

Abstract
BACKGROUND AND PURPOSE
Pentoxifylline is in clinical trials for non-alcoholic fatty liver disease and diabetic nephropathy. Metabolic and hepatic effects of pentoxifylline were assessed in a murine model of obesity and type 2 diabetes.
EXPERIMENTAL APPROACH
Pentoxifylline (100 mg·kg−1·day−1) was administered for 4 days or 3 weeks in lean and obese/diabetic ob/ob mice. Plasma lipids, glucose, other metabolites and relevant enzymes were measured by standard assays. Hepatic lipids in vivo were assessed with magnetic resonance spectroscopy and by histology. Hepatic extracts were also analysed with RT-PCR and Western blotting.
KEY RESULTS
Four days of pentoxifylline treatment slightly increased liver lipids in ob/ob mice. After 3 weeks, pentoxifylline exacerbated fatty liver and plasma transaminases in ob/ob mice but did not induce liver steatosis in lean mice. Plasma glucose was highest in fed, but not fasted, ob/ob mice treated with pentoxifylline. During the first 10 min of an oral glucose tolerance test, blood glucose increased more rapidly in pentoxifylline-treated mice. Jejunal expression of glucose transporter 2 isoform was increased in pentoxifylline-treated obese mice. Hepatic activity of carbohydrate response element binding protein (ChREBP) increased after pentoxifylline in ob/ob, but not lean, mice. Hepatic expression of lipogenic enzymes was highest in pentoxifylline-treated ob/ob mice. However, pentoxifylline reduced markers of oxidative stress and inflammation in ob/ob liver.
CONCLUSION AND IMPLICATIONS
Pentoxifylline exacerbated fatty liver in ob/ob mice through enhanced intestinal glucose absorption, increased postprandial glycaemia and activation of hepatic lipogenesis. Long-term treatment with pentoxifylline could worsen fatty liver in some patients with pre-existing hyperglycaemia.
Keywords: drug-induced liver injury, fatty liver, obesity, pentoxifylline, glucose, ChREBP, methylxanthine
Introduction
Pentoxifylline is a non-selective phosphodiesterase inhibitor and an antagonist of adenosine receptors (Windmeier and Gressner, 1997; Kim et al., 2002). This methylxanthine derivative is commonly prescribed for peripheral vascular disorders and intermittent claudication by improving blood flow and circulation (Windmeier and Gressner, 1997). Moreover, as an inhibitor of TNF-α synthesis, this drug has been tested successfully in different disease states involving this pro-inflammatory cytokine such as alcoholic hepatitis and cirrhosis (Lebrec et al., 2010; Stickel and Seitz, 2010). Importantly, TNF-α also plays a key role in the pathophysiology of non-alcoholic steatohepatitis (NASH), a liver disease observed in 10% to 20% of obese patients (Begriche et al., 2006; Fabbrini et al., 2010). Hence, clinical trials have recently been undertaken to determine whether long-term treatment with pentoxifylline (1200–1600 mg·day−1) could alleviate NASH (Adams et al., 2004; Satapathy et al., 2007; Lee et al., 2008a). Although pentoxifylline ameliorated NASH in most individuals, it worsened steatosis, inflammation and fibrosis in a few patients, although it was unclear whether this was related to pentoxifylline or other factors (Adams et al., 2004; Satapathy et al., 2007).
We previously reported that pentoxifylline (100 mg·kg−1·day−1) prevented hepatic apoptosis in obese and diabetic ob/ob mice in a TNF-α-dependent manner, after repeated ethanol consumption for four consecutive days (Robin et al., 2005). In addition, pentoxifylline significantly reduced liver triglycerides in ethanol-intoxicated ob/ob mice (Robin et al., 2005). However, hepatic triglycerides were slightly enhanced in naive obese animals treated for 4 days with pentoxifylline, although this effect was not statistically significant (Robin et al., 2005). Moreover, a 2 week treatment with 300 mg·kg−1·day−1 of pentoxifylline unexpectedly enhanced liver triglycerides in mice fed a methionine and choline-deficient diet (a murine model of NASH), while serum alanine aminotransferase (ALT) activity and hepatic inflammation were concomitantly alleviated (Koppe et al., 2004).Thus, pentoxifylline could have detrimental effects on liver lipid homeostasis despite its anti-inflammatory action.
In this study, ob/ob mice were treated for 4 days or 3 weeks with pentoxifylline. Our results indicated that pentoxifylline aggravated fatty liver in ob/ob mice in a time-dependent manner, whereas liver lipid deposition was not observed in lean mice. Moreover, our investigations suggested that pentoxifylline could promote lipid synthesis in ob/ob liver through a ChREBP-dependent pathway, possibly activated by pentoxifylline-induced hyperglycaemia in the fed state.
Methods
Animals and treatment
All experiments were performed according to national guidelines for the use of animals in biomedical research and approved by the local Ethics Committee in Animal Experiment of Rennes 1 University.
Five-week-old male C57BL/6J-ob/ob mice (also referred to as obese mice), weighing 26 to 30 g, and C57BL/6J-+/+ mice (wild-type, also referred to as lean mice), weighing 17 to 20 g, were purchased from Janvier (Le-Genest-St-Isle, France). All mice were fed ad libitum on a normal diet containing 2820 kcal per kg (A04 biscuits; UAR, Villemoisson-sur-Orge, France). After 1 week of acclimatization, the groups of lean and obese mice were further split into two subgroups that were treated with 100 mg·kg−1·d−1 of pentoxifylline (Sigma-Aldrich, St. Quentin-Fallavier, France) or placebo for 4 days, or 3 weeks. This dose of pentoxifylline corresponds to ∼8 mg·kg−1·d−1 in patients taking into consideration the difference of body surface areas between both species (Reagan-Shaw et al., 2007). Pentoxifylline was administered i.p. for the 4 day treatment and provided in the drinking water during the 3 week experiment. The quantity of pentoxifylline added in the drinking water was calculated on the basis of daily liquid consumption in both groups (∼5–6 and 8–9 mL in lean and obese mice, respectively), so that treated mice received each day 100 mg·kg−1 of pentoxifylline. Significantly, the daily liquid intake was unaffected by the treatment. Consumption of food and body weight was also evaluated on a regular basis. At the end of the treatment, mice were killed in the morning (around 10:00 a.m.) by cervical dislocation, and livers quickly removed. Most of the liver fragments were immediately frozen in liquid nitrogen but some were rapidly processed for appropriate histological staining. In some animals, the jejunum was also removed, rinsed and the mucosa was scraped off and immediately frozen in liquid nitrogen. Collected tissues frozen in liquid nitrogen were subsequently stored at −80°C until use.
Liver histology, total lipids and triglycerides
In order to evaluate necrosis and inflammation, liver fragments from fed animals were fixed in 10% neutral formalin and embedded in paraffin. Then, 4 µm thick sections were cut and stained with haematoxylin-eosin (H&E). For the detection of neutral lipids, 10 µm thick cryosections were stained with 0.3% Oil Red O for 10 min at room temperature and counterstained with haematoxylin. For each mouse, assessment of the area occupied by the lipid vacuoles was performed on five different fields at 100× magnification, using ImageJ software (National Institutes of Health, Bethesda, MD). This area of lipid vacuoles was expressed as a percentage of the total surface of these different fields.
Total lipids in the livers were measured gravimetrically as described previously (Robin et al., 2005). Lipids were subsequently resuspended in isopropanol (approximately 10 mg·mL−1). After removal of phospholipids with aluminum hydroxide hydrate, triglycerides were determined colorimetrically using Nash's reagent (containing acetylacetone, ammonium acetate and isopropanol) and periodate.
Liver magnetic resonance spectroscopy (MRS) and body composition
Liver MRS was performed using a 4.7 T horizontal shielded magnet (47/40 USR Bruker Biospec) and running Topspin 1.5 and Paravision 4.0 imaging software (Bruker Biospin MRI, Wissembourg, France). Animals were anaesthetized with ketamine (100 mg·kg−1) and xylazine (10 mg·kg−1), given i.p.. Localized MR spectra were acquired without water suppression (PRESS, TR/TE = 2500/20 ms and voxel size = 3 × 3 × 3 mm3) on the upper right lobe of the liver (Bottomley, 1987). During the experiment, the respiratory gating system allowed the synchronization of the acquisitions with the respiratory frequency to avoid motion artifacts. The peaks of water and total lipids were integrated, and lipid-to-water ratio (LWR) was calculated for each spectrum (Delgado et al., 2009; Marsman et al., 2010). Preliminary investigations in ob/ob and db/db mice showed a good correlation (r = 0.90, P = 0.02) between LWR and the lipid droplet areas determined after Oil Red O staining.
Body fat mass and lean mass were determined by dual-energy X-ray absorptiometry using a Piximus® apparatus (Lunar Corporation, Madison, WI), as previously described (Igoudjil et al., 2007).
Plasma studies
Blood was collected from the retro-orbitary sinus, before death, for the measurement of a set of plasma parameters. Blood was always collected in the morning (around 9:00 a.m.) either in the post-absorptive state (referred to as the fed state) or after an overnight period of fast (referred to as the fasted state). Plasma ALT and aspartate aminotransferase (AST) activities, total cholesterol, triglycerides, glucose, non-esterified fatty acids (NEFAs), glycerol and β-hydroxybutyrate were measured on an automatic analyzer AU400 (Olympus Diagnostics, Rungis, France). Plasma ALT, AST, total cholesterol, triglycerides and glucose were determined with the Olympus commercial kits OSR6107, OSR6109, OSR6116, OSR6133 and OSR6121 respectively. NEFAs, glycerol and β-hydroxybutyrate, were measured with kits FA115, GY105 and RB1007, respectively, purchased from RANDOX Laboratories (Montpellier, France). Insulin was determined with the mouse/rat elisa kit from Millipore (Molsheim, France), while leptin, adiponectin and IGF-1 were assessed with mouse elisa kits from R&D Systems (Lille, France). Insulin resistance was estimated using the homeostatic model assessment of insulin resistance (HOMA-IR) with the formula (glucose × insulin) / 22.5.
Hepatic 14C-oleic acid uptake and activity of microsomal triglyceride transfer protein
Hepatic uptake of 14C-oleate was assessed in obese mice as previously described (Rinella et al., 2008). Briefly, each mouse received an intragastric bolus of 200 µL olive oil containing 2 µCi of 14C-oleic acid (PerkinElmer, Courtaboeuf, France) after an overnight fast. Four hours after the bolus, the liver was removed, and approximately 250 mg of liver tissue was homogenized in 1 mL of Dulbecco's PBS. The resulting liver homogenate (0.8 mL) was then added to a mixture of 1 mL of chloroform and 2 mL of methanol. The sample was vortexed for 1 min, and 1 mL of distilled water and 1 mL of chloroform were successively added. The mixture was vortexed again and then centrifuged at 400×g for 5 min. The resultant lipid phase (1 mL) was dried and the 14C radioactivity measured.
Activity of microsomal triglyceride transfer protein (MTP) in liver was determined with a commercial kit (Roar Biomedical, New York, NY), as previously described (Lettéron et al., 2003). Assessment of MTP activity is based on the MTP-mediated transfer of a self-quenched fluorescent neutral lipid from the core of a donor particle to an acceptor particle.
Glucose and insulin tolerance tests
Oral and i.p. glucose tolerance test (OGTT and IPGTT, respectively) was performed in mice after a 6 h fast (Andrikopoulos et al., 2008). d-glucose was administered orally (0.5 and 1 g·kg−1 body weight for obese and lean mice, respectively), or injected i.p. (1.5 g·kg−1 body weight for both lean and obese mice), and blood was collected by tail bleeding at 0, 5, 10, 15, 30, 45, 60, 90 and 120 min for measurement of blood glucose by using an One-touch Accu-Check Glucometer® (Roche, Paris, France). I.p. insulin tolerance test (IPITT) was performed in mice after a 4 h fast. Human insulin (Actrapid®) purchased from Novo Nordisk (Chartres, France) was injected i.p. (0.5 and 1 U·kg−1 body weight in lean and obese mice, respectively), and blood was collected by tail bleeding with the timing detailed above for GTT. Areas under the curve (AUC) during GTT and IPITT were then calculated by the linear trapezoidal method (Begriche et al., 2008a,b).
RNA isolation and real-time quantitative PCR analysis
Total hepatic RNA was isolated by acid guanidinium thiocyanate–phenol–chloroform extraction. RNA integrity was assessed with the RNA 6000 nano LabChip kit (Agilent, Waldbronn, Germany). cDNAs were prepared by reverse transcription of 1 µg of total RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Courtaboeuf, France). cDNAs were amplified with specific primers using the Power SYBR Green PCR Master Mix (Applied Biosystems), in a StepOne Plus instrument (Applied Biosystems). The PCR conditions were one cycle at 94°C for 3 min, followed by 40 cycles at 94°C for 30 s and 60°C for 1 min. Amplification of specific transcripts was confirmed by melting curve profiles generated at the end of each run. Moreover, PCR specificity was further ascertained with an agarose gel electrophoresis by checking the length of the PCR products. Expression of the mouse ribosomal protein S6 (S6) was used as reference, and the 2–ΔΔCt method was used to express the relative expression of each selected gene (Begriche et al., 2008a,b). Sequences of the primers used in this study are available on request.
Hepatic DNA-binding activity of carbohydrate response element binding protein (ChREBP) and sterol regulatory element binding protein-1 (SREBP-1)
Preparation of liver nuclear extracts was adapted from the method described by Zhou et al. (1999). DNA-binding activity of ChREBP and SREBP-1 was then determined by using the Cayman's ChREBP and SREBP-1 Transcription Factor Assay elisa Kits (Cayman Chemical Company, Ann Arbor, MI), in accordance with the manufacturer's recommendations.
Western blot analysis and parameters of oxidative stress
To assess expression of total and phosphorylated acetyl-CoA carboxylase (ACC and pACC), total and phosphorylated AMP-activated protein kinase (AMPK and pAMPK) and fatty acid synthase (FAS) in liver, and that of Na+-dependent glucose transporter-1 (SGLT-1) and glucose transporter 2 isoform (GLUT2) in jejunum, ∼50 µg of hepatic proteins or 100 µg of jejunal proteins were separated by SDS-polyacrylamide electrophoresis. Proteins were then transferred to nitrocellulose membranes (Hybond ECL, Amersham Biosciences, Saclay, France) and immunoblotted with antibodies against FAS, GLUT2 (Santa Cruz Biotechnology, Santa Cruz, CA), ACC, pACC (Ser79) (Upstate, Lake Placid, NY), AMPKα, pAMPKα (Thr172) (Cell Signaling Technology Inc., Danvers, MA) or SGLT-1 (Millipore, Temecula, CA). Blots were subsequently incubated with appropriate secondary antibodies, and protein bands were revealed by enhanced chemiluminescence. β-Actin was used to normalize protein loadings.
Reduced glutathione (GSH) levels were determined as previously described (Robin et al., 2005). Lipid peroxidation was assessed by measuring thiobarbituric acid reactants (TBARs) (Ohkawa et al., 1979). TBAR contents were expressed as nanomoles of malondialdehyde (MDA) equivalents per milligram of protein. Activity of aconitase, an enzyme highly sensitive to oxidative damage and located both in cytosol and mitochondria, was assessed as previously described (Begriche et al., 2008b).
Culture of primary mouse adipocytes and HepaRG cells
The isolation of mouse adipocytes from C57BL/6J-+/+ mice and assessment of lipolysis were as previously described (Begriche et al., 2008b). After 48 h of culture without treatment, adipocytes were incubated with 1 and 10 µM pentoxifylline or 1 µM isoprenaline (Sigma-Aldrich), and fresh solutions of these compounds were added after 24 h. The culture medium was thus taken 24 h later for the determination of glycerol using an Olympus analyzer. Released glycerol after 48 h of treatment with pentoxifylline or isoprenaline was expressed as µmol per mg of protein.
A two-step procedure was used to obtain differentiated HepaRG (Cerec et al., 2007). Briefly, cells were cultured in Williams’ E medium supplemented with 10% fetal bovine serum, 100 units·mL−1 penicillin, 100 µg·mL−1 streptomycin, 5 µg·mL−1 insulin, 2 mM glutamine and 50 µM hydrocortisone hemisuccinate and incubated in 5% CO2 and 95% air at 37°C for 2 weeks. Cells were subsequently cultured in the presence of 2% dimethylsulphoxide (DMSO) for 2 additional weeks (Cerec et al., 2007). Hepatocyte-like cells were then selectively detached using mild trypsinization (Cerec et al., 2007). Cells from the pellets were washed in medium, plated at high density (1.5 × 105 cm−2) without serum and DMSO and treated 48 h later with 1, 10 and 50 µM pentoxifylline for 24, 48 h and 7 days. Fresh medium with or without pentoxifylline was added every day. After incubation, cells were observed under a contrast phase microscope to observe cell morphology. Cell viability was assessed using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) colorimetric assay, while intracellular ATP content was determined with the CellTiter-Glo Luminescent Cell Viability Assay kit (Promega, Madison, WI), as previously described (Dumont et al., 2010).
Statistical analyses
Data are presented as means ± SEM. When four groups were compared, two-way anova with the factors of genotype (wild-type or ob/ob) and pentoxifylline treatment was performed in order to assess statistical significances. When the anova indicated a significant interaction between factors, individual means were compared with least significant difference (LSD) post hoc test. In experiments with only two sets of data, the Student's t-test was used.
Results
Body parameters, liver lipids and triglycerides
Mice were first treated with pentoxifylline for 4 days. Liver triglycerides were slightly increased by 10% in treated obese mice, thus confirming our previous data (Robin et al., 2005). In contrast, this effect was not observed in lean mice. Indeed, liver triglycerides were 10.9 ± 1.6, 11.4 ± 2.2, 109.3 ± 5.8 and 120.3 ± 5.2 mg·g−1 liver in untreated lean, pentoxifylline-treated lean, untreated ob/ob and pentoxifylline-treated ob/ob mice (10–12 mice per group).
After 3 weeks of treatment, liver lipids and triglycerides were unchanged in lean mice but significantly augmented in treated obese mice by 24% and 30% respectively (Figure 1A,B). Pentoxifylline did not modify body fat mass, lean mass and body weight in lean and ob/ob mice during this period (data not shown). Consequently, pentoxifylline significantly increased the liver weight-to-body weight ratio (Figure 1C).
Figure 1.
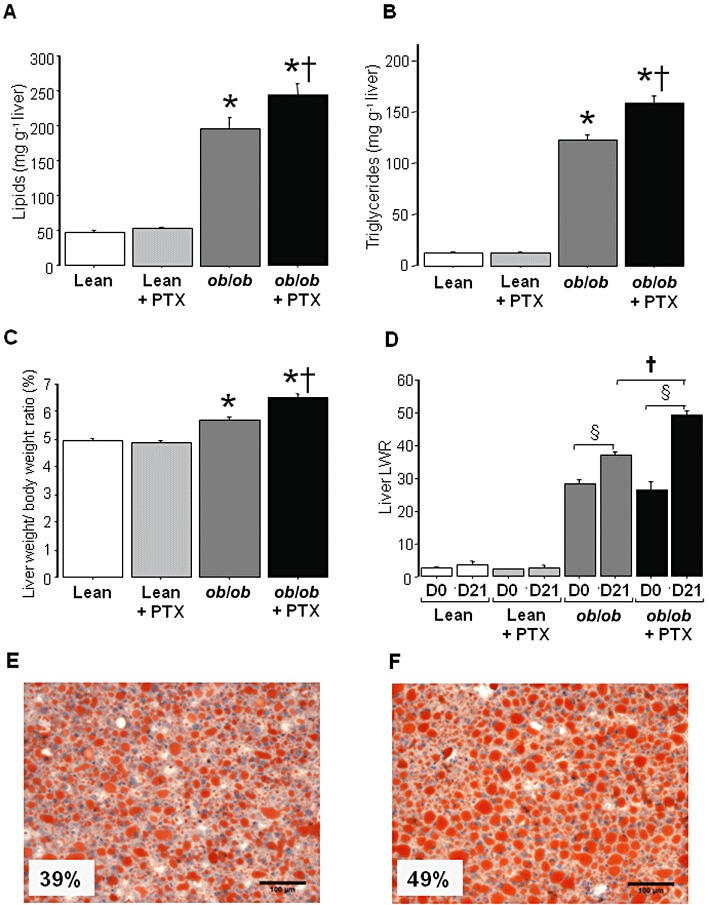
Liver lipids and triglycerides in lean and obese mice treated with pentoxifylline (PTX) or placebo for 3 weeks. (A) Liver lipids. (B) Liver triglycerides. (C) Liver weight-to-body weight ratio. (D) MRS liver lipids-to-water ratio (LWR) before experiment (D0) and after 3 weeks (D21) in untreated or pentoxifylline-treated mice. Results are means ± SEM for 10–12 mice per group, except for MRS data (four to six mice per group). * significantly different from lean mice (P < 0.05); † significantly different from untreated mice (P < 0.05); § significantly different from D0 (P < 0.05). (E) Hepatic steatosis evaluated with Oil Red O staining in an untreated ob/ob mouse. (F) Hepatic steatosis evaluated with Oil Red O staining in an ob/ob mouse treated with pentoxifylline for 3 weeks. The percentage indicated within photographs E and F represents the area of lipid vacuoles (stained in red) determined on five different fields at 100× magnification.
Liver lipids were also assessed by MRS (Figure 1D). Liver LWR was determined in untreated and pentoxifylline-treated mice on the day of the initiation of pentoxifylline treatment (D0) and then after 3 weeks (D21). This allowed us to calculate for each mouse the evolution of liver lipids over this period. Pentoxifylline treatment significantly enhanced LWR in obese mice but not in lean mice (Figure 1D). Finally, areas of the lipid vacuoles were assessed only in obese mice (four to six mice per group) since fat vacuoles were absent in lean animals. Areas of fat deposition were significantly higher (P < 0.05) in pentoxifylline-treated ob/ob mice (46 ± 2%) than in untreated obese mice (38 ± 2%). In untreated and pentoxifylline-treated obese mice, liver lipids accumulated mainly as large vacuoles with only a few small fat droplets within the hepatocytes (Figure 1E,F). The moderate necro-inflammation normally observed in ob/ob mice (Begriche et al., 2008b) was unchanged by pentoxifylline (data not shown).
Plasma biochemical markers
After 3 weeks, a mild but significant elevation in plasma ALT was observed in mice treated with pentoxifylline, whereas AST was not significantly increased (Table 1). Pentoxifylline treatment significantly enhanced plasma glycerol and NEFAs in the fed state, but only in lean mice (Table 1). This observation suggested a specific pentoxifylline-induced activation of peripheral lipolysis in the wild-type genotype. However, this effect was not observed in the fasted state (Table 1), characterized by a physiological stimulation of lipolysis (Begriche et al., 2006). Pentoxifylline treatment also significantly enhanced plasma β-hydroxybutyrate in lean mice but only in the fed state (Table 1). Thus, increased NEFA delivery to the liver induced by pentoxifylline could have promoted mitochondrial fatty acid oxidation (FAO) and ketogenesis. Obese ob/ob mice presented a slight hypotriglyceridaemia (Table 1), as already described in several studies (Lindström, 2007; Fromenty et al., 2009). Pentoxifylline significantly reduced plasma triglycerides in mice, but only in the fasted state, whereas it induced hyperglycaemia in the fed state, but not after fasting (Table 1). The insulin resistance index HOMA-IR was unchanged by pentoxifylline regardless of the genotype (Table 1).
Table 1.
Plasma parameters in lean and obese (ob/ob) mice treated with pentoxifylline (PTX) or placebo for 3 weeks
| Lean | Lean + PTX | ob/ob | ob/ob+ PTX | |
|---|---|---|---|---|
| Fed state | ||||
| Insulin (ng·mL−1) | 0.86 ± 0.13 | 0.92 ± 0.08 | 40.11 ± 8.15* | 33.77 ± 6.10* |
| Adiponectin (µg·mL−1) | 9.32 ± 0.57 | 8.58 ± 0.28 | 11.63 ± 0.63* | 13.60 ± 0.71* |
| IGF1 (ng·mL−1) | 182 ± 10 | 193 ± 11 | 226 ± 25 | 175 ± 18 |
| Leptin (ng·mL−1) | 2.9 ± 1.2 | 1.7 ± 0.2 | UD | UD |
| Glucose (mM) | 7.7 ± 0.3 | 8.5 ± 0.3† | 9.9 ± 0.6* | 12.0 ± 0.8*† |
| Triglycerides (mM) | 1.41 ± 0.10 | 1.52 ± 0.10 | 1.28 ± 0.14* | 0.99 ± 0.12* |
| Total cholesterol (mM) | 2.56 ± 0.08 | 2.50 ± 0.10 | 4.40 ± 0.12* | 4.59 ± 0.26* |
| Glycerol (µM) | 501 ± 25 | 655 ± 49† | 622 ± 52 | 677 ± 52 |
| NEFAs (mM) | 0.42 ± 0.04 | 0.56 ± 0.05† | 0.35 ± 0.03* | 0.33 ± 0.03* |
| β-Hydroxybutyrate (mM) | 0.08 ± 0.01 | 0.17 ± 0.01† | 0.11 ± 0.12 | 0.10 ± 0.26 |
| AST (UI·L−1) | 89 ± 16 | 116 ± 26 | 279 ± 17* | 348 ± 29* |
| ALT (UI·L−1) | 96 ± 26 | 135 ± 19† | 193 ± 27* | 288 ± 33*† |
| Fasted state | ||||
| Insulin (ng·mL−1) | 0.49 ± 0.16 | 0.33 ± 0.08 | 3.95 ± 0.14* | 4.36 ± 0.42* |
| Glucose (mM) | 5.45 ± 0.70 | 4.74 ± 0.38 | 7.71 ± 0.60* | 8.15 ± 0.91* |
| HOMA-IR | 0.11 ± 0.03 | 0.08 ± 0.02 | 1.40 ± 0.23* | 1.47 ± 0.27* |
| Triglycerides (mM) | 2.30 ± 0.28 | 1.84 ± 0.13† | 1.52 ± 0.04* | 1.33 ± 0.06*† |
| Total cholesterol (mM) | 2.78 ± 0.06 | 2.75 ± 0.14 | 5.72 ± 0.17* | 5.66 ± 0.09* |
| Glycerol (µM) | 839 ± 41 | 778 ± 33 | 849 ± 22 | 795 ± 19 |
| NEFAs (mM) | 1.66 ± 0.18 | 1.44 ± 0.14 | 1.26 ± 0.08* | 1.23 ± 0.08* |
| β-Hydroxybutyrate (mM) | 1.39 ± 0.12 | 1.21 ± 0.08 | 2.21 ± 0.24* | 2.41 ± 0.33* |
Blood was drawn in the fed or the fasted state, as specified in the Table. Results are means ± SEM for 12 mice per group, except for insulin, adiponectin, leptin and IGF1 (n = 7–10 mice per group).
Significantly different from lean mice (P < 0.05).
Significantly different from untreated mice (P < 0.05).
Insulin and glucose tolerance tests
IPITT was first performed to confirm the HOMA-IR data. Although the data confirmed insulin resistance in ob/ob mice, pentoxifylline treatment did not significantly modify the IPITT profiles (Figure 2A), nor AUCs (Figure 2B), in lean and obese mice. Together with the HOMA-IR data (Table 1), these results indicated that pentoxifylline did not favour systemic insulin resistance.
Figure 2.
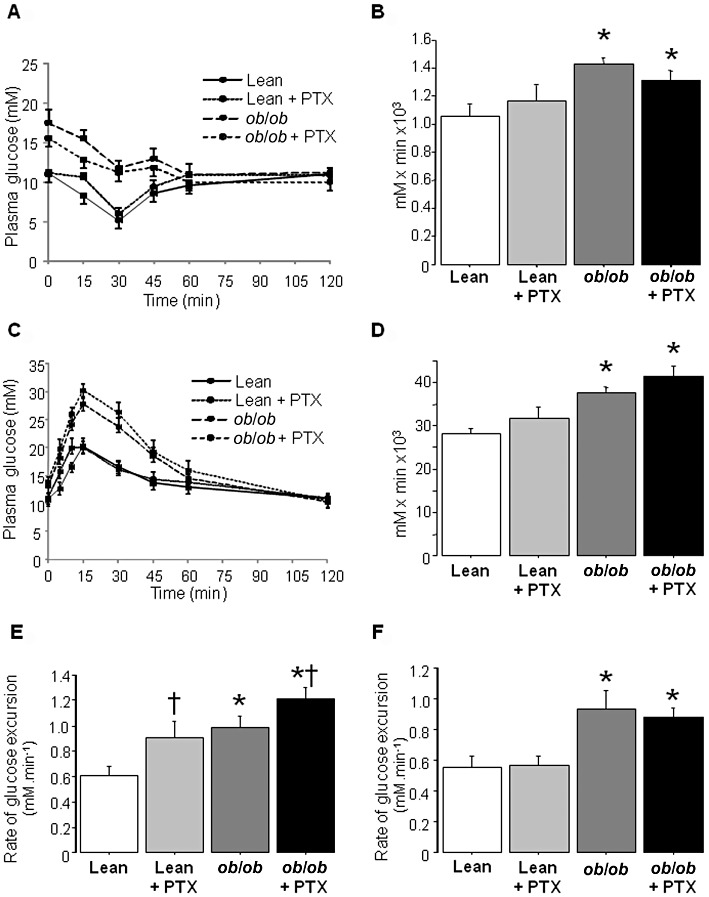
IPITT, OGTT and IPGTT performed in lean and obese mice treated with pentoxifylline (PTX) or placebo for 3 weeks. (A) IPITT in the different groups of mice. (B) AUC calculated from the IPITT data for each group of mice. (C) OGTT in the different groups of mice. (D) AUC calculated from the OGTT data for each group of mice. (E) Rate of glucose excursion in blood calculated during the first 10 min of the OGTT. (F) Rate of glucose excursion in blood calculated during the first 10 min of the IPGTT. Results are means ± SEM for six mice per group. * significantly different from lean mice (P < 0.05); † significantly different from untreated mice (P < 0.05).
Since pentoxifylline augmented glycaemia in fed animals (Table 1), an OGTT was performed to determine whether pentoxifylline could alter glucose tolerance. As expected, ob/ob mice showed overt glucose intolerance (Figure 2C). Ten minutes after glucose loading, glycaemia was 16.6 ± 0.6, 19.9 ± 1.5, 24.1 ± 0.5 and 26.0 ± 1.0 mM in lean, pentoxifylline-treated lean, obese and pentoxifylline-treated obese mice (P < 0.05 for both genotype and pentoxifylline treatment) (Figure 2C). After 15 and 30 min, glycaemia in pentoxifylline-treated ob/ob mice tended to increase, respectively, by 10% and 12% (P = 0.08) when compared with untreated ob/ob mice (Figure 2C). Calculation of AUCs indicated no significant difference between untreated and pentoxifylline-treated animals regardless of the genotype (Figure 2D). However, when blood glucose excursion was determined over the first 10 min of the OGTT, it was significantly increased in pentoxifylline-treated mice in both genotypes (Figure 2E). IPGTT also showed glucose intolerance in ob/ob mice when compared with lean animals, but glycaemia was not enhanced by pentoxifylline (data not shown). Moreover, pentoxifylline treatment did not affect the rate of blood glucose excursion during the first 10 min of IPGTT (Figure 2F). Thus, the higher glycaemia in the fed state, induced by pentoxifylline, could be related to increased intestinal glucose absorption.
Expression of GLUT2 and SGLT-1 in jejunum
Dietary glucose is absorbed in the small intestine, in particular in the jejunum. Two transporters are involved at the apical membrane of enterocytes, namely the high-affinity, low-capacity transporter SGLT-1 and the low-affinity, high-capacity transporter GLUT2. Once in the enterocytes, some of this glucose is used for metabolism, while the remaining glucose crosses the basolateral membrane into the circulation via GLUT2 (Wright et al., 2003; Kellett et al., 2008). Pentoxifylline significantly enhanced the mRNA expression of jejunal GLUT2 in lean and obese mice, but that of SGLT-1 was unchanged (Figure 3A). A similar change in GLUT2 protein levels was also observed, whereas SGLT-1 protein was not modified (Figure 3B). Interestingly, a comparable profile was observed between jejunal GLUT2 expression (Figure 3B) and the initial rate of glucose excursion during OGTT (Figure 2E). This observation suggested that obesity and pentoxifylline could combine to impair jejunal GLUT2 expression, thus leading to the highest rate of intestinal glucose absorption in pentoxifylline-treated ob/ob mice. Finally, GLUT2 mRNA and protein expression was also assessed in the liver. Although hepatic GLUT2 expression was significantly enhanced in obese mice, pentoxifylline did not cause a further increase regardless of the genotype (Figure 4). High hepatic mRNA expression of GLUT2 has been previously reported in ob/ob mice (Ferber et al., 1994). Taken together, our data suggests that pentoxifylline treatment enhanced GLUT expression in a tissue-specific manner.
Figure 3.
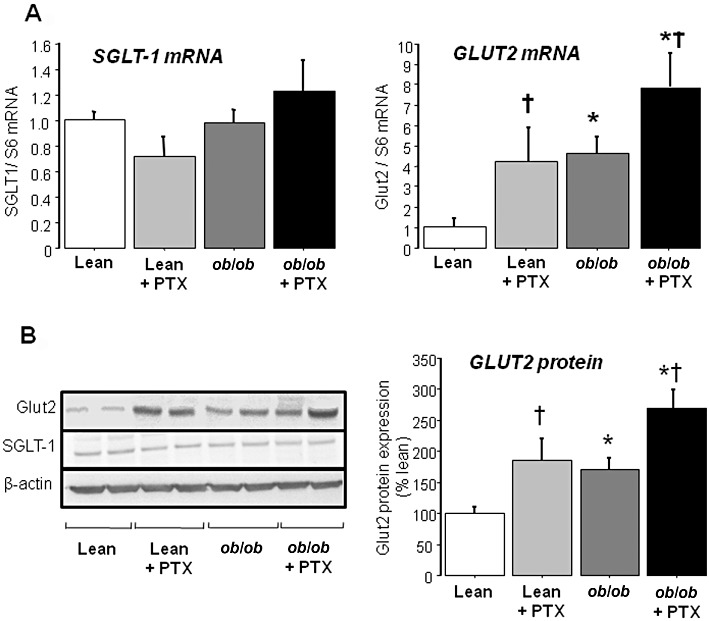
mRNA and protein expression of GLUT2 and SGLT-1 in the jejunum of lean and obese mice treated with pentoxifylline (PTX) or placebo for 3 weeks. (A) mRNA expression of GLUT2 and SGLT-1 in the jejunum assessed by real-time quantitative PCR analysis. Results are means ± SEM for six to eight mice per group. (B) Protein expression of GLUT2 and SGLT-1 in the jejunum assessed by Western blotting. Bar graph represents GLUT2 protein expression in six different mice per group. * significantly different from lean mice (P < 0.05); † significantly different from untreated mice (P < 0.05).
Figure 4.
mRNA and protein expression of GLUT2 in liver of lean and obese mice treated with pentoxifylline (PTX) or placebo for 3 weeks. A) mRNA expression of GLUT2 in liver assessed by real-time quantitative PCR analysis. Results are means ± SEM for eight mice per group. (B) Protein expression of GLUT2 in liver assessed by Western blotting. Bar graph represents GLUT2 protein expression in four different mice per group. * significantly different from lean mice (P < 0.05).
Hepatic ChREBP, SREBP1 and genes involved in glycolysis, lipogenesis and FAO
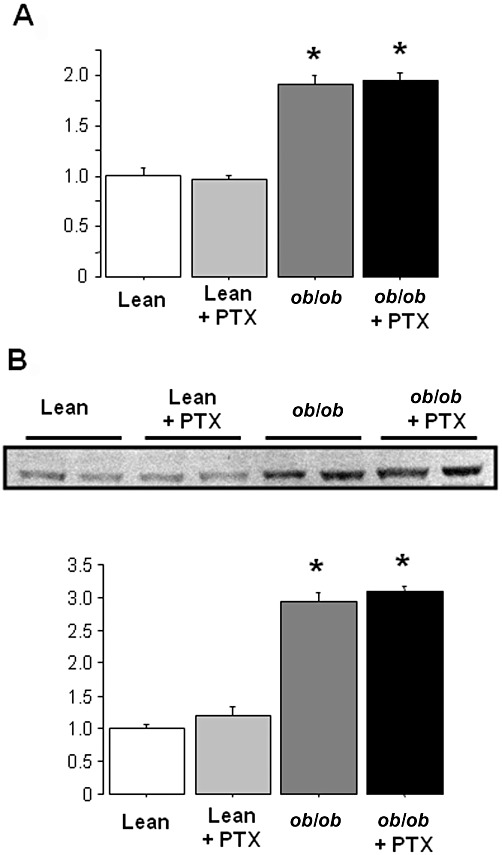
ChREBP and SREBP1 are transcription factors activated in lipogenic tissues by hyperglycaemia and hyperinsulinaemia respectively. Once activated, ChREBP and SREBP1 enhance the transcription of several glycolytic and lipogenic enzymes, thus allowing a tight coupling between glucose utilization and fatty acid synthesis (Uyeda and Repa, 2006; Begriche et al., 2009). Both hepatic mRNA levels and DNA-binding activity of ChREBP were significantly increased in pentoxifylline-treated ob/ob mice (Figure 5A), but not observed for SREBP1c (data not shown). Thus, ChREBP could be activated in the livers of pentoxifylline-treated ob/ob mice as a consequence of exacerbated hyperglycaemia.
Figure 5.
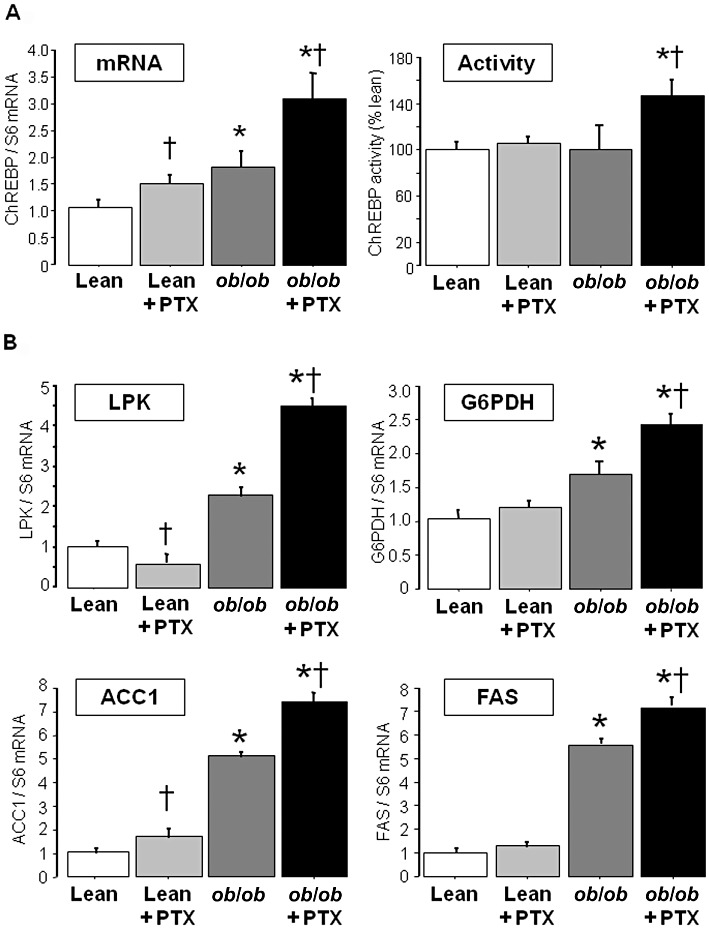
Hepatic ChREBP and mRNA expression of glycolytic and lipogenic enzymes in lean and obese mice treated with pentoxifylline (PTX) or placebo for 3 weeks. (A) Hepatic ChREBP mRNA expression and DNA-binding activity. (B) Hepatic mRNA expression of glycolytic and lipogenic enzymes. Results are means ± SEM for 8–12 mice per group. * significantly different from lean mice (P < 0.05); † significantly different from untreated mice (P < 0.05).
Next, liver mRNA expression of genes involved in glycolysis, lipogenesis and FAO was assessed. Pentoxifylline treatment increased expression of liver-type pyruvate kinase (L-PK), ACC1 and FAS, particularly in ob/ob mice (Figure 5B). Moreover, pentoxifylline administration in ob/ob mice also induced a high expression of glucose-6-phosphate dehydrogenase (G6PDH) (Figure 5B), an enzyme generating the NADPH required for lipogenesis. However, pentoxifylline treatment did not modify glucokinase and stearoyl-CoA desaturase-1 expression regardless of the genotype (data not shown). In this study, we noticed that the basal hepatic expression of G6PDH, L-PK, ACC1 and FAS was enhanced in untreated ob/ob mice, whilst ChREBP activity was unchanged. These observations suggest that, without pentoxifylline treatment, other transcription factors could be involved in the up-regulation of these glycolytic and lipogenic genes. For instance, while LPK expression is regulated by HNF4α (Stoffel and Duncan, 1997), FAS and ACC1 expression is controlled by LXR and XBP1 (Joseph et al., 2002; Lee et al., 2008b).
Pentoxifylline treatment in ob/ob mice also enhanced the expression of PPAR-α and liver carnitine palmitoyltransferase-1, whereas medium-chain acyl-CoA dehydrogenase expression was unchanged (data not shown). Enhanced hepatic expression of PPAR-α and FAO genes could be a compensatory adaptation in response to abnormal fat deposition (Chan et al., 2008; Begriche et al., 2009; Fromenty et al., 2009).
At the protein level, pentoxifylline significantly increased the hepatic expression of ACC in obese mice, although it did not modify its phosphorylation status (Figure 6). In keeping with this result, pentoxifylline did not change the expression of AMPKα and pAMPKα (data not shown). Besides ACC, pentoxifylline also enhanced hepatic FAS expression in obese mice (Figure 6).
Figure 6.
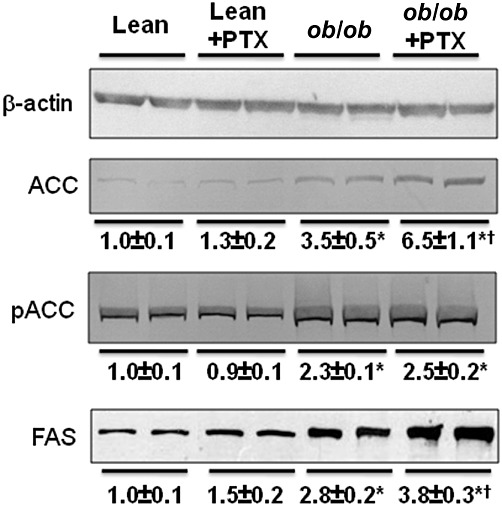
Hepatic protein expression of the lipogenic enzymes ACC and FAS in lean and obese mice treated with pentoxifylline (PTX) or placebo for 3 weeks. β-Actin was used as a loading control for Western blotting and for calculation of the relative expression of FAS, ACC and its phosphorylated form (pACC). Relative expression ratio of these proteins was set to 1 in untreated lean animals. Data below the blots are mean ± SEM for six mice per group. * significantly different from lean mice (P < 0.05); † significantly different from untreated mice (P < 0.05).
Finally, we determined whether pentoxifylline could modify the hepatic entry of fatty acids or the secretion of very low density lipoproteins (VLDL). MTP activity was assessed since it is rate-limiting in VLDL assembly and secretion (Lettéron et al., 2003; Begriche et al., 2006). Hepatic MTP activity was significantly increased by 58% in ob/ob mice compared with lean mice, thus confirming previous data (Bartels et al., 2002). However, pentoxifylline did not change this activity regardless of the genotype (100 ± 4, 101 ± 5, 158 ± 4 and 156 ± 3% in lean, pentoxifylline-treated lean, obese and pentoxifylline-treated obese mice respectively; n = 7–8 animals per group). In contrast, pentoxifylline treatment significantly reduced by 67% the hepatic 14C-oleate uptake in ob/ob mice (191 ± 39 and 63 ± 15 dpm·g−1 of liver in untreated and pentoxifylline-treated ob/ob mice, respectively; four animals per group). Thus, enhanced hepatic fat accumulation in pentoxifylline-treated ob/ob mice was not due to increased hepatic entry of fatty acids nor to reduced VLDL secretion.
Hepatic oxidative stress and inflammation
Some markers of hepatic oxidative stress and inflammation were assessed in obese mice. Total GSH was unchanged between both groups and TBARs were significantly reduced in pentoxifylline-treated ob/ob mice (Table 2). Two other markers were also assessed, namely the activity of aconitase, an enzyme highly sensitive to oxidative damage, and mRNA expression of insulin-like growth factor binding protein-1 (IGFBP-1), which can be enhanced after a variety of metabolic stresses (Fromenty et al., 2009). However, aconitase activity was increased and IGFBP-1 expression was decreased in pentoxifylline-treated ob/ob mice (Table 2). Finally, pentoxifylline treatment in ob/ob mice lowered hepatic expression of TNF-α and IL-1β (Table 2), two cytokines playing a key role in inflammation.
Table 2.
Hepatic markers of oxidative stress and inflammation in obese (ob/ob) mice treated with pentoxifylline (PTX) or placebo for 3 weeks
| ob/ob | ob/ob+ PTX | |
|---|---|---|
| Total hepatic GSH (nmol·mg−1·protein) | 32.4 ± 2.3 | 36.9 ± 2.0 |
| TBARs (nmol·mg−1·protein) | 11.1 ± 1.6 | 6.6 ± 0.9† |
| Aconitase activity (mU·mg−1·protein) | 7.2 ± 0.4 | 9.0 ± 0.4† |
| IGFBP-1 mRNA expression | 1.00 ± 0.07 | 0.34 ± 0.08† |
| TNF-α mRNA expression | 1.00 ± 0.08 | 0.53 ± 0.13† |
| IL-1β mRNA expression | 1.00 ± 0.15 | 0.66 ± 0.10† |
Results are means ± SEM for 6–10 mice per group.
Significantly different from untreated obese mice.
Effects of pentoxifylline on mouse primary adipocytes and human HepaRG cells
We assessed whether pentoxifylline could activate triglyceride hydrolysis in primary mouse adipocytes since our data suggested enhanced lipolysis in pentoxifylline-treated lean mice, in the fed state (Table 1). A 48 h incubation with 10 µM pentoxifylline significantly increased glycerol release from treated cells (550 ± 53 µmol·mg−1·protein) compared with control cells (285 ± 45 µmol·mg−1·protein). Glycerol release induced by a 48 h incubation with 1 µM isoprenaline was similar (495 ± 63 µmol·mg−1·protein), whereas it was not significantly enhanced with 1 µM pentoxifylline (379 ± 111 µmol·mg−1·protein).
Finally, HepaRG cells were used to determine whether pentoxifylline could be directly cytotoxic since our data indicated that pentoxifylline moderately (but significantly) increased plasma ALT in treated mice (Table 1). Whereas 1 µM pentoxifylline presented no effect in HepaRG cells, 10 µM significantly lowered intracellular ATP levels after 7 days of culture (Table 3). At 50 µM, pentoxifylline significantly reduced both ATP levels and cell viability after 7 days (Table 3). Thus, relatively low concentrations of pentoxifylline can induce some toxicity in HepaRG cells but only after 1 week. Further investigation is required to determine whether pentoxifylline-induced mitochondrial dysfunction is responsible for ATP shortage and subsequent cell death in HepaRG cells.
Table 3.
Effect of pentoxifylline (PTX) on cell viability and intracellular ATP content in HepaRG cells
| Cell viability (% control) | Intracellular ATP content (% control) | |||||
|---|---|---|---|---|---|---|
| 24 h | 48 h | 7 d | 24 h | 48 h | 7 d | |
| PTX 1 µM | 101 ± 4 | 99 ± 3 | 97 ± 3 | 98 ± 2 | 99 ± 3 | 97 ± 1 |
| PTX 10 µM | 101 ± 6 | 97 ± 2 | 94 ± 2 | 104 ± 2 | 100 ± 1 | 85 ± 1† |
| PTX 50 µM | 99 ± 1 | 100 ± 3 | 92 ± 2† | 98 ± 2 | 97 ± 2 | 82 ± 5† |
Results are means ± SEM for 3 independent cultures carried out in triplicate.
Significantly different from untreated cells (P < 0.05).
Discussion
Pentoxifylline is in clinical trials as a potential treatment for NASH (Adams et al., 2004; Satapathy et al., 2007; Lee et al., 2008a), based on the ability of this drug to reduce the synthesis of several potent pro-inflammatory cytokines including TNF-α and IL-1β (Windmeier and Gressner, 1997; Taha et al., 2009). Furthermore, pentoxifylline has antioxidant and anti-fibrotic properties (Windmeier and Gressner, 1997; Hepgül et al., 2010), which could be beneficial in the context of NASH treatment (Farrell and Larter, 2006; Begriche et al., 2009). The anti-inflammatory actions and favourable haemo-rheological effects of pentoxifylline have meant that it has also been tested for the management of diabetic nephropathy (Navarro-Gonzalez et al., 2009). Thus, long-term pentoxifylline treatment is a likely clincal option for an increasing number of obese and diabetic patients suffering from diseases linked to inflammation, oxidative stress and vascular complications.
In this study, we showed for the first time that a 3 week treatment with pentoxifylline aggravated fatty liver in ob/ob mice, which is a widely used murine model of obesity, type 2 diabetes and steatosis (Lindström, 2007; Schattenberg and Galle, 2010). In addition, pentoxifylline further enhanced plasma ALT activity in ob/ob mice, which reflects an aggravation of liver injury despite a lower expression of TNF-α and IL-1β and decreased lipid peroxidation in ob/ob liver. Moreover, our investigations in HepaRG cells indicated that relatively low concentrations of pentoxifylline caused some cytotoxicity and a moderate reduction of intracellular ATP content after 1 week of treatment. Thus, higher plasma ALT levels in treated mice could reflect pentoxifylline-induced direct hepatotoxicity. However, we cannot rule out the possibility that the mild aggravation of liver injury in pentoxifylline-treated obese mice could have been caused by higher deposition of deleterious lipid intermediates such as saturated free fatty acids (Begriche et al., 2006; Cazanave et al., 2009). It is noteworthy that triglyceride accumulation in hepatocytes could protect against necroinflammation and fibrosis as esterification of free fatty acids prevents their deleterious effects (Choi and Diehl, 2008). Thus, while pentoxifylline-induced hepatic triglyceride accumulation in ob/ob mice may have limited the injury to the liver to some extent, complete protection was not observed because plasma transaminases were concomitantly enhanced. Interestingly, rare cases of hepatotoxicity have been reported in patients treated with pentoxifylline (Biour et al., 2004), thus suggesting the role of predisposing factors.
Although the mechanism(s) whereby pentoxifylline aggravated fatty liver in ob/ob mice might be complex, our data suggested that ChREBP-dependent augmentation of lipogenesis could play a significant role. Moreover, hepatic ChREBP could have been specifically activated as the consequence of an aggravation of hyperglycaemia in pentoxifylline-treated ob/ob mice. Indeed, ChREBP may be regulated by enhanced glucose flux into hepatocytes through the generation of xylulose 5-phosphate, a signalling molecule activating protein phosphatase 2A (PP2A). PP2A activation in turn induces dephosphorylation of ChREBP, thus promoting its nuclear translocation and transcription of several glycolytic and lipogenic genes (Uyeda and Repa, 2006). Hence, activation of ChREBP by hyperglycaemia allows the channelling of glycolytic end-products into lipid synthesis. Interestingly, hepatic ChREBP activation in pentoxifylline-treated ob/ob mice was also associated with an increase in its mRNA expression. Since high glucose enhances mRNA levels of ChREBP (He et al., 2004; Dentin et al., 2005), activation of this transcription factor could favour its own expression directly, or indirectly. ChREBP was activated only in pentoxifylline-treated ob/ob mice but not in untreated obese mice when compared with lean mice. ChREBP is activated only by high intracellular concentrations of glucose (He et al., 2004; Ishii et al., 2004), and so pentoxifylline treatment could cause such high glucose levels in ob/ob hepatocytes.
Our OGTT data indicated that pentoxifylline could favour intestinal glucose absorption after an overload, which could explain why glycaemia was increased in the fed state but not after fasting. Whereas further investigation is required in order to determine the precise mechanism of enhanced intestinal glucose entry, higher jejunal expression of GLUT2 was found in pentoxifylline-treated mice. Significantly, GLUT2 is not only involved in glucose absorption at the enterocyte apical membrane but also in its basolateral transport into the circulation (Wright et al., 2003; Kellett et al., 2008). To our knowledge, there are no data reporting the effect of pentoxifylline on intestinal glucose absorption. However, two methylxanthine derivatives, theophylline and 3-isobutyl-1-methylxanthine, rapidly stimulated glucose transport across jejunal enterocytes through a mechanism that could involve cAMP (Reymann et al., 1985; Sharp and Debnam, 1994). Hence, phosphodiesterase inhibitors could favour intestinal glucose absorption through a direct pathway involving increased cAMP levels and another mechanism requiring enhanced GLUT2 expression. However, pentoxifylline treatment augmented GLUT2 expression in a tissue-specific manner since hepatic GLUT2 mRNA and protein were unchanged in pentoxifylline-treated lean and ob/ob mice.
Increased cAMP levels in adipocytes triggers triglyceride hydrolysis (i.e. lipolysis) into glycerol and free fatty acids through PKA-dependent activation of hormone-sensitive lipase. Although this effect has not been previously reported for pentoxifylline, several methylxanthines such as theophylline and caffeine can stimulate lipolysis by inhibiting phosphodiesterase (Scotini et al., 1983; Dallas et al., 2008). In this study, pentoxifylline enhanced plasma glycerol and NEFAs in lean mice in the fed state, suggesting lipolysis. This effect was confirmed in mouse adipocytes incubated with pentoxifylline, which released higher amounts of glycerol. In contrast, there was no apparent lipolysis in pentoxifylline-treated ob/ob mice. Interestingly, pentoxifylline increased plasma β-hydroxybutyrate in lean mice but not in obese animals. Altogether, our results indicated that pentoxifylline produced different effects depending on the metabolic/genetic status.
In this study, 10 µM pentoxifylline significantly increased glycerol release in mouse adipocytes and reduced ATP levels in human hepatoma HepaRG cells. Moreover, our data in mice also indicated that pentoxifylline was able to induce lipolysis and hepatic cytolysis. In treated patients, the mean plasma concentrations of pentoxifylline can reach values between 3 and 5 µM after a single i.v. treatment (600 mg) and repeated oral administration (1200 mg per day), with even higher levels in some individuals (Beermann et al., 1985; Nicklasson et al., 2002). Hence, our investigations could potentially be relevant to the clinical situation, at least in some obese and diabetic patients treated with PTX.
Our data also suggested that high basal glycaemia could be a major determinant in the pentoxifylline-induced aggravation of fatty liver in ob/ob mice. Indeed, hepatic activity of the glucose-activated transcription factor ChREBP was specifically induced in pentoxifylline-treated ob/ob mice, which presented the highest levels of glycaemia in the fed state. Accordingly, pre-existing type 2 diabetes in ob/ob mice (rather than obesity by itself) could have played a significant role in pentoxifylline-induced higher hepatic lipogenesis through ChREBP activation. Further investigation is required to elucidate the metabolic and hepatic effects of pentoxifylline in other experimental models of obesity and type 2 diabetes, including high-fat diet-induced obesity. However, it must be pointed out that high-fat diets in rodents do not always trigger frank hyperglycaemia and type 2 diabetes even after several months of feeding, and despite the occurrence of obesity and insulin resistance (Burcelin et al., 2002; Buettner et al., 2007; Begriche et al., 2008a).
This study may have several significant clinical implications. First, it would be important to determine whether pentoxifylline could enhance glycaemia in some obese and diabetic patients, in particular in the postprandial state. Indeed, postprandial hyperglycaemia in type 2 diabetics augments the risk of microvascular and cardiovascular complications (Vergès, 2002; Paolisso et al., 2003). Second, pentoxifylline could aggravate pre-existent liver injury in some obese and diabetic patients. Although pentoxifylline appeared to be effective in a majority of patients with NASH, it did not afford any hepatic benefit in some individuals or it may even have worsened steatosis, inflammation and fibrosis in a few others (Adams et al., 2004; Satapathy et al., 2007). From our results and previous data (Koppe et al., 2004), we believe that further clinical studies are warranted to determine whether long-term treatment with pentoxifylline could pose a risk to some obese and diabetic patients, in particular regarding gluco-lipid homeostasis and liver function. Finally, similar concerns could apply to other methylxanthine derivatives, as some of them have been shown to stimulate jejunal glucose transport (Reymann et al., 1985; Sharp and Debnam, 1994) and to induce hepatotoxicity (Biour et al., 2004).
Acknowledgments
This work was supported by INSERM (Institut Nationale de la Santé et de la Recherche Médicale). Julie Massart was a recipient of a grant from Ministère de L'Education Nationale, de la Recherche et de la Technologie. We are grateful to Pascale Bellaud from the Histopathology group for her excellent technical support.
Glossary
- ACC
acetyl-CoA carboxylase
- ALT
alanine aminotransferase
- AMPK
AMP-activated protein kinase
- AST
aspartate aminotransferase
- ChREBP
carbohydrate response element binding protein
- FAO
fatty acid oxidation
- FAS
fatty acid synthase
- GLUT2
glucose transporter 2 isoform
- GSH
reduced glutathione
- HOMA-IR
homeostatic model assessment of insulin resistance
- IPGTT
intraperitoneal glucose tolerance test
- IPITT
intraperitoneal insulin tolerance test
- MRS
magnetic resonance spectroscopy
- MTP
microsomal triglyceride transfer protein
- NEFAs
non-esterified fatty acids
- OGTT
oral glucose tolerance test
- SGLT-1
Na(+)-dependent glucose transporter-1
- TBARs
thiobarbituric acid reactants
Conflict of interest
The authors declare no conflict of interest.
References
- Adams LA, Zein CO, Angulo P, Lindor KD. A pilot trial of pentoxifylline in nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:2365–2368. doi: 10.1111/j.1572-0241.2004.40064.x. [DOI] [PubMed] [Google Scholar]
- Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab. 2008;295:E1323–E1332. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- Bartels ED, Lauritsen M, Nielsen LB. Hepatic expression of microsomal triglyceride transfer protein and in vivo secretion of triglyceride-rich lipoproteins are increased in obese diabetic mice. Diabetes. 2002;51:1233–1239. doi: 10.2337/diabetes.51.4.1233. [DOI] [PubMed] [Google Scholar]
- Beermann B, Ings R, Mansby J, Chamberlain J, McDonald A. Kinetics of intravenous and oral pentoxifylline in healthy subjects. Clin Pharmacol Ther. 1985;37:25–28. doi: 10.1038/clpt.1985.6. [DOI] [PubMed] [Google Scholar]
- Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Begriche K, Lettéron P, Abbey-Toby A, Vadrot N, Robin MA, Bado A, et al. Partial leptin deficiency favors diet-induced obesity and related metabolic disorders in mice. Am J Physiol Endocrinol Metab. 2008a;294:E939–E951. doi: 10.1152/ajpendo.00379.2007. [DOI] [PubMed] [Google Scholar]
- Begriche K, Massart J, Abbey-Toby A, Igoudjil A, Lettéron P, Fromenty B. β-aminoisobutyric acid prevents diet-induced obesity in mice with partial leptin deficiency. Obesity. 2008b;16:2053–2067. doi: 10.1038/oby.2008.337. [DOI] [PubMed] [Google Scholar]
- Begriche K, Knockaert L, Massart J, Robin MA, Fromenty B. Mitochondrial dysfunction in nonalcoholic steatohepatitis (NASH): are there drugs able to improve it. Drug Discov Today Dis Mech. 2009;6:e11–e23. [Google Scholar]
- Biour M, Ben Salem C, Chazouillères O, Grangé JD, Serfaty L, Poupon R. Drug-induced liver injury; fourteenth updated edition of the bibliographic database of liver injuries and related drugs. Gastroenterol Clin Biol. 2004;28:720–759. doi: 10.1016/s0399-8320(04)95062-2. [DOI] [PubMed] [Google Scholar]
- Bottomley PA. Spatial localization in NMR spectroscopy in vivo. Ann N Y Acad Sci. 1987;508:333–348. doi: 10.1111/j.1749-6632.1987.tb32915.x. [DOI] [PubMed] [Google Scholar]
- Buettner R, Schölmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity. 2007;15:798–808. doi: 10.1038/oby.2007.608. [DOI] [PubMed] [Google Scholar]
- Burcelin R, Crivelli V, Dacosta A, Roy-Tirelli A, Thorens B. Heterogeneous metabolic adaptation of C57BL/6J mice to high-fat diet. Am J Physiol Endocrinol Metab. 2002;282:E834–E842. doi: 10.1152/ajpendo.00332.2001. [DOI] [PubMed] [Google Scholar]
- Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009;284:26591–26602. doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerec V, Glaise D, Garnier D, Morosan S, Turlin B, Drenou B, et al. Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology. 2007;45:957–967. doi: 10.1002/hep.21536. [DOI] [PubMed] [Google Scholar]
- Chan MY, Zhao Y, Heng CK. Sequential responses to high-fat and high-calorie feeding in an obese mouse model. Obesity. 2008;16:972–978. doi: 10.1038/oby.2008.32. [DOI] [PubMed] [Google Scholar]
- Choi SS, Diehl AM. Hepatic triglyceride synthesis and non-alcoholic fatty liver injury. Curr Opin Lipidol. 2008;19:295–300. doi: 10.1097/MOL.0b013e3282ff5e55. [DOI] [PubMed] [Google Scholar]
- Dallas C, Gerbi A, Tenca G, Juchaux F, Bernard FX. Lipolytic effect of a polyphenolic citrus dry extract of red orange, grapefruit, orange (SINETROL) in human body fat adipocytes. Mechanism of action by inhibition of cAMP-phosphodiesterase (PDE) Phytomedicine. 2008;15:783–792. doi: 10.1016/j.phymed.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Delgado TC, Pinheiro D, Caldeira M, Castro MM, Geraldes CF, López-Larrubia P, et al. Sources of hepatic triglyceride accumulation during high-fat feeding in the healthy rat. NMR Biomed. 2009;22:310–317. doi: 10.1002/nbm.1327. [DOI] [PubMed] [Google Scholar]
- Dentin R, Benhamed F, Pégorier JP, Foufelle F, Viollet B, Vaulont S, et al. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J Clin Invest. 2005;115:2843–2854. doi: 10.1172/JCI25256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont J, Jossé R, Lambert C, Anthérieu S, Le Hegarat L, Aninat C, et al. Differential toxicity of heterocyclic aromatic amines and their mixture in metabolically competent HepaRG cells. Toxicol Appl Pharmacol. 2010;245:256–263. doi: 10.1016/j.taap.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic and clinical implications. Hepatology. 2010;51:679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2 Suppl 1):S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- Ferber S, Meyerovitch J, Kriauciunas KM, Kahn CR. Vanadate normalizes hyperglycemia and phosphoenolpyruvate carboxykinase mRNA levels in ob/ob mice. Metabolism. 1994;43:1346–1354. doi: 10.1016/0026-0495(94)90026-4. [DOI] [PubMed] [Google Scholar]
- Fromenty B, Vadrot N, Massart J, Turlin B, Barri-Ova N, Lettéron P, et al. Chronic ethanol consumption lessens the gain of body weight, liver triglycerides, and diabetes in obese ob/ob mice. J Pharmacol Exp Ther. 2009;331:23–34. doi: 10.1124/jpet.109.155168. [DOI] [PubMed] [Google Scholar]
- He Z, Jiang T, Wang Z, Levi M, Li J. Modulation of carbohydrate response element-binding protein gene expression in 3T3-L1 adipocytes and rat adipose tissue. Am J Physiol Endocrinol Metab. 2004;287:E424–E430. doi: 10.1152/ajpendo.00568.2003. [DOI] [PubMed] [Google Scholar]
- Hepgül G, Tanrikulu S, Unalp HR, Akguner T, Erbil Y, Olgaç V, et al. Preventive effect of pentoxifylline on acute radiation damage via antioxidant and anti-inflammatory pathways. Dig Dis Sci. 2010;55:617–625. doi: 10.1007/s10620-009-0780-x. [DOI] [PubMed] [Google Scholar]
- Igoudjil A, Abbey-Toby A, Begriche K, Grodet A, Chataigner K, Peytavin G, et al. High doses of stavudine induce fat wasting and mild liver damage without impairing mitochondrial respiration in mice. Antivir Ther. 2007;12:389–400. [PubMed] [Google Scholar]
- Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci U S A. 2004;101:15597–15602. doi: 10.1073/pnas.0405238101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, et al. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J Biol Chem. 2002;277:11019–11025. doi: 10.1074/jbc.M111041200. [DOI] [PubMed] [Google Scholar]
- Kellett GL, Brot-Laroche E, Mace OJ, Leturque A. Sugar absorption in the intestine: the role of GLUT2. Annu Rev Nutr. 2008;28:35–54. doi: 10.1146/annurev.nutr.28.061807.155518. [DOI] [PubMed] [Google Scholar]
- Kim SA, Marshall MA, Melman N, Kim HS, Müller CE, Linden J, et al. Structure-activity relationships at human and rat A2B adenosine receptors of xanthine derivatives substituted at the 1-, 3-, 7-, and 8-positions. J Med Chem. 2002;45:2131–2138. doi: 10.1021/jm0104318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppe SW, Sahai A, Malladi P, Whitington PF, Green RM. Pentoxifylline attenuates steatohepatitis induced by the methionine choline deficient diet. J Hepatol. 2004;41:592–598. doi: 10.1016/j.jhep.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Lebrec D, Thabut D, Oberti F, Perarnau JM, Condat B, Barraud H, et al. Pentoxifylline does not decrease short-term mortality but does reduce complications in patients with advanced cirrhosis. Gastroenterology. 2010;138:1755–1762. doi: 10.1053/j.gastro.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Lee YM, Sutedja DS, Wai CT, Dan YY, Aung MO, Zhou L, et al. A randomized controlled pilot study of pentoxifylline in patients with non-alcoholic steatohepatitis (NASH) Hepatol Int. 2008a;2:196–201. doi: 10.1007/s12072-008-9058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008b;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettéron P, Sutton A, Mansouri A, Fromenty B, Pessayre D. Inhibition of microsomal triglyceride transfer protein: another mechanism for drug-induced steatosis in mice. Hepatology. 2003;38:133–140. doi: 10.1053/jhep.2003.50309. [DOI] [PubMed] [Google Scholar]
- Lindström P. The physiology of obese-hyperglycemic mice [ob/ob mice] ScientificWorld Journal. 2007;7:666–685. doi: 10.1100/tsw.2007.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman HA, van Werven JR, Nederveen AJ, Ten Kate FJ, Heger M, Stoker J, et al. Noninvasive quantification of hepatic steatosis in tats using 3.0 T 1H-magnetic resonance spectrosopy. J Magn Reson Imaging. 2010;32:148–154. doi: 10.1002/jmri.22064. [DOI] [PubMed] [Google Scholar]
- Navarro-González JF, Jarque A, Muros M, Mora C, García J. Tumor necrosis factor-α as a therapeutic target for diabetic nephropathy. Cytokine Growth Factor Rev. 2009;20:165–173. doi: 10.1016/j.cytogfr.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Nicklasson M, Björkman S, Roth B, Jönsson M, Höglund P. Stereoselective metabolism of pentoxifylline in vitro and in vivo in humans. Chirality. 2002;14:643–652. doi: 10.1002/chir.10121. [DOI] [PubMed] [Google Scholar]
- Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues with thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- Paolisso G, Rizzo MR, Barbieri M, Manzella D, Ragno E, Maugeri D. Cardiovascular risk in type 2 diabetics and pharmacological regulation of mealtime glucose excursions. Diabetes Metab. 2003;29:335–340. doi: 10.1016/s1262-3636(07)70044-7. [DOI] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2007;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Reymann A, Braun W, Woermann C. Response of rat small intestinal active aldohexose transport to elevation of mucosal cyclic AMP by forskolin and 3-isobutyl-1-methylxanthine in vitro. Naunyn Schmiedebergs Arch Pharmacol. 1985;331:384–392. doi: 10.1007/BF00500824. [DOI] [PubMed] [Google Scholar]
- Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM, et al. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res. 2008;49:1068–1076. doi: 10.1194/jlr.M800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin MA, Demeilliers C, Sutton A, Paradis V, Maisonneuve C, Dubois S, et al. Alcohol increases tumor necrosis factor α and decreases nuclear factor-κB to activate hepatic apoptosis in genetically obese mice. Hepatology. 2005;42:1280–1290. doi: 10.1002/hep.20949. [DOI] [PubMed] [Google Scholar]
- Satapathy SK, Sakhuja P, Malhotra V, Sharma BC, Sarin SK. Beneficial effects of pentoxifylline on hepatic steatosis, fibrosis and necroinflammation in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22:634–638. doi: 10.1111/j.1440-1746.2006.04756.x. [DOI] [PubMed] [Google Scholar]
- Schattenberg JM, Galle PR. Animal models of non-alcoholic steatohepatitis: of mice and man. Dig Dis. 2010;28:247–254. doi: 10.1159/000282097. [DOI] [PubMed] [Google Scholar]
- Scotini E, Carpenedo F, Fassina G. New derivatives of methyl-xanthines: effect of thiocaffeine, thiotheophylline and 8-phenyltheophylline on lipolysis and on phosphodiesterase activities. Pharmacol Res Commun. 1983;15:131–143. doi: 10.1016/s0031-6989(83)80055-1. [DOI] [PubMed] [Google Scholar]
- Sharp PA, Debnam ES. The role of cyclic AMP in the control of sugar transport across the brush-border and basolateral membranes of rat jejunal enterocytes. Exp Physiol. 1994;79:203–214. doi: 10.1113/expphysiol.1994.sp003753. [DOI] [PubMed] [Google Scholar]
- Stickel F, Seitz HK. Alcoholic steatohepatitis. Best Pract Res Clin Gastroenterol. 2010;24:683–693. doi: 10.1016/j.bpg.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Stoffel M, Duncan SA. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4alpha regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad Sci U S A. 1997;94:13209–13214. doi: 10.1073/pnas.94.24.13209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha H, Grochot-Przeczek A, Was H, Kotlinowski J, Kozakowska M, Marek A, et al. Modulation of inflammatory response by pentoxifylline is independent of heme oxygenase-1 pathway. J Physiol Pharmacol. 2009;60:3–12. [PubMed] [Google Scholar]
- Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006;4:107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Vergès B. The impact of prandial glucose regulation in practice. Diabetes Nutr Metab. 2002;15(6 Suppl):28–32. [PubMed] [Google Scholar]
- Windmeier C, Gressner AM. Pharmacological aspects of pentoxifylline with emphasis on its inhibitory actions on hepatic fibrogenesis. Gen Pharmacol. 1997;29:181–196. doi: 10.1016/s0306-3623(96)00314-x. [DOI] [PubMed] [Google Scholar]
- Wright EM, Martín MG, Turk E. Intestinal absorption in health and disease-sugars. Best Pract Res Clin Gastroenterol. 2003;17:943–956. doi: 10.1016/s1521-6918(03)00107-0. [DOI] [PubMed] [Google Scholar]
- Zhou D, Brown SA, Yu T, Chen G, Barve S, Kang BC, et al. A high dose of ionizing radiation induces tissue-specific activation of nuclear factor-κB in vivo. Radiat Res. 1999;151:703–709. [PubMed] [Google Scholar]