152/ajpgi.00409.2005. [DOI] [PubMed] [Google Scholar]
- 47.Summers R, et al. Trichuris suis seems to be safe and possibly effective in the treatment of inflammatory bowel disease. Am J Gastroenterol. 2003;98:2034–2041. doi: 10.1111/j.1572-0241.2003.07660.x. [DOI] [PubMed] [Google Scholar]
- 48.Luong TV. Prevention of Intestingal Worm Infections through Improved Sanitation and Hygiene Unicef East Asia & Pacific Regional Office. 2002 Oct;2002 [Google Scholar]
- 49.Juby LD, et al. Lactulose/mannitol test: An ideal screen for celiac disease. Gastroenterology. 1989;96:79–85. doi: 10.1016/0016-5085(89)90767-1. [DOI] [PubMed] [Google Scholar]
- 50.Camilleri M, et al. Understanding measurements of intestinal permeability in healthy humans with urine lactulose and mannitol excretion. Neurogastroenterology and motility. 2010;22:e15–e26. doi: 10.1111/j.1365-2982.2009.01361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lunn PG, et al. Intestinal permeability, mucosal injury, and growth faltering in Gambian infants. Lancet. 1991;338:907–910. doi: 10.1016/0140-6736(91)91772-m. [DOI] [PubMed] [Google Scholar]
- 52.Campbell DI, et al. Growth faltering in rural Gambian infants is associated with impaired small intestinal barrier function, leading to endotoxemia and systemic inflammation. J Nutr. 2003;133:1332–1338. doi: 10.1093/jn/133.5.1332. [DOI] [PubMed] [Google Scholar]
- 53.Lunn PG. The impact of infection and nutrition on gut function and growth in childhood. Proc Nutr Soc. 2000;59:147–154. doi: 10.1017/s0029665100000173. [DOI] [PubMed] [Google Scholar]
- 54.Tibble JA, et al. A simple method for assessing intestinal inflammation in Crohn’s disease. Gut. 2000;47:506–513. doi: 10.1136/gut.47.4.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tibble JA, et al. Use of surrogate markers of inflammation and rome criteria to distinguish organic from nonorganic intestinal disease. Gastroenterology. 2002;123:450–460. doi: 10.1053/gast.2002.34755. [DOI] [PubMed] [Google Scholar]
- 56.Penalva JC, et al. A Study of intestinal permeability in relation to the inflammatory response and plasma endocab IgM levels in patients with acute pancreatitis. J Clin Gastroenterol. 2004;38:512–7. doi: 10.1097/01.mcg.0000129060.46654.e0. [DOI] [PubMed] [Google Scholar]
- 57.Crenn P, et al. Plasma citrulline: A marker of enterocyte mass in villous atrophy-associated small bowel disease. Gastroenterology. 2003;124:1210–1219. doi: 10.1016/s0016-5085(03)00170-7. [DOI] [PubMed] [Google Scholar]
- 58.Papadia C, et al. Plasma citrulline as a quantitative biomarker of HIV-associated villous atrophy in a tropical enteropathy population. Clin Nutr. 2010;29:795–800. doi: 10.1016/j.clnu.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 59.Lima A, et al. Intestinal Barrier Function and Weight Gain in Malnourished Children Taking Glutamine Supplemented Enteral Formula. J Pediatr Gastroenterol Nutr. 2005;40:28–35. doi: 10.1097/00005176-200501000-00006. [DOI] [PubMed] [Google Scholar]
- 60.Sullivan PB. Studies of the small intestine in persistent diarrhea and malnutrition: the Gambian experience. J Pediatr Gastroenterol Nutr. 2002;34:S11–3. doi: 10.1097/00005176-200205001-00003. [DOI] [PubMed] [Google Scholar]
- 61.Sheehy TW, et al. Hookworm disease and malabsorption. Gastroenterology. 1962;42:148–156. [PubMed] [Google Scholar]
- 62.Pickering AJ, et al. Efficacy of alcohol-based hand sanitizer on hands soiled with dirt and cooking oil. J Water Health. 2011;9:429–433. doi: 10.2166/wh.2011.138. [DOI] [PubMed] [Google Scholar]
- 63.Dobe M, et al. Sanitation: The hygienic means of promoting health. Indian Journal of Public Health. 2011;55:49–51. doi: 10.4103/0019-557X.82557. [DOI] [PubMed] [Google Scholar]
- 64.Costa LB, et al. Cryptosporidium-malnutrition interactions: mucosal disruption, cytokines, and TLR signaling in a weaned murine model. J Parasitol. 2011;97:1113–1120. doi: 10.1645/GE-2848.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang W, et al. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci. 2000;113:4435–40. doi: 10.1242/jcs.113.24.4435. [DOI] [PubMed] [Google Scholar]
- 66.Fasano A, et al. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. 2000;355:1518–1519. doi: 10.1016/S0140-6736(00)02169-3. [DOI] [PubMed] [Google Scholar]
- 67.Wood GM, et al. Small bowel morphology in British Indian and Afro-Carribean subjects: evidence of tropical enteropathy. Gut. 1991;32:256–259. doi: 10.1136/gut.32.3.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Green PHR. The Many Faces of Celiac Disease: Clinical Presentation of Celiac Disease in the Adult Population. Gastroenterology. 2005;128:S74–S78. doi: 10.1053/j.gastro.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 69.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mondal, et al. Contribution of enteric infection, altered intestinal barrier function, and maternal malnutrition to infant malnutrition in Bangladesh. Clin Infect Dis. 2012;54:185–192. doi: 10.1093/cid/cir807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mao R, et al. Fecal calprotectin in predicting relapse of inflammatory bowel diseases: A meta-analysis of prospective studies. Inflamm Bowel Dis. 2012 doi: 10.1002/ibd.22861. ( http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)1536-4844) [DOI] [PubMed]
- 72.Sidler MA, et al. Fecal S100A12 and fecal calprotectin as noninvasive markers for inflammatory bowel disease in children. Inflamm Bowel Dis. 2008;14:359–366. doi: 10.1002/ibd.20336. [DOI] [PubMed] [Google Scholar]
- 73.Foell D, et al. Neutrophil derived human S100A12 (EN-RAGE) is strongly expressed during chronic active inflammatory bowel disease. Gut. 2003;52:847–853. doi: 10.1136/gut.52.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaiser T, et al. Faecal S100A12 as a non-invasive marker distinguishing inflammatory bowel disease from irritable bowel syndrome. Gut. 2007;56:1706–1713. doi: 10.1136/gut.2006.113431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nylund CM, et al. Granulocyte Macrophage-Colony-stimulating Factor Autoantibodies and Increased Intestinal Permeability in Crohn Disease. J Pediatr Gastroenterol Nutr. 2011;52:542–548. doi: 10.1097/MPG.0b013e3181fe2d93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fuchs D, et al. Urinary neopterin in the diagnosis and follow-up of neoplasia: a biochemical parameter to detect cell-mediated immune. Tumour Biol. 1984;5:199–209. [PubMed] [Google Scholar]
- 77.Chu CC, et al. Pretreatment with alanyl-glutamine suppresses T-helper-cell-associated cytokine expression and reduces inflammatory responses in mice with acute DSS-induced colitis. J Nutr Biochem. 2011 doi: 10.1016/j.jnutbio.2011.06.002. http://dx.doi.org/10.1016/j.bbr.2011.03.031. [DOI] [PubMed]
- 78.Peterson KM, et al. The expression of REG1A and REG1B is increased during acute amebic colitis. Parasitol Int. 2011;60:296–300. doi: 10.1016/j.parint.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Planas R, et al. Regenerating gene Ia is a biomarker for diagnosis and monitoring of celiac disease: a preliminary study. Trans Res. 2011;158:140–145. doi: 10.1016/j.trsl.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 80.Guerrant RL, et al. Measurement of fecal lactoferrin as a marker for fecal leukocytes. J Clin Microbiol. 1992;30:1238–42. doi: 10.1128/jcm.30.5.1238-1242.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Steiner TL, et al. Fecal lactoferrin, interleukin-1beta, and interleukin-8 are elevated in patients with severe Clostridium difficile colitis. Clin Diagn Lab Immunol. 1997;4:719–22. doi: 10.1128/cdli.4.6.719-722.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alcantara CS, et al. Interleukin-8, tumor necrosis factor-alpha, and lactoferrin in immunocompetent hosts with experimental and Brazilian children with acquired cryptosporidiosis. Am J Trop Med Hyg. 2003;68:325–328. [PubMed] [Google Scholar]
- 83.Humphrey JH. Child undernutrition, tropical enteropathy, toilets, and handwashing. Lancet. 2009;374:1032–1035. doi: 10.1016/S0140-6736(09)60950-8. [DOI] [PubMed] [Google Scholar]
- 84.Garcia S. Malabsorption and Malnutrition in Mexico. Am JClin Nutr. 1968;21:1066–76. doi: 10.1093/ajcn/21.9.1066. [DOI] [PubMed] [Google Scholar]