

Abstract
BACKGROUND AND PURPOSE
Independent studies in experimental models of Trypanosoma cruzi appointed different roles for endothelin-1 (ET-1) and bradykinin (BK) in the immunopathogenesis of Chagas disease. Here, we addressed the hypothesis that pathogenic outcome is influenced by functional interplay between endothelin receptors (ETAR and ETBR) and bradykinin B2 receptors (B2R).
EXPERIMENTAL APPROACH
Intravital microscopy was used to determine whether ETR/B2R drives the accumulation of rhodamine-labelled leucocytes in the hamster cheek pouch (HCP). Inflammatory oedema was measured in the infected BALB/c paw of mice. Parasite invasion was assessed in CHO over-expressing ETRs, mouse cardiomyocytes, endothelium (human umbilical vein endothelial cells) or smooth muscle cells (HSMCs), in the presence/absence of antagonists of B2R (HOE-140), ETAR (BQ-123) and ETBR (BQ-788), specific IgG antibodies to each GPCRs; cholesterol or calcium-depleting drugs. RNA interference (ETAR or ETBR genes) in parasite infectivity was investigated in HSMCs.
KEY RESULTS
BQ-123, BQ-788 and HOE-140 reduced leucocyte accumulation in HCP topically exposed to trypomastigotes and blocked inflammatory oedema in infected mice. Acting synergistically, ETAR and ETBR antagonists reduced parasite invasion of HSMCs to the same extent as HOE-140. Exogenous ET-1 potentiated T. cruzi uptake by HSMCs via ETRs/B2R, whereas RNA interference of ETAR and ETBR genes conversely reduced parasite internalization. ETRs/B2R-driven infection in HSMCs was reduced in HSMC pretreated with methyl-β-cyclodextrin, a cholesterol-depleting drug, or in thapsigargin- or verapamil-treated target cells.
CONCLUSIONS AND IMPLICATIONS
Our findings suggest that plasma leakage, a neutrophil-driven inflammatory response evoked by trypomastigotes via the kinin/endothelin pathways, may offer a window of opportunity for enhanced parasite invasion of cardiovascular cells.
LINKED ARTICLE
This paper is commented on by D'Orléans-Juste et al., pp. 1330–1332 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2011.01636.x
Keywords: bradykinin, cardiomyopathy, Chagas disease, cruzipain, endothelin, oedema, GPCRs, invasion, kininogens, microcirculation, Trypanosoma cruzi
Introduction
Chagas disease, the chronic infection caused by the intracellular protozoan Trypanosoma cruzi, is still a major public health burden in Latin America (Coura and Dias, 2009; Tanowitz et al., 2009). Natural infection is initiated when a bloodsucking triatomine insect releases the infective metacyclic trypomastigotes onto vulnerable skin surfaces or mucous membranes. T. cruzi infection may also be transmitted by blood transfusion, by organ transplantation, from mother to fetus (congenital) and via the oral route (Coura, 2006). Trypomastigotes invade cells and are initially confined to a parasitophorous vacuole. After escaping to the host cell cytoplasm, the parasites transform into amastigotes, the replicating forms. After several cycles of binary division, the amastigotes transform into mammalian-stage trypomastigotes. Upon host cell death, the trypomastigotes invade adjacent uninfected cells or are carried by the blood and lymphatics to various organs. During the early stages of infection, innate immunity is triggered by microbial-derived ligands of toll-like receptors (TLRs) (Almeida and Gazzinelli, 2001; Medeiros et al., 2007). Inflammation is intensified by the vasoactive action of proteolytically derived endogenous peptides, such as bradykinin (BK) (Monteiro et al., 2006, 2007). With the onset of adaptive immunity, the acute manifestations (blood parasitaemia and systemic inflammation) gradually subside. Thereafter, the disease enters a clinically undetermined stage characterized by paucity of bloodstream parasites and positive serology. Lasting several years, chronic infection causes a progressive form of myocardiopathy in 15–30% of the patients. Characterized by fibrosis, myocytolysis and cardiac myocyte hypertrophy, the clinical manifestations vary but may include congestive heart failure, thromboembolic events and dysrhythmias (Rassi Jr et al., 2009). Although not excluding the participation of autoimmunity (Gutierrez et al., 2011), studies in humans and experimental animals suggest that infection-associated immunopathology and microvascular abnormalities are crucial factors in the pathogenesis of chronic heart disease (Higuchi et al., 1999; Tarleton, 2003; Tanowitz et al., 2005).
Studies on the mechanisms underlying infection-associated vasculopathy revealed that the vasoconstrictor endothelin-1 (ET-1) is strongly up-regulated in cardiovascular tissues of T. cruzi-infected mice (Tanowitz et al., 1999; Petkova et al., 2000). Preproendothelins are processed by furin-like proteases to form big endothelin intermediates that are further cleaved to three physiologically active endothelin peptides (ET-1, ET-2 and ET-3) by endothelin-converting enzymes. ETs (21 amino acids) are secreted by endothelial cells, cardiac myocytes and cardiac fibroblasts (Goto, 2001; Kedzierski and Yanagisawa, 2001) and act by signalling via two endothelin GPCR subtypes, ETAR and ETBR (for review, see Dhaun et al., 2007). Apart from its powerful vasoconstrictor effects, ET-1 induces plasma exudation (Filep et al., 1993; Sampaio et al., 2000), stimulates cytokine production (Speciale et al., 1998; Sampaio et al., 2000), regulates neutrophil adhesion and migration (Zouki et al., 1999; Sampaio et al., 2000), modulates the expression of leucocyte adhesion molecules on endothelial cells and on fibroblast-like synovial cells (Schwarting et al., 1996) and participates in zymosan-induced arthritis (Conte et al., 2008). Concerning Chagas disease, systematic studies have linked infection-associated vasculopathy to dysregulated activation of the endothelin pathway (Tanowitz et al., 2005). The finding that plasma levels of ET-1 are increased both in chagasic patients and mice (Petkova et al., 2000; Salomone et al., 2001) suggests that ET-1-driven vasospasm might cause myocardial ischaemia and myonecrosis (Tanowitz et al., 2005). Notably, cardiac remodelling was ameliorated in T. cruzi-infected mice in which the ET-1 gene was deleted only from cardiac myocytes (Tanowitz et al., 2005). These results have causally linked the endothelin pathway to chronic chagasic cardiomyopathy, raising the possibility that ETR targeting may serve as adjunctive therapy in Chagas disease (Mukherjee et al., 2004; Tanowitz et al., 2005).
While research on endothelins progressed, studies focusing on the kinin system (Scharfstein and Andrade, 2011) revealed that tissue culture trypomastigotes (TCTs) rely on the enzymatic versatility of the major cysteine protease (cruzipain) to generate peptide ligands (i.e. kinins) which enhance parasite infectivity through the signalling of G protein-coupled BK receptors (Scharfstein et al., 2000; Todorov et al., 2003). In follow-up studies, it was demonstrated that BK strongly stimulates cellular immunity (Monteiro et al., 2007), suggesting that activation of the kinin system translates into mutual benefits for the host–parasite relationship (Scharfstein and Andrade, 2011). Analysis of the dynamics of the inflammatory response evoked by TCTs revealed that the generation of vasoactive kinins in peripheral sites of infection is a downstream response conditioned by tissue culture-derived trypomastigotes (TLR2)/chemokine (C-X-C motif) receptor 2 (CXCR2)-dependent extravasation of plasma proteins (including kininogens) (Monteiro et al., 2006; Schmitz et al., 2009). Briefly, the inflammatory cascade is initiated when innate sentinel cells, such as macrophages, sense mucin-linked glycosylphosphatidylinositol (GPI)-anchors shed by trypomastigotes (Almeida and Gazzinelli, 2001; Campos et al., 2001) via TLR2. Translated as secretion of CXC chemokines, the TLR2-driven innate response activates endothelium/neutrophils via CXCR2 (Schmitz et al., 2009), thereby increasing vascular permeability. As a result of plasma extravasation, there is a rapid increase in the levels of blood-borne kininogens in peripheral sites of infection. Linking TLR2-dependent innate immunity to inflammation, cruzipain liberates BK/LBK from kininogens bound to the extracellular matrix (Del Nery et al., 1997; Lima et al., 2002; Monteiro et al., 2006). Further downstream, the vasoactive kinins further intensify interstitial oedema through the triggering of the endothelium bradykinin B2 receptor (B2R). As the inflammation escalates, the released kinins activate immature dendritic cells via B2R, converting these specialized antigen presenting cells into Th1 inducers (Aliberti et al., 2003; Monteiro et al., 2006, 2007). Based on these studies, angiotensin converting enzyme (ACE/kininase II), a kinin-degrading metallopeptidase, emerged as a modulator of the TLR2/CXCR2/B2R inflammatory pathway (Monteiro et al., 2006; Scharfstein et al., 2008).
In a striking example of biological versatility, here, we demonstrate that T. cruzi can further exploit the structural diversification of the GPCR family to infect mammalian cells via ETRs, sometimes involving the co-operation of B2R. Beyond their involvement in parasite infectivity, we present evidence that infection-associated alterations in the microcirculation depend on the functional interplay between endothelin and kinin pathways.
Methods
Parasites
T. cruzi Dm28c TCTs were harvested from the supernatants of infected rhesus monkey kidney cell line cultures maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 2% fetal calf serum (FCS) (Scharfstein et al., 2000). Suspensions of freshly released parasites were washed twice with excess HBSS before being tested in vitro or in vivo.
Cell cultures
Human primary umbilical vein endothelial cells (HUVECs) were obtained by treatment of umbilical veins with a 0.1% (wt/vol) collagenase IV solution. Primary HUVECs were seeded in 25 cm2 flasks coated with 2% porcine skin gelatin and grown in M199 medium supplemented with 2 mM glutamine, 2.5 µg·mL−1 amphotericin B, 100 µg·mL−1 penicillin, 100 µg·mL−1 gentamycin, 0.13% sodium bicarbonate and 20% FCS. Cells were maintained at 37°C in a humidified 5% CO2 atmosphere until confluence. After treatment with 0.02% trypsin/0.02% EDTA, the HUVECs were seeded into 24-well plates with gelatin-coated glass coverslips, and cultivated in M199-inactivated FCS (20%) at 37°C for 2 days before being washed and used (sub-confluent conditions) in invasion assays. Human smooth muscle cells (HSMCs) of gastric stomach origin [Cell Bank from Hospital Universitário Clementino Fraga Filho, Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro] were seeded on coverslips laid in 24-well plates and cultivated in DMEM-inactivated FCS (10%) at 37°C for 24 h before being washed and used in invasion assays. The phenotypic distinction between short-term HSMC cultures (<20 passages) and long-term cultures of myofibroblasts was carried out by labelling the monolayers with an anti-α-actin antibody (1:100; Sigma, St. Louis, MO, USA) that identifies muscle stress fibres (absent in myofibroblasts). CHO cells transfected with ETA receptor cDNA (CHO-ETAR) or ETB (CHO-ETBR) (from Euroscreen, Belgium, 2006) and mock cell (CHO-K1 control transfected with pcDNA3 plasmid) were cultivated in 5% CO2 in Ham's F12 medium (Sigma) containing 10% FCS at 37°C. Primary cultures of mice embryonic cardiomyocytes obtained from ventricular muscle as previously described (Meirelles et al., 1986). The purified heart cells were plated for 20 min in 24-well culture plates previously coated with 0.01% to allow the adhesion of fibroblasts. Next, the culture wells were seeded with the cardiomyocyte-enriched supernatant (5 × 105 cells mL−1). Cultures were maintained at 37°C in 5% CO2 atmosphere in DMEM supplemented with 10% FCS to get synchronized contractility.
Invasion assays with T. cruzi
Monolayers of CHO-ETRs, HUVECs or HSMCs were prepared on 13 mm round coverslips at a density of 5 × 104 cells per coverslip using the appropriated culture medium (see earlier discussion) supplemented with 10% inactivated FCS and further cultivated in 24-well plates for 24 or 48 h at 37°C in a 5% CO2 atmosphere. Before being incubated with parasites, the monolayers were washed with HBSS and kept in the appropriate serum-free culture medium containing 1 mg·mL−1 human serum albumin (HSA) (Baxter Pharmaceutical, Deerfield, IL, USA) and/or 25 µM of lisinopril (ACE inhibitor; Sigma) diluted in saline. Parasite–host cell interaction took place in a final volume of 300 µL per well. Before addition of TCTs (ratio of 10:1 parasite : host cell or as indicated), the culture medium was supplemented, or not, with the B2R antagonist HOE-140 (Sigma), the ETBR antagonist or the ETAR antagonist BQ-123 (Calbiochem, La Jolla, San Diego, CA, USA). Stock solutions of HOE-140 and BQ-123 were prepared in apyrogenic water, whereas BQ-788 was dissolved in dimethyl sulphoxide (DMSO). Dose–response curves were used to define the optimal working concentrations of HOE-140 (100 nM), BQ-788 (10 µM) and BQ-123 (1 µM) (Supporting Information Figure S1). As a control for BQ-788 vehicle, DMSO (0.6%) was added to the complete culture medium (DMEM-HSA or DMEM-FCS). In some invasion assays, the medium was supplemented with suboptimal concentrations of these drugs, tested alone or in combination. Host–parasite interactions proceeded for 3 h (except when otherwise informed in legends of figures) at 37°C in a humidified incubator, in a 5% CO2 atmosphere. In assays using neutralizing antibodies, the monolayers were pre-incubated with goat IgG antibodies against B2R, rabbit anti-ETAR (rabbit) or anti-ETBR (Santa Cruz Biotechnology, Santa Cruz, CA, USA), at a final concentration of 10 µg·mL−1, and washed twice with HBSS before incubation with parasites. Purified goat or rabbit control IgG was used as control. To study the role of [Ca2+]i -dependent pathways in parasite uptake, HSMC monolayers were either pre-incubated with 0.5 µM of thapsigargin (Sigma) for 30 min or with 100 µM of verapamil (Sigma) in DMEM-FCS 10%. The drugs were removed by extensive washing in HBSS, and the cells were incubated with DMEM-HSA. Before addition of TCTs (ratio of 10:1 parasite : host cell for 1 h), the culture medium was supplemented, or not, with BQ-788 (10 µM) or BQ-123 (1 µM). Lipid raft involvement was studied by pretreating HSMCs with 2 mM of methyl-β-cyclodextrin (MβCD; Sigma) for 30 min in DMEM-HSA or with MβCD + cholesterol (3.6 mM) complex (molar ratio 1:1.8; Tortelote et al., 2004), at 37°C before addition of TCTs. After 3 h of incubation, the infected monolayers were washed three times with HBSS, once with PBS, followed by fixation with Bouin and staining with Giemsa. The determination of infection index was obtained by counting the number of intracellular parasites in a total of 100 cells per coverslip.
Interference RNA of ETR
HSMCs (5 × 105 cells per sample) were electroporated with 3.5 pmol siRNA oligos to ETAR or to ETBR silencer, pre-designed (inventoried) siRNA or with scrambled silencer negative control siRNA (ID: AM4635) (Applied Biosystems, Foster City, CA, USA). ETAR (siRNA ID: s4467) sequence 5′-3′ sense: CGAUGUGAAUUACUUAGUUtt; antisense: AACUAAGUAAUUCACAUCGgt and ETBR (siRNA ID: s4469) sequence 5′-3′ sense: CUGUUGGUAUUGGACUAUAtt; antisense: UAUAGUCCAAUACCAACAGaa. HSMCs were washed, detached from culture flasks by incubation with trypsin and washed once in medium FCS before transfection. Cells were resuspended in 100 µL of electroporation solution (Nucleofactor kit V, Lonza, Basel, Switzerland), incubated with the corresponding oligos and electroporated in suspension using a Nucleofactor II device (Amaxa Nucleofector II – ADD-1001S) and programme T-28. Cells were immediately plated at 1.5 × 104 density and cultured overnight in DMEM-FCS before analyses. After electroporation, 500 µL of pre-warmed DMEM-10% FCS culture medium was added to the cuvette, and the cells were gently transferred into a 24-well plate (for invasion assays) or a 6-well plate (for Western blotting experiments). After 5 h, the non-adherent cells were removed by washing twice in serum-free medium, and the remaining cells were cultured at 37°C, in a humidified 5% CO2 atmosphere for 24–48 h, before the assays. Fluorescence microscopy of cells transfected with 2 µg of pmax-GFP control vector at 24 h revealed that transfection efficiency of HSMCs was ∼50% (90% cell viability). At the indicated times, the cultures were either lysed and subjected to Western blotting (as described later) or tested in conventional invasion assays (as described earlier). The densitometry of the reactive bands was measured using the Image J Software (http://imagej.nih.gov/ij/) (National Institutes of Health, Bethesda, MD, USA). Mock transfected cells were set as 100%.
Western blotting analysis
Cells (106) were washed twice with ice-cold PBS and then lysed in 100 µL of lysis buffer (50 mM Tris–HCl, pH 7.5; 5 mM EDTA; 10 mM EGTA; 50 mM NaF; 250 mM NaCl; 0.1% Triton X-100) with 1:1000 dilution protease inhibitor cocktail II (Calbiochem, Gibbstown, NJ, USA). Total proteins were determined with DC protein assay (Bio-Rad, Hercules, CA, USA) according to the manufacturer's protocol. Then, 40 µg of protein was diluted in SDS-PAGE sample buffer [8% SDS; 5% 2-β-mercaptoethanol; 20% glycerol; 0.05% bromophenol blue; 0.125 M Tris–HCL (pH 6.8)], boiled for 5 min and subjected to electrophoresis in 10% SDS-polyacrylamide gels and transferred to a nitrocellulose membrane (Bio-Rad). The membranes were blocked with 5% albumin in Tris-buffered saline containing 0.10% Tween (TBS-T) for 1 h at room temperature, followed by incubation overnight with sheep polyclonal anti-ETBR antibody (Abcam, Cambridge, UK, 1:3000). The membranes were washed three times with TBS-T for 10 min and incubated with horseradish peroxidase-labelled goat anti-sheep IgG (Abcam, 1:15 000). Visualization of the ETBR reactive band (50 kDa) was achieved by chemiluminescence (ECL kit; Amersham Bioscience, GE Healthcare, Buckinghamshire, UK). ETBR normalization was performed by stripping the membrane for 30 min at 50°C in stripping buffer (100 mM of 2-β-mercaptoethanol, 2% SDS, 62.5 mM Tris–HCl, pH 6.8). After stripping, the membrane was washed with TBS-T, blocked with 5% BSA TBS-T and incubated overnight with mouse monoclonal anti-β-actin (1:10 000 Sigma). Detection was performed as described earlier.
Immunofluorescence assays
HSMCs were plated on 13 mm round coverlips at a density of 3 × 104 cells per coverslip in the appropriate culture medium (see earlier discussion). After 24 h, we added (or not) TCTs to the monolayers for 1 h. The monolayers were washed twice in PBS and fixed in PBS containing 4% paraformaldehyde for 30 min at room temperature (RT) and then washed twice in PBS. After blocking non-specific binding with PBS-bovine serum albumin (3%) for 30 min, the HSMCs monolayers were incubated with primary antibodies rabbit anti-ETBR (1:100; Santa Cruz Biotechnology) or anti-ETAR (1:100) or normal rabbit IgG control, diluted in the blocking buffer for 1 h. After two washes in PBS, the coverslips were incubated with Alexa-Fluo488-conjugated anti-rabbit antibody (1:400; Invitrogen, Eugene, OR, USA) for 1 h at RT, washed in PBS and 4′-6-Diamidino-2-phenylindole (DAPI) (Sigma) stained for 5 min. The monolayers were laser dissected 13 times in 0.25 µm slices from bottom to top. The figures represent slice 5 (top) (Figure 6B and Supporting Information Figure S5). For the visualization of the cytoskeleton (Supporting Information Figure S4B), the fixed monolayers were permeabilized with Triton X-100 0.5% for 15 min at 4°C and then incubated with 546-Alexa-phalloidin from Molecular Probes (Invitrogen, Carlsbad, CA, USA) (1:50) for 90 min at RT. The results are representative of 30 fields of two independent experiments, performed in duplicate. The monolayers were DAPI stained and observed under a Zeiss LSM710 confocal laser scan microscope with an oil immersion 40× objective.
Endocytosis assays
HSMCs maintained in DMEM-10%FCS were infected (or not) with TCTs in the presence of 1 mg·mL−1 fluorescein isothiocyanate – dextran (FITC)-Dextran40 (Sigma) for 1 h at 37°C. In some assays, we added BQ-123 (1 µM) or BQ-788 (10 µM) to the culture medium. Monolayers kept at 4°C were used as controls, to assess the degree of non-specific surface binding of the FITC-Dextran to HSMCs. Following the incubation, the cellular uptake of the fluorescent probe was ceased by the addition of ice-cold PBS, and the monolayers were then washed and fixed with 1% paraformaldeyde for 30 min and then scraped out of the wells before being subjected to flow cytometry (FACScan) analysis (Supporting Information Figure S4A).
Oedema measurements
Experiments were conducted with normal BALB/C (males) (University of Campinas, Sao Paulo), 2–3 months old, each weighing 20–25 g, housed in micro-isolators (Alesco CO., São Paulo) for a week at 22 ± 2°C with a 12 h light–dark cycle. The animals under light ether anaesthesia were injected with 10 µL s.c. injection of Dm28c TCTs (106) in PBS, as previously described (Monteiro et al., 2006; Schmitz et al., 2009). ETR antagonists (see later discussion) were added to the parasite suspensions shortly before footpad injection. As a control, the contralateral paw received the same volume of PBS. Dose–response experiments in Swiss mice revealed that ETR antagonists (100 µg·kg−1) effectively reduced TCT-evoked oedema, this dose being similar to those used in other animal models (Piovezan et al., 2004; Fujita et al., 2008; Motta et al., 2009; Verri et al., 2009). The B2R antagonist HOE-140 (100 µg·kg−1) was injected (s.c.) 1 h before parasite injection in the animal's scruff, as previously described (Schmitz et al., 2009). Oedema volume differences (3 h p.i.) were measured with the aid of a plethysmometer; the data were expressed in microlitres (volume difference between the test and control paws). As control for BQ-788 vehicle, DMSO (0.6%) was added to the parasite suspension. Also, parasite motility (in complete culture medium) was not affected at the DMSO concentration indicated. All animal care and experimental procedures were in accordance with current guidelines for experiments in conscious animals and approved by the ethical committees of UFRJ (code number: IBCCF 101).
Intravital digital microscopy
Hamsters (male), 3 months old, each weighing 110–120 g, were maintained and anaesthetized according to regulations by the local ethical committee (IBCCF, license protocol number 014). Altogether, we used 27 hamsters (Anilab, São Paulo, Brazil) with a mean weight of 117 g ± 15. Hamsters were anaesthetized by i.p. injection of sodium pentobarbital supplemented with i.v. a-chloralose (2.5% W/V, solution in saline) through a femoral vein catheter as previously described (Monteiro et al., 2006.; Schmitz et al., 2009; Svensjöet al., 2009). Briefly, the microcirculation of the hamster cheek pouch (HCP) was analysed by epiluminescence using an Axioskop 40 microscope, objective 4× and oculars 10× (Carl Zeiss, Germany), equipped with appropriate filters (490/520 nm, FITC-Dextran; 540/580 nm, rhodamine). A digital camera, AxioCam HRC and a computer with the AxioVision 4.4 software program (Carl Zeiss) were used for image analysis of arteriolar diameter and total fluorescence in a representative rectangular area (5 mm2) of the prepared HCP, as recently described (Svensjöet al., 2009). The blood flow was normal and there was no evidence of leakage of FITC-Dextran before application of TCTs. Leucocytes were labelled in vivo by i.v. injection of rhodamine 100 µg·kg−1 body weight at 10 min prior to parasite application and, thereafter,10 µg·kg−1 body wt every 5 min for 45 min. Two images were recorded every 5 min interval during the entire experiment; one was used for arteriolar diameter and the second was used for measurement of the total amount of leucocytes in the circulation, rolling, adherent and migrated in the observed area (5 mm2). HCPs were topically exposed to 500 µL saline (controls) or to the same volume of a suspension of TCTs in PBS. The effect of ETR antagonists on parasite-induced leucocyte accumulation in the microvasculature of the HCP was tested by applying 500 µL of BQ-123 (10 µM), BQ-788 (10 µM) or vehicle controls, after interrupted superfusion or HOE-140 (0.5 µM final concentration, added to the continuous superfusion). Four minutes later, we added the TCTs to the HCP for another 5 min before resumption of superfusion. At 60 min after topical application of TCTs or saline, the HCPs were exposed to histamine (4 µM) or BK (0.25 µM) for 5 min as an internal control to confirm that the reactivity of the microvasculature was preserved. Hamsters with no response to histamine or BK were excluded. The experimental groups included n = 4–8 hamsters.
Statistical analyses
Statistically significant differences for all experimental data were determined by anova. When the mean values of the groups showed a significant difference, pair-wise comparison was performed by using the Bonferroni test. Statistical evaluation of hamster results was performed with anova followed by Student's t-test (between groups). A P-value of 0.05 or less was considered to indicate a statistically significant difference. Data are presented as means ± SD.
Results
TCTs evoke interstitial oedema via activation of ETRs and B2R
Unlike the situation prevailing in the acute infection, the release of parasites from infected heart cells is a sporadic event during the course of chronic chagasic infection because the parasitism in myocardial tissues is kept at very low levels by the selective pressure of anti-parasite effector T cells. Given the technical obstacles to investigate the dynamics of infection-associated inflammation in the cardiac microcirculation of T. cruzi-infected animals, in previous studies, we have used a combination of two models to study the pro-inflammatory role of the kinin pathway: measuring parasite-evoked plasma leakage in the microcirculation of the HCP and monitoring swelling responses (inflammatory oedema) in the footpad of infected B2R-deficient mice (Monteiro et al., 2006; Schmitz et al., 2009). Here, we determined whether the endothelin pathway participates in the B2R-driven inflammatory cascade evoked by Dm28c TCTs. First, we checked if ETR antagonists could attenuate the swelling responses, which TCT evoke in the footpad of BALB/c mice. Our results (Figure 1) showed a marked decrease in footpad swelling in mice injected with a TCT suspension supplemented with optimal concentrations of BQ-123 (highly selective ETAR antagonist) or BQ-788 (highly selective ETBR antagonist). Noteworthily, the anti-inflammatory activity of ETR antagonists was as effective as HOE-140 (B2R antagonist), used here as an internal control. We then examined if ETR and B2R antagonists when added to the HCP could reduce leucocyte accumulation in microvascular beds topically exposed to TCTs. Positive controls showed leucocyte accumulation 15 min after TCT application and this response reached plateau levels within 60 min (Figure 2). Pharmacological interventions revealed that the maximum values of leukocyte accumulation observed after TCT application (100%) were significantly reduced by BQ-788 (to 25%; P < 0.01) and HOE-140 (to 25%; P < 0.01), that is, 75% inhibition (Figure 2). BQ-123 reduced leucocyte accumulation to 66% of the maximal TCT-induced response (P < 0.05) (Figure 2). Baseline levels of leucocyte accumulation were not different in HCPs exposed to vehicle controls, that is, saline-DMSO (0.4%) or saline (data not shown). BQ-788 reduced leucocyte accumulation below (32%) saline control values, perhaps reflecting ETBR-dependent influence on endothelium interaction with leucocytes in the steady state. In the absence of ACE inhibitors, TCTs only elicit a modest (albeit significant) increase in plasma leakage (15%, P < 0.05) in HCPs sensitized with Dm28c TCTs (Monteiro et al., 2006; Schmitz et al., 2009), as compared with the saline control group. Initially, this discrete leakage response was insensitive to HOE-140. However, the inhibitory activity of the B2R antagonist became significant as the experiment progressed (up to 60 min; data not shown). Collectively, these studies suggest that ETRs contribute to the TLR2/CXCR2/B2R-dependent cascade type of inflammatory response elicited by kinin-releasing strains of T. cruzi (Monteiro et al., 2006; Schmitz et al., 2009).
Figure 1.
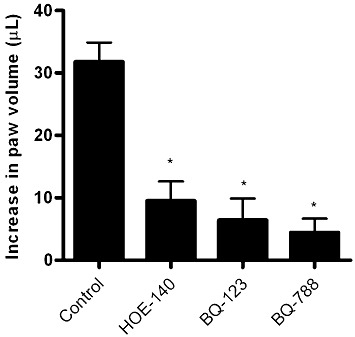
TCT evokes footpad swelling via activation of ETAR, ETBR and B2R. BALB/c mice (n = 5/group) were injected with 10 µL of Dm28c TCTs (106) in PBS. As a control, the contralateral paw received the same volume of PBS. The specificity of B2R responses was examined by injecting HOE-140 (100 µg·kg−1) s.c. 1 h before parasite injection. Involvement of ETR in the induction of footpad oedema was examined by injecting TCT suspensions supplemented with BQ-123 (100 µg·kg−1) and BQ-788 (100 µg·kg−1). Oedema was measured with the aid of a plethysmometer 3 h after the injections and was expressed in µL (volume difference between the test and control paws). The results (media ± SD) are representative of three independent experiments (n = 5 mice/group). *P < 0.01.
Figure 2.
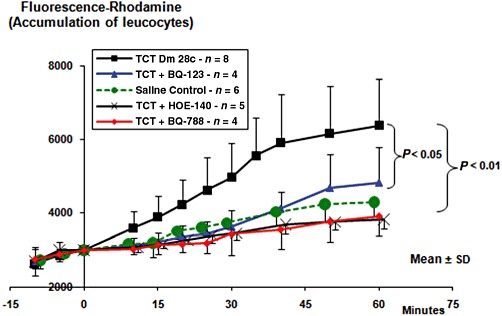
Intravital microscopy reveals the accumulation of rhodamine-labelled leucocytes in the hamster cheek pouch microcirculation. After labelling the leucocytes with i.v. injection of rhodamine, the superfusion of the cheek pouch was interrupted. After removing the superfusate, we applied 500 µL of BQ-123 (10 µM), or BQ-788 (10 µM) or the corresponding drug vehicle. In a separate group of hamsters, HOE-140 (0.5 µM) was added directly to the superfusion solution. Four minutes later, TCTs (3.107) or saline control were applied topically to the cheek pouch, and the incubation was prolonged up to 9 min total time. Controls (not shown) for drug vehicle were done by adding saline or saline-DMSO (0.4%) to the pouch for 4 min, prior to the application of TCTs. Results represent the mean ± SD of rhodamine fluorescence units in a 5 mm2 area of the cheek pouch. The results are representative of five series of experiments (n = 4–8 hamsters per group). P < 0.05; P < 0.01, as indicated).
Host cell invasion is influenced by T. cruzi interplay between ETAR, ETBR and B2R
Given the precedent that T. cruzi invasion is enhanced by activation of G protein-coupled BK receptors (Scharfstein et al., 2000; Todorov et al., 2003), and the evidence that ET-1 expression is up-regulated in parasite-infected cardiovascular cells (Tanowitz et al., 2005), we reasoned that TCTs may have developed competence to persistently infect heart cells through the signalling of ETRs. As a starting point, we checked the outcome of Dm28c TCT interaction (3 h) with CHO cells over-expressing individual ETRs (ratio of 10:1 parasite vs. CHO) in serum-free HAM's medium supplemented with HAS. Using a parasite : host cell ratio of 10:1, our data (Figure 3) show that the extent of TCT invasion was significantly increased in CHO cells expressing either ETAR or ETBR (60%; P < 0.05), as compared to CHO-K1 (controls). As shown in Figure 3, BQ-123 (1 µM) significantly reduced TCT invasion of CHO-ETAR (35%; P < 0.05), whereas BQ-788 (10 µM) did not interfere at all with the extent of invasion of CHO-ETAR (Figure 3). Conversely, BQ-788 (but not BQ-123) selectively reduced parasite uptake by ETBR-transfected CHO cells (Figures 3 and 35%; P < 0.05), whereas BQ-123 did not significantly alter parasite uptake by CHO-ETBR or CHO-K1 (Figure 3). Noteworthily, the inhibitory effects of these ETR antagonists were more pronounced when the invasion assays were performed at lower parasite : host cell ratio (5:1) (data not shown). Reminiscent of the phenotype of CHO-B2R and CHO-B1R (Scharfstein et al., 2000; Todorov et al., 2003), the current results indicated that TCTs (Dm28) can aptly invade mammalian cells over-expressing both subtypes of ETRs.
Figure 3.
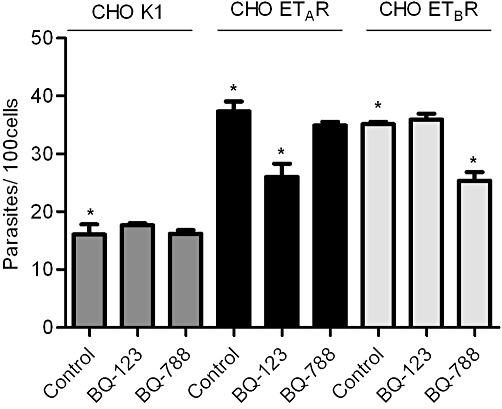
T. cruzi invades CHO cells over-expressing ETAR or ETBR in a subtype receptor-specific manner. Transfected CHO cells over-expressing endothelin receptors (i) CHO ETAR, (ii) CHO ETBR or (iii) CHO K1 (mock control) were incubated with TCTs at parasite : cell ratio of 10:1 for 3 h in HAM's F12 medium supplemented with HAS. Where indicated, the medium was supplemented with BQ-788 (10 µM) or BQ-123 (1 µM) immediately before addition of parasites. Controls for drug vehicle involved addition of equivalent volume of HBSS or HBSS-DMSO (0.6%) to HSMC cultures treated with TCTs (not shown). After 3 h of host–parasite interaction, the monolayers were washed in HBSS, fixed with Bouin for 24 h and stained with Giemsa. The infection index represents the number of intracellular parasites per 100 cells (in triplicate) (mean ± SD). The results are representative of two independent experiments. *P < 0.05.
Next, we examined if TCTs could invade human cells that naturally express ETRs. We chose to work with HSMCs as they express ETAR, ETBR (Kitsukawa et al., 1994) and B2R (Bachvarov et al., 1995; Knox et al., 2001). Titration assays (Supporting Information Figure S1) were performed to determine the optimal concentration of GPCR antagonists that protect HSMCs from parasite invasion. Based on these results, we used BQ-123 (1 µM), BQ-788 (10 µM) and HOE-140 (100 nM) in subsequent studies. Invasion assays run under these conditions showed that parasite uptake was markedly inhibited either by BQ-123 (65% of inhibition; P < 0.05), BQ-788 (60% of inhibition; P < 0.05) or HOE-140 (50%, P < 0.05) (Figure 4A). Extending this analysis to mouse cardiomyocytes, we observed that parasite invasion was partially, yet significantly inhibited by each of these drugs (Supporting Information Figure S2). As endothelial cells express ETBR but not ETAR (Kedzierski and Yanagisawa, 2001), we took advantage of this differential functional property to double-check the selectivity of our subtype-specific ETR antagonists in invasion assays with HUVECs. As predicted, parasite infectivity was partially inhibited by BQ-788 (Supporting Information Figure S3A; 53%, P < 0.05). In contrast to HSMCs or cardiomyocytes, HUVECs were not protected from infection by BQ-123 or HOE-140, thus adding credence to the specificity profile of these GPCR-antagonists. As predicted (Scharfstein et al., 2000), we restored the functional role of B2R in parasite infectivity by adding lisinopril (ACE/kininase II inhibitor) to the HUVEC culture medium (Supporting Information Figure S3B). Under these particular conditions, the half-life of kinin agonists (liberated by cruzipain) is strongly increased, allowing for increased engagement of the B2R/[Ca2+]i pathway of invasion in endothelial cells (Scharfstein et al., 2000). Strikingly, BQ-788 abolishes parasite uptake via the kinin/B2R pathway. These results imply that ACE dictates whether T. cruzi uptake by HUVECs is co-ordinated by ETBR (without B2R) or via ‘cross-talk’ between B2R/ETBR.
Figure 4.
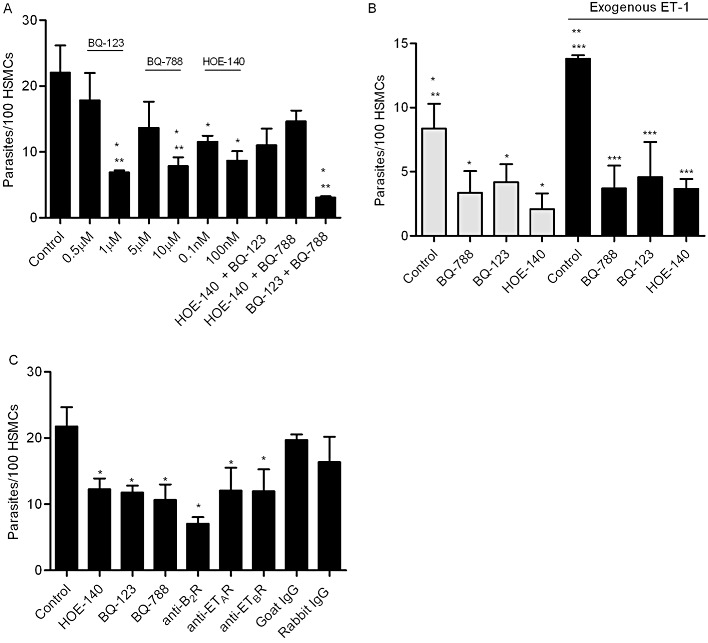
GPCR antagonists specifically reduce TCT uptake by mammalian cells that naturally express ETRs and B2R. (A) HSMCs were incubated with TCTs (parasite : cell ratio 10:1) for 3 h in DMEM-HAS. Where indicated, the cultures received either BQ-788 (5 µM or 10 µM), BQ-123 (0.5 µM or 1 µM) or HOE-140 (0.1 nM or 100 nM). At the right side of the panel, the host–parasite interaction occurred in the presence of the combination of two drugs, each one tested at suboptimal concentrations (i.e. 0.5 µM BQ-123, 5 µM BQ-788 and 0.1 nM HOE-140). (B) HSMCs were pre-incubated or not with 100 nM of ET-1 in DMEM-HAS for 20 min prior to the addition of TCTs (parasite : cell ratio 5:1 – suboptimal ratio). Where indicated, the ET-1 treated medium was supplemented with either BQ-788 (10 µM), BQ-123 (1 µM) or HOE-140 (100 nM). (C) HSMCs were pre-incubated with 10 µg·mL−1 of IgG anti-B2R, anti-ETAR, anti-ETBR or the respective rabbit or goat IgG controls. Thirty minutes later, the HSMCs were washed twice and incubated with TCTs (parasite : cell ratio 10:1) for 3h in DMEM-HAS. Positive controls for ETR and B2R-driven parasite uptake were performed in the presence of BQ-788 (10 µM) or BQ-123 (1 µM) or HOE-140 (100 nM). The cells were then fixed with Bouin and stained with Giemsa. The infection index represents the number of intracellular parasites per 100 cells (in triplicate) (mean ± SD). The results are representative of three independent experiments. *P < 0.05. **, *** < 0.05 indicate significant difference between controls marked with the same symbols.
We next determined whether ETR subtypes act co-operatively. This was checked using suboptimal concentrations of BQ-123 (0.5 µM) and BQ-788 (5 µM) in invasion assays performed with HSMCs (Figure 4A). These experiments showed that the combination of BQ-123 and BQ-788 effectively inhibited (82% of control levels; P < 0.01) parasite uptake by HSMCs. Based on these results, we concluded that ETAR- and ETBR-driven intracellular pathways may converge, optimizing the efficiency of the invasion process. We then extended this analysis to HOE-140 (0.1 nM). Our results showed that HSMCs exposed to suboptimal concentrations of [HOE-140 plus BQ-123] or [HOE-140 plus BQ-788] were not protected from infection (Figure 4A). Thus, these data suggest that Dm28c trypomastigotes drive HSMC invasion via mechanisms involving co-operation between ETAR and ETBR.
Next, we checked if exogenous ET-1 could potentiate parasite uptake by HSMCs. Indeed, using a lower ratio of parasite/HSMCs (5:1), we found that ET-1 significantly increased the efficiency of infection (Figure 4B). As predicted, these effects were reverted by BQ-123 or BQ-788 (baseline levels) (Figure 4B). Strikingly, the potentiating effects of exogenous ET-1 were also nullified by HOE-140, thus suggesting that B2R and ETRs induce trypomastigotes uptake via interdependent mechanisms. Noteworthily, the results of assays performed at optimal concentrations of these GPCR antagonists (data not shown) revealed that (i) HOE-140 alone protects HSMCs from parasite invasion to the same extent as each ETR antagonist; and (ii) the combination of HOE-140 to BQ-123 or to BQ-788 did not increase the protection over the levels achieved with the individual ETR blocker. Collectively, these results suggest that B2R and ETRs, acting interdependently, promote parasite uptake by HSMCs through the signalling of common intracellular pathways.
Seeking for independent criteria to validate these pharmacological data, we next verified whether neutralizing IgG antibodies (10 µg·mL−1) specifically recognizing individual GPCRs (B2R and ETAR or ETBR) could interfere with parasite invasion. Indeed, our results showed that the receptor-specific antibodies reduced cellular invasion to a similar extent as their respective antagonists (Figure 4C).
To further confirm the involvement of the ETR gene products in the invasion of HSMCs, we repeated these experiments using RNA interference methods. Results from Western blots performed 24 and 48 h after nucleofection revealed that ETBR silencer siRNA reduced ETBR expression over control gene expression (Figure 5A), but only 48 h after electroporation. This notwithstanding, at the early (24 h) time point, the percentage of infected HSMCs was significantly reduced as compared to cells electroporated with scrambled silencer control siRNA (Figure 5B and 57%, P < 0.05) or mock-electroporated HSMCs (24 h) (45%, P < 0.05). The phenotypic effects were only marginal at the 48 h time point, most likely reflecting preferential expansion of non-infected nucleofected HSMCs. The densitometry of the reactive bands was measured using the Image J Software. Mock transfected cells were set as 100% (Figure 5A, right panel).
Figure 5.
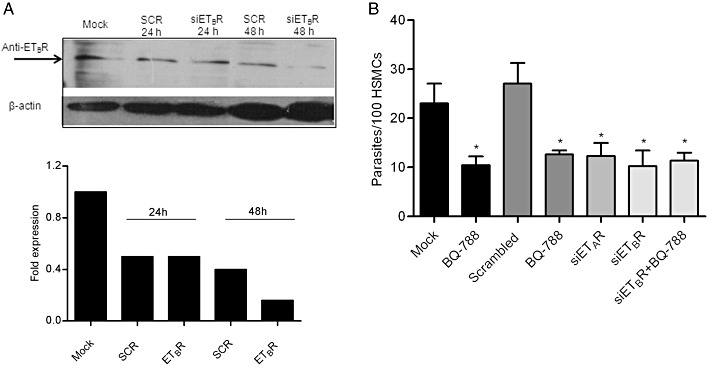
Interference RNA indicates that TCTs invade HSMCs in an ETR-dependent manner. HSMCs were either nucleofected with siRNA ETBR-silencer selected siRNA, ETAR-silencer selected siRNA, scrambled-silencer selected siRNA (SCR) or nucleofected without siRNA (mock treatment) as controls. After 24 h (A), the cell lysates derived from the earlier mentioned HSMCs cultures were boiled in complete sample buffer and then subjected to electrophoresis in 10% SDS-PAGE. After transferring the proteins to a nitrocellulose membrane, they were probed with sheep anti-ETBR antibody. The immunoblotted ETBR protein (50 kDa) was identified using horseradish peroxidase-labelled goat anti-sheep IgG followed by ECL. Lower panel: the optical density of mock transfected cells was set as 1. The graph represents the relative intensity in relation to mock cells (24 h). (B) HSMCs subjected to ETR silencing were incubated with TCTs (parasite : cell ratio 10:1) in DMEM-HSA. Where indicated, these cultures also received BQ-788 (10 µM) or BQ-123 (1 µM). The infection index represents the intracellular parasites per 100 cells (in triplicate) (mean ± SD). The results are representative of two independent experiments. *P < 0.05.
Since G protein-coupled B2R and ETR compartmentalize in lipid rafts/caveolae (De Weerd and Leeb-Lundberg, 1997; Bremnes et al., 2000; Okamoto et al., 2000; Harada et al., 2002; Ostrom, 2002), we then examined whether MβCD, a cholesterol-depleting drug, could impair the functional co-operation between B2R/ETR, rendering HSMCs refractory to in vitro infection. We found (Figure 6A) that parasite invasion was significantly reduced (85%, P < 0.01) by the MβCD treatment. Of note, the host protective effects imparted by MβCD was not an artefact resulting from TCT toxicity since we were able to fully restore parasite infectivity (and conversely, their sensitivity to ETR/B2R antagonists) by adding an exogenous source of cholesterol (86%, P < 0.01) to the MβCD-treated parasite/HMSCs cultures (Figure 6A). Collectively, these results suggest that ETBR/B2R co-operation may depend on their segregation in cholesterol-rich domains, such as rafts/caveolae. Immunofluorescence assays using anti-ETAR or anti-ETBR showed clusters of ETR at parasite–cell interaction site as compared to non-infected cells or IgG control (Figure 6B and Supporting Information Figure S5).
Figure 6.
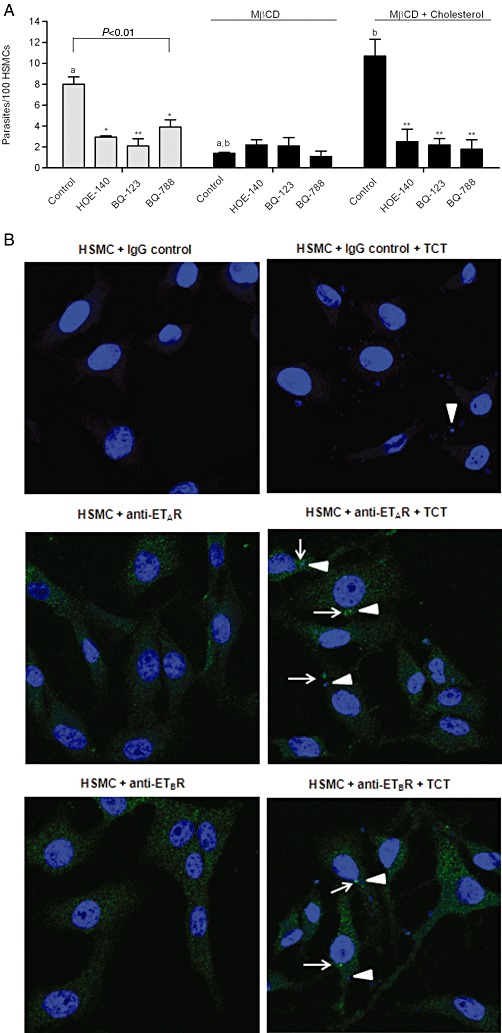
Integrity of lipid rafts/caveolae is crucial for B2R/ETR-driven uptake of T. cruzi. (A) HSMCs were incubated or not with 2 mM of methyl-β-cyclodextrin (MβCD) for 30 min in DMEM-HAS or with MβCD + cholesterol [3.6 mM] complex (molar ratio 1:1.8) incubated together the day before the onset of invasion assays. After 30 min, the monolayers were washed in HBSS and incubated with TCTs (ratio 10:1 target cells) for 1 h in DMEM-HSA. Where indicated, BQ-788, BQ-123 or HOE-140 were added the medium. After 1 h of host–parasite interaction, the infected monolayers were washed in HBSS, fixed with Bouin for 24 h and stained with Giemsa. The infection index represents the intracellular parasites per 100 cells (in triplicate) (mean ± SD). The results are representative of three independent experiments. *P < 0.05; **,a,bP < 0.01. (B) Surface immunolabelling of ETAR or ETBR in HSMCs incubated (or not) with TCTs (parasite : cell ratio 20:1) for 1 h. After fixing the monolayers to the glass coverslips with 4% paraformaldehyde for 30 min, they were incubated with primary IgG (rabbit) anti-ETBR (1:100) or anti-ETAR (1:100) or rabbit IgG isotype control (1:100) for 1 h, followed by incubation with Alexa 488-conjugated secondary anti-rabbit IgG antibody. After staining the monolayers with DAPI, the fluorescent-labelled preparations were analysed by Zeiss LSM710 confocal laser scan microscope with oil immersion 40× objective. White arrow heads indicate TCTs, and the white arrows indicate clusters of the respective receptors.
Calcium has differential effects on parasite uptake by HSMCs
Since trypomastigotes mobilize intracellular calcium stores during the cellular invasion process (Caler et al., 2000), we examined whether ETR-mediated parasite internalization depends on [Ca2+]i. To this end, HSMCs were pretreated, or not, for 30 min with 0.5 µM of thapsigargin (Thastrup et al., 1990). Our results (Figure 7A) show that depletion of [Ca2+]i stores by thapsigargin significantly reduced parasite uptake (50%; P < 0.05). Strikingly, parasite infectivity was further reduced when we added either BQ-123 or BQ-788 to thapsigargin-treated HSMCs (Figure 7A; black bars; P < 0.05). In parallel experiments performed with HUVECs, we found that the invasion of thapsigargin-treated endothelial cells was abolished (data not shown). Thus, our results suggest that ETR-dependent signalling pathways synergize with [Ca2+]i-inducing responses controlled by hitherto uncharacterized receptor(s).
Figure 7.
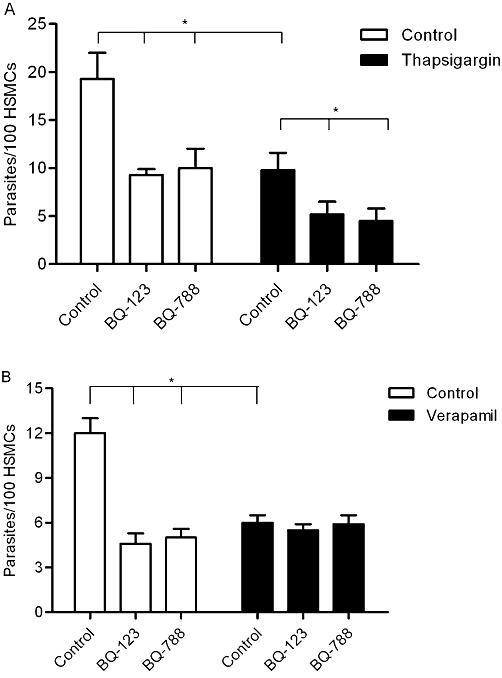
Influence of calcium on ETR-driven pathways of parasite uptake by HSMCs. (A) HSMCs were pre-incubated (or not) at 37°C with 0.5 µM of thapsigargin for 30 min or (B) with 100 µM of verapamil (or not) for 4 h in DMEM-FCS (10%). After removal of the drugs, the monolayers of HSMCs were washed and incubated with TCTs (ratio 1:10) for 1 h in DMEM-HSA. Where indicated, the medium contained BQ-788 or BQ-123. The infected monolayers were washed in HBSS, and then fixed and stained as previously described. The infection index represents the intracellular parasites per 100 cells (in triplicates) (mean ± SD). The results are representative of two independent experiments. *P < 0.05.
Since ETBR activates calcium channels in the plasma membrane (Kawanabe and Nauli, 2005), we examined the outcome of parasite interaction with HSMCs pretreated with the calcium channel blocker verapamil (100 µM in Ca+2-free medium for 4 h) or with the calcium chelating agent EGTA (5 mM in Ca+2-free medium for 20 min). The results with verapamil-treated HSMCs (Figure 7B) indicate that extracellular calcium is indeed required for parasite uptake (inhibition of 42%, P < 0.05). Likewise, EGTA-treated HSMCs were also protected from parasite invasion (data not shown). Of note, the addition of BQ-123 or BQ-788 to verapamil-treated HSMCs did not further increase host cell susceptibility to infection (Figure 7B). Similar results were observed when we studied the role of ETBR in the invasion of verapamil-treated HUVECs (data not shown). Collectively, the results obtained with verapamil suggest that ETR-mediated uptake of TCTs depends on calcium influx through voltage-operated calcium channel (Goto et al. 1989; Iwamuro et al. 1998).
Discussion and conclusions
In the first part of this study, we presented evidence that TCTs induce leucocyte accumulation in the HCP through activation pathways mediated by ETAR, ETBR and B2R. Extending these studies to a previously described subcutaneous model of T. cruzi infection, our data implicated the endothelin/kinin pathways in the mechanisms underlying oedematogenic inflammation. So far, it is unclear how the endothelin pathway integrates the TLR2/CXCR2/B2R-driven inflammatory cascade elicited by Dm28 trypomastigotes. Although not explored in the present work, it is conceivable that trypomastigotes released from ruptured pseudocysts are sensed by endothelin-positive tissue-resident mast cells (Ehrenreich et al., 1992). The possibility that these innate sentinel cells might recognize T. cruzi at early stages of infection is worth exploring in the light of precedent findings indicating that Mycobacterium tuberculosis activates mast cells via the TLR2 pathway (Carlos et al., 2009). Once released by mast cells, ET-1 acts as an autocrine mediator, inducing secretion of vasoactive mediators such as histamine, LTC4 (Yamamura et al., 1994) and TNF-α (Coulombe et al., 2002). The findings that T. cruzi-induced oedema is blunted in cromolyn-treated BALB/c mice (data not shown) are consistent with the notion that mast cell-derived endothelins may link TLR2-dependent innate responses to the cruzipain-driven proteolytic cascades that amplify inflammation at the expense of kinin system activation.
In spite of the wealth of information on the biochemical make-up of T. cruzi, it is still unclear how these pathogens persistently infect cardiovascular tissues of immunocompetent individuals. In the current study, we demonstrated that T. cruzi trypomastigotes (Dm28c strain) infect a broad range of non-phagocytic host cells (including endothelial cells and cardiomyocytes) through the co-operative signalling of ETRs and B2R. Albeit limited to the in vitro settings, the previous findings are of potential relevance to pathogenesis research for the following reasons: firstly, there is evidence that the endothelin pathway is up-regulated in parasite-infected heart tissues (Tanowitz et al., 2005). Thus, it is conceivable that this regulatory dysfunction may render cardiomyocytes more sensitive to ETR signalling relayed by extracellular trypomastigotes navigating through the heart parenchyma. Secondly, it has been shown that ET-1 levels are increased in the blood of Chagasic patients and T. cruzi-infected animals (Petkova et al., 2000; Salomone et al., 2001). This pathological response may be advantageous for T. cruzi because the trypomastigotes rely on the co-operative activities of pro-oedematogenic molecules, for example, glycosylphosphatidylinositol-anchored mucin-like glycoproteins from T. cruzi trypomastigotes (tGPIm) and cruzipain, to promote the rapid diffusion of blood-borne ET-1 (along with kininogens) through post-capillary venules (see scheme; Figure 8). In this hypothetical scenario, we predict that ET-1 levels (along with those of vasoactive kinins) might gradually rise in the trypomastigotes-laden interstitial spaces. Further downstream, T. cruzi may use this window of opportunity to infect cardiomyocytes via the ETR/B2R ‘cross-talk’ in ways that recapitulate the in vitro findings described in this paper.
Figure 8.
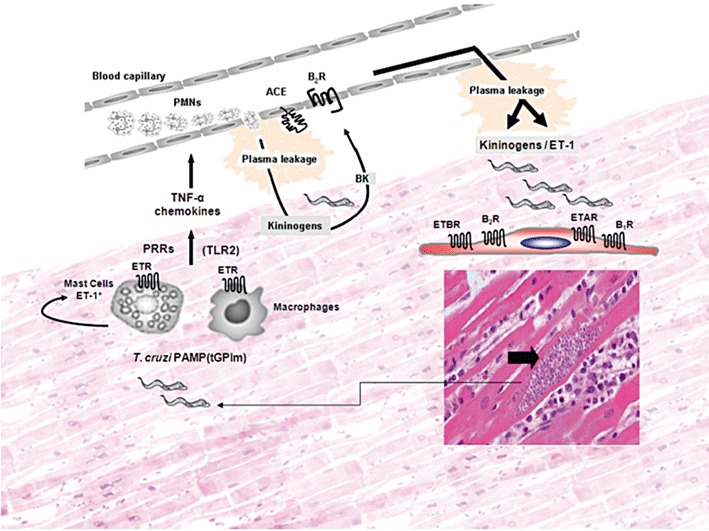
Interstitial oedema elicited by extracellular trypomastigotes might offer a window of opportunity for infection of heart cells via the endothelin/kinin pathways. Despite the scanty presence of parasitized heart cells in the myocardium of chronically infected Chagasic patients, we may predict that these pseudocysts occasionally burst (right/bottom side of panel), releasing high numbers of pro-inflammatory trypomastigotes into the surrounding interstitial spaces. On the left side of the panel, innate sentinel cells, such as macrophages and endothelin-positive mast cells, initiate inflammation through the sensing of microbial TLR2 ligands shed by extracellular TCTs. Following TLR2 signalling, activated mast cells may release ET-1, which then amplifies inflammation in an autocrine manner through ETR-driven secretion of histamine and TNF-α. Acting synergistically with CXC chemokines, these vasoactive mediators activate CXCR2 expressed by neutrophils/endothelium (Schmitz et al., 2009). We hypothesize that the ETR/B2R-driven leakage of plasma increases the concentrations of blood-borne kininogens and ET-1 (present at high levels in the serum of Chagasic patients) in the trypomastigote-laden infection sites (upper side of panel). T. cruzi trypomastigotes may then exploit this window of opportunity to invade cardiovascular cells via interdependent activation of ETRs and B2R and/or engaging the inducible B1R pathway (Todorov et al., 2003). The extracellular parasites may also take advantage of the up-regulated activation of ET-1/ETR pathway in parasite-infected cardiomyocytes (Petkova et al., 2000) to persistently infect cardiomyocytes in chronic Chagasic patients.
Beyond the potential significance to pathogenesis research, our studies provide a convenient model to investigate the molecular basis of host cell susceptibility to T. cruzi (Scharfstein and Andrade, 2011). Albeit involving different host cell/T. cruzi strain combinations, it was previously reported that cholesterol-depleting drugs disrupt the microdomain-associated signalling complexes that steer parasite penetration in non-phagocytic cells (Fernandes et al., 2007). In the current study, we demonstrated that (i) MβCD drastically reduced parasite entry in HSMCs; and (ii) addition of exogenous cholesterol to MβCD-HSMCs restored ETAR/ETBR/B2R-dependent pathways of T. cruzi invasion (Figure 6A). Collectively, these results suggest that co-operative interaction between these subtype-specific GPCRs may critically depend on the integrity of lipid rafts/caveolae. These results were not unexpected because ETR subtypes are co-expressed in a variety of natural cell types, and physiological responses depend on intracellular cross-talk of ETRs (Ozaki et al., 1997). According to some groups, the ET-1 ligand is a bridge that forges ETR heterodimerization through the binding to the N-terminal domain of ETAR and to the C-terminal portion of ETBR (Sakamoto et al., 1993; Harada et al., 2002). More recently, Evans and Walker (2008) used more refined FRET techniques to analyse ETR function in HEK293-transfected cells. These authors reported that the combination of ETAR and ETBR subtype-selective antagonists (but not the individual antagonists) inhibited ET-induced conformational changes on ETR heterodimers as well as the sustained intracellular Ca2+ signalling responses transduced by these heterodimers. In contrast, the transient intracellular Ca2+ increase, which ET-1 induces via triggering of ETR homodimers was selectively inhibited by the appropriate receptor subtype-selective antagonist, that is, BQ-123 (for ETAR homodimers) or BQ-788 (for ETBR homodimers). Based on these observations, it was suggested that blockade of physiopathological responses controlled by ETR heterodimers present in native tissues might be achieved through the combined treatment with both subtype-receptor antagonists or through the use of antagonists with mixed ETAR/ETBR specificities (Evans and Walker, 2008). Admittedly, the pharmacological profile of our GPCR antagonists do not meet the requirements of the heterodimer paradigm because (i) parasite uptake by HSMCs and cardiomyocytes was significantly inhibited by either BQ-123, BQ-788 or HOE-140; (ii) the combined treatment of the cultures with ETAR and ETBR subtype-selective antagonists (and for that matter, HOE-140 plus BQ-788 or HOE-140 plus BQ-123) failed to decrease parasite infectivity over values induced by the individual drugs; (iii) TCTs efficiently invade CHO-ETAR or CHO-ETBR, in a receptor subtype-specific manner. Collectively, these results indicate that the formation of ETR heterodimers is not an absolute requirement for parasite entry in transfected mammalian cells. Considering that the density of transfected ETRs displayed at the cell surface of CHO-transfected cells is probably higher than in cardiovascular cells, predictions from the space-filling model (Park et al., 2004) suggest that ETAR/ETBR and/or B2R/ETR heterodimers may act as functional units. Our findings that ETAR or ETBR are identified as clusters in the proximity of parasite attachment sites (Figure 6B) suggest that ETRs may be assembled within the secluded sites (synapses) formed by juxtaposition of host–parasite plasma membranes (Tardieux et al., 1992; Woolsey et al., 2003; Tyler et al., 2005). Future studies are needed to clarify whether these ETRs are physically associated with B2R at the cell surface, or into signalling complexes segregated into cholesterol-rich domains. Alternatively, some of these GPCR subtypes may assemble into single functional units further downstream in the endocytic pathway, perhaps controlling the phosphatidylinositol 3-kinase (PI3K)-dependent remodelling responses that are critically required for T. cruzi retention in susceptible host cells (Woolsey and Burleigh, 2004). Pertinently, the results from the invasion assays performed in the presence of suboptimal concentrations of BQ-123 and BQ-788 (Figure 4A) suggest that parasite invasion depends on converging signals transduced by ETBR and ETAR. These data strongly suggest that ETBR and ETAR have distinct roles during the spatiotemporal progression of the penetration process.
Collectively, our studies suggest that parasite-evoked inflammatory oedema might render myocardial tissues hypersensitive to infection via the ETR/B2R pathway. These findings provide a new mechanistic framework to characterize molecular determinants of susceptibility in Chagas heart disease.
Acknowledgments
This research was supported by funds from the Instituto Nacional de Biologia Estrutural e Bio-Imagem do CNPq; PRONEX (26/110.562/2010), FAPERJ; CNPq. Financed in part by National Institutes of Health (NIH) Grant AI-076248 (HBT), Daniele Andrade was supported in part by a Fogarty International Center-NIH Training Grant (D43-TW007129). We wish to thank Dr Marcelo Einicker Lamas and Dr Técia M Carvalho for helpful discussions and methodological advice. This work was performed with the skilful technical assistance of Leila Faustino, Daniela O. Faustino and Alda F. Alves.
Glossary
- B2R
bradykinin B2 receptor
- BK
bradykinin
- cruzipain
the major cysteine protease of T. cruzi
- CXCR2
CXCR2 chemokine (C-X-C motif) receptor 2
- ET-1
endothelin-1, ETR, endothelin receptors
- HSA
human serum albumin
- HSMCs
human smooth muscle cells
- HUVECs
primary human umbilical vein endothelial cells
- MβCD
methyl-β-cyclodextrin
- PI3K
phosphatidylinositol 3-kinase
- TCTs
tissue culture trypomastigotes
- TLR2
toll-like 2 receptors
Conflict of interests
The authors declare that there is no conflict of interest in the publication of this study.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Dose response of GPCR antagonists inHSMC invasion assays. (A) Human smooth muscle cells (HSMCs) wereincubated with TCTs (parasite: cell ratio 10:1) for 3 h inDMEM-HSA. Where indicated, the cultures received either BQ-788(0.1, 0.5, 1, 5 or 10 μM), BQ-123 (0.1, 0.5, 1, 5 or 10 μM);(B) HOE-140 (0.1–100 nM). After 3 h, the monolayers werewashed in HBSS, fixed with Bouin for 24 h and stained with Giemsa.The infection index represents the intracellular parasites per 100cells (in triplicate) (media ± SD). The results arerepresentative of three independent experiments. *P < 0.05.
Figure S2 GPCR antagonists specifically reduceTCT uptake by cardiomyocyte cells that naturally express ETRs andB2R. Primary mouse cardiomyocytes infected with TCTs, asdescribed in Material and Methods. In all assays, the infectionprocess was halted by washing the monolayers with HBSS. The cellswere then fixed with Bouin and stained with Giemsa. The infectionindex represents the number of intracellular parasites per 100cells (in triplicate) (media ± SD). The results arerepresentative of three independent experiments. *P < 0.05.
Figure S3 ACE inhibitors forge a‘cross-talk’ between B2R/ETBR inHUVECs. (A,B) HUVECs were incubated with TCTs (ratio 10:1) inM199-HAS in the presence or absence of the ACE inhibitor lisinopril(25 μM) (grey bars). The infection index represents theintracellular parasites per 100 cells (in triplicate) (media± SD). The results are representative of three independentexperiments. *P < 0.05; **P < 0.01.
Figure S4 Assessment of the specificityactivity of GPCR antagonists on endocytosis and cytoskeletonorganization of HSMCs. (A) HSMCs were infected (or not) with TCTDm28 and incubated with 1 mg·mL−1FITC-Dextran for 1 h, in the presence or absence of BQ-123 orBQ-788 at 37°C. Incubation at 4°C was included as a controlto monitor the extent of non-specific surface binding of thefluorescent probe. After stopping the interaction process by theaddition of ice-cold PBS, the monolayers were fixed withparaformaldehyde 1% for 30 min and then scraped out of wells formeasurement of FITC-Dextran labelling by flow cytometry (FACScan).(B) HSMCs were incubated or not with TCTs (parasite: cell ratio20:1) for 1 h. After fixing with 4% paraformaldehyde for 30 min,the monolayers were permeabilized before being incubated with546-alexa-phalloidin (1:50) for 90 min. After washing themonolayers three times with PBS, the preparations were stained withDAPI for 5 min and then analysed by confocal laser scan microscopeZeiss LSM710 with oil immersion 40× objective. White arrow head indicate TCTs and the white arrows indicate actin rearrangement.
Figure S5 ETRs clusters at adhesion sitesbetween T. cruzi and HSMCs. Surface immunolabelling ofETAR or ETBR in HSMCs incubated with TCTs(parasite : cell ratio 20:1) for 1 h. After fixing the monolayerswith 4% paraformaldehyde for 30 min, coverslips were incubated withanti-ETBR (1:100) or anti-ETAR (1:100) for 1h, followed by incubation with Alexa 488-conjugated secondaryanti-rabbit IgG antibody. The monolayers were DAPI stained andobserved under a Zeiss LSM710 confocal laser scan microscope withan oil immersion 40x objective. (A) Slice 5 (top) withanti-ETAR antibodies (C) Slice 5 (top) withanti-ETBR antibodies. (B,D) The monolayers were laserdissected 10–13 times in 0.25 μm slices from bottom totop. White arrows indicate that surface ETRs are recruited fromsurrounding areas to the T. cruzi adhesion site.Z-section show surface co-localization of the parasites withthe receptor.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aliberti J, Viola VP, Vieira-de-Abreu A, Bozza PT, Sher A, Scharfstein J. Cutting edge: bradykinin induces IL-12 production by dendritic cells: a danger signal that drives Th1 polarization. J Immunol. 2003;170:5349–5353. doi: 10.4049/jimmunol.170.11.5349. [DOI] [PubMed] [Google Scholar]
- Almeida IC, Gazzinelli RT. Proinflammatory activity of glycosylphosphatidylinositol anchors derived from Trypanosoma cruzi: structural and functional analyses. J Leukoc Biol. 2001;70:467–477. [PubMed] [Google Scholar]
- Bachvarov DR, Saint-Jacques E, Larrivée JF, Levesque L, Rioux F, Drapeau G, et al. Cloning and pharmacological characterization of the rabbit bradykinin B2 receptor. J Pharmacol Exp Ther. 1995;275:1623–1630. [PubMed] [Google Scholar]
- Bremnes T, Paasche JD, Mehlum A, Sandberg C, Bremnes B, Attramadal H. Regulation and intracellular trafficking pathways of the endothelin receptors. J Biol Chem. 2000;275:17596–17604. doi: 10.1074/jbc.M000142200. [DOI] [PubMed] [Google Scholar]
- Caler EV, Morty RE, Burleigh BA, Andrews NW. Dual role of signaling pathways leading to Ca2+ and cyclic AMP elevation in host cell invasion by Trypanosoma cruzi. Infect Immun. 2000;68:6602–6610. doi: 10.1128/iai.68.12.6602-6610.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos MA, Almeida IC, Takeuchi O, Akira S, Valente EP, Procópio DO, et al. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol. 2001;167:416–423. doi: 10.4049/jimmunol.167.1.416. [DOI] [PubMed] [Google Scholar]
- Carlos D, Frantz FG, Souza-Júnior DA, Jamur MC, Oliver C, Ramos SG, et al. TLR2- dependent mast cell activation contributes to the control of Mycobacterium tuberculosis infection. Microbes Infect. 2009;11:770–778. doi: 10.1016/j.micinf.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Conte FdeP, Barja-Fidalgo C, Verri WA, Jr, Cunha FQ, Rae GA, Penido C, et al. Endothelins modulate inflammatory reaction in zymosan-induced arthritis: participation of LTB4, TNF-alpha, and CXCL-1. J Leukoc Biol. 2008;84:652–660. doi: 10.1189/jlb.1207827. [DOI] [PubMed] [Google Scholar]
- Coulombe M, Battistini B, Stankova J, Pouliot P, Bissonnette EY. Endothelins regulate mediator production of rat tissue-cultured mucosal mast cells. Up-regulation of Th1 and inhibition of Th2 cytokines. J Leukoc Biol. 2002;71:829–836. [PubMed] [Google Scholar]
- Coura JR. Transmission of chagasic infection by oral route in the natural history of Chagas disease. Rev Soc Bras Med Trop. 2006;39(Suppl. 3):113–117. [PubMed] [Google Scholar]
- Coura JR, Dias JC. Epidemiology, control and surveillance of Chagas disease: 100 years after its discovery. Mem Inst Oswaldo Cruz. 2009;104(Suppl. 1):31–40. doi: 10.1590/s0074-02762009000900006. [DOI] [PubMed] [Google Scholar]
- De Weerd WF, Leeb-Lundberg LM. Bradykinin sequesters B2 bradykinin receptors and the receptor-coupled Galpha subunits Galphaq and Galphai in caveolae in DDT1 MF-2 smooth muscle cells. J Biol Chem. 1997;272:17858–17866. doi: 10.1074/jbc.272.28.17858. [DOI] [PubMed] [Google Scholar]
- Del Nery E, Juliano MA, Lima APCA, Scharfstein J, Juliano J. Kininogenase activity by the major cysteinl proteinase (cruzipain) from Trypanosoma cruzi. J Biol Chem. 1997;272:25713–25718. doi: 10.1074/jbc.272.41.25713. [DOI] [PubMed] [Google Scholar]
- Dhaun N, Pollock DM, Goddard J, Webb DJ. Selective and mixed endothelin receptor antagonism in cardiovascular disease. Trends Pharmacol Sci. 2007;28:573–579. doi: 10.1016/j.tips.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Ehrenreich H, Burd PR, Rottem M, Hültner L, Hylton JB, Garfield M, et al. Endothelins belong to the assortment of mast cell-derived and mast cell-bound cytokines. New Biol. 1992;4:147–156. [PubMed] [Google Scholar]
- Evans NJ, Walker JW. Endothelin receptor dimers evaluated by FRET, ligand binding, and calcium mobilization. Biophys J. 2008;95:483–492. doi: 10.1529/biophysj.107.119206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes MC, Cortez M, Geraldo-Yoneyama KA, Straus AH, Yoshida N, Mortara RA. Novel strategy in Trypanosoma cruzi cell invasion: implication of cholesterol and host cell microdomains. Int J Parasitol. 2007;37:1431–1441. doi: 10.1016/j.ijpara.2007.04.025. [DOI] [PubMed] [Google Scholar]
- Filep JG, Sirois MG, Foldes-Filep E, Rousseau A, Plante GE, Fournier A, et al. Enhancement by endothelin-1 of microvascular permeability via the activation of ETA receptors. Br J Pharmacol. 1993;109:880–886. doi: 10.1111/j.1476-5381.1993.tb13657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, Andoh T, Saiki I, Kuraishi Y. Involvement of endothelin and ET(A) endothelin receptor in mechanical allodynia in mice given orthotopic melanoma inoculation. J Pharmacol Sci. 2008;106:257–263. doi: 10.1254/jphs.fp0072051. [DOI] [PubMed] [Google Scholar]
- Goto K. Basic and therapeutic relevance of endothelin-mediated regulation. Biol Pharm Bull. 2001;24:1219–1230. doi: 10.1248/bpb.24.1219. [DOI] [PubMed] [Google Scholar]
- Goto K, Kasuya Y, Matsuki N, Takuwa Y, Kurihara H, Ishikawa T, et al. Endothelin activates the dihydropyridine-sensitive, voltage-dependent Ca2+ channel in vascular smooth muscle. Proc Natl Acad Sci USA. 1989;86:3915–3918. doi: 10.1073/pnas.86.10.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez FR, Mariano FS, Oliveira CJ, Pavanelli WR, Guedes PM, Silva GK, et al. Regulation of Trypanosoma cruzi-induced myocarditis by programmed death cell receptor 1 (PD-1) Infect Immun. 2011;79:1873–1881. doi: 10.1128/IAI.01047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Himeno A, Shigematsu K, Sumikawa K, Niwa M. Endothelin-1 binding to endothelin receptors in the rat anterior pituitary gland: possible formation of an ETA-ETB receptor heterodimer. Cell Mol Neurobiol. 2002;22:207–226. doi: 10.1023/a:1019822107048. [DOI] [PubMed] [Google Scholar]
- Higuchi ML, Fukasawa S, De Brito T, Parzianello LC, Bellotti G, Ramires JA. Different microcirculatory and intersticial matrix patterns in idiopathic dilated cardiomyopathy and Chagas’ disease: a three dimensional confocal microscopy study. Heart. 1999;82:279–285. doi: 10.1136/hrt.82.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamuro Y, Miwa S, Minowa T, Enoki T, Zhang XF, Ishikawa M, et al. Activation of two types of Ca2+-permeable nonselective cation channel by endothelin-1 in A7r5 cells. Br J Pharmacol. 1998;124:1541–1549. doi: 10.1038/sj.bjp.0701984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanabe Y, Nauli SM. Involvement of extracellular Ca2+ influx through voltage-independent Ca2+ channels in endothelin-1 function. Cell Signal. 2005;17:911–916. doi: 10.1016/j.cellsig.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol. 2001;41:851–876. doi: 10.1146/annurev.pharmtox.41.1.851. [DOI] [PubMed] [Google Scholar]
- Kitsukawa Y, Gu ZF, Hilderbrand P, Jensen RT. Gastric smooth muscle cells possess two classes of endothelin receptors but only one alters contraction. Am J Physiol. 1994;266(4 Pt 1):G713–G721. doi: 10.1152/ajpgi.1994.266.4.G713. [DOI] [PubMed] [Google Scholar]
- Knox AJ, Corbett L, Stocks J, Holland E, Zhu YM, Pang L. Human airway smooth muscle cells secrete vascular endothelial growth factor: up-regulation by bradykinin via a protein kinase C and prostanoid-dependent mechanism. FASEB J. 2001;15:2480–2488. doi: 10.1096/fj.01-0256com. [DOI] [PubMed] [Google Scholar]
- Lima AP, Almeida IC, Tersariol ILS, Schmitz V, Schmaier AH, Juliano L, et al. Heparan sulfate modulates kinin release by Trypanosoma cruzi through the activity of cruzipain. J Biol Chem. 2002;277:5875–5881. doi: 10.1074/jbc.M108518200. [DOI] [PubMed] [Google Scholar]
- Medeiros MM, Peixoto JR, Oliveira AC, Cardilo-Reis L, Koatz VL, Van Kaer L, et al. Toll-like receptor-4 (TLR4)-dependent proinflammatory and immunomodulatory properties of the glycoinositolphospholipid (GIPL) from Trypanosoma cruzi. J Leukoc Biol. 2007;82:488–496. doi: 10.1189/jlb.0706478. [DOI] [PubMed] [Google Scholar]
- Meirelles MN, de Araujo-Jorge TC, Miranda CF, de Souza W, Barbosa HS. Interaction of Trypanosoma cruzi with heart muscle cells: ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur J Cell Biol. 1986;41:198–206. [PubMed] [Google Scholar]
- Monteiro AC, Schmitz V, Svensjö E, Gazzinelli RT, Almeida IC, Todorov A, et al. Cooperative activation of TLR2 and bradykinin B2 receptor is required for induction of type 1 immunity in a mouse model of subcutaneous infection by Trypanosoma cruzi. J Immunol. 2006;177:6325–6335. doi: 10.4049/jimmunol.177.9.6325. [DOI] [PubMed] [Google Scholar]
- Monteiro AC, Schmitz V, Morrot A, de Arruda LB, Nagajyothi F, Granato A, et al. Bradykinin B2 Receptors of dendritic cells, acting as sensors of kinins proteolytically released by Trypanosoma cruzi, are critical for the development of protective type-1 responses. PLoS Pathog. 2007;3:e185. doi: 10.1371/journal.ppat.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta EM, Chichorro JG, D'Orléans-Juste P, Rae GA. Roles of endothelin ETA and ETB receptors in nociception and chemical, thermal and mechanical hyperalgesia induced by endothelin-1 in the rat hindpaw. Peptides. 2009;30:918–925. doi: 10.1016/j.peptides.2009.01.011. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Huang H, Petkova SB, Albanese C, Pestell RG, Braunstein VL, et al. Trypanosoma cruzi infection activates extracellular signal-regulated kinase in cultured endothelial and smooth muscle cells. Infect Immun. 2004;72:5274–5282. doi: 10.1128/IAI.72.9.5274-5282.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y, Ninomiya H, Miwa S, Masaki T. Cholesterol oxidation switches the internalization pathway of endothelin receptor type A from caveolae to clathrin-coated pits in Chinese hamster ovary cells. J Biol Chem. 2000;275:6439–6446. doi: 10.1074/jbc.275.9.6439. [DOI] [PubMed] [Google Scholar]
- Ostrom RS. New determinants of receptor-effector coupling: trafficking and compartmentation in membrane microdomains. Mol Pharmacol. 2002;61:473–476. doi: 10.1124/mol.61.3.473. [DOI] [PubMed] [Google Scholar]
- Ozaki S, Ohwaki K, Ihara M, Ishikawa K, Yano M. Coexpression studies with endothelin receptor subtypes indicate the existence of intracellular cross-talk between ET(A) and ET(B) receptors. J Biochem. 1997;121:440–447. doi: 10.1093/oxfordjournals.jbchem.a021608. [DOI] [PubMed] [Google Scholar]
- Park PS, Filipek S, Wells JW, Palczewski K. Oligomerization of G protein-coupled receptors: past, present, and future. Biochemistry. 2004;43:15643–15656. doi: 10.1021/bi047907k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova SB, Tanowitz HB, Magazine HI, Factor SM, Chan J, Pestell RG, et al. Myocardial expression of endothelin-1 in murine Trypanosoma cruzi infection. Cardiovasc Pathol. 2000;9:257–265. doi: 10.1016/s1054-8807(00)00045-4. [DOI] [PubMed] [Google Scholar]
- Piovezan AP, D'Orléans-Juste P, Frighetto M, Souza GE, Henriques MG, Rae GA. Endothelins contribute towards nociception induced by antigen in ovalbumin-sensitised mice. Br J Pharmacol. 2004;141:755–763. doi: 10.1038/sj.bjp.0705663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassi A, Jr, Rassi A, Marin-Neto JÁ. Chagas heart disease: pathophysiologic mechanisms, prognostic factors and risk stratification. Mem Inst Oswaldo Cruz. 2009;104(Suppl. 1):152–158. doi: 10.1590/s0074-02762009000900021. [DOI] [PubMed] [Google Scholar]
- Sakamoto A, Yanagisawa M, Sawamura T, Enoki T, Ohtani T, Sakurai T, et al. Distinct subdomains of human endothelin receptors determine their selectivity to endothelin A-selective antagonist and endothelin B-selective agonists. J Biol Chem. 1993;268:8547–8553. [PubMed] [Google Scholar]
- Salomone OA, Caciro TF, Madoery RJ, Amichastegui M, Juri D, Kask JC. High plasma immunoreactive endothelin levels in patients with Chagas’ cardiomyopathy. Am J Cardiol. 2001;87:1217–1220. doi: 10.1016/s0002-9149(01)01502-8. [DOI] [PubMed] [Google Scholar]
- Sampaio AL, Era GA, Henriques MG. Participation of endogenous endothelins in delayed eosinophil and neutrophil recruitment in mouse pleurisy. Inflamm Res. 2000;49:170–176. doi: 10.1007/s000110050577. [DOI] [PubMed] [Google Scholar]
- Scharfstein J, Andrade D. Infection-associated vasculopathy in Experimental Chagas Disease: pathogenic roles of endothelin and kinin pathways. In: Weiss LM, Tanowitz HB, editors. Advances in Parasitology. Chagas Disease: Trypanosoma Cruzi, the First Hundred Years. London, UK: Academic Press: Elsevier Limited; 2011. pp. 101–120. [DOI] [PubMed] [Google Scholar]
- Scharfstein J, Schmitz V, Morandi V, Capella MM, Lima AP, Morrot A, et al. Host cell invasion by Trypanosoma cruzi is potentiated by activation of bradykinin B2 receptors. J Exp Med. 2000;192:1289–1300. doi: 10.1084/jem.192.9.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfstein J, Monteiro AC, Schmitz V, Svensjö E. Angiotensin-converting enzyme limits inflammation elicited by Trypanosoma cruzi cysteine proteases: a peripheral mechanism regulating adaptive immunity via the innate kinin pathway. Biol Chem. 2008;389:1015–1024. doi: 10.1515/BC.2008.126. [DOI] [PubMed] [Google Scholar]
- Schmitz V, Svensjö E, Serra RR, Teixeira MM, Scharfstein J. Proteolytic generation of kinins in tissues infected by Trypanosoma cruzi depends on CXC chemokine secretion by macrophages activated via Toll-like 2 receptors. J Leukoc Biol. 2009;85:1005–1014. doi: 10.1189/jlb.1108693. [DOI] [PubMed] [Google Scholar]
- Schwarting A, Schlaak J, Lotz J, Pfers I, Meyer zum Buschenfelde KH, Mayet WJ. Endothelin-1 modulates the expression of adhesion molecules on fibroblast-like synovial cells (FLS) Scand J Rheumatol. 1996;25:246–256. doi: 10.3109/03009749609069994. [DOI] [PubMed] [Google Scholar]
- Speciale L, Roda K, Saresella M, Taramelli D, Ferrante P. Different endothelins stimulate cytokine production by peritoneal macrophages and microglial cell line. Immunology. 1998;93:109–114. doi: 10.1046/j.1365-2567.1998.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensjö E, Saraiva EM, Bozza MT, Oliveira SM, Lerner EA, Scharfstein J. Salivary gland homogenates of Lutzomyia longipalpis and its vasodilatory peptide maxadilan cause plasma leakage via PAC1 receptor activation. J Vasc Res. 2009;46:435–446. doi: 10.1159/000197866. [DOI] [PubMed] [Google Scholar]
- Tanowitz HB, Wittner M, Morris SA, Zhao W, Weiss LM, Hatcher SA, et al. The putative mechanistic basis for modulatory role of endothelin-1 in the altered vascular tone induced by Trypanosoma cruzi. Endothelium. 1999;6:217–230. doi: 10.3109/10623329909053412. [DOI] [PubMed] [Google Scholar]
- Tanowitz HB, Huang H, Jelicks LA, Chandra M, Loredo ML, Weiss LM, et al. Role of endothelin 1 in the pathogenesis of chronic chagasic heart disease. Infect Immun. 2005;73:2496–2503. doi: 10.1128/IAI.73.4.2496-2503.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanowitz HB, Machado FS, Jelicks LA, Shirani J, de Carvalho AC, Spray DC, et al. Perspectives on Trypanosoma cruzi-induced heart disease (Chagas disease) Prog Cardiovasc Dis. 2009;51:524–539. doi: 10.1016/j.pcad.2009.02.001. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieux I, Webster P, Ravesloot J, Boron W, Lunn JA, Heuser JE, et al. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell. 1992;71:1117–1130. doi: 10.1016/s0092-8674(05)80061-3. [DOI] [PubMed] [Google Scholar]
- Tarleton RL. Chagas disease: a role for autoimmunity? Trends Parasitol. 2003;19:447–451. doi: 10.1016/j.pt.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorov AG, Andrade D, Pesquero JB, Araujo RDC, Bader M, Stewart J, et al. Trypanosoma cruzi induces edematogenic responses in mice and invades cardiomyocytes and endothelial cells in vitro by activating distinct kinin receptor (B1/B2) subtypes. FASEB J. 2003;17:73–75. doi: 10.1096/fj.02-0477fje. [DOI] [PubMed] [Google Scholar]
- Tortelote GG, Valverde RH, Lemos T, Guilherme A, Einicker-Lamas M, Vieyra A. The plasma membrane Ca2+ pump from proximal kidney tubules is exclusively localized and active in caveolae. FEBS Lett. 2004;576:31–35. doi: 10.1016/j.febslet.2004.08.055. [DOI] [PubMed] [Google Scholar]
- Tyler KM, Luxton GW, Applewhite DA, Murphy SC, Engman DM. Responsive microtubule dynamics promote cell invasion by Trypanosoma cruzi. Cell Microbiol. 2005;7:1579–1591. doi: 10.1111/j.1462-5822.2005.00576.x. [DOI] [PubMed] [Google Scholar]
- Verri WA, Jr, Cunha TM, Magro DA, Guerrero AT, Vieira SM, Carregaro V, et al. Targeting endothelin ETA and ETB receptors inhibits antigen-induced neutrophil migration and mechanical hypernociception in mice. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:271–279. doi: 10.1007/s00210-008-0360-1. [DOI] [PubMed] [Google Scholar]
- Woolsey AM, Burleigh BA. Host cell actin polymerization is required for cellular retention of Trypanosoma cruzi and early association with endosomal/lysosomal compartments. Cell Microbiol. 2004;6:829–838. doi: 10.1111/j.1462-5822.2004.00405.x. [DOI] [PubMed] [Google Scholar]
- Woolsey AM, Sunwoo L, Petersen CA, Brachmann SM, Cantley LC, Burleigh BA. Novel PI 3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J Cell Sci. 2003;116:3611–3622. doi: 10.1242/jcs.00666. [DOI] [PubMed] [Google Scholar]
- Yamamura H, Nabe T, Kohno S, Ohata K. Endothelin-1 induces release of histamine and leukotriene C4 from mouse bone marrow-derived mast cells. Eur J Pharmacol. 1994;257:235–242. doi: 10.1016/0014-2999(94)90134-1. [DOI] [PubMed] [Google Scholar]
- Zouki C, Baron C, Fournier A, Filep JG. Endothelin-1 enhances neutrophil adhesion to human coronary artery endothelial cells: role of ET(A) receptors and platelet-activating factor. Br J Pharmacol. 1999;127:969–979. doi: 10.1038/sj.bjp.0702593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.