

Abstract
The past several years has seen increasing appreciation for plasticity of higher-level chromatin folding. Four distinct “30 nm” chromatin fiber structures have been identified, while new in situ imaging approaches have questioned the universality of 30 nm chromatin fibers as building blocks for chromosome folding in vivo. 3C-based approaches have provided a non-microscopic, genomic approach to investigating chromosome folding while uncovering a plethora of long-distance cis interactions difficult to accommodate in traditional hierarchical chromatin folding models. Recent microscopy based studies have suggested complex topologies co-existing within linear interphase chromosome structures. These results call for a reappraisal of traditional models of higher-level chromatin folding.
Introduction
For several decades, the path towards understanding higher order organization of chromatin appeared clear: solve the 30 nm chromatin fiber structure and then determine its path within interphase and mitotic chromosomes. Underlying these two goals, however, were two major assumptions- first, that a unique structure exists for “the” 30 nm chromatin fiber and second, that the 30 nm chromatin fiber as it exists in vitro is the basic structural motif underlying further chromatin folding in vivo.
These two long-standing assumptions increasingly have been challenged in recent years. A family of 30 nm chromatin fiber structures has emerged together with speculation concerning alternative pathways for chromatin compaction in vivo that bypass the 30 nm fiber. 3C (chromosome conformation capture) molecular methodologies and new microscopy approaches suggest a surprising plasticity of chromatin folding challenging conventional ideas for how chromatin folds into chromosomes.
Higher order folding of chromatin into 30 nm fibers in vitro and in situ
Salt-dependent folding in vitro of “beads on a string” oligonucleosomes into a roughly 30 nm diameter fiber led to the natural assumption of a unique “30 nm”, higher-order chromatin folding motif. The solenoid model, the first proposed for its structure, postulated a one-start, helical folding of the nucleofilament with the linker DNA bent between adjacent nucleosomes. At low ionic strength, unfolded oligonucleosomes typically show a zig-zag appearance, leading to two alternative model classes, the twisted ribbon model in which the zig-zag folds to form the fiber such that the zig-zag is along the fiber trajectory versus the crossed-linker model in which a two-start helical folding is created through extension of the straight linker DNA across the fiber cross-section (reviewed in [1]). Interestingly, inter-nucleosome cross-linking patterns suggest both solenoid and zig-zag model features can co-exist within a single 30 nm fiber [2*].
Electron microscopy (EM) of oligonucleosomes reconstituted with linker histone on tandem arrays of nucleosome positioning DNA sequences with variable linker lengths now reveals a family of higher-order chromatin fibers. A nucleosome repeat length (NRL) of 167 bp produces a 21 nm diameter ladder-like crossed-linker pattern [3] consistent with previous X-ray crystallography of a nucleosome tetramer (167 NRL) [4] and predictions from cross-linking experiments (177 NRL) [5]. NRLs of 177, 187, 197, and 207 bp produced 34 nm diameter chromatin fibers, but a jump to a 44 nm diameter was seen for NRLs of 217, 227, and 237 bp [6]. The invariance of fiber diameter over a 177-207 NRL range suggests an important role for specific inter-nucleosome contacts, consistent with the strong disruption of compaction observed for 202 NRL arrays reconstituted with histone H4 acetylated at K16 [7]. H4K16 is predicted to contact the acidic patch on histone H2A on adjacent nucleosomes to stabilize higher-order folding [8]. While linker histone plays an important role in modulating higher order folding, the exact location of the linker histone relative to the nucleosome and linker DNA remains unknown.
Surprisingly, whereas the 21 nm fiber has a density of 6.1 nucleosomes/11 nm [3], the density of the compacted 34 nm fiber is nearly double at 11.2 nucleosomes/11 nm [6], suggesting inter-digitation of adjacent nucleosome gyres and a much higher compaction than previous estimates.
In contrast, recent cryo-electron microscopy tomography of avian erythrocyte nuclei [9**] (212 bp NRL) demonstrates an ~32 nm diameter fiber formed by a left-handed two start helical folding of nucleosomes with ~6.5 nucleosomes/11 nm density, closer to previous estimates of nucleosome density. Density within the fiber interior suggests linker DNA crossing the fiber cross-section. Docking of nucleosome structure within the EM reconstruction and molecular modeling leads to a geometry similar to the crossed-linker model. There is a prominent face-to-face orientation of nucleosomes and a more homogenous radial distribution of nucleosomes, not unlike that predicted in a solenoid model, yet nucleosomes remain separated rather than close packed and no inter-digitation of nucleosomes is observed. Chromatin within intact erythrocyte nuclei is compacted into dense chromocenters. This study used isolated nuclei and buffer conditions that presumably decondensed these chromocenters into dispersed 30 nm chromatin fibers. The compaction of these 30 nm chromatin fibers relative to their conformation in vivo therefore remains unclear.
Thus we already are now at four distinct “30 nm” structures (Fig. 1). Interestingly, imposing just a few, simple geometrical constraints was shown to produce a family of possible “30 nm” chromatin fiber structures [10], as now more recently demonstrated by molecular dynamics simulations [11*,12*].
Figure 1. Four different higher-order chromatin structures and counting.
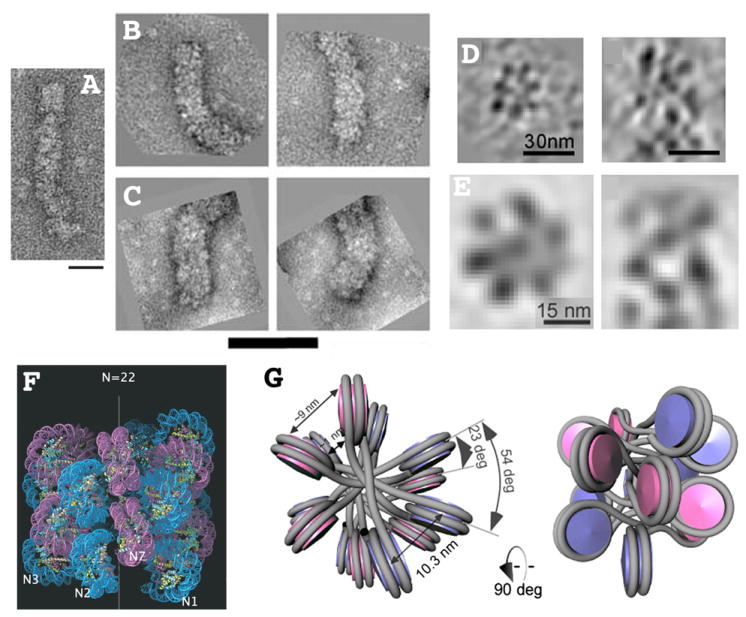
(A) 21 nm diameter zig-zag structure formed after reconstitution with both core and linker histones using a 167 bp nucleosome repeat length. Reprinted from Fig. 4, reference [3], Copyright 2008 National Academy of Sciences, USA. (B-C) 33 nm diameter (B) versus 44 nm diameter (C) higher-order fibers formed after reconstitution with both core and linker histones but with nucleosome repeat lengths of 197 bp (B) or 207 (C). Reprinted from Fig. 1, reference [6], Copyright 2006 National Academy of Sciences, USA. (D-E) Transverse (left) or longitudinal (right) sections through individual (D) or averaged (E) tomograms of 32 nm diameter higher-order fibers visualized within cryo-sections of avian erythrocyte nuclei [9]. These fibers contain ~6.5 nucleosomes per 11 nm length of fiber. Reprinted from Figs. 1&2, reference [9]. (F) Molecular model [6] of one start helical structure proposed for 33 nm diameter reconstituted fiber (B). Inter-digitation of neighboring gyres of helix results in high nucleosome fiber density of ~ 11 nucleosomes per 11 nm fiber length. Reprinted from Fig. 3, reference [6], Copyright 2006 National Academy of Sciences, USA. (G) Molecular model for two-start, cross-linker type structure proposed for 32 nm fiber visualized by cryo-EM (D-E). The approximately 6.5 nucleosomes per 11 nm of fiber length corresponds more closely to previous estimates of fiber compaction. Reprinted from Fig. 3, reference [9], Copyright 2011 National Academy of Sciences, USA. Scale bars = 50 nm (A), 100 nm (B-C), 30 nm (D), or 15 nm (E).
Chromatin array oligomerization- an in vitro model for in vivo compaction
FISH analysis reveals most genome regions in higher metazoan somatic nuclei are condensed well above the 30 nm fiber. Interestingly, polycation concentrations which preserve large-scale chromatin folding within isolated nuclei [13] are similar to those causing inter-fiber associations and chromatin aggregation in vitro [14], suggesting oligomerization of chromatin arrays as an in vitro model for chromatin compaction in vivo [15]. Oligomerization is cooperative and, like higher order chromatin folding, dependent on linker histone and histone tails, particularly H4. However, its dependence on histone tails appears to be largely through nonspecific, charge interactions [16*]. This contrasts with specific interactions, in particular between H4K16 and an acidic patch on H2A [7,17], implicated in higher order folding. Whereas H2A variant H2ABbd has a reduced acidic patch and reduced higher order folding but increased oligomerization, histone variant H2A.Z with an increased acidic patch has the opposite behavior, suggesting competition between chromatin inter-fiber and intra-fiber interactions [18]. Moreover a histone mutation mimicking acetylation at H3K56 has no effect on higher order chromatin compaction but increases oligomerization of chromatin arrays with nucleosome free regions, which simulates sites of nucleosome assembly where this mark is enriched [19*]. In contrast, p300 mediated histone acetylation at the HIV promoter leads to both higher order chromatin decondensation and inhibition of array oligomerization [20**]. Which effect is most closely related to the accompanying in vitro transcriptional activation remains unclear.
Higher order chromatin folding within nuclei and chromosomes
The assumption of 30 nm chromatin fibers as the basic building block of further chromosome folding is derived from a relatively small number of observations. These include visualization of 30 nm chromatin fibers in isolated mitotic chromosomes swollen by low salt isolation buffers [23,24], the clearly distinct and regular packing of well spaced 30 nm chromatin fibers within intact nuclei of a few specialized cell types such as starfish sperm nuclei [25], and the clear presence of 30 nm chromatin fibers at specific, less compacted chromosomal loci, for example in polytene chromosome puffs after inhibition of transcription [26]. Given these in situ observations, more condensed chromosome regions whose substructure cannot be easily visualized were assumed to be formed by these same fibers.
New experimental approaches now question this assumption [21*,22*]. First, cryo-EM of mitotic chromosomes fails to reveal any substructure above 11 nm packing of nucleosomes, leading to a “melt” chromosome model in which strong inter-fiber nucleosome interactions of comparable strength to intra-fiber interactions cause mixing of nucleosomes into a uniform mass [27]. Second, using conventional chemical fixation and embedding but energy spectroscopic imaging (ESI) for improved chromatin contrast, Bazzet-Jones and colleagues fail to observe any hint of 30 nm chromatin structure in a range of cell types, with no 30 nm type fiber structures visualized within more compact chromatin regions [28*,29*].
Reconciling the absence of any apparent substructure above 11 nm in cryo-EM images of mitotic chromosomes with the observation of distinct mitotic chromosome substructure above 30 nm by conventional light microscopy in fixed and live cells [30] and now by super-resolution light microscopy [31**] remains difficult. Fixation by high pressure freezing and other cryo-methods producing vitreous ice is generally considered ideal, yet estimated millisecond freezing rates are still slow relative to FRET FCS measurements of microsecond trinucleosome array dynamics and sub-millisecond trinucleosome decompaction [32**]. Possible effects of temperature dependent changes in DNA pitch during freezing are unknown. Meanwhile, the ESI studies described above used formaldehyde fixation and immunostaining procedures previously shown to significantly perturb chromatin ultrastructure [33]. Considerable cell extraction is observed in these ESI images, particularly in the ES cells where unfolded nucleosomal filaments (defined here as “10 nm chromatin fibers”) were predominately observed. Conventional TEM of ES cells using standard fixation conditions which do not cause cell extraction show extensive regions of condensed chromatin, including at the nuclear periphery [34]. Repeating this work using stronger fixation conditions and eliminating the pre-embedding immunostaining would be desirable.
Still, reconciling the ~240 nm (~25-50 kb) persistence length estimated from molecular simulations of 30 nm fiber folding [1] with the highly compacted chromatin prevalent in most nuclei and mitotic chromosomes remains difficult. The possibility that a major fraction of the genome is compacted in a non-canonical 30 nm fiber structure is significant, while EM tomography of chromosomes assembled in a Xenopus oocyte extract demonstrates the ability to achieve high compaction without canonical 30 nm fibers in such a reconstituted system [35].
Lessons from 3C
A similar challenge to the concept of further chromosome compaction through folding of 30 nm chromatin fibers now comes indirectly from molecular biology based 3C methods. In 3C experiments, intact nuclei are fixed with formaldehyde to preserve physical interactions between chromatin loci. Cross-linked chromatin is then cut with restriction enzymes and this is followed by inter-molecular DNA ligation of neighboring cross-linked fragments. The capability of 3C to map chromatin interactions has been expanded by combination with high-throughput sequencing approaches and newer derivatives of 3C, including 4C [36,37], 5C [38,39*], Hi-C [40**], e4C [41**] and ChIA-PET [42**,43**]. These derivatives allow mapping the interactions between one locus to the whole genome [36,37], mapping interactions between many foci within a chromatin domain [38,39*], identifying, at lower resolution, genome-wide interactions [40**], and/or detecting interactions mediated by specific proteins [41**,42**,43**].
An emerging theme from these experiments has been the high frequency of observed interactions between distal regulatory regions (promoters, enhancers, insulators, and DNase I HS sites) separated by several to thousands of kb, leading to a chromatin “hub” model of organization [44*]. While attention has focused on the high frequency of long-range interactions, multiple interactions between regulatory elements separated less than the estimated ~25-50 kb 30 nm fiber persistence length are equally surprising. 30 nm chromatin fiber looping over such short DNA stretches would be unlikely without local kinking or fiber discontinuities, perhaps related to local chromatin structure, and/or buckling of 30 nm fibers induced by strong inter-nucleosome interactions. In vitro studies have revealed disruption of both higher order folding and oligomerization of nucleosome arrays over several kb surrounding local recruitment of the p300 HAT [20**], raising the possibility of stretches of 10 nm structure, encompassing regulatory regions, acting as flexible connectors facilitating looping of adjacent 30 nm chromatin fiber segments.
Recent studies have employed computational modeling to extrapolate the comprehensive datasets generated from 3C-based assays into estimated 3D structures of large-scale chromatin folding [39*,40**,45*,46*]. 3C interaction frequencies are used as distance constraints for model reconstruction. A model generated for a 500kb region surrounding the alpha-globin locus predicts its folding into 1 or 2 highly compact chromatin domains, consisting of rosettes of 50- 60kb chromatin loops, in its transcriptionally inactive state. A less compact structure is predicted in cells where the alpha-globin locus is expressed, with active genes clustered towards the globule center.
On a still larger scale, Hi-C methods have pointed to spatial segregation of Mbp domains separating active and inactive genomic regions in a functional compartmentalization of the genome [40**]. Interestingly, these Hi-C identified domains match closely with comparable size genomic domains with distinctive early versus late replication timing [48**].
Interpretation of these 3C-based results is tempered by uncertainty regarding key assumptions regarding this methodology. First, is the question of whether rare cross-linking events represent temporal fluctuations occurring within a large fraction of the cell population versus rare but stable conformation heterogeneity within the cell population. Second, is the question of what is the actual nature of the cross-linking events. Whereas the 3C ligation event typically is depicted as between two naked DNA elements cross-linked at a single molecular complex, even at 20-fold reduced formaldehyde fixation levels than commonly used for 3C-based methods internucleosome cross-linking events are frequent within nucleosome arrays [2*]. UV irradiation, a similar short range cross-linking method, produces up to 20% inter-fiber nucleosome cross-linking events using oligomerized nucleosome arrays as a substrate [47*]. Extensive cross-linked protein/DNA networks therefore may be the actual template for 3C ligation events. This might explain why many loci connected by long-distance 3C interactions show microscopic versus molecular separation distances as measured by FISH [41**]. Taking this idea one step further, these cross-linked networks could include protein/RNA complexes from nuclear bodies, linking DNA fragments from different chromosomes attached to the same nuclear body. Understanding which of these possibilities occur in 3C experiments will be critical for deriving correct structural information from 3C data.
Direct visualization of large-scale chromatin organization and dynamics
FISH reveals interphase chromatin compaction for most higher metazoan species is significantly higher than that predicted for 30 nm chromatin folding in most tissue types and for most genomic loci, transcriptionally active or inactive. One exception is the Hox B locus which in its active form has a compaction on the order expected for an extended 30 nm chromatin fiber [49]. Both FISH [50,51] and DNA replication pulse-chase studies [52] have shown spatially distinct, localized chromatin domains, or “globules”, 100’s to 1000’s of kb in size, as predicted by 3C methodologies. At the EM level, compacted domains and fibers with diameters well above 30 nm have been observed, with locally decondensed segments showing suggestions of loose coiling of ~30 nm fibers [53]. Live cell microscopy shows similar compacted, fiber-like conformation for lac operator tagged engineered regions comprised of BAC trangenes with high transcriptional activity [54**], while in vivo immuno-gold labeling which avoids detergent extraction and prolonged staining procedures also reveals large-scale chromatin fibers for gene amplified chromosome regions [55].
While it is tempting to model the internal folding of these large-scale chromatin domains as compaction of 30 nm fibers, their structure remains an open question. The close-packing of chromatin and the inability to visualize linker DNA paths, makes extraction of fiber trajectories or distinguishing a short 30 nm fiber segment from two interdigitated 10 nm fibers or a single, supercoiled 10 nm fiber loop difficult.
Just as 3C interactions between sites several to tens of kb apart are difficult to reconcile with canonical 30 nm chromatin fiber folding, 3C interactions between loci at variable distances tens to several hundred kb apart are difficult to reconcile with hierarchical models of chromatin folding. For example, it is difficult to imagine interactions between loci separated by several hundred kb within extended large-scale chromatin fibers with a 1-3 Mbp / micron compaction level [56*]. Yet FISH analysis of several Mbp genomic regions in fact have suggested spatial segregation of transcriptionally active versus inactive regions within these genomic regions [51], and, on a larger scale, segregation of unique from repetitive sequences in the inactive X chromosome [57], suggesting that sequence distances along the DNA are not packed linearly along the interphase chromosome. Indeed, pairing of lac operator repeats over distances of several hundred kb in BAC transgene arrays suggests complex topological looping of DNA can coexist within linear large-scale chromatin fibers [58*].
Conclusions
The last several years has seen an important evolution of how we conceptualize higher order chromatin folding. A family of different “30 nm” fibers has emerged together with an increasing appreciation for higher-level compaction of chromatin above the 30 nm fiber and an increasing willingness to consider alternative models (summarized in Fig. 2) for this compaction. 3C-based methodologies may present outstanding opportunities for genomic analysis of higher levels of chromatin folding through molecular rather than microscopy approaches. However, major uncertainty still exists regarding experimental assumptions at the heart of deriving structural models from these 3C data sets. Moving forward, an important step would be to directly test some of these assumptions through a coordinated approach combining microscopy with 3C-derived and other genomic methodologies.
Figure 2. Models for interphase chromosome large-scale chromatin folding.
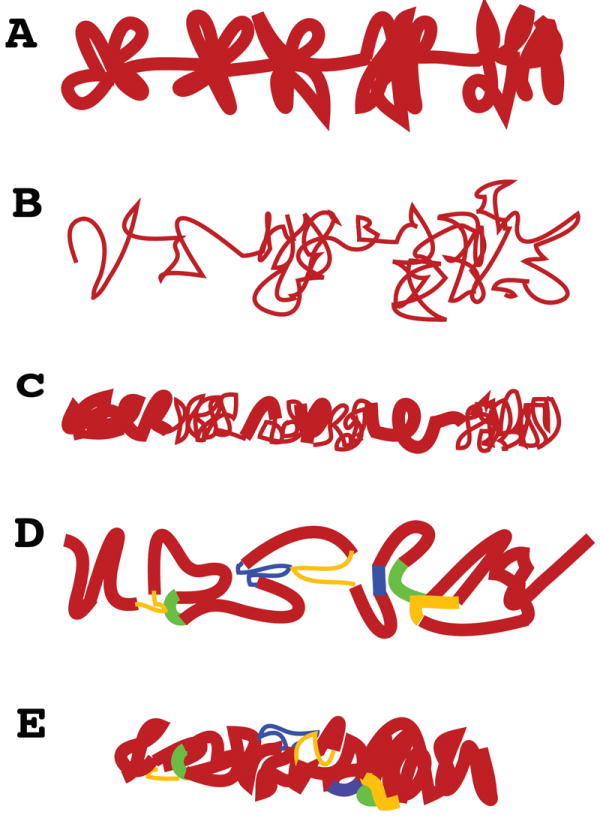
(A) Conventional model showing 30 nm higher order fibers forming clusters of loops. In older models these loop interactions are driven by poorly defined nuclear matrix interactions. This model is derived to a significant extent from extrapolation of radial loop models for mitotic chromosomes rather than direct visualization of interphase chromosomes. (B) Based on cryo-EM and ESI contrast for conventional EM, new models have suggested in vivo interphase chromosome structure consists nearly entirely of 10 nm fibers, locally dispersed or concentrated in compact local domains. (C) Chromonema fiber model, derived from a combination of light and electron microscopy, proposes existence of large-scale chromatin fibers on the order of 100 nm diameter. Irregular folding of 10 and/or 30 nm chromatin fibers underlies these large-scale fibers. Tight packing within these large-scale chromatin fibers has made it difficult to determine the substructure of these fibers and the relative ratio of 10 or 30 nm chromatin fibers. Discrete large-scale fiber segments are connected by less tightly coiled 10 and 30 nm fibers. (D) Chromatin hub model suggested by 3C experiments, in which looping interactions are formed through close interactions between various regulatory DNA elements. These loops are usually illustrated as composed of 30 nm fibers but could involve any combination of 10 and 30 nm fibers. (E) Hybrid chromonema / chromatin hub model in which complex topological looping interactions exist within large-scale chromatin fibers.
Acknowledgments
This work was supported by National Institutes of Health grant GM58460 to A.S.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Emanuel M, Radja NH, Henriksson A, Schiessel H. The physics behind the larger scale organization of DNA in eukaryotes. Physical biology. 2009;6:025008. doi: 10.1088/1478-3975/6/2/025008. [DOI] [PubMed] [Google Scholar]
- 2*.Grigoryev SA, Arya G, Correll S, Woodcock CL, Schlick T. Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13317–13322. doi: 10.1073/pnas.0903280106. Combined experimental and theoretical approaches support the idea of features corresponding to both solenoid and zig-zag models for 30 nm higher order chromatin structure co-existing within single fibers as studied with reconstituted oligonucleosome arrays. A novel formaldehyde, electron microscopy based assay is used to examine preferential internucleosome interactions within these fibers which differ depending on the model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Routh A, Sandin S, Rhodes D. Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:8872–8877. doi: 10.1073/pnas.0802336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 5.Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ. Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science. 2004;306:1571–1573. doi: 10.1126/science.1103124. [DOI] [PubMed] [Google Scholar]
- 6.Robinson PJ, Fairall L, Huynh VA, Rhodes D. EM measurements define the dimensions of the “30-nm” chromatin fiber: evidence for a compact, interdigitated structure. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6506–6511. doi: 10.1073/pnas.0601212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson PJ, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. Journal of molecular biology. 2008;381:816–825. doi: 10.1016/j.jmb.2008.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 9**.Scheffer MP, Eltsov M, Frangakis AS. Evidence for short-range helical order in the 30-nm chromatin fibers of erythrocyte nuclei. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16992–16997. doi: 10.1073/pnas.1108268108. Cryoelectron tomography on vitreous ice physical sections through isolated erythrocyte nuclei is used to examine 30 nm fiber ultrastructure. A left-handed, two start helical arrangement of nucleosomes is observed consistent with a cross-linker model with ~6.5 nuclesomes per 11 nm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodcock CL, Grigoryev SA, Horowitz RA, Whitaker N. A chromatin folding model that incorporates linker variability generates fibers resembling the native structures. Proc Natl Acad Sci U S A. 1993;90:9021–9025. doi: 10.1073/pnas.90.19.9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11*.Perisic O, Collepardo-Guevara R, Schlick T. Modeling studies of chromatin fiber structure as a function of DNA linker length. Journal of molecular biology. 2010;403:777–802. doi: 10.1016/j.jmb.2010.07.057. Molecular mesoscale modeling predicts different preferrred folding motifs for the 30 nm chromatin fiber as a function of nucleosome repeat length. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Boroudjerdi H, Naji A, Netz RR. Salt-modulated structure of polyelectrolyte-macroion complex fibers. The European physical journal E, Soft matter. 2011;34:72. doi: 10.1140/epje/i2011-11072-1. Using a minimalist model of a long, semi-flexible polyelectrolyte chain complexed with spherical macroions of the opposite charge, energy minimization is used to predict structures. Interestingly, for spheres and chains approximating the size and characteristics of DNA and histone octomers, fibers of diameters ~30 nm with zig-zag and solenoid folding motifs emerge as a function of salt concentrations, with a zig-zag pattern and 5-6 macroions per 11 nm predicted at physiological salt concentrations. [DOI] [PubMed] [Google Scholar]
- 13.Belmont AS, Braunfeld MB, Sedat JW, Agard DA. Large-scale chromatin structural domains within mitotic and interphase chromosomes in vivo and in vitro. Chromosoma. 1989;98:129–143. doi: 10.1007/BF00291049. [DOI] [PubMed] [Google Scholar]
- 14.Schwarz PM, Felthauser A, Fletcher TM, Hansen JC. Reversible oligonucleosome self-association: dependence on divalent cations and core histone tail domains. Biochemistry. 1996;35:4009–4015. doi: 10.1021/bi9525684. [DOI] [PubMed] [Google Scholar]
- 15.Lu X, Klonoski JM, Resch MG, Hansen JC. In vitro chromatin self-association and its relevance to genome architecture. Biochem Cell Biol. 2006;84:411–417. doi: 10.1139/o06-068. [DOI] [PubMed] [Google Scholar]
- 16*.McBryant SJ, Klonoski J, Sorensen TC, Norskog SS, Williams S, Resch MG, Toombs JA, 3rd, Hobdey SE, Hansen JC. Determinants of histone H4 N-terminal domain function during nucleosomal array oligomerization: roles of amino acid sequence, domain length, and charge density. The Journal of biological chemistry. 2009;284:16716–16722. doi: 10.1074/jbc.M109.011288. Tail swapping experiments indicate that the effect of the histone H4 N-terminal tail in mediating oligomerization of nucleosome arrays with increasing divalent cation concentration is largely through charge density rather than sequence specific effects. However, this predominant effect of the H4 N-terminal tail in mediating nucleosome array oligomerization is dependent on its association with the H4 histone fold as opposed to the histone fold of other core histones. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 18.Zhou J, Fan JY, Rangasamy D, Tremethick DJ. The nucleosome surface regulates chromatin compaction and couples it with transcriptional repression. Nature structural & molecular biology. 2007;14:1070–1076. doi: 10.1038/nsmb1323. [DOI] [PubMed] [Google Scholar]
- 19*.Watanabe S, Resch M, Lilyestrom W, Clark N, Hansen JC, Peterson C, Luger K. Structural characterization of H3K56Q nucleosomes and nucleosomal arrays. Biochimica et biophysica acta. 2010;1799:480–486. doi: 10.1016/j.bbagrm.2010.01.009. The H3K56 acetylation of histone H3 is associated with nucleosome assembly during DNA replication and repair. Analysis of chromatin assembled with H3 where K56 has been replaced with either glutamine to mimic constitutive acetylation or glutamate to simulate charge reversal suggests that the effect of H3K56 acetylation is mediated not on nucleosome structure or compaction of 30 nm chromatin fibers but rather through an increased tendency of chromatin with nucleosome free gaps to oligomerize through inter-fiber interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20**.Szerlong HJ, Prenni JE, Nyborg JK, Hansen JC. Activator-dependent p300 acetylation of chromatin in vitro: enhancement of transcription by disruption of repressive nucleosome-nucleosome interactions. The Journal of biological chemistry. 2010;285:31954–31964. doi: 10.1074/jbc.M110.148718. Activator dependent acetylation of chromatin mediated by p300 on in vitro reconstituted nucleosome arrays containing the HTLV-1 promoter leads to disruption of both array oligomerization through inter-fiber interactions as well as higher order chromatin compaction of the array itself and is correlated with increased transcription. These results suggest that localized histone acetylation near the promoter can lead to disruption of higher order chromatin folding over at least several kb and may have longer-range effects as well through disruption of inter-fiber interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21*.Maeshima K, Hihara S, Eltsov M. Chromatin structure: does the 30-nm fibre exist in vivo? Current opinion in cell biology. 2010;22:291–297. doi: 10.1016/j.ceb.2010.03.001. Review summarizing results from cyro-EM suggesting a “polymer melt” model in which interdigition of nuclesomes from adjacent chromatin fibers cause loss of 30 nm chromatin fiber organization in areas of high nucleosome density typical of in vivo conditions. 30 nm higher order folding is proposed as a possible transition state between highly decondensed chromatin associated for instance with transcriptional activation and the aggregated chromatin domains typical of many interphase and mitotic chromosomes, with these aggregated chromatin domains representing a special chromatin compaction state quite distinct functionally from 30 nm chromatin fibers. [DOI] [PubMed] [Google Scholar]
- 22*.Fussner E, Ching RW, Bazett-Jones DP. Living without 30nm chromatin fibers. Trends in biochemical sciences. 2011;36:1–6. doi: 10.1016/j.tibs.2010.09.002. Second review summarizing arguments against the common assumption that 30 nm chromatin fibers represent the prevalent structural motif within interphase and mitotic chromosomes. [DOI] [PubMed] [Google Scholar]
- 23.Rattner JB, Hamkalo BA. Higher order structure in metaphase chromosomes. II. The relationship between the 250 A fiber, superbeads and beads-on-a-string. Chromosoma. 1978;69:373–379. doi: 10.1007/BF00332140. [DOI] [PubMed] [Google Scholar]
- 24.Marsden MPF, Laemmli UK. Metaphase chromosome structure: evidence for a radial loop model. Cell. 1979;17:849–858. doi: 10.1016/0092-8674(79)90325-8. [DOI] [PubMed] [Google Scholar]
- 25.Woodcock CL. Chromatin fibers observed in situ in frozen hydrated sections. Native fiber diameter is not correlated with nucleosome repeat length. J Cell Biol. 1994;125:11–19. doi: 10.1083/jcb.125.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersson K, Mahr R, Bjorkroth B, Daneholt B. Rapid reformation of the thick chromosome fiber upon completion of RNA synthesis at the Balbiani ring genes in Chironomus tentans. Chromosoma. 1982;87:33–48. doi: 10.1007/BF00333508. [DOI] [PubMed] [Google Scholar]
- 27.Eltsov M, Maclellan KM, Maeshima K, Frangakis AS, Dubochet J. Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc Natl Acad Sci U S A. 2008;105:19732–19737. doi: 10.1073/pnas.0810057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Ahmed K, Dehghani H, Rugg-Gunn P, Fussner E, Rossant J, Bazett-Jones DP. Global chromatin architecture reflects pluripotency and lineage commitment in the early mouse embryo. PloS one. 2010;5:e10531. doi: 10.1371/journal.pone.0010531. Pronounced changes in global chromatin compaction and distribution relative to the nuclear periphery are seen during early embryonic stages, with pluripotent cell stages associated with a more highly dispersed chromatin compaction. Deletion of Oct4 and loss of pluripotency is associated with increased global chromatin compaction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Fussner E, Djuric U, Strauss M, Hotta A, Perez-Iratxeta C, Lanner F, Dilworth FJ, Ellis J, Bazett-Jones DP. Constitutive heterochromatin reorganization during somatic cell reprogramming. The EMBO journal. 2011;30:1778–1789. doi: 10.1038/emboj.2011.96. More dispersed chromatin is observed for H3K9me3 heterochromatin in mouse cell chromocenters after reprogramming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strukov YG, Wang Y, Belmont AS. Engineered chromosome regions with altered sequence composition demonstrate hierarchical large-scale folding within metaphase chromosomes. J Cell Biol. 2003;162:23–35. doi: 10.1083/jcb.200303098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31**.Matsuda A, Shao L, Boulanger J, Kervrann C, Carlton PM, Kner P, Agard D, Sedat JW. Condensed mitotic chromosome structure at nanometer resolution using PALM and EGFP- histones. PloS one. 2010;5:e12768. doi: 10.1371/journal.pone.0012768. This paper largely focuses on development of a PALM method for GFP and further improvements in PALM through the use of noise-reduction methods. Performing PALM on Drosophila embryonic mitotic chromosomes which contain the GFP labeled histone variant H2AvD revealed labeled filamentous chromatin blocks with ~70 nm diameter. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32**.Poirier MG, Oh E, Tims HS, Widom J. Dynamics and function of compact nucleosome arrays. Nature structural & molecular biology. 2009;16:938–944. doi: 10.1038/nsmb.1650. Using a FRET assay, dynamics of a trinucleosome array are measured over timescales of microseconds to seconds. In 1 mM Mg2+ arrays show fluctuations from the compacted state on a time scale of ~105 per second, while stopped-flow indicate magnesium induced partial compaction occurs faster than the 1 millisecond measurement resolution with full compaction occurring over ~2 seconds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rego A, Sinclair PB, Tao W, Kireev I, Belmont AS. The facultative heterochromatin of the inactive X chromosome has a distinctive condensed ultrastructure. J Cell Sci. 2008;121:1119–1127. doi: 10.1242/jcs.026104. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y, Sharov AA, McDole K, Cheng M, Hao H, Fan CM, Gaiano N, Ko MS, Zheng Y. Mouse B-Type Lamins Are Required for Proper Organogenesis But Not by Embryonic Stem Cells. Science. 2011 doi: 10.1126/science.1211222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konig P, Braunfeld MB, Sedat JW, Agard DA. The three-dimensional structure of in vitro reconstituted Xenopus laevis chromosomes by EM tomography. Chromosoma. 2007;116:349–372. doi: 10.1007/s00412-007-0101-0. [DOI] [PubMed] [Google Scholar]
- 36.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and inter-chromosomal interactions. Nat Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- 38.Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39*.Fraser J, Rousseau M, Shenker S, Ferraiuolo MA, Hayashizaki Y, Blanchette M, Dostie J. Chromatin conformation signatures of cellular differentiation. Genome Biol. 2009;10:R37. doi: 10.1186/gb-2009-10-4-r37. 5C analysis reveals generalized increased compaction of the HoxA locus and the formation of specific long-range contacts between neighboring repressed genes accompanying cell differentiation and gene silencing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40**.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. A genomic approach to 3C, Hi-C, is described which exploits high throughput DNA sequencing. Spatial proximity maps of the human genome at ~1 Mbp resolution were produced showing spatial segregation of open versus closed chromatin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet. 2010;42:53–61. doi: 10.1038/ng.496. Combination of 4C with chromatin immunoprecipitation and enrichment of ligated sequences using a biotin labeled PCR primer, termed e4C, was used to describe long-range cis and trans interactions of sequences with globin and other genes in erythroid cells. Coassociation of active genes with transcription factories, including specialized transcription factories enriched in the Klf4 transcription factor, was detected by both e4C and RNA FISH methods, with Klf4 regulated genes preferentially localizing with these Klf4 enriched specialized transcription factories. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. The development of ChIA-PET, using chromatin immunoprecipitation followed by ligation of DNA ends and high throughput paired end sequencing, is described and applied to study long-range DNA interactions involving the estrogen receptor. Most interactions are on the same chromosome and within 100 kb (85%) with a significant fraction (13%) ranging from 100-1000 kb with many sets of mutual interacting loci distributed over a similar 100-1000 kb range. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43**.Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet. 2011;43:630–638. doi: 10.1038/ng.857. ChIA-PET is applied to analyze chromosomal looping interactions mediated by CTCF. Five distinct classifications of chromatin domains were defined through these interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44*.Stadhouders R, Thongjuea S, Andrieu-Soler C, Palstra RJ, Bryne JC, van den Heuvel A, Stevens M, de Boer E, Kockx C, van der Sloot A, et al. Dynamic long-range chromatin interactions control Myb proto-oncogene transcription during erythroid development. The EMBO journal. 2011 doi: 10.1038/emboj.2011.450. 3C-Seq, which uses an inverse PCR method and high throughput sequencing to probe 3C interactions with a specific target sequence, was applied to probe long-distance interactions between enhancer sites in an intergenic region and the Myb gene. An active chromatin hub with long-distance contacts between enhancer and promoter sequences as well as the first intron containing a transcription elongation block site was proposed, with high concentrations of elongation factors at enhancer sites proposed as facilitating active transcription through long-distance interactions with the intron site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45*.Bau D, Sanyal A, Lajoie BR, Capriotti E, Byron M, Lawrence JB, Dekker J, Marti-Renom MA. The three-dimensional folding of the alpha-globin gene domain reveals formation of chromatin globules. Nat Struct Mol Biol. 2011;18:107–114. doi: 10.1038/nsmb.1936. 5C, a 3C method which uses a multiplex approach to interogate large numbers of long-distance interactions simultaneously over Mbp distances, is combined with distance constraint 3D modeling to derive an estimated structure for the human alpha-globin locus in expressing and nonexpressing cells. A globular or multi-globular conformation is predicted with central actively transcribed sequences and peripheral nontranscribed sequences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46*.Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, Noble WS. A three-dimensional model of the yeast genome. Nature. 2010;465:363–367. doi: 10.1038/nature08973. Another 3C interactin method combining a biotin pulldown to enrich for 3C ligated fragments and deep sequencing is used to produce a yeast genomic interactome map of cis and trans interactions. Many previously described features of yeast genome organization, including a Rabl conformation and clustering of tRNA genes, telomeres, and centromeres, are observed providing a validation of the method. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47*.Kan PY, Caterino TL, Hayes JJ. The H4 tail domain participates in intra- and internucleosome interactions with protein and DNA during folding and oligomerization of nucleosome arrays. Molecular and cellular biology. 2009;29:538–546. doi: 10.1128/MCB.01343-08. The histone H4 tail domain is dissected identifying regions involved in inter-fiber, intra-fiber interactions involved in higher order folding, and intranucleosome interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48**.Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome research. 2010;20:761–770. doi: 10.1101/gr.099655.109. The pattern of early versus late DNA replication domains is observed to be conserved between mouse and human cell lines of comparable differentiation state and to mirror the spatially segregated chromatin domains identified by Hi-C. Boundaries of early replicating domains are enriched in a number of histone marks associated with transcriptionally active chromatin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chambeyron S, Bickmore WA. Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 2004;18:1119–1130. doi: 10.1101/gad.292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muller WG, Rieder D, Kreth G, Cremer C, Trajanoski Z, McNally JG. Generic features of tertiary chromatin structure as detected in natural chromosomes. Mol Cell Biol. 2004;24:9359–9370. doi: 10.1128/MCB.24.21.9359-9370.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shopland LS, Lynch CR, Peterson KA, Thornton K, Kepper N, Hase J, Stein S, Vincent S, Molloy KR, Kreth G, et al. Folding and organization of a contiguous chromosome region according to the gene distribution pattern in primary genomic sequence. J Cell Biol. 2006;174:27–38. doi: 10.1083/jcb.200603083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol. 1992;116:1095–1110. doi: 10.1083/jcb.116.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Belmont AS, Bruce K. Visualization of G1 chromosomes: a folded, twisted, supercoiled chromonema model of interphase chromatid structure. J Cell Biol. 1994;127:287–302. doi: 10.1083/jcb.127.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54**.Hu Y, Kireev I, Plutz MJ, Ashourian N, Belmont AS. Large-scale chromatin structure of inducible genes- transcription on a linear template. J Cell Biol. 2009;185:87–100. doi: 10.1083/jcb.200809196. The large-scale chromatin structure of multi-copy BAC transgenes carrying 100-200 kb human or mouse genomic sequences is directly visualized through tagging the transgenes with lac operator repeats. Levels of transcription from these transgenes are within several fold of their endogenous counterparts but occur in the context of a condensed, linear template with a mean linear compaction ratio ~10-fold fold higher than a 30 nm fiber with no evidence of active regions looping out from these large-scale chromatin fibers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kireev I, Lakonishok M, Liu W, Joshi VN, Powell R, Belmont AS. In vivo immunogold labeling confirms large-scale chromatin folding motifs. Nat Methods. 2008;5:311–313. doi: 10.1038/nmeth.1196. [DOI] [PubMed] [Google Scholar]
- 56*.Belmont AS, Hu Y, Sinclair PB, Wu W, Bian Q, Kireev I. Insights into interphase large-scale chromatin structure from analysis of engineered chromosome regions. Cold Spring Harbor symposia on quantitative biology. 2010;75:453–460. doi: 10.1101/sqb.2010.75.050. Review of work with engineered chromosome regions leading to models for large-scale chromatin folding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clemson CM, Hall LL, Byron M, McNeil J, Lawrence JB. The X chromosome is organized into a gene-rich outer rim and an internal core containing silenced nongenic sequences. Proc Natl Acad Sci U S A. 2006;103:7688–7693. doi: 10.1073/pnas.0601069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58*.Sinclair P, Bian Q, Plutz M, Heard E, Belmont AS. Dynamic plasticity of large-scale chromatin structure revealed by self-assembly of engineered chromosome regions. J Cell Biol. 2010;190:761–776. doi: 10.1083/jcb.200912167. A surprising plasticity of large-scale chromatin folding is visualized with significant changes in folding patterns as a function of cell type and with complex topological looping interactions imposed by pairing of lac operator and prokarytoic vector sequences existing within the context of linear large-scale chromatin fibers. [DOI] [PMC free article] [PubMed] [Google Scholar]