

Abstract
BACKGROUND AND PURPOSE
The transactivation of the epidermal growth factor (EGF) receptor appears to be an important central transduction mechanism in mediating diabetes-induced vascular dysfunction. Angiotensin-(1-7) [Ang-(1-7)] via its Mas receptor can prevent the development of hyperglycaemia-induced cardiovascular complications. Here, we investigated whether Ang-(1-7) can inhibit hyperglycaemia-induced EGF receptor transactivation and its classical signalling via ERK1/2 and p38 MAPK in vivo and in vitro.
EXPERIMENTAL APPROACH
Streptozotocin-induced diabetic rats were chronically treated with Ang-(1-7) or AG1478, a selective EGF receptor inhibitor, for 4 weeks and mechanistic studies performed in the isolated mesenteric vasculature bed as well as in primary cultures of vascular smooth muscle cells (VSMCs).
KEY RESULTS
Diabetes significantly enhanced phosphorylation of EGF receptor at tyrosine residues Y992, Y1068, Y1086, Y1148, as well as ERK1/2 and p38 MAPK in the mesenteric vasculature bed whereas these changes were significantly attenuated upon Ang-(1–7) or AG1478 treatment. In VSMCs grown in conditions of high glucose (25 mM), an Src-dependent elevation in EGF receptor phosphorylation was observed. Ang-(1-7) inhibited both Ang II- and glucose-induced transactivation of EGF receptor. The inhibition of high glucose-mediated Src-dependant transactivation of EGF receptor by Ang-(1-7) could be prevented by a selective Mas receptor antagonist, D-Pro7-Ang-(1-7).
CONCLUSIONS AND IMPLICATIONS
These results show for the first time that Ang-(1-7) inhibits EGF receptor transactivation via a Mas receptor/Src-dependent pathway and might represent a novel general mechanism by which Ang-(1-7) exerts its beneficial effects in many disease states including diabetes-induced vascular dysfunction.
Keywords: diabetes, hyperglycaemia, vascular dysfunction, vascular smooth muscle cells, streptozotocin
Introduction
Candidate pathways that might play an important role in diabetes-induced cardiovascular dysfunction include GPCR-mediated signalling activated by angiotensin II (Ang II) and receptor tyrosine kinases such as epidermal growth factor (EGF) receptor. EGF receptor, a 175-kDa receptor tyrosine kinase, can be activated by several different EGF-like ligands including EGF and heparin binding-EGF (Ciardiello and Tortora, 2008) to induce receptor clustering and autophosphorylation. This subsequently leads to activation of multiple downstream signalling pathways such as the mitogenic Ras/Raf/ERK1/2, the p38 MAPK or the PI3-kinase/PKB (Akt) survival pathways that regulate cell growth, proliferation and differentiation (Ciardiello and Tortora, 2008; Cai et al., 2010; Avraham and Yarden, 2011). Alternatively, transactivation of EGF receptor can occur via GPCRs, such as angiotensin II (Ang II), thrombin, aldosterone and endothelin (Higuchi et al., 2007; Abdallah et al., 2010; Almendro et al., 2010; Liebmann, 2011; Smith et al., 2011). Depending on the specific cellular conditions, EGF receptor transactivation can occur via upstream kinases such as Src (a tyrosine-specific protein kinase encoded by the v-Src oncogene of Rous sarcoma virus), or involve metalloprotease and/or ADAM (a disintegrin and metalloprotease)-dependent shedding of cell-surface bound EGF-like ligands (Higuchi et al., 2007).
We previously showed that diabetes-induced vascular dysfunction was mediated by enhanced EGF receptor signalling, whereby chronic or acute treatment with AG1478, a selective inhibitor of EGF receptor tyrosine kinase, significantly corrected vascular dysfunction in the mesenteric bed, the renal vasculature and the carotid artery (Yousif et al., 2005; Benter et al., 2005a,b; 2009a). Gene expression profiling of the mesenteric vasculature showed that the correction in vascular dysfunction achieved by AG1478 was attained by blocking the up-regulation of the majority (∼85%) of the 1100+ genes whose expression had been altered in the diabetic mesenteric bed vasculature (Benter et al., 2009a). Betacellulin, a ligand for the ErbB family of receptors, when administered in mice also led to retinal vascular damage, thereby further implicating EGF receptor signalling in vascular dysfunction (Anand-Apte et al., 2010).Thus, the EGF receptor was proposed to be an important early detrimental pathway mediating vascular dysfunction in both an experimental model of type 1 diabetes (Benter et al., 2005a,b) and subsequently in a model of type 2 diabetes (Belmadani et al., 2008).
Strategies to inhibit formation and actions of Ang II are used extensively in clinical practice to prevent hypertension- or diabetes-induced end-organ damage. Angiotensin-(1-7) [Ang (1-7)] is a metabolite of Ang II that exhibits antihypertensive, antithrombotic and antiproliferative properties (Benter et al., 1993; 1995; Chappell, 2007; Gava et al., 2009; Tesanovic et al., 2010). Thus, Ang-(1-7) is thought to counterbalance or oppose the detrimental effects of Ang II (Iwai and Horiuchi, 2009; Ferrario et al., 2010; Hayashi et al., 2010). Ang-(1-7) functions through the G protein-coupled Mas receptor that is highly expressed in several tissues including the brain, heart, kidney and the vasculature (Gava et al., 2009). Ang-(1-7) produces relaxation of several vascular beds including coronary, cerebral, renal, pulmonary, femoral and mesenteric arteries (Ferrario et al., 2010). In humans, Ang-(1-7) attenuates Ang II-induced vasoconstriction in resistant vessels (Ueda et al., 2000). Recent studies have shown that treatment with Ang-(1-7) can attenuate end-organ damage in models of diabetes, hypertension and endothelial dysfunction (Benter et al., 2006, 2007; Oudit et al., 2010). In the case of diabetes-induced vascular dysfunction, we showed that the abnormal vascular reactivity to vasoactive agents such as noradrenaline, Ang II and carbachol observed in 4 weeks of experimental [streptozotocin (STZ) induced] diabetes was prevented by chronic Ang-(1-7) treatment (Benter et al., 2007) without a correction in hyperglycaemia in a manner analogous to that previously reported by us for AG1478, a selective inhibitor of EGF receptor signalling (Benter et al., 2005a,b). Ang-(1-7) inhibits NADPH oxidase and NF-κB in diabetic animals and attenuates oxidative stress (Benter et al., 2008; Al-Maghrebi et al., 2009). It is also known that Ang II signalling, including its transactivation of EGF receptor, is enhanced in diabetes. Thus, given the facts that both Ang-(1-7) administration and EGF receptor inhibition lead to a similar attenuation of diabetes-induced vascular dysfunction and that Ang-(1-7) counteracts the effects of Ang II, we hypothesized that Ang-(1-7) may counteract Ang II signalling by inhibiting EGF receptor transactivation in the diabetic vasculature. Hence, we investigated whether Ang-(1-7) can inhibit diabetes-induced EGF receptor transactivation and its classical signalling via ERK1/2 and p38 MAPK in the mesenteric vasculature of diabetic rats and both glucose- and Ang II-induced EGF receptor transactivation in cultured vascular smooth muscle cells (VSMCs). Indeed, our results show for the first time that Ang-(1-7) inhibits EGF receptor transactivation via a Mas receptor/Src-dependent pathway.
Methods
Drugs
AG1478 (N-(3-chlorophenyl)-6,7-dimethoxy-4-quinazolinanine hydrochloride) was purchased from Tocris (Bristol, UK). STZ, SU6656 ({2,3-dihydro-N, N-dimethyl-2-oxo-3-[{4,5,6,7-tetrahydro-1H-indol-2-yl}methylene]-1H-indole-5-sulphonamide), Losartan, Ang-(1-7) were all purchased from Sigma Chemical Co. (St Louis, MO, USA). D-Pro7-Ang-(1-7) was purchased from American Peptide Company (Sunnyvale, CA, USA).
In vivo studies
Male Wistar rats weighing about 300 g were used in this study and divided into the following groups (n = 8 per group). Group 1: non-diabetic (Control) animals; Group 2: STZ (55 mg·kg–1 body weight)-treated animals; Group 3: STZ + AG1478 (1 mg·kg–1 day–1 i.p.); Group 4: STZ + Ang-(1-7) (576 µg kg–1 day–1 i.p.). A total of 32 animals were used in this study. All animal care and experimental procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication no. 85–23, Revised 1985) as approved by Kuwait University Research Administration.
Induction of diabetes and treatment regimens
Diabetes was induced by a single i.p. injection of 55 mg·kg–1 body weight STZ dissolved in citrate buffer (pH 4.5). Age-matched control rats were injected with the citrate buffer vehicle used to dissolve STZ. Body weight and basal glucose levels were determined before the STZ injection, using an automated blood glucose analyzer (Glucometer Elite XL). Blood glucose concentrations were determined 48 h after STZ injection. Rats with a blood glucose concentration above 250 mg·dL–1 were declared diabetic. The animals’ body weights and the diabetic state were re-assessed after 4 weeks just before the animals were killed.
The regimen for drug administration [for AG1478 and Ang-(1-7)] was based on our previous studies in models of hypertension and/or diabetes (Benter et al., 1995; 2005a,b; 2006; 2007; 2009a; 2011). In all cases, the last dose of the pharmacological agents was administered at 4 pm the day before and animals were killed at 9 am the next day. This strategy leads to clinically relevant circulating levels of Ang-(1-7) in rats that are about 25-fold higher than basal (Benter et al., 2011).
Western blotting studies
Western blotting for total or phosphorylated forms of EGF receptor, Src, ERK1/2 and p38 MAPK was performed essentially as described by us previously (Petch et al., 2003; Hussain et al., 2004; Benter et al., 2005a,b; Hollins et al., 2007). Briefly, rat mesenteric vascular beds were isolated, snap-frozen in liquid nitrogen and stored at −80°C. The tissue samples were defrosted in ice then transferred to lysis buffer (pH 7.6) containing 50 mM Tris-base, 5 mM EGTA, 150 mM NaCl, 1% Triton 100, 2 mM Na3VO4, 50 mM NAF, 1 mM PMSF, 20 µM phenylarsine, 10 mM sodium molybdate, 10 µg·mL–1 leupeptin and 8 µg·mL–1 aprotinin, mixed three times with a sonicator probe (OMNI 2000, Omni International, Waterbury, CT, USA) at speed 2 for 20 s each time. The samples were left to lyse completely by incubation on ice for 30 min. Lysates were then centrifuged at 16 000×g for 20 min at 4°C and supernatants were collected and protein concentration estimated by Bio-Rad BCA protein assay (Hercules, CA, USA). Aliquots containing equal amounts of protein were subjected to SDS-PAGE and transferred onto nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany). Membranes were then incubated with either monoclonal antibodies (Cell Signaling, Danvers, MA, USA) to detect phosphorylated and total forms of EGF receptor (bands seen at approximately 175 kDa), Src (at approx. 60 kDa), ERK1/2 (at 42/44 kDa) or p38 MAPK (at 38 kDa) and subsequently with appropriate secondary antibodies conjugated to horseradish peroxidase (Amersham, Buckinghamshire, UK). Immunoreactive bands were detected with SuperSignal chemiluminescent substrate (Pierce, Cheshire, UK) using Kodak autoradiography film (G.R.I., Rayne, UK). To ensure equal loading of proteins, β-actin levels were detected using primary rabbit anti-human β-actin antibody followed by the secondary anti-rabbit IgG horse-radish peroxidase conjugated antibody (Cell Signaling). Images were finally analysed and quantified by densitometry and all data were normalized to β-actin levels.
VSMC studies
Primary rat aortic smooth muscle cell (VSMC) cultures were obtained by enzymatic dissociation of the thoracic aortas taken from untreated male Wistar rats essentially as described by us previously (Dhaunsi and Hassid, 1996; Muthalif et al., 1998). Briefly, four aortic fragments were thoroughly cleaned free of the adherent fatty tissue and the endothelium was removed by gently rubbing the lumen of the vessel in serum-free Dulbecco's modified Eagle's medium (DMEM; Sigma Chemical Co.). The fragments were then digested for 30 min at 37°C in 6 mL of DMEM that contained 1.5 mg·mL–1 BSA, 25 U mL−1 of pancreatic elastase (Sigma) and 200 U mL−1 collagenase (Type IX, Sigma) with gentle shaking in between. After the incubation period, the adventitial medial layer was removed, the fragments cut into small pieces and digested by incubation in 4 mL of digestion mixture for 45 min followed by washing twice with fresh DMEM and centrifugation. The resulting cell suspension was plated onto 25 cm2 culture flasks in DMEM-F12 HAM (Sigma) containing 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT, USA), 100 mg·mL–1 antibiotic-antimycotic (Invitrogen, Carlsbad, CA, USA) and 2 mL·L–1 insulin transferrin sodium selenate (Sigma), and in humidified conditions under 5% CO2. The VSMCs obtained were characterized as smooth muscle cells by morphology (multilayer sheets, ‘hills and valleys’) and immunostaining with monoclonal antibody specific for smooth muscle α-actin. Cells were passaged upon reaching confluence with 0.5% trypsin-containing 0.2% EDTA and utilized between passages 3 and 10. For the mechanistic studies, VSMCs were initially cultured in serum containing DMEM media until 60–70% confluence and then in serum-free DMEM media containing either normal (5 mM) or high (25 mM) d-glucose (or L-glucose as an osmotic control) and/or co-treated with different doses of the named drugs for 72 h. Cells were then lysed in cell lysis buffer, and total proteins were estimated and equivalent amounts of proteins were subjected to SDS-PAGE and immunoblotting as previously described for the mesenteric bed.
Statistical analysis
Data are presented as mean ± SEM of n number of experiments. Mean values were compared using analysis of variance followed by post hoc test (Bonferroni). Significant difference was considered when P value was less than 0.05.
Results
Hyperglycaemia and animals’ body weights
Induction of diabetes by STZ resulted in a significant increase in blood glucose concentration. Hyperglycaemia persisted in the diabetic animals and was 33.1 ± 1.5 mmol·L–1 after 4 weeks of diabetes as compared with 4.4 ± 0.8 mmol·L–1 in the non-diabetic control animals. Treatment with AG1478 (32.7 ± 0.9 mmol·L–1) or Ang-(1-7) (31.9 ± 1.1 mmol·L–1) did not significantly reduce blood glucose levels. There was a significant reduction of around 70 g in the weights of STZ-diabetic rats (154 ± 6 g) compared with the non-diabetic control animals (224 ± 4 g) after 4 weeks of diabetes, whereas AG1478 or Ang-(1-7) treatment significantly improved the weight of diabetic rats to 185 ± 8 g and 195 ± 10 g, respectively.
Ang-(1-7) inhibits hyperglycaemia-induced transactivation of EGF receptor in an animal model of diabetes
Four weeks of diabetes resulted in enhanced phosphorylation of EGF receptor at multiple tyrosine residues: Y992, Y1068, Y1086 and Y1148 that could be significantly attenuated by chronic treatment with Ang (1–7) or AG1478, a selective inhibitor of EGF receptor, in the mesenteric bed vasculature of STZ-induced diabetic rats (Figure 1A,C,D). Diabetes also induced increased expression of EGF receptor protein (Figure 1B) that could be prevented by Ang-(1-7) and AG1478 treatment (Figure 1A,B). Diabetes also enhanced phosphorylation of the downstream effectors ERK1/2 and p38 MAPK, an effect that was significantly attenuated upon chronic treatment with Ang (1–7) or AG1478 (Figure 2).
Figure 1.
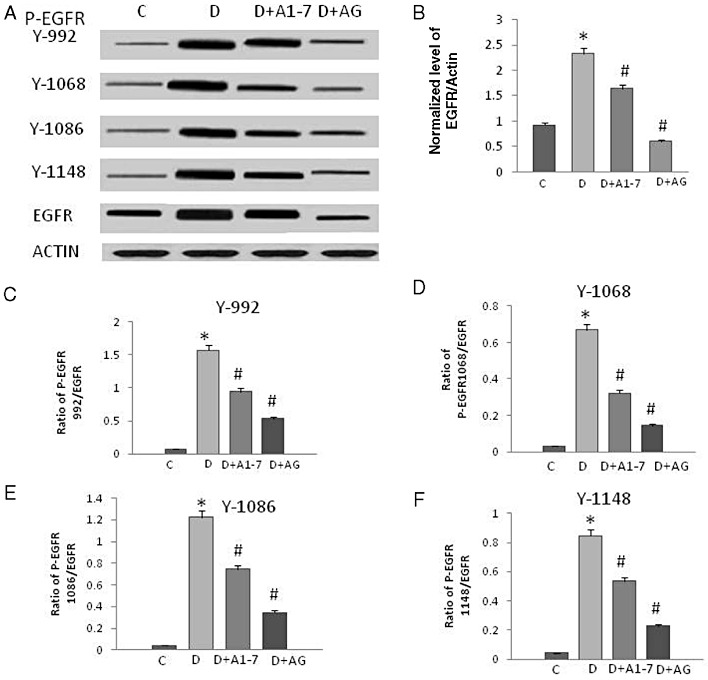
Diabetes-induced phosphorylation of EGF receptor occurs at multiple tyrosine residues that can be attenuated by chronic treatment with Ang-(1-7) or AG1478, a selective inhibitor of EGF receptor, in the mesenteric bed vasculature of STZ-induced diabetic rats. (A) A representative Western blot showing the levels of phosphorylated EGF receptor (P-EGFR) at the indicated tyrosines Y992, Y1068, Y1086 and Y1148, total EGF receptor (EGFR) and β-actin in the isolated mesenteric bed from normal controls (C), diabetic (D) and diabetic animals treated for 4 weeks with Ang-(1-7) (A1-7) or AG1478 (AG). (B–F) Densitometry histograms showing levels of total EGF receptor normalized to actin (B) and levels of phosphorylated EGF receptor at the stated tyrosine residue normalized to total EGF receptor (C–F). n = 6; mean ± SD. *Indicates significantly different (P < 0.05) mean values from normal non-diabetic rats (C), whereas # indicates significantly different mean values (P < 0.05) from diabetic rats (D).
Figure 2.
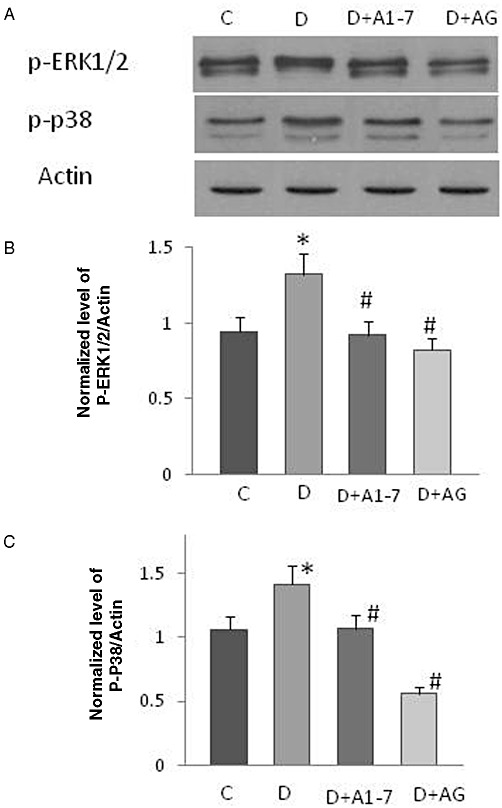
The effect of chronic treatment with Ang-(1-7) or AG1478 on ERK1/2 and p38 MAPK signalling in the isolated mesenteric bed of STZ-induced diabetic rats. (A) Representative Western blot showing the levels of phosphorylated ERK1/2 and p38 MAPK (p P38) in the isolated mesenteric bed from normal controls (C), diabetic (D) and diabetic animals treated for 4 weeks with Ang-(1-7) [A1-7]or AG1478 [AG]. (B–C) Densitometry histograms showing levels of phosphorylated (p-) ERK1/2 (B) and p38 MAPK (C) normalized to actin. n = 6; Mean ± SD. *Indicates significantly different (P < 0.05) mean values from normal non-diabetic rats (C), whereas # indicates significantly different mean values (P < 0.05) from diabetic rats (D).
Ang-(1-7) inhibits glucose-induced and Ang II-mediated transactivation of EGF receptor in VSMC
Culturing VSMC cells in 25 mM high glucose (Figure 3A,B), but not the osmotic control, L-glucose (Figure 3E,F) for 72 h led to significantly enhanced transactivation of EGF receptor as evidenced by phosphorylation of the receptor at Y1068 compared with those grown in normal glucose (5.5 mM). Glucose-induced EGF receptor transactivation was accompanied by enhanced phosphorylation of the downstream effectors, ERK1/2 and p38 MAPK (Figure 3A,C,D). Ang-(1-7) inhibited phosphorylation of EGF receptor, ERK1/2 and p38 MAPKs in a manner similar to that of the selective inhibitor of EGF receptor phosphorylation, AG1478 (Figure 3A–D).
Figure 3.
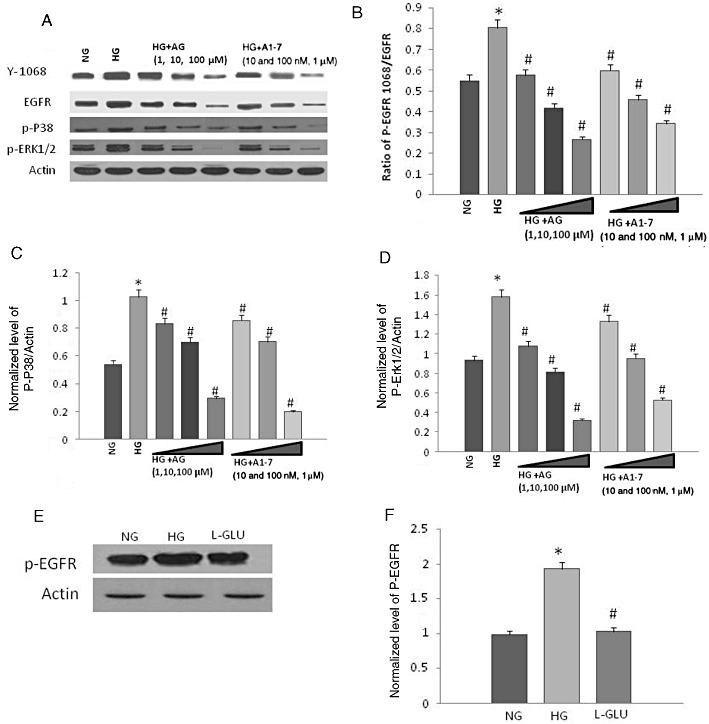
Ang-(1-7) is a dose-dependent inhibitor of high glucose-induced transactivation of epidermal growth factor (EGF) receptor at Y1068 and phosphorylation of p38 MAPK and ERK1/2 in a manner similar to AG1478 in VSMC. (A) Representative Western blot showing the levels of phosphorylated EGF receptor (Y1068), p38MAPK (p-P38) and ERK1/2 (p-ERK1/2) in VSMC grown in normal (5.5 mM) d-glucose (NG), high glucose (25.5 mM d-glucose; HG) or HG co-treated with increasing doses of AG1478 (AG) or Ang-(1-7) (labelled as A1-7). (B–D) Densitometry histograms showing levels of phosphorylated EGF receptor normalized to total EGF receptor (B) and levels of phosphorylated p38 MAPK (C) or ERK1/2 (D) normalized to actin. (E) and (F) Representative Western blot and histogram, respectively, showing that in contrast to 25 mM d-glucose (HG), the osmotic control, L-glucose (19.5 mM L-glucose + 5.5 mM d-glucose) had no significant effect on EGF receptor phosphorylation compared with normal glucose (5.5 mM glucose) in VSMC. n = 6; mean ± SD. *Indicates significantly different (P < 0.05) mean values from VSMC grown in normal 5.5 mM d-glucose (NG), whereas # indicates significantly different mean values (P < 0.05) from those grown in high glucose, 25.5 mM d-glucose (HG).
In VSMC cultured in normal glucose (5.5 mM), stimulation with Ang II led to transactivation of EGF receptor as evidenced by an enhanced phosphorylation at Y1068 as well as enhanced phosphorylation of ERK1/2 and p38MAPK (Figure 4). Ang-(1-7) inhibited Ang II-mediated phosphorylation of EGF receptor, ERK1/2 and p38 MAPKs in a manner similar to that of losartan, an Ang II type 1 receptor (AT1) receptor blocker, and AG1478 (Figure 4).
Figure 4.
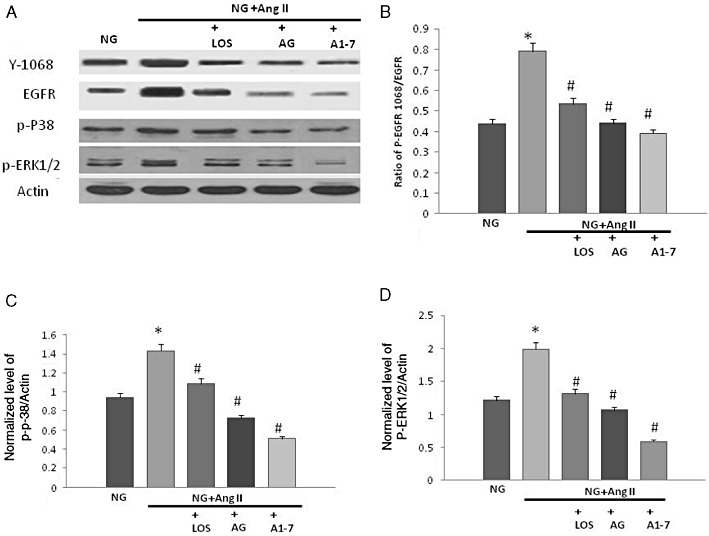
Ang-(1-7) inhibits Ang II-mediated transactivation of EGF receptor and signalling via p38 MAPK and ERK1/2 in VSMC in a manner similar to that of an AT1 receptor antagonist, losartan, and AG1478, a selective inhibitor of EGF receptor phosphorylation. (A) Representative Western blot showing the levels of phosphorylated EGF receptor (Y1068), p38MAPK (p-P38) and ERK1/2 (p-ERK1/2), total EGF receptor (EGFR) and actin in VSMC grown in normal (5.5 mM) d-glucose (NG), or treated with 1 µM Ang II, 1 µM losartan (LOS), 100 µM AG1478 (AG) or 1 µM Ang-(1-7) (labelled as A1-7); (B–D) Densitometry histograms showing levels of phosphorylated EGF receptor normalized to total EGF receptor (B) and levels of phosphorylated p38 MAPK (C) or ERK1/2 (D) normalized to actin. n = 6; mean ± SD. *Indicates significantly different (P < 0.05) mean values from VSMC grown in normal 5.5 mM d-glucose (NG), whereas # indicates significantly different mean values (P < 0.05) from those grown in high glucose 25.5 mM d-glucose (HG).
Ang-(1-7) via its Mas receptor inhibits glucose-induced EGF receptor transactivation by inhibiting Src phosphorylation in VSMC
High glucose (Figure 5A,B), but not the osmotic control L-glucose (Figure 5E,F), induced a significant enhancement in phosphorylation of Src (Y416) that was prevented in a dose-dependent manner by the selective Src inhibitor, SU6656 in VSMC (see Figure 5A,B). Inhibition of Src phosphorylation at Y416 by SU6656 also led to a dose-dependent inhibition of EGF receptor and ERK1/2 phosphorylation in VSMC (Figure 5C,D).
Figure 5.
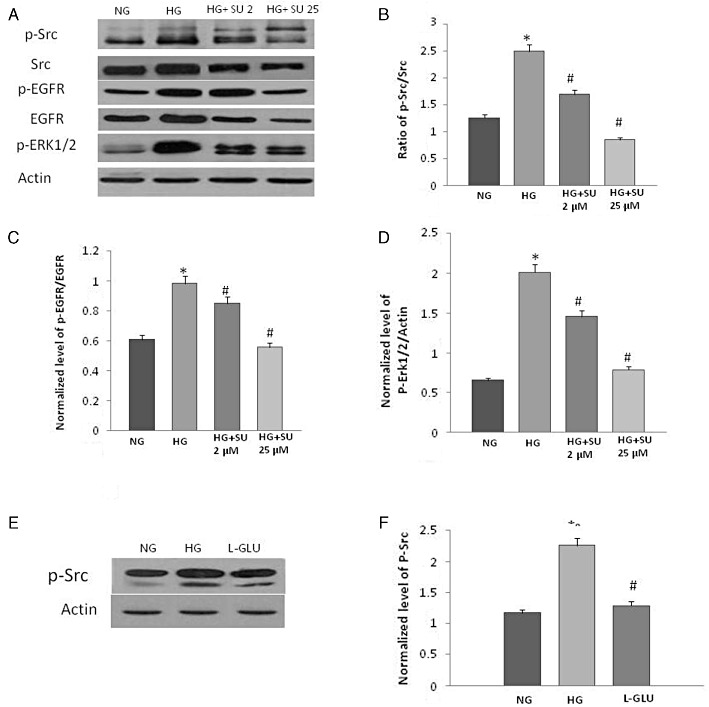
High glucose-mediated EGF receptor transactivation occurs via Src-depedent pathway. (A) Representative Western blot showing the levels of phosphorylated Src at Y436 (p-Src), total Src (Src), phosphorylated EGF receptor at Y1068 (p-EGFR), total EGF receptor (EGFR) and phosphorylated ERK1/2 in VSMC grown in normal (5.5 mM) d-glucose (NG), high glucose (25.5 mM) d-glucose (HG) or HG treated with increasing doses (2 and 25 µM) of Src selective inhibitor, SU6656 (lanes labelled as HG+ SU 2 and HG+ SU 25, respectively). (B–D) Densitometry histograms showing levels of phosphorylated Src (Y416) normalized to total Src (B), phosphorylated EGF receptor normalized to total EGF receptor (B) and levels of phosphorylated ERK1/2 (D) normalized to actin. (E) Representative Western Blot and (F) densitometry histogram showing that in contrast to 25 mM d-glucose (HG), the osmotic control, L-glucose (19.5 mM L-glucose + 5.5 mM d-glucose) had no significant effect on Src phosphorylation compared with normal glucose (5.5 mM glucose) in VSMC. n = 6; mean ± SD. *Indicates significantly different (P < 0.05) mean values from VSMC grown in normal 5.5 mM d-glucose (NG), whereas # indicates significantly different mean values (P < 0.05) from those grown in high glucose 25.5 mM d-glucose (HG).
Glucose-induced Src phosphorylation and subsequent signalling via the EGF receptor/ERK1/2 pathway could be inhibited by Ang-(1–7) (Figure 6). The inhibitory effect of Ang-(1-7) on Src/EGF receptor/ERK1/2 signalling could be blocked by the selective MAS receptor inhibitor [D-Pro7-Angiotensin-(1-7)] (Figure 6).
Figure 6.
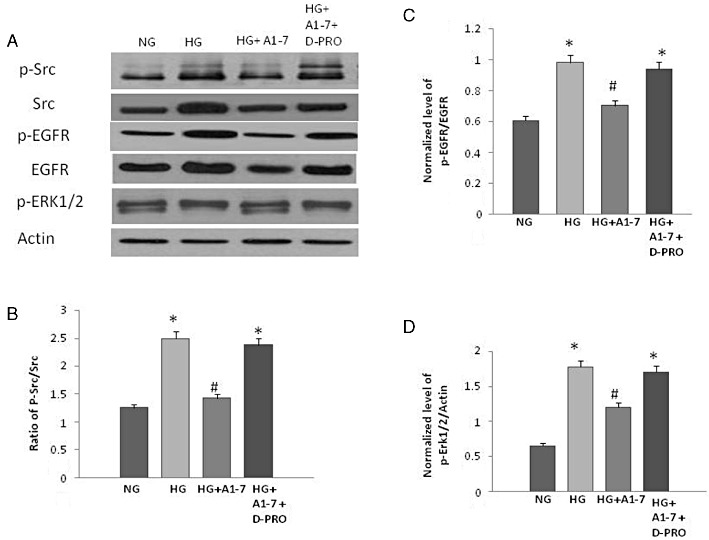
The effect of Ang-(1-7) and the selective Mas receptor inhibitor [D-Pro7-Ang-(1-7)] on glucose-induced Src phosphorylation and subsequent signalling via EGF receptor/ERK1/2 pathway in VSMC. (A) Representative Western blot showing the levels of phosphorylated Src at Y416 (p-Src), total Src (Src), phosphorylated EGF receptor at Y1068 (p-EGFR), total EGF receptor (EGFR) and phosphorylated ERK1/2 in VSMC grown in normal (5.5 mM) d-glucose (NG), high glucose (25.5 mM) d-glucose (HG) or HG treated with 1 µM Ang-(1-7) alone or together with 10 µM D-Pro7-Ang-(1-7) (lanes labelled as HG+A1-7 and HG+A1-7 + D-PRO, respectively). (B–D) Densitometry histograms showing levels of phosphorylated Src normalized to total Src (B), phosphorylated EGF receptor normalized to total EGF receptor (C), and levels of phosphorylated ERK1/2 (D) normalized to actin. n = 6; mean ± SD. *Indicates significantly different (P < 0.05) mean values from VSMC grown in normal 5.5 mM d-glucose (NG), whereas # indicates significantly different mean values (P < 0.05) from those grown in high glucose 25.5 mM d-glucose (HG).
Discussion
The major finding of the data presented in this study is that Ang-(1-7) is an inhibitor of EGF receptor transactivation and its subsequent signalling via ERK1/2 and p38 MAPK in vitro and in vivo.
We first showed that hyperglycaemia-induced EGF receptor phosphorylation occurs at multiple tyrosine residues in the diabetic mesenteric vascular bed. Chronic treatment of STZ-diabetic animals with Ang-(1-7) or AG1478, at doses and treatment regimens that prevented development of diabetes-induced vascular dysfunction in the mesenteric bed (Benter et al., 2005a; 2007) but did not correct hyperglycaemia, significantly inhibited EGF receptor phosphorylation at all of the sites studied with an associated reduction in phosphorylation of the downstream effectors, ERK1/2 and p38 MAPK. Part but not all of the enhanced phosphorylation of EGF receptor could be accounted for by an increased EGF receptor expression. Diabetes-induced elevation in EGF receptor expression could be inhibited by both Ang-(1-7) and AG1478 treatment (Figure 1). These data are consistent with our previous micro-array-based gene expression profiling study where diabetes significantly increased EGF receptor mRNA levels in the mesenteric vascular bed and this effect was prevented by AG148 treatment (Benter et al., 2009a).
To study this mechanism further, we next evaluated whether Ang-(1-7) could inhibit both glucose-induced and Ang II-induced EGF receptor transactivation in VSMC. Here we showed that both Ang-(1-7) and the selective EGF receptor inhibitor, AG1478, dose-dependently inhibited EGF receptor transactivation and subsequent downstream signalling via p38MAPK and ERK1/2 as a result of exposure to high glucose or stimulation with Ang II (Figures 3 and 4). These data imply that Ang-(1-7) can inhibit EGF receptor transactivation induced by multiple stimuli.
The EGF receptor transactivation by GPCRs appears to mediate several critical downstream signals and functions, such as ERK1/2 activation, and cell proliferation (Higuchi et al., 2007; Almendro et al., 2010; Liebmann, 2011). The exact mechanisms for EGF receptor transactivation by GPCRs are not entirely clear but can occur via multiple pathways including those involved in the elevation of intracellular Ca2+, activation of PKC and generation of reactive oxygen species (Higuchi et al., 2007; Almendro et al., 2010; Liebmann, 2011). Nonetheless, the two most widely reported mechanisms for EGF receptor transactivation are (i) via a metalloprotease-mediated ectodomain shedding of cell-surface bound ErbB family ligands or (ii) via phosphorylation of EGF receptor by a cytosolic non-receptor tyrosine kinase such as Src (Higuchi et al., 2007; Almendro et al., 2010). However, it is not clear whether Src is involved in high glucose-induced transactivation of EGF receptor.
We then showed for the first time that glucose-induced EGF receptor transactivation in VSMC occurs via activation of Src through its phosphorylation at Y416 and that Ang-(1-7) via its Mas receptor inhibits EGF receptor/ERK1/2 signalling through inhibition of Src phosphorylation (Figures 5 and 6). Dose-dependent inhibition of Src phosphorylation by SU6656, a selective c-Src tyrosine kinase inhibitor, led to inhibition of EGF receptor phosphorylation at Y1068 and phosphorylation of ERK1/2 confirming that Src phosphorylation was an upstream event of the EGF receptor/ERK1/2 signalling pathway. Further, we showed that Ang-(1-7)-mediated inhibition of Src/EGF receptor/ERK1/2 pathway could be completely reversed by Mas receptor blockade using the selective inhibitor, D-Pro7-Ang-(1-7), implying that Ang-(1-7)-mediated inhibition of Src-dependent EGF receptor phosphorylation occurs via its Mas receptor.
Figure 7 summarizes our current working hypothesis of the interplay between EGF receptor and Ang-(1-7) signalling in experimental type 1 diabetes and VSMC grown in high glucose. Diabetes or high glucose can induce Ang II-mediated transactivation of EGF receptor via an Src-dependent pathway that subsequently through signalling cascades involving p38 MAPK and ERK1/2 leads to vascular complications. It is likely that diabetes and/or hyperglycaemia might activate EGF receptor signalling via other pathways as well (as indicated in Figure 7). This is supported by data from recent clinical trials studies that have suggested that treatment of patients with Ang II (type 1A) receptor blockers such as losartan, as used in this study, cannot completely reverse vascular dysfunction (Izzo and Zion, 2011). Further, simply correction of hyperglycaemia per se also does not completely prevent the development of vascular complications in patients with diabetes (Gerstein et al., 2008), implying that other factors associated with the diabetic state could induce EGF receptor signalling – a possibility that needs further study. In any case, the findings of the present study show that Src-dependent EGF receptor transactivation can be inhibited by Ang-(1-7) acting through its Mas receptor (indicated in Figure 7) representing a novel mechanism of action for inhibiting EGF receptor and for counteracting the effects of Ang II.
Figure 7.
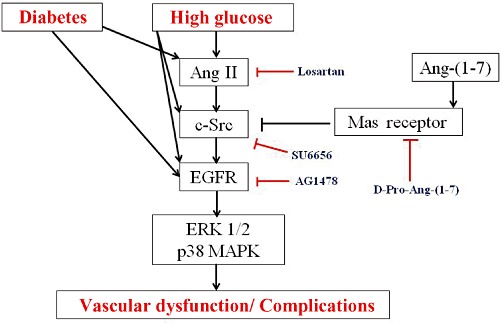
Model of how Ang-(1-7) via its Mas receptor might inhibit diabetes or high glucose-induced EGF receptor signalling and prevent vascular complications associated with diabetes and/or hyperglycaemia. The pharmacological inhibitors used in the present study are also indicated. (See text for further explanation.)
Taken together, these data support the notion that the beneficial effects of Ang-(1-7) on the diabetic vasculature involve inhibition of the detrimental EGF receptor/ERK1/2/p38MAPK pathway. Our findings lead us to speculate that this could represent a general mechanism by which Ang-(1-7) exerts its beneficial effects in many conditions especially where enhanced EGF receptor signalling has been implicated. In addition to its importance in diabetes-induced vascular complications, increased EGF receptor signalling is also thought to be an important mediator of kidney dysfunction in diabetes and in hypertension. Recent studies have confirmed that EGF receptor inhibition results in attenuation of kidney enlargement and albuminuria as well as preservation of podocytes and electrolyte homeostasis in models of diabetes (Wassef et al., 2004; Andrew et al., 2011; Gilbert et al., 2011; Panchapakesan et al., 2011). Our own studies have also suggested that enhanced EGF receptor and downstream Ras signalling are key mediators of kidney damage in an experimental model of hypertension (Benter et al., 2009b). The beneficial effects reported for Ang-(1-7) in diabetes and/or hypertension-induced vascular and renal complications (Benter et al., 2006; 2008; Dhaunsi et al., 2010; Ferrario et al., 2010; Giani et al., 2011), and possibly in other inflammatory conditions, are likely to be multifactorial but the data presented here suggest for the first time that, at least in part, they could involve attenuation of EGF receptor signalling.
Previous studies have shown that the beneficial effects of Ang-(1-7) in the cardiovascular system may involve modulation of ERK1/2, NADPH oxidase, Rho kinases, PKB and NF-kB activities that are also thought to be potential downstream effectors of the EGF receptor signalling cascade (Benter et al., 2008; 2009a; Al-Maghrebi et al., 2009; Ferrario et al., 2010; Gwathmey et al., 2011; Moon et al., 2011). In a recent study, Stegbauer et al. (2011) showed that chronic Ang-(1-7) treatment improves renal endothelial function via Mas receptors and this was associated with increased levels of endogenous NO and decreased NADPH levels in an experimental model of human cardiovascular disease. Although Ang-(1-7) may act through several mechanisms, our conclusion that Ang-(1-7) inhibits EGF receptor signalling via its Mas receptor is consistent with these previous findings and indeed, may offer a mechanistic explanation for the effects described.
Interestingly, chronic treatment with Ang-(1-7) or AG1478 significantly attenuated diabetes-induced weight loss to a similar extent. As to how the administration of Ang-(1-7) or EGF receptor inhibition mediates weight gain in diabetic animals is not clear as food intake was not monitored in this study. However, Kristensen and colleagues have reported that sustained administration of EGF, a ligand for EGF receptor, leads to reduced fat mass in rats despite an unaltered food intake (Kristensen et al., 1998; Pedersen et al., 2000). These authors proposed that EGF administration reduced body mass by enhancing activation of mitochondrial uncoupling proteins (UCP2 and UCP3) involved in increased energy expenditure (Pedersen et al., 2000) that would ultimately result in induction of lipolysis. Thus, attenuation of weight loss due to treatment with AG1478 or Ang-(1-7) during development of diabetes may be occurring via inhibition of the EGF/EGF receptor/UCP2,3-pathway and through inhibition of lipolysis. Furthermore, lipolysis, or more specifically, non-esterified fatty acid (NEFA) products of lipolysis, has an adverse effect on vascular reactivity (Egan et al., 1999) possibly via modulation of PKC activity and calcium homeostasis. NEFAs can lead to activation of PKC and have also been reported to directly activate EGF receptor (Vacaresse et al., 1999). Thus, it seems that EGF receptor-mediated signalling may be a key link between signal transduction pathways leading to weight loss and vascular dysfunction.
In conclusion, this study has highlighted a novel mechanism by which Ang-(1-7) may exert its many actions in the vasculature through inhibition of EGF receptor transactivation. Thus, inhibition of EGF receptor through activation of ACE2-Ang-(1-7)-Mas axis may offer an important alternative or additional approach to the use of ACE inhibitors or AT1 blockers in the treatment of diabetes-induced end-organ damage. Indeed, treatment with ACE inhibitors and/or AT1 blockers has already been shown to elevate levels of Ang-(1-7) probably due to increased Ang II levels and ACE2 expression. Moreover, the beneficial effects of ACE inhibitors and/or AT1 blockers are attenuated when given together with a Mas receptor antagonist (Iyer et al., 1998), implying that their beneficial effects may involve a contribution from Ang-(1-7) that, as suggested by this study, involves the inhibition of EGF receptor transactivation. Further, because ACE2 expression is decreased under diabetic conditions that reduce production of Ang-(1-7), it is possible that part of the reason for induction of vascular dysfunction in diabetes is removal of the inhibitory effects of ACE2 on EGF receptor transactivation. In addition, based on the present findings, Ang-(1-7) may offer an important alternative approach to the use of conventional tyrosine kinase inhibitors that exhibit considerable resistance problems such as in tumour patients.
Acknowledgments
This research project was funded by a Kuwait University Research Administration grant MR05/09.
Glossary
- AG1478
N-(3-chlorophenyl)-6,7-dimethoxy-4-quinazolinanine hydrochloride
- Ang
I, angiotensin I
- Ang
II, angiotensin II
- Ang
(1-7), angiotensin (1-7)
- DMEM
Dulbecco's modified Eagle's medium
- EGF
epidermal growth factor
- HB-EGF
heparin-binding EGF
- RAAS
renin-angiotensin-aldosterone system
- RTK
receptor tyrosine kinases
- STZ
streptozotocin
- SU6656
2,3-dihydro-N, N-dimethyl-2-oxo-3-[{4,5,6,7-tetrahydro-1H-indol-2-yl}methylene]-1H-indole-5-sulphonamide
- VSMC
vascular smooth muscle cell
Conflicts of interest
None.
References
- Abdallah RT, Keum JS, Lee MH, Wang B, Gooz M, Luttrell DK, et al. Plasma kallikrein promotes epidermal growth factor receptor transactivation and signaling in vascular smooth muscle through direct activation of protease-activated receptors. J Biol Chem. 2010;285:35206–35215. doi: 10.1074/jbc.M110.171769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Maghrebi M, Benter IF, Diz DI. Endogenous angiotensin-(1-7) reduces cardiac ischemia-induced dysfunction in diabetic hypertensive rats. Pharmacol Res. 2009;59:263–268. doi: 10.1016/j.phrs.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almendro V, García-Recio S, Gascón P. Tyrosine kinase receptor transactivation associated to G protein-coupled receptors. Curr Drug Targets. 2010;11:1169–1180. doi: 10.2174/138945010792006807. [DOI] [PubMed] [Google Scholar]
- Anand-Apte B, Ebrahem Q, Cutler A, Farage E, Sugimoto M, Hollyfield J, et al. Betacellulin induces increased retinal vascular permeability in mice. PLoS ONE. 2010;5:e13444. doi: 10.1371/journal.pone.0013444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew A, Kathryn JW, Alison JC, Yuan Z, Richard EG, Darren JK. Inhibition of the epidermal growth factor receptor preserves podocytes and attenuates albuminuria in experimental diabetic nephropathy. Nephrology (Carlton) 2011;16:573–581. doi: 10.1111/j.1440-1797.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–117. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- Belmadani S, Palen DI, Gonzalez-Villalobos RA, Boulares HA, Matrougui K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes. 2008;57:1629–1637. doi: 10.2337/db07-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benter IF, Diz DI, Ferrario CM. Cardiovascular actions of angiotensin-(1-7) Peptides. 1993;14:679–684. doi: 10.1016/0196-9781(93)90097-z. [DOI] [PubMed] [Google Scholar]
- Benter IF, Ferrario CM, Morris M, Diz DI. Antihypertensive actions of angiotensin-(1-7) in spontaneously hypertensive rats. Am J Physiol. 1995;269:H313–H319. doi: 10.1152/ajpheart.1995.269.1.H313. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MH, Griffiths SM, Benboubetra M, Akhtar S. Epidermal growth factor receptor tyrosine kinase-mediated signalling contributes to diabetes-induced vascular dysfunction in the mesenteric bed. Br J Pharmacol. 2005a;145:829–836. doi: 10.1038/sj.bjp.0706238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benter IF, Yousif MH, Hollins AJ, Griffiths SM, Akhtar S. Diabetes-induced renal vascular dysfunction is normalized by inhibition of epidermal growth factor receptor tyrosine kinase. J Vasc Res. 2005b;42:284–291. doi: 10.1159/000085904. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MH, Anim JT, Cojocel C, Diz DI. Angiotensin-(1-7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Am J Physiol Heart Circ Physiol. 2006;290:H684–H691. doi: 10.1152/ajpheart.00632.2005. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MH, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. Am J Physiol Heart Circ Physiol. 2007;292:H666–H672. doi: 10.1152/ajpheart.00372.2006. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MH, Dhaunsi GS, Kaur J, Chappell MC, Diz DI. Angiotensin-(1-7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am J Nephrol. 2008;28:25–33. doi: 10.1159/000108758. [DOI] [PubMed] [Google Scholar]
- Benter IF, Benboubetra M, Hollins AJ, Yousif MH, Canatan H, Akhtar S. Early inhibition of EGFR signaling prevents diabetes-induced up-regulation of multiple gene pathways in the mesenteric vasculature. Vascul Pharmacol. 2009a;51:236–245. doi: 10.1016/j.vph.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Benter IF, Canatan H, Benboubetra M, Yousif MH, Akhtar S. Global upregulation of gene expression associated with renal dysfunction in DOCA-salt-induced hypertensive rats occurs via signaling cascades involving epidermal growth factor receptor: a microarray analysis. Vascul Pharmacol. 2009b;51:101–109. doi: 10.1016/j.vph.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MH, Al-Saleh FM, Raghupathy R, Chappell MC, Diz DI. Angiotensin-(1-7) blockade attenuates captopril- or hydralazine-induced cardiovascular protection in spontaneously hypertensive rats treated with NG-nitro-L-arginine methyl ester. J Cardiovasc Pharmacol. 2011;57:559–567. doi: 10.1097/FJC.0b013e31821324b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Zhang H, Liu J, Berezov A, Murali R, Wang Q, et al. Targeting erbB receptors. Semin Cell Dev Biol. 2010;21:961–966. doi: 10.1016/j.semcdb.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS receptor axis: more than regulation of blood pressure? Hypertension. 2007;50:596–599. doi: 10.1161/HYPERTENSIONAHA.106.076216. [DOI] [PubMed] [Google Scholar]
- Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- Dhaunsi GS, Hassid A. Atrial and C-type natriuretic peptides amplify growth factor activity in primary aortic smooth muscle cells. Cardiovasc Res. 1996;31:37–47. [PubMed] [Google Scholar]
- Dhaunsi GS, Yousif MH, Akhtar S, Chappell MC, Diz DI, Benter IF. Angiotensin-(1-7) prevents diabetes-induced attenuation in PPAR-gamma and catalase activities. Eur J Pharmacol. 2010;638:108–114. doi: 10.1016/j.ejphar.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan BM, Lu G, Greene EL. Vascular effects of non-esterified fatty acids: implications for the cardiovascular risk factor cluster. Prostaglandins Leukot Essent Fatty Acids. 1999;60:411–420. doi: 10.1016/s0952-3278(99)80022-2. [DOI] [PubMed] [Google Scholar]
- Ferrario CM, Ahmad S, Joyner J, Varagic J. Advances in the renin angiotensin system focus on angiotensin-converting enzyme 2 and angiotensin-(1-7) Adv Pharmacol. 2010;59:197–233. doi: 10.1016/S1054-3589(10)59007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gava E, Samad-Zadeh A, Zimpelmann J, Bahramifarid N, Kitten GT, Santos RA, et al. Angiotensin-(1-7) activates a tyrosine phosphatase and inhibits glucose-induced signalling in proximal tubular cells. Nephrol Dial Transplant. 2009;24:1766–1773. doi: 10.1093/ndt/gfn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein HC, Miller ME, Byington RP, Goff DC, Jr, Bigger JT, Buse JB, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giani JF, Muñoz MC, Pons RA, Cao G, Toblli JE, Turyn D, et al. Angiotensin-(1-7) reduces proteinuria and diminishes structural damage in renal tissue of stroke-prone spontaneously hypertensive rats. Am J Physiol Renal Physiol. 2011;300:F272–F282. doi: 10.1152/ajprenal.00278.2010. [DOI] [PubMed] [Google Scholar]
- Gilbert RE, Huang Q, Thai K, Advani SL, Lee K, Yuen DA, et al. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 2011;79:1312–1321. doi: 10.1038/ki.2011.39. [DOI] [PubMed] [Google Scholar]
- Gwathmey TM, Pendergrass KD, Reid SD, Rose JC, Diz DI, Chappell MC. Angiotensin-(1-7)-angiotensin-converting enzyme 2 attenuates reactive oxygen species formation to angiotensin II within the cell nucleus. Hypertension. 2011;55:166–171. doi: 10.1161/HYPERTENSIONAHA.109.141622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi N, Yamamoto K, Ohishi M, Tatara Y, Takeya Y, Shiota A, et al. The counterregulating role of ACE2 and ACE2-mediated angiotensin 1-7 signaling against angiotensin II stimulation in vascular cells. Hypertens Res. 2010;33:1182–1185. doi: 10.1038/hr.2010.147. [DOI] [PubMed] [Google Scholar]
- Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci (Lond) 2007;112:417–428. doi: 10.1042/CS20060342. [DOI] [PubMed] [Google Scholar]
- Hollins AJ, Omidi Y, Benter IF, Akhtar S. Toxicogenomics of drug delivery systems: exploiting delivery system-induced changes in target gene expression to enhance siRNA activity. J Drug Target. 2007;15:83–88. doi: 10.1080/10611860601151860. [DOI] [PubMed] [Google Scholar]
- Hussain M, Shchepinov M, Sohail M, Benter IF, Hollins AJ, Southern EM, et al. A novel anionic dendrimer for improved cellular delivery of antisense oligonucleotides. J Control Release. 2004;99:139–155. doi: 10.1016/j.jconrel.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1-7)-Mas receptor axis. Hypertens Res. 2009;32:533–536. doi: 10.1038/hr.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SN, Ferrario CM, Chappell MC. Angiotensin-(1-7) contributes to the antihypertensive effects of blockade of the renin-angiotensin system. Hypertension. 1998;31:356–361. doi: 10.1161/01.hyp.31.1.356. [DOI] [PubMed] [Google Scholar]
- Izzo JL, Jr, Zion AS. Value of angiotensin receptor blocker therapy in diabetes. J Clin Hypertens (Greenwich) 2011;13:290–295. doi: 10.1111/j.1751-7176.2011.00447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen K, Pedersen SB, Vinter-Jensen L, Flyvbjerg A, Richelsen B. Systemic administration of epidermal growth factor reduces fat mass in rats: effects on the hormone-sensitive-lipase, lipoprotein lipase and leptin. Horm Res. 1998;50:292–296. doi: 10.1159/000023293. [DOI] [PubMed] [Google Scholar]
- Liebmann C. EGF receptor activation by GPCRs: a universal pathway reveals different versions. Mol Cell Endocrinol. 2011;331:222–231. doi: 10.1016/j.mce.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Moon JY, Tanimoto M, Gohda T, Hagiwara S, Yamazaki T, Ohara I, et al. Attenuating effect of angiotensin-(1-7) on angiotensin II-mediated NAD(P)H oxidase activation in type 2 diabetic nephropathy of KK-Ay/Ta mice. Am J Physiol Renal Physiol. 2011 doi: 10.1152/ajprenal.00065.2010. doi: 10.1152/ajprenal.00065.2010. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Muthalif MM, Benter IF, Karzoun N, Fatima S, Harper J, Malik KU. 20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin-dependent protein kinase II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Proc Natl Acad Sci USA. 1998;95:12701–12706. doi: 10.1073/pnas.95.21.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudit GY, Liu GC, Zhong J, Basu R, Chow FL, Zhou J, et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes. 2010;59:529–538. doi: 10.2337/db09-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchapakesan U, Pollock C, Saad S. Renal epidermal growth factor receptor: its role in sodium and water homeostasis in diabetic nephropathy. Clin Exp Pharmacol Physiol. 2011;38:84–88. doi: 10.1111/j.1440-1681.2010.05472.x. [DOI] [PubMed] [Google Scholar]
- Pedersen SB, Kristensen K, Bruun JM, Flyvbjerg A, Vinter-Jensen L, Richelsen B. Systemic administration of epidermal growth factor increases UCP3 mRNA levels in skeletal muscle and adipose tissue in rats. Biochem Biophys Res Commun. 2000;279:914–919. doi: 10.1006/bbrc.2000.4022. [DOI] [PubMed] [Google Scholar]
- Petch AK, Sohail M, Hughes MD, Benter I, Darling J, Southern EM, et al. Messenger RNA expression profiling of genes involved in epidermal growth factor receptor signalling in human cancer cells treated with scanning array-designed antisense oligonucleotides. Biochem Pharmacol. 2003;66:819–830. doi: 10.1016/s0006-2952(03)00407-6. [DOI] [PubMed] [Google Scholar]
- Smith NJ, Chan HW, Qian H, Bourne AM, Hannan KM, Warner FJ, et al. Determination of the exact molecular requirements for type 1 angiotensin receptor epidermal growth factor receptor transactivation and cardiomyocyte hypertrophy. Hypertension. 2011;57:973–980. doi: 10.1161/HYPERTENSIONAHA.110.166710. [DOI] [PubMed] [Google Scholar]
- Stegbauer J, Potthoff S, Quack I, Mergia E, Clasen T, Friedrich S, et al. Chronic treatment with angiotensin-(1-7) improves renal endothelial dysfunction in apolipoproteinE-deficient mice. Br J Pharmacol. 2011;163:974–983. doi: 10.1111/j.1476-5381.2011.01295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesanovic S, Vinh A, Gaspari TA, Casley D, Widdop RE. Vasoprotective and atheroprotective effects of angiotensin-(1-7) in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2010;30:1606–1613. doi: 10.1161/ATVBAHA.110.204453. [DOI] [PubMed] [Google Scholar]
- Ueda S, Masumori-Maemoto S, Ashino K, Nagahara T, Gotoh E, Umemura S, et al. Angiotensin-(1-7) attenuates vasoconstriction evoked by angiotensin II but not by noradrenaline in man. Hypertension. 2000;35:998–1001. doi: 10.1161/01.hyp.35.4.998. [DOI] [PubMed] [Google Scholar]
- Vacaresse N, Lajoie-Mazenc I, Augé N, Suc I, Frisach MF, Nègre-Salvayre A. Activation of epithelial growth factor receptor pathway by unsaturated fatty acids. Circ Res. 1999;85:892–899. doi: 10.1161/01.res.85.10.892. [DOI] [PubMed] [Google Scholar]
- Wassef L, Kelly DJ, Gilbert RE. Epidermal growth factor receptor inhibition attenuates early kidney enlargement in experimental diabetes. Kidney Int. 2004;66:1805–1814. doi: 10.1111/j.1523-1755.2004.00955.x. [DOI] [PubMed] [Google Scholar]
- Yousif MH, Benter IF, Akhtar S. The role of tyrosine kinase-mediated pathways in diabetes-induced alterations in responsiveness of rat carotid artery. Auton Autacoid Pharmacol. 2005;25:69–78. doi: 10.1111/j.1474-8673.2004.00333.x. [DOI] [PubMed] [Google Scholar]