

Abstract
BACKGROUND AND PURPOSE
Many cells express proteinase activated receptor 2 (PAR2) on their plasma membrane. PAR2 is activated by proteolytic enzymes, such as trypsin and tryptase that cleave the receptor N-terminus, inititating signalling to intracellular G proteins. Studies on PAR2 have relied heavily upon activating effects of proteases and peptide agonists that lack stability and bioavailability in vivo.
EXPERIMENTAL APPROACH
A novel small molecule agonist GB110 and an antagonist GB88 were characterized in vitro against trypsin, peptide agonists, PAR2 antibody, PAR1 agonists and flow cytometry,in seven cell lines using intracellular Ca2+ mobilization and examined in vivo against PAR2- and PAR1-induced rat paw oedema.
KEY RESULTS
GB110 is a potent non-peptidic agonist activating PAR2-mediated Ca2+ release in HT29 cells (EC50∼200 nM) and six other human cell lines, inducing PAR2 internalization. GB88 is a unique PAR2 antagonist, inhibiting PAR2 activated Ca2+ release (IC50∼2 µM) induced by native (trypsin) or synthetic peptide and non-peptide agonists. GB88 was a competitive and surmountable antagonist of agonist 2f-LIGRLO-NH2, a competitive but insurmountable antagonist of agonist GB110, and a non-competitive insurmountable antagonist of trypsin. GB88 was orally active and anti-inflammatory in vivo, inhibiting acute rat paw oedema elicited by agonist GB110 and proteolytic or peptide agonists of PAR2 but not by corresponding agonists of PAR1 or PAR4.
CONCLUSIONS AND IMPLICATIONS
The novel PAR2 agonist and antagonist modulate intracellular Ca2+ and rat paw oedema, providing novel molecular tools for examining PAR2-mediated diseases.
Keywords: proteinase activated receptor 2, agonist, antagonist, anti-inflammatory
Introduction
PAR2 is one of four known proteinase activated receptors (PARs) (Bohm et al., 1996; MacFarlane et al., 2001; Coughlin and Camerer, 2003; Steinhoff et al., 2005; Barry et al., 2006; receptor nomenclature follows Alexander et al., 2011), which are functionally unlike other membrane-spanning GPCRs (Blakeney et al., 2007) in being activated by serine proteases. There are no endogenous ligands known for PAR2. However, nM concentrations of trypsin-like serine proteases can cleave the receptor N-terminus to expose a membrane-anchored signalling sequence (SLIGKV, SLIGRL), which perturbs the membrane-spanning domain of PAR2 and directs intracellular signalling via coupled G proteins (Bohm et al., 1996; MacFarlane et al., 2001; Coughlin and Camerer, 2003; Steinhoff et al., 2005; Barry et al., 2006). Hexapeptides corresponding to the new N-terminus of the receptor (SLIGKV-NH2, SLIGRL-NH2) can also activate PAR2 at µM concentrations (Hollenberg et al., 1996), while some hexapeptides with heterocycles at the N-terminus are PAR2 agonists at high nM concentrations (Kawabata et al., 2004; Kanke et al., 2005; Barry et al., 2007).
PAR2 has been proposed to be important in inflammation (Niu et al., 2008; Shin et al., 2009) and proliferation (Nishibori et al., 2005; Arisawa et al., 2007; Matej et al., 2007; Iwaki et al., 2008). Based mainly on the properties of these exogenous peptide agonists, together with some knockout mouse studies, activation of PAR2 has been purported to be pro-inflammatory in arthritis (Ferrell et al., 2003; Kelso et al., 2007; Lam, 2007; McIntosh et al., 2007), pancreatitis (Maeda et al., 2005; Hirota et al., 2006), gastric (Hansen et al., 2005; Cottrell et al., 2007; Kawabata et al., 2008) and kidney (Moussa et al., 2007; Vesey et al., 2007a,b) cells, but anti-inflammatory in airways inflammation (Cocks et al., 1999; D'Agostino et al., 2007). Increased expression of PAR2 associated with proliferation has been consistently found in many types of cancer cells, with reports on PAR2-induced cancer progression in breast (Matej et al., 2007), colon (Nishibori et al., 2005), gastric (Arisawa et al., 2007) and pancreatic cancers (Iwaki et al., 2008). However, roles for PAR2 in vivo are still somewhat speculative, in part due to the unavailability of very potent, in vivo stable and bioavailable PAR2 agonists and antagonists as physiologically useful tools. Use of siRNA, blocking antibodies and gene deletion has met with limited success (Ferrell et al., 2003) in clarifying roles for PAR2 in physiology and disease, partly due to poor selectivity or inbuilt redundancies. Recently (Suen et al., 2010), we profiled effects of PAR2 activation on gene expression in HEK293 cells, a commonly studied cell line in PAR2 research, by examining intersecting up- or down-regulation induced by both an endogenous protease PAR2 activator (trypsin) and a PAR2 activating hexapeptide (2f-LIGRLO-NH2). From a 19 000 human gene set, there were clear associations between PAR2 activation and cell metabolism (1000 genes), the cell cycle, the MAPK pathway, HDAC and sirtuin enzymes, inflammatory cytokines, anti-complement function and cancer, as well as support for developing both antagonists and agonists of PAR2 as potential disease-modifying therapeutic agents.
Only an extremely weak non-specific antagonist (ENMD-1068, IC50 5 mM) (Kelso et al., 2006), and a peptidomimetic antagonist (K-14585, IC50 10 µM) (Kanke et al., 2009) that does not inhibit trypsin-activation of PAR2, are currently available to interrogate PAR2 function in vivo. Here we report some properties of two new, low MW, non-peptidic compounds that regulate PAR2 activity in vitro and in vivo. The agonist GB110 was equipotent in vitro with the best synthetic peptide agonist reported for PAR2, 2-furoyl-LIGRLO-NH2 (2f-LIGRLO-NH2), while the antagonist GB88 is a member of the first class of PAR2 antagonists (Barry et al., 2010) that inhibits the functions of both endogenous and synthetic PAR2 agonists, but not PAR1 agonists, at low µM concentrations. GB88 was an orally active inhibitor of PAR2 activation in vivo with anti-inflammatory activity in the rat.
Methods
Cell culture
Seven human cell lines (HT29, A549, Panc-1, MKN45, MKN1, HUVEC and MDA-MB-231) and one rat cell line were used to study the effect of PAR2 compounds. Cell lines were cultured in medium at 37°C and 5% CO2 based on information provided by ATCC (Manassas, VA, USA). During cell culture passage, cell dissociation solution (Sigma Aldrich, St Louis, MO, USA) was used to dissociate cells from the surface of culture flasks.
Intracellular calcium mobilization
Cells were grown to 80% confluence. Before an experiment, cells were seeded overnight in 96-well black-walled, clear bottom, plates at ∼4 × 105 cells per well. On the day of experiment, supernatant was removed and cells were incubated in dye loading buffer (HBSS with 4 µM Fluo-3, 25 µL pluronic acid, 1% fetal bovine serum and 2.5 mM probenecid) for 1 h at 37°C. Cells were then washed twice with HBSS and transferred to a Polarstar spectrofluorimeter (BMG, Durham, NC, USA) for agonist injection and fluorescence measurements. PAR2 agonists at various concentrations were added 10 s after reading commenced and fluorescence was measured in real time from the bottom of the plate using excitation at λ= 480 nm and emission at 520 nm. HBSS was prepared in-house. All other reagents and calcimycin (A23187) were purchased from Invitrogen (Carlsbad, CA, USA), the latter for measuring maximum fluorescence. Plates were purchased from DKSH (Zurich, Switzerland).
Antagonist surmountability
Cells were prepared as for the calcium mobilization assay. After 1 h incubation with dye loading buffer, cells were incubated with different concentrations of antagonist for 15 min. The plate was then transferred to the Polarstar spectrofluorimeter and examined for concentration-dependent effects on the activity of agonists in the presence of different concentrations of antagonist.
Receptor internalization
Cells were grown to 90% confluence in a tissue culture flask, then removed and suspended at 5 × 106 cells·mL−1 in HBSS. Aliquots of cells were incubated with various concentrations of agonists (30 min, 37°C) then placed on ice (5 min), centrifuged and re-suspended in assay buffer (PBS, 0.1% w/v BSA, 0.01% w/v NaN3, pH 7.4). Cells were treated with goat anti-PAR2 antibody (N-19, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:50 dilution (30 min on ice), washed twice with buffer and incubated with anti-goat IgG conjugate with Alexa Fluor 488 (A-11078, Invitrogen, 1:500) for 30 min on ice. Upon completion, cells were centrifuged and resuspended in PBS containing 1% paraformaldehyde. Samples were analysed by flow cytometry (FACScalibur, BD Biosciences, Frankin Lakes, NJ, USA).
PAR2-induced paw oedema
All animal care and experimental procedures complied with the Guidelines of the Australian National Health and Medical Research Council and were approved by the animal ethics committee of The University of Queensland. Male Wistar rats (8–9 weeks) were injected with 100 µL of isotonic saline containing 2f-LIGRLO-NH2 (350 µg per paw), trypsin (20 µg per paw), SLIGRL-NH2 (2 mg per paw), GB110 (350 µg per paw), thrombin (5 U per paw), TFLLR-NH2 (2 mg per paw) or AYPGKF-NH2 (2 mg per paw) into the plantar surface of the right hind paw pad using a 30G needle, the left hind paw receiving 100 µL saline alone, as described earlier (Vergnolle et al., 1999; Kelso et al., 2006). GB88 (5 or 10 mg·kg−1 in 500 µL olive oil) was administered 120 min before paw injections by oral gavage, while control animals received olive oil only by gavage. Paw thickness and width were measured using digital calipers (WPI, Sarasota, FL, USA) at 0, 0.5, 1 and 2 h after PAR2 agonist administration. Hind paw size was expressed as % change in area from baseline, right (affected) paw compared with left (control) paw.
Statistical analysis
Data are presented as means ± SEM. Data were analysed in GraphPad Prism using anova or Student's t-test. Differences between means with a P-value <0.05 were considered significant.
Materials
All cell culture reagents were purchased from Invitrogen and Sigma Aldrich. PAR2-activating agonist peptide 2f-LIGRLO-NH2, agonist GB110 and antagonist GB88, PAR1-activating peptide TFLLR-NH2, PAR4 activating peptide AYPGKF-NH2 were synthesized in-house. Goat anti-PAR2 antibody was purchased from Santa Cruz Biotechnology and anti-goat antibody conjugated to Alexa-Fluor 488 was purchased from Invitrogen. The rat cell line NRK-52e was a gift from Dr David Vesey from the Princess Alexandra Hospital, Brisbane, Australia.
Results
GB110 is a novel non-peptidic PAR2 agonist
GB110 comprises an N-terminal isoxazole, unnatural (cyclohexylalanine) and natural (isoleucine) amino acids, a para-carboxymethyl aniline, and a C-terminal aminomethyl piperidine (Figure 1A). Its design and chemical synthesis have been recently reported by our group (Barry et al., 2010). This compound does not possess a proteolytically cleavable peptide bond and is serum stable, thus sharing characteristics of other non-peptidic, low MW organic compounds. GB110 is selective for PAR2 (Barry et al., 2010) and failed to induce a response in a PAR2-negative cell line (Supporting Information Figure S1). In an intracellular Ca2+ (iCa2+) mobilization assay using HT29 colon cancer cells (Figure 1B), GB110 (EC50 240 ± 20 nM; pEC50 6.7 ± 0.05) was equipotent with the peptide agonist 2f-LIGRLO-NH2 (EC50 210 ± 30 nM; pEC50 6.6 ± 0.05), 10-fold more potent than SLIGRL-NH2 (Barry et al., 2010), but ∼35-fold less potent than trypsin (EC50 6 ± 0.5 nM; pEC50 8.2 ± 0.8). Despite similar agonist potencies in this assay, GB110 was an order of magnitude less potent than 2f-LIGRLO-NH2 in inducing PAR2 internalization (Figure 1C), as detected by anti-PAR2 antibody using FACS. This suggests different mechanisms for G-protein activation and (β-arrestin-dependent) receptor internalization. However, cells treated with either GB110 or 2f-LIGRLO-NH2 re-expressed PAR2 on the cell surface at a similar rate (Figure 1D), indicating that post-activation recovery of receptor sensitivity is unaffected by differences in agonists.
Figure 1.
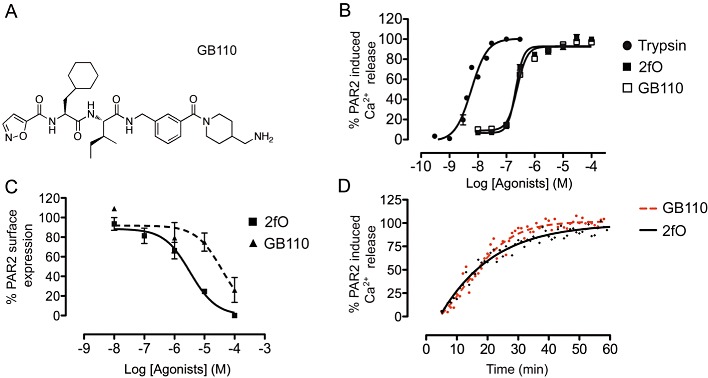
Profile of a non-peptide PAR2 agonist, GB110 in HT29 cells. (A) Structure of non-peptide PAR2 agonist, GB110. (B) Comparison of PAR2 agonist-induced intracellular Ca2+ (iCa2+) release by trypsin (pEC50 8.2 ± 0.08), 2f-LIGRLO-NH2 (2fO; pEC50 6.7 ± 0.05) and GB110 (pEC50 6.6 ± 0.05). GB110 is equipotent with the known PAR2 activating peptide, 2f-LIGRLO-NH2. (C) Comparison of PAR2 agonist-induced receptor internalization by 2f-LIGRLO-NH2 and GB110. (D) Recovery of PAR2-induced Ca2+ release after PAR2 desensitization by 5 µM 2f-LIGRLO-NH2 and 5 µM GB110. Data shown are means (+SEM) of ≥3 experiments in triplicate.
GB110 also behaves in a similar manner to trypsin and 2f-LIGRLO-NH2 in activating iCa2+ release in HUVEC and five human cancer cell lines (A549, Panc-1, MDA-MB-231, MKN1 and MKN45) (Figure 2). Similar to HT-29, these cell lines all expressed PAR2 at significant levels (Supporting Information Figure S2). All three agonists were two orders of magnitude less potent in MKN45 cells. Despite an almost 100-fold variation in EC50 values between cell lines (200 nM to 20 µM), GB110 consistently showed the same level of activity as 2f-LIGRLO-NH2 but one to two orders of magnitude lower potency than trypsin in all cell lines tested.
Figure 2.
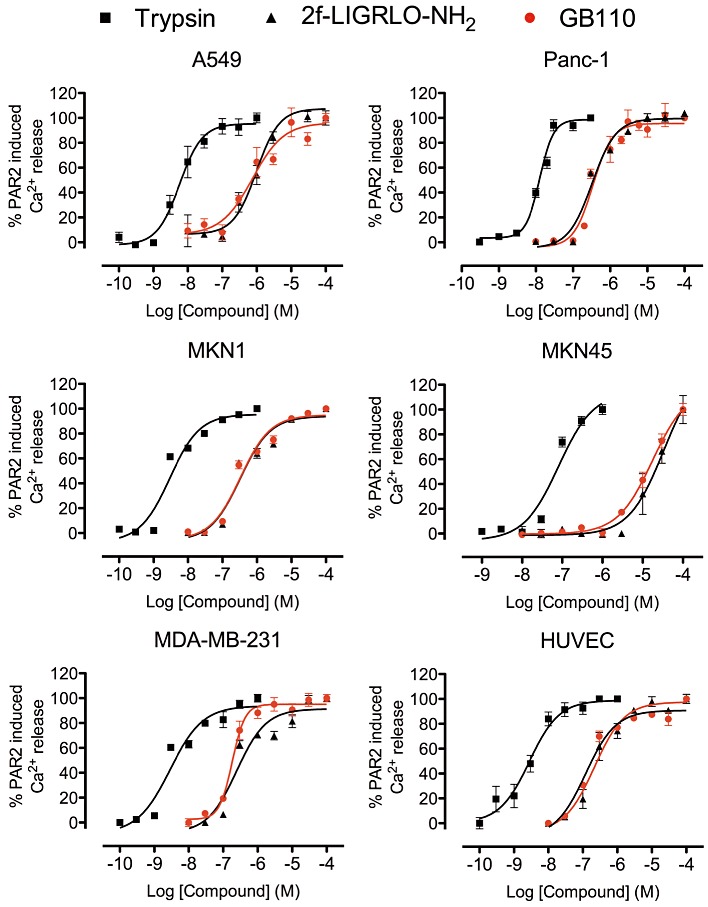
Intracellular Ca2+ mobilization by PAR2 agonists in six human cell lines. Concentration-dependent curves for iCa2+ mobilization by PAR2 agonists, trypsin, 2f-LIGRLO-NH2 and GB110 in six cell lines (A549, Panc-1, MKN1, MKN45, MDA-MB231, HUVEC). 2f-LIGRLO-NH2 and GB110 were equipotent. Data shown are means (+SEM) of ≥3 experiments in triplicate.
GB88 is a non-peptide antagonist of PAR2
GB88 is a novel analogue of GB110, with the same N-terminal isoxazole, L-cyclohexylalanine and L-isoleucine, but with a bulky C-terminal spiroindenepiperidine that confers PAR2 antagonism at low µM concentrations (Figure 3). This antagonist GB88 had comparable potency in inhibiting three structurally and mechanistically different PAR2 agonists (trypsin, 2f-LIGRLO-NH2, GB110) in HT29 cells (Figure 3A, IC50 2–9 µM, pIC50 5–6, n≥ 3), but not certain other GPCRs (Supporting Information Figure S3).
Figure 3.
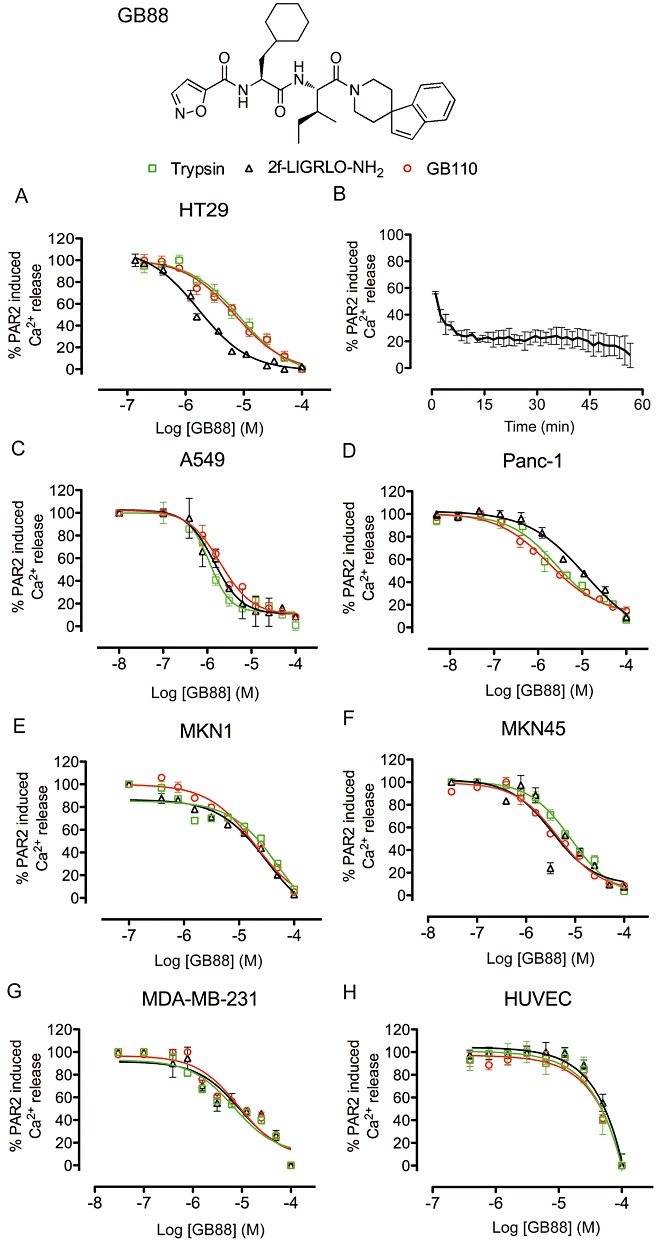
Profile of a non-peptide PAR2 antagonist, GB88, in human cell lines. (A) PAR2 antagonist GB88 inhibits iCa2+ release induced in HT29 by trypsin, 2f-LIGRLO-NH2 and GB110. (B) Maximum GB88 inhibition (10 µM) of PAR2 activation after 15 min and stable for at least 1 h. (C-H) PAR2 antagonist GB88 inhibits iCa2+ release induced in all six cell types (A549, Panc-1, MKN1, MKN45, MDA-MB231, HUVEC) by trypsin, 2f-LIGRLO-NH2 and GB110, with IC50 < 10 µM except HUVEC. Data shown are means (+SEM) of ≥3 experiments in triplicate.
A temporal study of PAR2 antagonism by GB88 revealed that the compound has a slow on-rate, requiring up to 15 min incubation to fully saturate PAR2 (Figure 3B) and the inhibitory effect of GB88 was stable up to 1 h after application in vitro. GB88 was also an equipotent antagonist of all three structurally and functionally different PAR2 agonists (trypsin, GB110, 2f-LIGRLO-NH2) when tested against their EC80, in each of seven different human cell lines (Figure 3C–H). While the antagonist had the same effect on the three agonists in each cell line, the potency of the antagonist varied between cell types, from IC50 1–10 µM (pIC50 5–6; HT29, A549, Panc-1, MKN45, MDA-MB-231) to >10 µM (pIC50 < 5; MKN1) to >80 µM in HUVEC cells (pIC50 < 4.1; Figure 3C–H).
Antagonism by GB88 is agonist dependent
The mechanism of antagonism of GB88 was found to be agonist dependent. This compound was a surmountable and reversible antagonist against hexapeptide agonist 2f-LIGRLO-NH2, as increasing concentrations of antagonist resulted in a horizontal shift in the agonist concentration-response curves (Figure 4A). The absence of a significant reduction of the maximum response indicated that GB88 is completely displaced by high concentrations (1 mM) of agonist 2f-LIGRLO-NH2. This is further supported by a Schild plot with a linear slope of 0.99, establishing competitive antagonism (Figure 4D) and a calculated pA2 6.3 ± 0.29.
Figure 4.
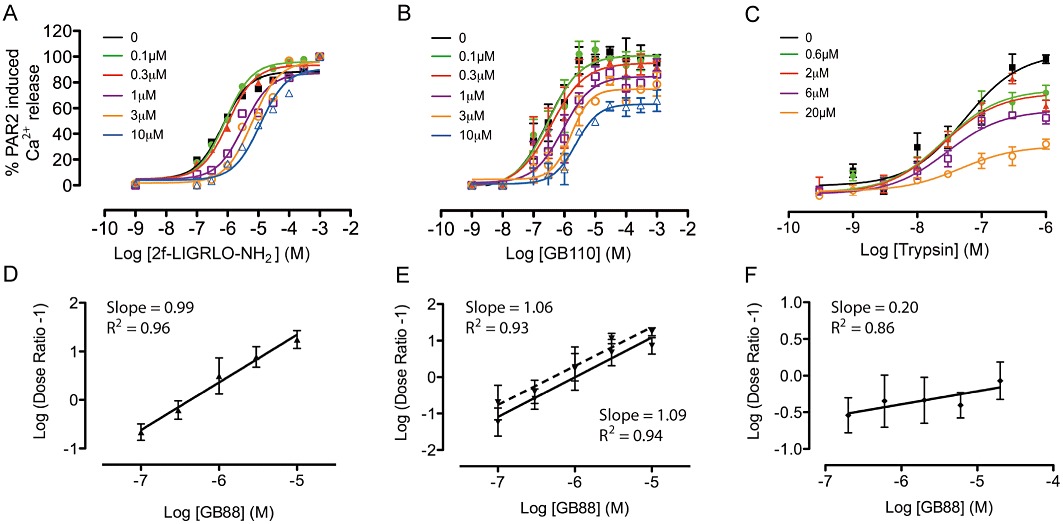
Mechanism of PAR2 antagonism. (A) GB88 is a surmountable PAR2 antagonist against 2f-LIGRLO-NH2, showing concentration-dependent (0–10 µM) inhibition of iCa2+ release in HT29 cells induced by varying concentrations of PAR2 agonist 2f-LIGRLO-NH2. (B) GB88 is a competitive yet insurmountable antagonist against GB110 (0–10 µM). (C) GB88 is a non-competitive and insurmountable antagonist against trypsin (0–20 µM). (D–F), Schild plot for antagonist GB88 against (D) 2f-LIGRLO-NH2, (E) GB110 and (F) trypsin. (E) Data were fitted to both a competitive (solid line) and non-competitive insurmountable (dotted line) model for Schild analysis. Calculated pA2 values for GB88 were 6.3 ± 0.29 against 2f-LIGRLO-NH2; and 6.0 ± 0.31 (competitive) or 6.3 ± 0.4 (insurmountable) against GB110. Data shown are means (+SEM) of 3 experiments in triplicate.
However, when similar studies were performed with the structurally different agonists GB110 (Figure 4B) or trypsin (Figure 4C), GB88 appeared to be an insurmountable antagonist, indicated by a vertical reduction in the agonist-induced response. A shift in IC50 was observed for PAR2 agonist GB110, but absent for trypsin. Even at higher concentrations (µM – mM), neither trypsin nor GB110 were able to attain full receptor activation, indicating that they were unable to completely displace GB88 binding to PAR2. This result is in marked contrast to that for 2f-LIGRLO-NH2. A Schild plot for antagonism by GB88 of agonist GB110 was linear with slope 1.08, consistent with competitive antagonism (Figure 4E) and a calculated pA2 of 6.0 ± 0.31. When the data were fitted to a non-competitive model using low agonist concentrations (i.e. EC20), the calculated pA2 was 6.3 ± 0.40. Antagonism by GB88 of trypsin, however, was clearly non-competitive (Figure 4F) and consistent with results expected for a tethered ligand.
In summary, GB88 displayed different antagonism against each of the three agonists tested, surmountable and competitive (2f-LIGRLO-NH2), insurmountable and competitive (GB110), and insurmountable and non-competitive (trypsin), likely reflecting slightly different binding modes in PAR2 for the three agonists.
GB88 inhibits PAR2-induced acute inflammation in vivo
To investigate whether antagonist GB88 might be anti-inflammatory in vivo, we examined it in a model of acute inflammation in the rat. PAR2 peptide agonists such as 2f-LIGRLO-NH2, trypsin and SLIGRL-NH2 reportedly induce acute paw oedema in rodents (Vergnolle et al., 1999; Kelso et al., 2006). Before testing GB88 in vivo, we examined the potencies of antagonist GB88 against two agonists (2f-LIGRLO-NH2 and trypsin) on the rat epithelial cell line NRK-52e (Supporting Information Figure S4). GB88 showed activity similar to that observed for human cells expressing PAR2, suggesting that it was likely to have comparable activity in a rodent model. Therefore, we administered GB88 (10 mg·kg−1) to rats by oral gavage in olive oil on a prophylactic dosing schedule, 120 min before intra-plantar (i.pl.) injection of either 2f-LIGRLO-NH2 (350 µg per paw, Figure 5A), trypsin (20 µg per paw, Figure 5B), SLIGRL-NH2 (2 mg per paw, Figure 5B) or GB110 (350 µg per paw, Figure 5B). All four PAR2 agonists alone induced substantial paw oedema that maximized after about 30 min, while an identical set of rats orally pretreated with the PAR2 antagonist GB88 had significantly reduced (≤80%) paw swelling (Figure 5). These results clearly demonstrated that the antagonist GB88 was both orally active and anti-inflammatory in vivo, with specific antagonist activity against four structurally and mechanistically different PAR2 agonists. Furthermore, GB88 had no effect on oedema similarly induced by two structurally different PAR1 agonists (the protease thrombin, 5 U per paw; the hexapeptide TFLLR-NH2, 2 mg per paw) or a PAR4 hexapeptide agonist (AYPGKF-NH2, 2 mg per paw) (Figure 5B), confirming the selectivity of GB88 against PAR2 over PAR1 and PAR4 in vivo.
Figure 5.
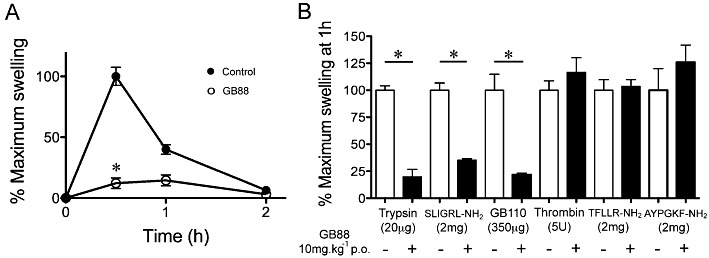
GB88 is a PAR2-selective antagonist of PAR2-induced inflammation in rat paws. (A) GB88 attenuates the acute paw oedema induced by PAR2 agonists. Intraplantar (i.pl.) administration of 2f-LIGRLO-NH2 (350 µg per paw in 100 µL of saline control) induces paw oedema (% area change from baseline) after 30 min (reducing to baseline after 24 h) that is inhibited (≤80%) by prophylactic GB88 (10 mg·kg−1, p.o. in olive oil) given orally 120 min prior to 2f-LIGRLO-NH2 (n = 5 per group, *P < 0.01, significant effect of GB88; anova). (B) GB88 blocks PAR2, but not PAR1, agonist-induced paw oedema. GB88 (10 mg·kg−1 p.o.) significantly reduces paw swelling induced by PAR2 agonists trypsin (20 µg per paw), SLIGRL-NH2 (2 mg per paw) or GB110 (350 µg per paw), but not by oedema induced by PAR1 agonists thrombin (5 U per paw) or TFLLR-NH2 (2 mg per paw) or by PAR4 agonist AYPGKF-NH2 (2 mg per paw). Data (means + SEM) normalized to maximal swelling of controls. *P < 0.01, significant effect of GB88; n = 6 per group.
Discussion and conclusions
GPCRs are the most prevalent signalling proteins on the cell surface, mediating a wide variety of physiological responses that can be targeted by drugs (Blakeney et al., 2007). PARs are a unique class of GPCRs that are activated through irreversible modification by extracellular proteases (Bohm et al., 1996; MacFarlane et al., 2001; Coughlin and Camerer, 2003; Steinhoff et al., 2005; Barry et al., 2006). PAR2 is specifically activated by a range of serine proteases, such as trypsin, but not by the PAR1 agonist thrombin. Most PAR research to date has focused on PAR1 due to its importance in cardiovascular disease. In this study, we have evaluated the new PAR2 agonist GB110 and the new PAR2 antagonist GB88 in vitro in seven human cell lines, as well as the effect of the antagonist in vivo in an rat model of acute inflammation, induced, by PAR2 activation.
Recently we showed that GB110 is a selective agonist for PAR2 over PAR1 in a receptor desensitization assay, exhibiting no agonist activity when PAR2 was desensitized by other PAR2 agonists (Barry et al., 2010). The agonist GB110 is equipotent with the most potent known peptide agonist, 2f-LIGRLO-NH2, in inducing iCa2+ release in seven human cell lines (Blakeney et al., 2007; Barry et al., 2010). All seven cell lines have been previously used to show the role of PAR2 in cancer and inflammation (Miyata et al., 2000; Darmoul et al., 2004; Shimamoto et al., 2004; Feistritzer et al., 2005; Kajikawa et al., 2007; Moriyuki et al., 2009; Ryden et al., 2010). Significant differences in agonist potency were observed between the various cell lines, but not between these two agonists in each cell type. The finding that GB110 was capable of temporarily desensitizing PAR2 receptors and inducing PAR2 internalization, but of lower potency in these properties than 2f-LIGRLO-NH2, suggests that the conformational changes in the receptor that are required for iCa2+ mobilization may be different from those required for β-arrestin binding to PAR2 (Stalheim et al., 2005; Kumar et al., 2007). This distinction at the molecular level between low MW ligand structures that activate the same receptor, even the same isoform, could be a valuable clue to the design of compounds that target different downstream signalling events mediated by the same GPCR. In this case, 2f-LIGRLO-NH2 appears to be able to stabilize, better than GB110, a conformation of the receptor that can couple to β-arrestins, reflected in the 10-fold enhancement in the potency that is specifically related to receptor internalization.
This study also assessed a new PAR2 antagonist, GB88, which was more effective than any PAR2 antagonist reported to date. In our recent description of the chemical synthesis of PAR2 ligands, we reported a structurally related analogue GB83 (with a spiroindanepiperidine) that inhibited iCa2+ mobilization induced in HT29 cells by both trypsin and 2f-LIGRLO-NH2 at their EC80 in a concentration-dependent fashion (Barry et al., 2010). GB88 has comparable potency and, along with GB83, is the first PAR2 antagonist to inhibit responses from both trypsin and synthetic agonists in this low concentration range. In contrast to the antagonist K14585 which does not inhibit trypsin-induced PAR2 activation (Kanke et al., 2009), GB88 exerted almost 100% inhibition against responses induced by both endogenous protease activators (trypsin) and synthetic peptide agonists (2f-LIGRLO-NH2, SLIGRL-NH2) at low µM concentrations. Indeed, GB88 is around 1000-fold more potent as a PAR2 antagonist than another compound, ENMD-1068, reported to antagonize trypsin-induced PAR2 activation at mM concentrations (Kelso et al., 2006). GB88 also antagonized PAR2 activation by the new non-peptide agonist GB110. Interestingly, despite the protease trypsin being a more potent PAR2 agonist than either GB110 or 2f-LIGRLO-NH2, GB88 was an equipotent antagonist against all three PAR2 agonists. Moreover, GB88 maintained relative antagonist potency against all three agonists at their EC80 in seven human cell lines, as measured by iCa2+ mobilization.
The potencies of all three agonists were substantially reduced for the gastric carcinoma cell line MKN45, but GB88 showed no significant change in its IC50 values, despite relatively high concentrations of agonists used (trypsin – 300 nM; 2f-LIGRLO-NH2, GB110 – 30 µM). By contrast, for HUVEC cells, the activities of the three agonists were unaffected but the antagonist potency of GB88 was greatly reduced by ∼10-fold. These differences may be due to polymorphisms in PAR2 between cell lines, something that is quite common for GPCRs in different human or mammalian cell lines. Even very slight changes in the amino acid composition of a GPCR can profoundly affect ligand activity and signalling characteristics, and this should be carefully considered in future evaluation and ligand development for PAR2 and other similar receptors.
Interestingly, the nature of the antagonism observed for GB88 was dependent on the PAR2 agonist being evaluated. Our data are consistent with GB88 being a competitive and surmountable antagonist against 2f-LIGRLO-NH2, but an insurmountable antagonist against GB110 and trypsin. Of these three agonists, only 2f-LIGRLO-NH2 at high concentrations (1 mM) was able to completely reduce the potency of GB88, which we attribute to a competitive interaction. However, due to the short time frame of the iCa2+ assay, it is entirely possible that true equilibrium may not have been attained and this could explain the depression of the maxima (Kenakin et al., 2006; Charlton and Vauquelin, 2010) observed when GB110 was employed as the agonist. In consideration of this, we re-analysed the data in an insurmountable model by constructing a Schild plot with a dose-ratio generated at low agonist concentrations. The adjusted pA2 value showed only a small difference (6.3 vs. 6.0) and was very similar to 2f-LIGRLO-NH2. The finding that GB88 was an insurmountable and non-competitive antagonist against trypsin-induced PAR2 activation was not so surprising, as trypsin is really an indirect agonist that induces PAR2 activation via remote proteolytic cleavage of the N-terminus. The vertical suppression of trypsin-induced concentration-response curves by GB88 is typical for antagonism of an indirect agonist (Kenakin, 1997).
The novel PAR2 antagonist was also tested for its in vivo activity in an acute model of inflammation in rats. Various roles for PAR2 in inflammation have been previously suggested, and our discovery of a new PAR2 antagonist that is the first to be potent and effective against several types of agonists (protease, peptide, non-peptide) provided an excellent opportunity to identify whether PAR2 antagonism is indeed anti-inflammatory in vivo. Furthermore, the structure of GB88 obeys the rule-of-five (two hydrogen bond donors, five hydrogen bond acceptors, log P 3.3) (Lipinski et al., 1997), as well as having acceptable polar surface area (110 Å2) and rotatable bond (nine rotatable bonds) limits (Veber et al., 2002), suggesting that it might be orally active. Given orally, GB88 was indeed able to reduce the severity of paw oedema induced by each of the four different PAR2 agonists administered i.pl.. This result suggests that GB88 could potentially be an effective systemic anti-inflammatory agent after oral administration.
This study has associated PAR2 (and, by implication, its activating proteolytic enzymes) on several human cell lines, including cancer cells, with certain intracellular signalling pathways in vitro and with inflammation in vivo. In vitro, the new non-peptide agonist GB110 was equipotent with the most potent known hexapeptide agonist in activating PAR2 to release iCa2+, but 10-fold less potent in promoting PAR2 internalization and desensitization. At 10 µM, GB110 caused full receptor activation and desensitization, but only 20% receptor internalization. There was no difference between GB110 and peptide agonists in the rate of receptor re-expression or re-sensitization on the cell surface. This finding suggests that PAR2 ligands can differentially regulate activation, desensitization and internalization (Stalheim et al., 2005; Kumar et al., 2007; Ricks and Trejo, 2009), and such selective ligands could be valuable probes for dissecting signalling pathways associated with different specific diseased states. An important finding was that GB88 was orally active in vivo and an equipotent but mechanistically distinct antagonist in vitro against three structurally different agonists (a protease, a hexapeptide and a non-peptide), making it a valuable tool to analyse inhibition of PAR2. Our results suggest that PAR2 agonists can differentially affect intracellular pathways, that a PAR2 antagonist can block receptor activation via different mechanisms and that PAR2 antagonists may be beneficial in controlling inflammatory and proliferative disease. Further efforts should now be made to investigate the effects of novel PAR2 modulators in specific intracellular signalling pathways that can be associated with specific diseases and in a range of animal models of inflammatory diseases and cancer.
Acknowledgments
This work was supported by grants from the Australian Research Council (FF668733, DP1093245) and the National Health and Medical Research Council of Australia (569595, APP1000745). DPF acknowledges a Federation Fellowship from the Australian Research Council.
Glossary
- A549
adenocarcinomic human alveolar basal epithelial cells
- ATCC
American Tissue Culture Collection
- FBS
fetal bovine serum
- HPRT
hypoxanthine-guanine phosphoribosyl transferase
- HT-29
human colon adenocarcinoma grade II cell line
- HUVEC
human umbilical vein endothelial cell
- iCa2+
intracellular Ca2+
- i.pl.
intra-plantar
- MDA-MB-231
human M.D. Anderson – metastatic breast cancer cells
- MKN45
MKN1, human gastric adenocarcinoma cells
- NRK-52e
rat renal promximal tubule cells
- Panc-1
human exocrine pancreas carcinoma cells
- PAR2
protease activated receptor 2
- U937
human monocytic cells from histiocytic lymphoma
Conflict of interest
JYS, GDB and DPF were inventors on patent AU2010903378 on PAR2 agonists and antagonists owned by University of Queensland. There are no other competing interests.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 PAR2 agonists do not induceintracellular Ca2+ release in U937 cells. Human U937 cells weretreated with C5a (•), 2f-LIGRLO-NH2 (♦;), GB110(○) and trypsin (□) in the fluorescence intracellularcalcium assay. Up to 100 μM of 2f-LIGRLO-NH2 or GB110 and up to10 μM trypsin caused no significant iCa2+ release,whereas the hormone C5a did induce iCa2+ release at nMconcentrations. Error bars represented ± SEM with n≥ 3.
Figure S2 PAR2 mRNA expression correlates withPAR2 surface activity on multiple human cell lines. QuantitativeRT-PCR examination of PAR2 mRNA expression in eight human celllines is shown in comparison with human housekeeping genehypoxanthine guanine phosphoribosyl transferase (HPRT). PAR2 mRNAexpression (clear) on each cell line is compared with maximumPAR2-induced intracellular calcium release (filled). Calciumrelease is expressed as a percentage of that induced by calcimycin(A-23187) and is used as an indicator of PAR2 activity on cellsurfaces. PAR2 mRNA levels and its activities were comparable inthe cell lines tested. Error bars represented ± SEM withn ≥ 3.
Figure S3 GB88 does not antagonize two otherGPCRs. Human U937 cells were treated with either 30 nM C5a(•) or 300 nM C3a (▪) in an intracellular calciumassay. Up to 100 μM GB88 showed no significant reduction iniCa2+ release in these cells. Error bars represent± SEM with n ≥ 3.
Figure S4 Profile of PAR2 agonist(2f-LIGRLO-NH2) and antagonist (GB88) on rat NRK-52e cells. (A) Concentration-dependent curve for iCa2+ mobilization by2f-LIGRL-NH2. EC50 210 nM (pEC50 6.7 ±0.07). (B) PAR2 antagonist GB88 inhibits iCa2+ releaseinduced in NRK-52e by 1 μM 2f-LIGRLO-NH2. IC50 20μM (pIC50 4.7 ± 0.2). Data points = means of three experiments in triplicate, bars = SE.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arisawa T, Tahara T, Shibata T, Nagasaka M, Nakamura M, Kamiya Y, et al. Promoter hypomethylation of protease-activated receptor 2 associated with carcinogenesis in the stomach. J Gastroenterol Hepatol. 2007;22:943–948. doi: 10.1111/j.1440-1746.2007.04847.x. [DOI] [PubMed] [Google Scholar]
- Barry GD, Le GT, Fairlie DP. Agonists and antagonists of protease activated receptors (PARs) Curr Med Chem. 2006;13:243–265. doi: 10.2174/092986706775476070. [DOI] [PubMed] [Google Scholar]
- Barry GD, Suen JY, Low HB, Pfeiffer B, Flanagan B, Halili M, et al. A refined agonist pharmacophore for protease activated receptor 2. Bioorg Med Chem Lett. 2007;17:5552–5557. doi: 10.1016/j.bmcl.2007.08.026. [DOI] [PubMed] [Google Scholar]
- Barry GD, Suen JY, Le GT, Cotterell A, Reid RC, Fairlie DP. Novel agonists and antagonists for human protease activated receptor 2. J Med Chem. 2010;53:7428–7440. doi: 10.1021/jm100984y. [DOI] [PubMed] [Google Scholar]
- Blakeney JS, Reid RC, Le GT, Fairlie DP. Nonpeptidic ligands for peptide-activated G protein-coupled receptors. Chem Rev. 2007;107:2960–3041. doi: 10.1021/cr050984g. [DOI] [PubMed] [Google Scholar]
- Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, et al. Molecular cloning, expression and potential functions of the human proteinase-activated receptor 2. Biochem J. 1996;314:1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton SJ, Vauquelin G. Elusive equilibrium: the challenge of interpreting receptor pharmacology using calcium assays. Br J Pharmacol. 2010;161:1250–1265. doi: 10.1111/j.1476-5381.2010.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocks TM, Fong B, Chow JM, Anderson GP, Frauman AG, Goldie RG, et al. A protective role for protease-activated receptors in the airways. Nature. 1999;398:156–160. doi: 10.1038/18223. [DOI] [PubMed] [Google Scholar]
- Cottrell GS, Amadesi S, Pikios S, Camerer E, Willardsen JA, Murphy BR, et al. Protease-activated receptor 2, dipeptidyl peptidase I, and proteases mediate Clostridium difficile toxin A enteritis. Gastroenterology. 2007;132:2422–2437. doi: 10.1053/j.gastro.2007.03.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest. 2003;111:25–27. doi: 10.1172/JCI17564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Agostino B, Roviezzo F, De Palma R, Terracciano S, De Nardo M, Gallelli L, et al. Activation of protease-activated receptor-2 reduces airways inflammation in experimental allergic asthma. Clin Exp Allergy. 2007;37:1436–1443. doi: 10.1111/j.1365-2222.2007.02793.x. [DOI] [PubMed] [Google Scholar]
- Darmoul D, Gratio V, Devaud H, Laburthe M. Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem. 2004;279:20927–20934. doi: 10.1074/jbc.M401430200. [DOI] [PubMed] [Google Scholar]
- Feistritzer C, Lenta R, Riewald M. Protease-activated receptors-1 and -2 can mediate endothelial barrier protection: role in factor Xa signaling. J Thromb Haemost. 2005;3:2798–2805. doi: 10.1111/j.1538-7836.2005.01610.x. [DOI] [PubMed] [Google Scholar]
- Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, et al. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest. 2003;111:35–41. doi: 10.1172/JCI16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KK, Sherman PM, Cellars L, Andrade-Gordon P, Pan Z, Baruch A, et al. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc Natl Acad Sci USA. 2005;102:8363–8368. doi: 10.1073/pnas.0409535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota M, Ohmuraya M, Baba H. The role of trypsin, trypsin inhibitor, and trypsin receptor in the onset and aggravation of pancreatitis. J Gastroenterol. 2006;41:832–836. doi: 10.1007/s00535-006-1874-2. [DOI] [PubMed] [Google Scholar]
- Hollenberg MD, Saifeddine M, al-Ani B. Proteinase-activated receptor-2 in rat aorta: structural requirements for agonist activity of receptor-activating peptides. Mol Pharmacol. 1996;49:229–233. [PubMed] [Google Scholar]
- Iwaki K, Shibata K, Ohta M, Endo Y, Uchida H, Tominaga M, et al. A small interfering RNA targeting proteinase-activated receptor-2 is effective in suppression of tumor growth in a Panc1 xenograft model. Int J Cancer. 2008;122:658–663. doi: 10.1002/ijc.23123. [DOI] [PubMed] [Google Scholar]
- Kajikawa H, Yoshida N, Katada K, Hirayama F, Handa O, Kokura S, et al. Helicobacter pylori activates gastric epithelial cells to produce interleukin-8 via protease-activated receptor 2. Digestion. 2007;76:248–255. doi: 10.1159/000113041. [DOI] [PubMed] [Google Scholar]
- Kanke T, Ishiwata H, Kabeya M, Saka M, Doli T, Hattori Y, et al. Binding of a highly potent protease-activated receptor-2 (PAR2) activating peptide, [3H]2-furoyl-LIGRL-NH2, to human PAR2. Br J Pharmacol. 2005;145:255–263. doi: 10.1038/sj.bjp.0706189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanke T, Kabeya M, Kubo S, Kondo S, Yasuoka K, Tagashira J, et al. Novel antagonists for proteinase-activated receptor 2: inhibition of cellular and vascular responses in vitro and in vivo. Br J Pharmacol. 2009;158:361–371. doi: 10.1111/j.1476-5381.2009.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A, Kanke T, Yonezawa D, Ishiki T, Saka M, Kabeya M, et al. Potent and metabolically stable agonists for protease-activated receptor-2: evaluation of activity in multiple assay systems in vitro and in vivo. J Pharmacol Exp Ther. 2004;309:1098–1107. doi: 10.1124/jpet.103.061010. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Matsunami M, Sekiquchi F. Gastrointestinal roles for proteinase-activated receptors in health and disease. Br J Pharmacol. 2008;153(Suppl. 1):S230–S240. doi: 10.1038/sj.bjp.0707491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso EB, Lockhart JC, Hembrough T, Dunning L, Plevin R, Hollenberg MD, et al. Therapeutic promise of proteinase-activated receptor-2 antagonism in joint inflammation. J Pharmacol Exp Ther. 2006;316:1017–1024. doi: 10.1124/jpet.105.093807. [DOI] [PubMed] [Google Scholar]
- Kelso EB, Ferrell WR, Lockhart JC, Elias-Jones I, Hembrough T, Dunning L, et al. Expression and proinflammatory role of proteinase-activated receptor 2 in rheumatoid synovium. Arthritis Rheum. 2007;56:765–771. doi: 10.1002/art.22423. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Competitive antagonism. In: McLaughlin MA, editor. Pharmacologic Analysis of Drug-Receptor Interaction. Philadelphia, PA: Lippincott-Raven Publishers; 1997. pp. 369–371. [Google Scholar]
- Kenakin T, Jenkinson S, Watson C. Determining the potency and molecular mechanism of action of insurmountable antagonists. J Pharmacol Exp Ther. 2006;319:710–723. doi: 10.1124/jpet.106.107375. [DOI] [PubMed] [Google Scholar]
- Kumar P, Lau CS, Mathur M, Wang P, DeFea KA. Differential effects of beta-arrestins on the internalization, desensitization and ERK1/2 activation downstream of protease activated receptor-2. Am J Physiol Cell Physiol. 2007;293:C346–C357. doi: 10.1152/ajpcell.00010.2007. [DOI] [PubMed] [Google Scholar]
- Lam FF. Role of protease-activated receptor 2 in joint inflammation. Arthritis Rheum. 2007;56:3514–3517. doi: 10.1002/art.22935. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- MacFarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- Maeda K, Hirota M, Kimura Y, Ichihara A, Ohmuraya M, Sugita H, et al. Proinflammatory role of trypsin and protease-activated receptor-2 in a rat model of acute pancreatitis. Pancreas. 2005;31:54–62. doi: 10.1097/01.mpa.0000163178.37050.0d. [DOI] [PubMed] [Google Scholar]
- Matej R, Mandakova P, Netikova I, Pouckova P, Olejar T. Proteinase-activated receptor-2 expression in breast cancer and the role of trypsin on growth and metabolism of breast cancer cell line MDA MB-231. Physiol Res. 2007;56:475–484. doi: 10.33549/physiolres.930959. [DOI] [PubMed] [Google Scholar]
- McIntosh KA, Plevin R, Ferrell WR, Lockhart JC. The therapeutic potential of proteinase-activated receptors in arthritis. Curr Opin Pharmacol. 2007;7:334–338. doi: 10.1016/j.coph.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Miyata S, Koshikawa N, Yasumitsu H, Miyazaki K. Trypsin stimulates integrin alpha(5)beta(1)-dependent adhesion to fibronectin and proliferation of human gastric carcinoma cells through activation of proteinase-activated receptor-2. J Biol Chem. 2000;275:4592–4598. doi: 10.1074/jbc.275.7.4592. [DOI] [PubMed] [Google Scholar]
- Moriyuki K, Sekiguchi F, Matsubara K, Nishikawa H, Kawabata A. Proteinase-activated receptor-2-triggered prostaglandin E(2) release, but not cyclooxygenase-2 upregulation, requires activation of the phosphatidylinositol 3-kinase/Akt/nuclear factor-kappaB pathway in human alveolar epithelial cells. J Pharmacol Sci. 2009;111:269–275. doi: 10.1254/jphs.09155fp. [DOI] [PubMed] [Google Scholar]
- Moussa L, Apostolopoulos J, Davenport P, Tchonque J, Tipping PG. Protease-activated receptor-2 augments experimental crescentic glomerulonephritis. Am J Pathol. 2007;171:800–808. doi: 10.2353/ajpath.2007.061155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishibori M, Mori S, Takahashi HK. Physiology and pathophysiology of proteinase-activated receptors (PARs): PAR-2-mediated proliferation of colon cancer cell. J Pharmacol Sci. 2005;97:25–30. doi: 10.1254/jphs.fmj04005x5. [DOI] [PubMed] [Google Scholar]
- Niu QX, Chen HQ, Chen ZY, Fu YL, Lin JL, He SH. Induction of inflammatory cytokine release from human umbilical vein endothelial cells by agonists of proteinase-activated receptor-2. Clin Exp Pharmacol Physiol. 2008;35:89–96. doi: 10.1111/j.1440-1681.2007.04755.x. [DOI] [PubMed] [Google Scholar]
- Ricks TK, Trejo J. Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J Biol Chem. 2009;284:34444–34457. doi: 10.1074/jbc.M109.048942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryden L, Grabau D, Schaffner F, Jonsson PE, Ruf W, Belting M. Evidence for tissue factor phosphorylation and its correlation with protease-activated receptor expression and the prognosis of primary breast cancer. Int J Cancer. 2010;126:2330–2340. doi: 10.1002/ijc.24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto R, Sawada T, Uchima Y, Inoue M, Kimura K, Yamashita Y, et al. A role for protease-activated receptor-2 in pancreatic cancer cell proliferation. Int J Oncol. 2004;24:1401–1406. [PubMed] [Google Scholar]
- Shin K, Nigrovic PA, Crish J, Boilard E, McNeil HP, Larabee KS, et al. Mast cells contribute to autoimmune inflammatory arthritis via their tryptase/heparin complexes. J Immunol. 2009;182:647–656. doi: 10.4049/jimmunol.182.1.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalheim L, Ding Y, Gullapalli A, Paing MM, Wolfe BL, Morris DR, et al. Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol Pharmacol. 2005;67:78–87. doi: 10.1124/mol.104.006072. [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, et al. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev. 2005;26:1–43. doi: 10.1210/er.2003-0025. [DOI] [PubMed] [Google Scholar]
- Suen JY, Gardiner B, Grimmond S, Fairlie DP. Profiling gene expression induced by protease-activated receptor 2 (PAR2) activation in human kidney cells. PLoS ONE. 2010;5:e13809. doi: 10.1371/journal.pone.0013809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Vergnolle N, Hollenberg MD, Sharkey KA, Wallace J. Characterization of the inflammatory response to proteinase-activated receptor-2 (PAR2)-activating peptides in the rat paw. Br J Pharmacol. 1999;127:1083–1090. doi: 10.1038/sj.bjp.0702634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesey DA, Hooper JD, Gobe GC, Johnson DW. Potential physiological and pathophysiological roles for protease-activated receptor-2 in the kidney. Nephrology. 2007a;12:36–43. doi: 10.1111/j.1440-1797.2006.00746.x. [DOI] [PubMed] [Google Scholar]
- Vesey DA, Kruger WA, Poronnik P, Gobe GC, Johnson DW. Proinflammatory and proliferative responses of human proximal tubule cells to PAR-2 activation. Am J Physiol Renal Physiol. 2007b;293:F1441–F1449. doi: 10.1152/ajprenal.00088.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.