

Abstract
BACKGROUND AND PURPOSE
TASK1 (K2P3.1) two-pore-domain K+ channels contribute substantially to the resting membrane potential in human pulmonary artery smooth muscle cells (hPASMC), modulating vascular tone and diameter. The endothelin-1 (ET-1) pathway mediates vasoconstriction and is an established target of pulmonary arterial hypertension (PAH) therapy. ET-1-mediated inhibition of TASK1 currents in hPASMC is implicated in the pathophysiology of PAH. This study was designed to elucidate molecular mechanisms underlying inhibition of TASK1 channels by ET-1.
EXPERIMENTAL APPROACH
Two-electrode voltage clamp and whole-cell patch clamp electrophysiology was used to record TASK1 currents from hPASMC and Xenopus oocytes.
KEY RESULTS
ET-1 inhibited TASK1-mediated IKN currents in hPASMC, an effect attenuated by Rho kinase inhibition with Y-27632. In Xenopus oocytes, TASK1 current reduction by ET-1 was mediated by endothelin receptors ETA (IC50= 0.08 nM) and ETB (IC50= 0.23 nM) via Rho kinase signalling. TASK1 channels contain two putative Rho kinase phosphorylation sites, Ser336 and Ser393. Mutation of Ser393 rendered TASK1 channels insensitive to ETA- or ETB-mediated current inhibition. In contrast, removal of Ser336 selectively attenuated ETA-dependent TASK1 regulation without affecting the ETB pathway.
CONCLUSIONS AND IMPLICATIONS
ET-1 regulated vascular TASK1 currents through ETA and ETB receptors mediated by downstream activation of Rho kinase and direct channel phosphorylation. The Rho kinase pathway in PASMC may provide a more specific therapeutic target in pulmonary arterial hypertension treatment.
Keywords: endothelin-1, background potassium current, pulmonary arterial hypertension, Rho kinase, K2P channel, leak current, membrane potential
Introduction
Pulmonary arterial hypertension (PAH) is characterized by a progressive increase in pulmonary vascular resistance leading to right ventricular failure. Pulmonary vasoconstriction is mediated by multiple signalling mechanisms including endothelin-1 (ET-1), many of which exert their effects by altering electrical activity (Gurney and Manoury, 2009). Activation of the ET-1 system has been demonstrated in plasma samples and lung tissue of PAH patients and in corresponding animal models (Archer and Rich, 2000; Lüscher and Barton, 2000; Galiéet al., 2004). ET-1 promotes vasoconstriction in pulmonary arteries by targeting two G protein-coupled receptor subtypes, ETA and ETB (receptor nomenclature follows Alexander et al., 2011). ETA receptors are found exclusively on smooth muscle cells of large human pulmonary arteries, whereas ETB receptors were detected on smooth muscle cells of capillaries and airways as well as on alveolar endothelial cells (Lippton et al., 1993; Seo et al., 1994; Dupuis et al., 1996).
Two-pore domain potassium (K2P) channels stabilize resting membrane potential and promote action potential repolarization (Goldstein et al., 2001; Thomas et al., 2008). The sensitivity of K2P channels to a wide range of physiological signals such as polyunsaturated fatty acids, pH and O2, suggests significance for regulation of vascular tone, particularly in small pulmonary resistance vessels (Gurney et al., 2003; Gardener et al., 2004; Olschewski et al., 2006). Indeed, TASK1 channels (TWIK-related acid sensitive K+ channels; K2P3.1) contribute substantially to the resting membrane potential in human pulmonary artery smooth muscle cells (hPASMC) (Olschewski et al., 2006). Inhibition of vascular TASK1 channels by ET-1 has previously been implicated in PAH (Tang et al., 2009). The present study was designed to assess the role of ETA and ETB receptor subtypes and associated signal transduction pathways in order to identify potential therapeutic targets downstream of ET receptors. We reveal that ET-1 regulates vascular TASK1 currents through ETA and ETB receptors, mediated by activation of Rho kinase and phosphorylation at residues S336 and S393. The Rho kinase pathway may represent a novel drug target for future treatment of PAH.
Methods
Molecular biology
ETA and ETB receptor complementary DNAs (GenBank accession numbers NM001957 and L06623, respectively) were obtained from Dr H. Ninomiya (Yonago, Japan) and the human TASK1 clone (GenBank accession number AF065163) was kindly provided by Prof Dr J. Daut (Marburg, Germany). Complementary RNAs were prepared from the corresponding cDNA in the pcDNA3 plasmid with the mMESSAGE mMACHINE in vitro transcription kit (Ambion, Austin, TX, USA) using T7 polymerase. Transcripts were quantified using a spectrophotometer and by comparison with control samples separated by agarose gel electrophoresis. Rho kinase-dependent phosphorylation sites (amino acid motif R/K-XX-S/T) were identified in TASK1 using PPSearch software (EMBL-EBI, European Bioinformatics Institute, Cambridge, UK). S336A and S393A amino acid point mutations where generated by polymerase chain reaction with synthetic mutant oligonucleotide primers using the QuickChange II site-directed mutagenesis kit (Stratagene, La Jolla, San Diego, CA, USA), and resulting cDNAs were verified by sequencing (SeqLab, Göttingen, Germany).
Oocyte preparation
All animal care and experimental procedures were in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health. Ovarian lobes were surgically removed with aseptic techniques from female Xenopus laevis frogs anaesthetized with 1 g L−1 tricaine solution (pH = 7.5) as previously described (Gierten et al., 2008). Frogs were not fed on the day of surgery to avoid emesis during anaesthesia. After surgery, the frogs were allowed to recover consciousness, followed by at least 2 months of recovery period. Oocyte collection was alternated between left and right ovaries, and no more than three surgeries were performed on one individual frog. After the final taking of oocytes, the anaesthetized frog was killed by decerebration and pithing. Following collagenase treatment, stage V-VI defolliculated oocytes were manually isolated under a stereomicroscope. Injection of cRNA (46 nL per oocyte) into stage V and VI defolliculated oocytes was performed using a Nanoject automatic injector (Drummond, Broomall, PA, USA). For co-expression of endothelin receptors and TASK1 channels, cRNA was mixed prior to injection in a 2:1 ratio. Measurements were made 1 to 3 days after injection.
Cell culture
hPASMC (PromoCell, Heidelberg, Germany) were handled according to the manufacturer's instructions as previously reported (Staudacher et al., 2011). Briefly, cells were maintained in growth medium (PromoCell) in an atmosphere of 95% humidified air and 5% CO2 at 37°C and passaged every 24–48 h. For electrophysiological recordings, hPASMC were seeded on glass coverslips 24–72 h before use. On the day of the experiments, cells were transferred into a recording chamber and were continuously superfused with the bath solution.
Electrophysiology
Current recordings from hPASMC were performed using the whole-cell patch clamp configuration as previously reported (Kiesecker et al., 2006). Currents were measured with an RK-400 amplifier (Bio-Logic SAS, Claix, France), stored on hard disk, and analysed with pCLAMP (Axon Instruments, Foster City, CA, USA) and Origin 6 (OriginLab, Northampton, MA, USA) software. Pipettes pulled from borosilicate glass (1B120F-4; World Precision Instruments, Berlin, Germany) were fabricated on a Flaming/Brown micropipette puller P-87 (Sutter Instruments, Novato, CA, USA) and fire polished to give a final resistance of 3–7 MΩ. To isolate IKN from other voltage-dependent K+ currents, cells were clamped at 0 mV for at least 5 min (Gurney et al., 2003) at the beginning of each experiment. Application of additional ion channel blockers was not required (Olschewski et al., 2006).
The two-microelectrode voltage clamp configuration was used to record currents from Xenopus laevis oocytes (Thomas et al., 1999). Data were low-pass filtered at 1–2 kHz (–3 dB, four-pole Bessel filter) before digitalization at 5–10 kHz. Recordings were performed using a commercially available amplifier (Warner OC-725C, Warner Instruments, Hamden, CT, USA) and pCLAMP (Axon Instruments) or Origin 6 (OriginLab) software for software for data acquisition and analysis. No leak subtraction was performed during the experiments. Recordings with less than 10% leak current were considered for data analysis.
Solutions and drug administration
Patch clamp recordings from hPASMC were performed at room temperature in a bath solution containing (in mM) 5.5 KCl, 140.5 NaCl, 1.5 CaCl2, 1 MgCl2, 10 glucose, 0.5 Na2HPO4, 0.5 KH2PO4 and 10 HEPES (pH 7.3 with NaOH). The pipette solution contained (in mM) 20 KCl, 135 K-methanesulphonate (to suppress Cl- currents), 1 MgCl2, 2 Na2ATP, 1 EGTA and 20 HEPES (pH 7.2 with KOH). Two-microelectrode voltage clamp measurements of Xenopus oocytes were carried out in a K+ solution containing (in mM) 5 KCl, 100 NaCl, 1.5 CaCl2, 2 MgCl2 and 10 HEPES (pH 7.4). Current and voltage electrodes were filled with 3 M KCl solution. All measurements were carried out at room temperature. ET-1 (Sigma-Aldrich, Steinheim, Germany) was dissolved in dimethylsulphoxide (DMSO) to a 20 µM stock solution and stored at −20°C. Aliquots of the stock solution were diluted to the desired concentration with the bath solution. The maximum concentration of DMSO in the bath had no electrophysiological effect (data not shown).
Data analysis and statistics
Concentration–response relationships for drug-induced block were fit with a Hill equation of the following form: Idrug/Icontrol= 1/[1 + (D/IC50)n], where I indicates current, D is the drug concentration, n is the Hill coefficient and IC50 is the concentration necessary for 50% block. Data are expressed as mean ± SEM. We used paired and unpaired Student's t-tests (two-tailed tests) to compare the statistical significance of the results. P < 0.05 was considered statistically significant.
Results
ET-1 reduces TASK1 (IKN) currents in hPASMC
The effects of ET-1 on native TASK1 currents were investigated in hPASMC. Endogenous TASK1 channels produce a non-inactivating background K+ current (IKN) in hPASMC (Gurney et al., 2003; Olschewski et al., 2006). IKN was elicited by test pulses from −80 mV to +60 mV in 10 mV increments (400 ms) from a holding potential of −80 mV (Figure 1A). Under control conditions, cells displayed time-independent outward K+ currents (Figure 1A). Current amplitudes recorded after 20 min in bath solution were stable (I20 min= 100.9 ± 4.7%; n = 5; Figure 1D, E). After exposure to ET-1 (10 nM) for 20 min, outward currents were markedly reduced (Figure 1B–E). In a series of 10 experiments, 10 nM ET-1 blocked steady-state outward currents recorded at +30 mV by 64 ± 8% (P < 0.0001). At 0 mV membrane voltage, ET-1 reduced IKN by 52 ± 10% (n = 5; P < 0.001). Subsequent analyses were performed at +30 mV. We found that co-application of a Rho kinase inhibitor (Y-27632; 10 µM) for 20 min significantly reduced the endothelin effect after 20 min (n = 10; Figure 1D, E). The TASK1 blocker anandamide (Maingret et al., 2001) was applied at the end of each experiment to confirm that TASK1 was the molecular basis of IKN. The degree of anandamide block was virtually indistinguishable from endothelin-induced inhibition, indicating that the hormone caused virtually complete reduction of ITASK1 in hPASMC (Figure 1D).
Figure 1.
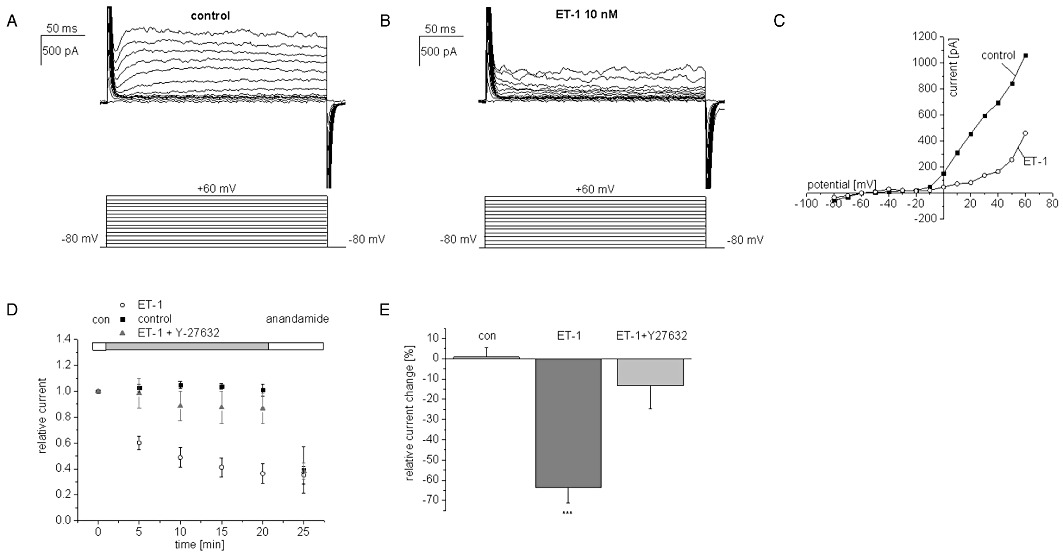
Endothelin-1 (ET-1) inhibition of TASK1/IKN currents in human pulmonary artery smooth muscle cells. Representative experiments before (A) and after exposure to 10 nM ET-1 (B) are displayed. The corresponding current voltage relationship is shown in (C). (D) the time course of inhibition upon ET-1 exposure (10 nM) was monitored over a period of 20 min and is shown together with the respective current measurements under control conditions, after co-application of ET-1 (10 nM) and Y-27632 (10 µM) and following additional application of anandamide (10 µM) at the end of each experiment. (E) Summary of current changes after 20 min of drug exposure. Control values after incubation in bath solution (20 min) are not significantly different from results after application of ET-1 (10 nM) and Y-27632 (10 µM). In contrast, the effect of ET-1 (10 nM) was significantly different from control values; ***P < 0.001 vs. untreated controls.
ET-1 targets ETA and ETB receptor subtypes to inhibit TASK1 channels in Xenopus oocytes
To dissect molecular mechanisms of TASK1 inhibition by ET-1 the Xenopus laevis oocyte system was used. Human TASK1 channels expressed heterologously in Xenopus oocytes gave rise to potassium currents with characteristic outward rectification (Figure 2A, C). A standardized voltage protocol was employed to measure TASK1 currents. Test pulses to potentials ranging from −120 mV to +30 mV with 400 ms duration were applied in 10 mV-increments. The holding potential was −80 mV. Steady-state outward currents were determined at +30 mV to quantify functional effects. This voltage protocol and a standardized observation period of 30 min were applied during all TASK1 current recordings from Xenopus oocytes in this study to allow for ready comparison of results. First, specificity of endothelin receptor subtypes ETA and ETB was studied. Under control conditions, TASK1 currents exhibited a run-up of 9 ± 5% during an observation period of 30 min (n = 8). Incubation of oocytes with ET-1 (20 nM) in the absence of heterologously expressed endothelin receptors had no influence on TASK1 currents, revealing a run-up of 24 ± 3% (n = 4; data not shown) similar to control conditions. In contrast, application of ET-1 (20 nM) reduced TASK1 currents by 74 ± 6% (n = 7; P < 0.0001) upon co-expression of cloned human ETA receptors with the channels (Figure 2A–D). The onset of block is illustrated in Figure 2D. ETB receptors coupled to TASK1 channel as well. ET-1 (20 nM) lead to a reduction of TASK1 currents by 60 ± 8% (n = 5; P < 0.0001) with similar time course, compared with ETA (Figure 2E–H). The difference between ETA- and ETB-mediated TASK1 inhibition was not significant (P = 0.27). Dose–response relationships were analysed for endothelin receptors under conditions described earlier, yielding low IC50 values for ETA (0.08 ± 0.04 nM; n = 5–9; Figure 3A) and ETB receptors (0.23 ± 0.05 nM; n = 5–9; Figure 3B) with Hill coefficients nH of 0.9 ± 0.2 for ETA receptors and 0.9 ± 0.1 for ETB receptors respectively.
Figure 2.
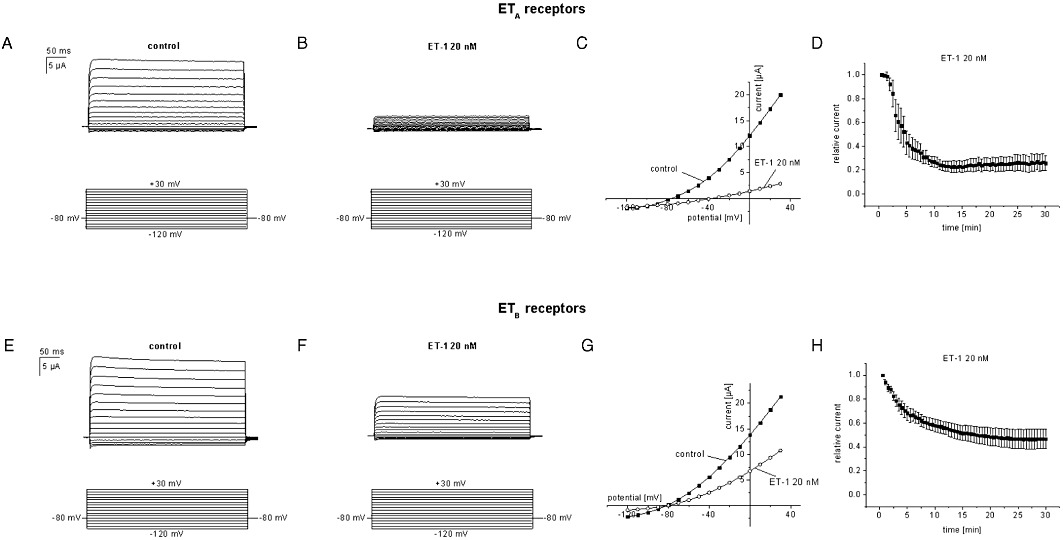
Endothelin-1 (ET-1) inhibits TASK1 channels in Xenopus oocytes upon co-expression with endothelin receptors. Representative current recordings before and after exposure to 20 nM ET-1 are displayed for ETA (A, B) and ETB (E, F) receptors. (C, G) corresponding current voltage (I/V) relationships. (D, H) time course of TASK1 current blockade by 20 nM endothelin in the presence of ETA (D; n = 7) and ETB receptors (H; n = 7).
Figure 3.
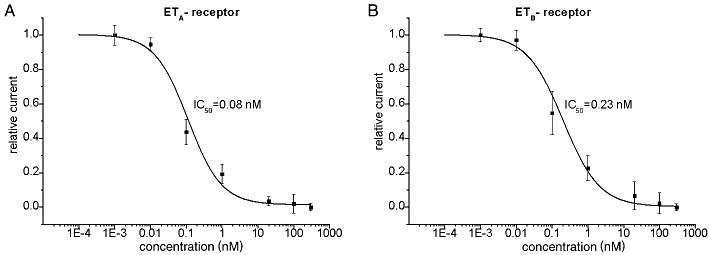
Dose-dependent inhibition of TASK1 channels in oocytes. The IC50 values of the inhibitory effect of endothelin-1 on TASK1 currents were 0.08 ± 0.04 nM for ETA receptor activation (A) and 0.23 ± 0.05 nM for ETB receptor stimulation (B). Five to nine cells were studied at each concentration.
TASK1 channel inhibition by ETA receptor activation is Rho kinase dependent
To identify signalling pathways involved in TASK1 current inhibition by endothelin, channels were co-expressed with ETA receptors, and ET-1 (20 nM) was applied with several small molecule protein kinase inhibitors. Incubation with the specific Rho kinase inhibitor Y-27632 (10 µM) for 30 min markedly attenuated the inhibitory regulation of TASK1 currents by ET-1 (Figure 4A); n = 5; P = 0.03) compared with the effect of ET-1 in the absence of kinase inhibitors, indicating that Rho kinase activation mediated ETA receptor-mediated TASK1 current inhibition.
Figure 4.
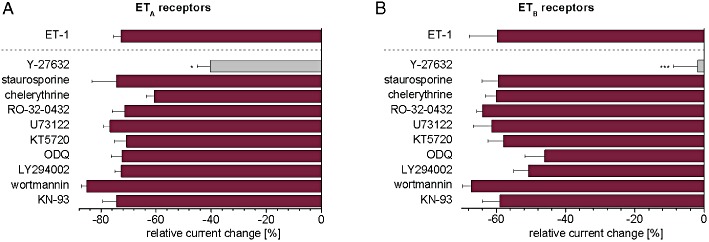
Intracellular signalling kinases associated with ET-1 regulation of TASK1 in oocytes, co-expressing TASK1 channels and ETA or ETB receptors. In order to investigate the signalling pathways, a range of low MW protein kinase inhibitors were co-applied together with 20 nM endothelin-1. Results for ETA receptors (A) and ETB receptors (B) are displayed. For comparison, the ET-1 effect in the absence of inhibitors is shown separated by a dashed line. The following kinase inhibitors were used: Y-27632 (10 µM) to inhibit Rho kinase; staurosporine (1 µM), chelerythrine (10 µM) or RO-32–0432 (3 µM) to inhibit protein kinase C; U73122 (10 µM) to inhibit phospholipase C; KT5720 (2.5 µM) to inhibit protein kinase A; ODQ (10 µM) to inhibit cGMP-dependent signalling; LY294002 (30 µM) and wortmannin (10 µM) to inhibit PI3kinase and myosin light chain kinase; and KN-93 (10 µM) to inhibit CamKII. Data are given as mean ± SEM; *P < 0.05; ***P < 0.001 versus ET-1 alone.
We extended our pharmacological screening to other pathways that have been linked to endothelin receptor signalling (Figure 4A). In a previous report, TASK1 current regulation by endothelin via PKC-dependent pathways was shown in primary hPASMC (Tang et al., 2009). In oocytes, however, co-application of PKC inhibitors, staurosporine (1 µM), chelerythrine (10 µM) or RO-32–0432 (3 µM), with ET-1 did not affect TASK1 inhibition. In order to exclude a direct effect of PLC, the PLC inhibitor U73122 was used. Endothelin-induced current reduction following co-application with 10 µM U73122 (n = 6) exhibited no significant difference compared with ET-1 alone, ruling out direct effects mediated by PLC activity. Furthermore, neither the specific PKA inhibitor KT5720 (2.5 µM) nor the cGMP inhibitor ODQ (10 µM) altered TASK1 current inhibition by 20 nM ET-1 (n = 6–9). In addition, the PI3-kinase (PI3K) inhibitor LY294002 (30 µM), the PI3K and myosin light chain kinase (MLCK) inhibitor wortmannin (10 µM) and the calmodulin kinase (CaMKII) inhibitor KN-93 (10 µM) did not affect current decrease induced by endothelin (n = 5–7). In summary, only inhibition of Rho kinase reduced ETA receptor-dependent TASK1 current inhibition by ET-1. Other low MW kinase inhibitors tested did not significantly affect the action of ET-1.
ETB receptors activate Rho kinase signalling to inhibit TASK1 currents
Next, we examined intracellular signalling pathways underlying ETB receptor-dependent TASK1 current reduction, employing the approach described for ETA. Similar to the results obtained from ETA receptors, the Rho kinase inhibitor Y-27632 (10 µM) attenuated the inhibitory regulation of TASK1 currents through ETB receptors and, after 30 min, the effect of ET-1 was almost abolished, suggesting a critical role for Rho kinase in ETB signalling (Figure 4B; n = 5; P = 0.008).
Co-application of the PKA inhibitor KT5720 (2.5 µM) or the PKC inhibitors staurosporine (1 µM), chelerythrine (10 µM) or RO-32–0432 (3 µM), with ET-1 (20 nM) had no effects (n = 6–7). The PLC inhibitor U73122 (10 µM), the cGMP inhibitor ODQ, the MLCK inhibitor wortmannin (10 µM), the PI3K inhibitor LY294002 (30 µM) or the CamKII inhibitor KN-93 similarly did not affect the regulation of TASK1 through ETB receptor activation (n = 7–10). These data are shown in Figure 4B.
Rho kinase activation by the GTPase RhoA is involved in TASK1 inhibition
Rho kinase is a downstream effector of small GTPases. The family of upstream Rho GTPases consists of three subfamilies, Rho (RhoA, RhoB and RhoC), Rac (Rac1, Rac2 and Rac3) and CDC42 (CDC42Hs, G25K) (Oleksy et al., 2006; Ridley, 2006; Boureux et al., 2007). RhoA has been shown to be the primary activator of Rho kinase (Miyazaki et al., 2006; Sato and Iiri, 2006). However, Rac1 may serve to activate Rho kinase as well (Vincent and Settleman, 1997). Therefore, we investigated the role of RhoA and Rac1 in the inhibition of TASK1 by ET-1 in the Xenopus oocyte expression system. Clostridium botulinum C3 exoenzyme inactivates RhoA via ADP ribosylation. As C3 is not cell permeable, we injected the enzyme 3 h prior to electrophysiological experiments to ensure sufficient RhoA inhibition prior to endothelin application. In oocytes co-expressing TASK1 channels and ETA receptors, pre-treatment with C3 to inactivate RhoA resulted in significant attenuation of TASK1 current decrease induced by 20 nM ET-1 (Figure 5A; n = 4; P = 0.03) compared with the effect of ET-1 in the absence of inhibitors. Similarly, C3 injection diminished the effect of 20 nM ET-1 through ETB receptors (Figure 5B; n = 5; P = 0.04), compared with the effect of ET-1 alone.
Figure 5.
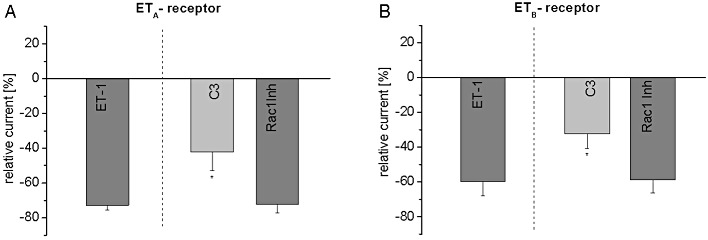
Upstream Rho kinase signalling in oocytes with TASK1 channels and ETA or ETB receptors. The small molecule Rac1 inhibitor (50 µM) was applied together with 20 nM ET-1. In addition, C3 toxin, an inhibitor of RhoA, was injected 3 h prior to the experiments (1 ng per oocyte) with ET-1 20 nM. Panel (A) shows the results for ETA receptors, and data obtained from ETB receptors are displayed in (B). Data are shown as mean ± SEM; *P < 0.05 versus ET-1 alone.
Rac1 was inactivated by Rac1 inhibitor, a membrane-permeable small molecule compound which was co-applied with ET-1 (20 nM) in according to the experimental procedure described above. Rac1 inhibitor (50 µM) did not significantly affect the ETA or ETB receptor-associated current reduction (Figure 5A, B; n = 5–6). These results indicate that RhoA but not Rac1 was involved in activation of Rho kinase signalling to regulate TASK1 channels.
TASK1 inhibition by ET receptor activation is mediated by channel phosphorylation
We detected two Rho kinase consensus sites located in the C terminus of TASK1, Ser336 and Ser393. To investigate whether Rho kinase-dependent phosphorylation was required for endothelin regulation, we generated TASK1 clones where serine residues were replaced by the alanine to prevent channel phosphorylation (TASK1-S336A; TASK1-S393A). The mutant channels were co-expressed with ETA receptors in Xenopus oocytes and found to exhibit current amplitudes and biophysical characteristics comparable to those of wild type channels (data not shown). In oocytes with TASK1-S336A channels, the inhibitory effect of ET-1 (20 nM) was significantly attenuated (Figure 6A; n = 7; P < 0.0001) compared with the effect in wild type channels. Moreover, the mutation of Ser393 in TASK1 channels (TASK1-S393A) completely abolished the effect of endothelin (Figure 6A; n = 10; P < 0.0001). These results indicate an essential role of Ser393 and an additional minor role of Ser336 in ETA-dependent TASK1 regulation. Next, we co-expressed the mutant channels with ETB receptors and found that (Figure 6B) in cells with TASK1-S336A channels, the effects of ET-1 (20 nM) were not different from those with wild type channels (n = 5; P = 0.95). In contrast, the inhibitory effect of ET-1 was abolished in cells with ETB receptors and TASK1-S393A channels (n = 7). Hence, the Ser393 residue was essential for the regulation of TASK1 channels through ETB receptors, but Ser-336 was not functionally relevant.
Figure 6.
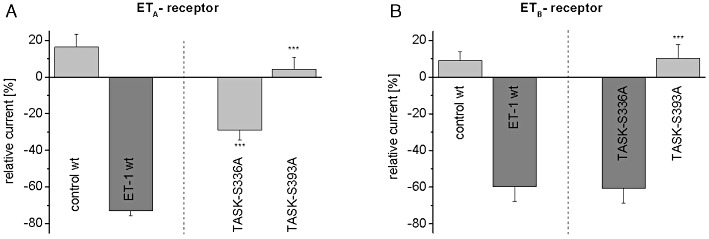
Downstream Rho kinase signalling in oocytes with mutant TASK1 channels and ETA or ETB receptors. Mutant TASK1 channels lacking the consensus sites for Rho kinase phosphorylation (TASK1-S336A; TASK1-S393A) were generated. Effects of ET-1 (20 nM) on mutant TASK1 channels in the presence of ETA receptors (A) or ETB receptors (B) are displayed. In each panel, control recordings (30 min) and the effect of ET-1 (20 nM) on TASK1 wild type channels are shown for comparison. Data are given as mean ± SEM; ***P < 0.001 versus wild type TASK1.
Discussion
ET-1 inhibits TASK1 (IKN) currents
TASK1 background potassium currents were inhibited by ET-1 in hPASMCs and in Xenopus oocytes. ETA and ETB receptors functionally coupled to TASK1 channels in the oocyte expression system. The IC50 values were 0.08 nM for ETA receptors and 0.23 nM for ETB receptors respectively. Endothelin receptors may activate multiple downstream signal transduction routes including protein kinase C- and CaMKII-dependent mechanisms (Rockman et al., 2002). We found that native and recombinant TASK1 current modulation by ET-1 was mediated through Rho kinase signalling that was not previously reported to affect TASK1 channels. In contrast, pharmacological screening with a wide range of low MW kinase inhibitors showed that activation of protein kinases A, C, CamKII, PI3 kinase, and myosin light chain kinase was not required for TASK1 current inhibition by ET-1. Detailed signal transduction mechanisms downstream of endothelin receptor activation are delineated below.
TASK1 channel regulation was described earlier in freshly isolated hPASMC (Tang et al., 2009). In these cells, ETA receptor activation was shown to inhibit TASK1 channels via PKC-dependent pathways. However, PKC inhibition did not affect TASK1 current modulation by ET-1 in our mechanistic study. Differences in experimental results may be attributed to the cell type (i.e. freshly isolated vs. cultured pulmonary artery smooth muscle cells) and to specific properties of protein kinase inhibitors used. Specifically, Tang et al. (2009) applied Ro318220, Gö6983 and rottlerin to inhibit PKC. Ro318220 is recognized as less specific protein kinase inhibitor with additional effects on a G protein-coupled receptor kinase, GRK-5, which could affect Rho kinase signalling. Furthermore, rottlerin did not fully prevent ET-1 inhibition of TASK1, suggesting additional regulation that may be explained by Rho kinase activation described in the present study.
Molecular mechanisms of TASK1 current inhibition by ET-1
Intracellular mechanisms of TASK1 inhibition by ET-1 were studied using the Xenopus oocyte system to specifically address signalling molecules in isolation (Figure 7). Stimulation of both ETA and ETB receptors reduced TASK1 currents involving Rho kinase activation. Rho kinase is a member of the Ras superfamily of small GTP-binding proteins (Van Aelst and D'Souza-Schorey, 1997). The kinase is activated by the small GTPase, RhoA. We describe a critical role of RhoA in endothelin-dependent TASK1 regulation. Inhibition of RhoA consistently attenuated TASK1 inhibition after ETA or ETB receptor activation. However, the inhibitory effect of ET-1 was not suppressed completely because of experimental difficulties associated with the use of the C3 exoenzyme as a RhoA inhibitor. C3 application required pre-injection as the enzyme is not cell permeable, resulting in a short effective time window between onset of effect and progressive enzyme degradation. Furthermore, we identified Rho kinase phosphorylation of C-terminal consensus sites as downstream effects of ET-1 signalling. Mutation of Ser393 (TASK1-S393A) rendered TASK1 channels insensitive to both ETA- and ETB- mediated current inhibition. Similar wild type and TASK1-S393A current levels in the absence of ET-1 indicated negligible baseline phosphorylation at this amino acid residue. In addition, mutation of Ser336 selectively attenuated ETA-dependent TASK1 regulation without affecting the ETB pathway, suggesting a specific role of Ser336 following ETA receptor activation.
Figure 7.
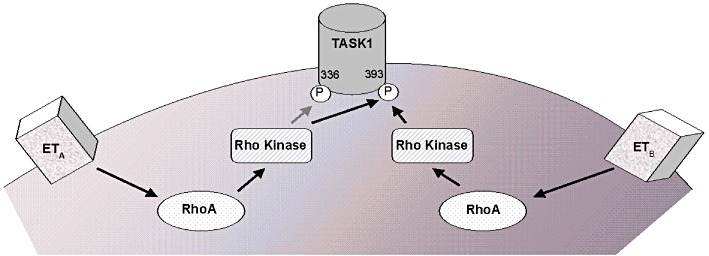
Overview of ET-dependent regulation of TASK1 channels. See text for details.
Regulation of TASK1 channels by Gq protein-coupled receptors has been reported previously. Upon ligand binding, the receptors activate Gq proteins that stimulate PLC. PLC then hydrolyses phosphatidylinositol 4,5-bisphosphate (PIP2) to produce two second messengers, inositol 1,4,5-trisphosphate (IP3) and DAG. PKC, a downstream target of DAG, suppresses murine ITASK1 through phosphorylation at Thr381 (Lopes et al., 2000; Barbuti et al., 2002; Besana et al., 2004). In addition, activation of Gq-coupled metabotropic glutamate receptors inhibits TASK1 channels through PIP2 depletion and IP3 liberation (Chemin et al., 2003), while intracellular signalling associated with α1A adrenoceptor-dependent TASK1 blockade remains to be elucidated (Putzke et al., 2007). Intriguingly, angiotensin II AT1a as well as M1 muscarinic acetylcholine receptors reduce TASK1 currents independent of the second messengers IP3 and DAG, respectively, suggesting additional signalling pathways (Czirják et al., 2000; 2001). While there is evidence for direct regulation of TASK1 by PIP2 (Lopes et al., 2005), we hypothesize that Rho kinase contributes to previously described modulation of ITASK1 by Gq protein-coupled receptors. This notion is further supported by the observation that Gq-associated thyrotropin-releasing hormone (TRH) R1 receptors inhibit TASK1 channels in a PIP2- and PKC-independent fashion (Talley et al., 2000; Chen et al., 2006).
Clinical significance and conclusions
PAH is associated with high pulmonary vascular resistance, pulmonary vascular remodelling and altered vascular reactivity (Priest et al., 1998; Nagaoka et al., 2005). There is an increasing body of evidence suggesting that the TASK1 (IKN) potassium current determines the resting membrane potential in hPASMC, thereby modulating vascular tone and diameter (Olschewski et al., 2006). ET-1 is a strong vasoconstrictor that contributes to human PAH. In addition, recent studies have suggested a role for Rho-associated serine/threonine kinase (Rho kinase) in the development of PAH (McMurtry et al., 2003). Furthermore, the physiological Rho-kinase activator, RhoA, is stimulated by a variety of pulmonary vasoconstrictors, including ET-1, phenylephrine and 5-HT (Nagaoka et al., 2005). RhoA has been identified as a regulator of smooth muscle contraction (Janssen et al., 2001; Sakurada et al., 2001; Wang et al., 2001). Thus, we conclude that TASK1 inhibition by ET-1, RhoA and Rho kinase revealed in this work contributes to PAH at the molecular level. In summary, we have demonstrated that ET-1 regulated vascular TASK1 currents through activation of ETA and ETB receptors. This effect was mediated by Rho kinase signalling with an essential role of phosphorylation sites in the C terminus of TASK1 channels. The Rho kinase pathway may represent a novel therapeutic target for the treatment of PAH.
Acknowledgments
We thank Ramona Bloehs for excellent technical assistance. This study was supported in part by a postdoctoral fellowship of the Medical Faculty of the University of Heidelberg (to CS), and by research grants from the Deutsche Forschungsgemeinschaft (Ka 1714/1–2 to CK; Zi 1177/1–1 to EZ; FRONTIERS program to DT), the ADUMED foundation (to DT), and the German Heart Foundation/German Foundation of Heart Research (to DT).
Glossary
- ET-1
endothelin-1
- K2P
two-pore-domain K+ channel
- PAH
pulmonary arterial hypertension
- TASK
TWIK-related acid sensitive K+ channel
- TWIK
tandem of P domains in a weak inward rectifying K+ channel
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research ‘Work in progress’. Circulation. 2000;102:2781–2791. doi: 10.1161/01.cir.102.22.2781. [DOI] [PubMed] [Google Scholar]
- Barbuti A, Ishii S, Shimizu T, Robinson RB, Feinmark SJ. Block of the background K(+) channel TASK-1 contributes to arrhythmogenic effects of platelet-activating factor. Am J Physiol Heart Circ Physiol. 2002;282:H2024–H2030. doi: 10.1152/ajpheart.00956.2001. [DOI] [PubMed] [Google Scholar]
- Besana A, Barbuti A, Tateyama MA, Symes AJ, Robinson RB, Feinmark SJ. Activation of protein kinase C epsilon inhibits the two-pore domain K+ channel, TASK-1, inducing repolarization abnormalities in cardiac ventricular myocytes. J Biol Chem. 2004;279:33154–33160. doi: 10.1074/jbc.M403525200. [DOI] [PubMed] [Google Scholar]
- Boureux A, Vignal E, Faure S, Fort P. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol Biol Evol. 2007;24:203–216. doi: 10.1093/molbev/msl145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Girard C, Duprat F, Lesage F, Romey G, Lazdunski M. Mechanisms underlying excitatory effects of group I metabotropic glutamate receptors via inhibition of 2P domain K+ channels. EMBO J. 2003;22:5403–5411. doi: 10.1093/emboj/cdg528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Talley EM, Patel N, Gomis A, McIntire WE, Dong B, et al. Inhibition of a background potassium channel by Gq protein alpha-subunits. Proc Natl Acad Sci USA. 2006;103:3422–3427. doi: 10.1073/pnas.0507710103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czirják G, Fischer T, Spät A, Lesage F, Enyedi P. TASK (TWIK-related acid-sensitive K+ channel) is expressed in glomerulosa cells of rat adrenal cortex and inhibited by angiotensin II. Mol Endocrinol. 2000;14:863–874. doi: 10.1210/mend.14.6.0466. [DOI] [PubMed] [Google Scholar]
- Czirják G, Petheo GL, Spät A, Enyedi P. Inhibition of TASK-1 potassium channel by phospholipase C. Am J Physiol Cell Physiol. 2001;281:C700–C708. doi: 10.1152/ajpcell.2001.281.2.C700. [DOI] [PubMed] [Google Scholar]
- Dupuis J, Goresky CA, Fournier A. Pulmonary clearance of circulating endothelin-1 in dogs in vivo: exclusive role of ETB receptors. J Appl Physiol. 1996;81:1510–1515. doi: 10.1152/jappl.1996.81.4.1510. [DOI] [PubMed] [Google Scholar]
- Galié N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res. 2004;61:227–237. doi: 10.1016/j.cardiores.2003.11.026. [DOI] [PubMed] [Google Scholar]
- Gardener MJ, Johnson IT, Burnham MP, Edwards G, Heagerty AM, Weston AH. Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br J Pharmacol. 2004;142:192–202. doi: 10.1038/sj.bjp.0705691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierten J, Ficker E, Bloehs R, Schlömer K, Kathöfer S, Scholz E, et al. Regulation of two-pore-domain K2P potassium leak channels by the tyrosine kinase inhibitor genistein. Br J Pharmacol. 2008;154:1680–1690. doi: 10.1038/bjp.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein SA, Bockenhauer D, O'Kelly I, Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci. 2001;2:175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- Gurney A, Manoury B. Two-pore potassium channels in the cardiovascular system. Eur Biophys J. 2009;38:305–318. doi: 10.1007/s00249-008-0326-8. [DOI] [PubMed] [Google Scholar]
- Gurney AM, Osipenko ON, MacMillan D, McFarlane KM, Tate RJ, Kempsill FEJ. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ Res. 2003;93:957–964. doi: 10.1161/01.RES.0000099883.68414.61. [DOI] [PubMed] [Google Scholar]
- Janssen LJ, Lu-Chao H, Netherton S. Excitation-contraction coupling in pulmonary vascular smooth muscle involves tyrosine kinase and Rho kinase. Am J Physiol Lung Cell Mol Physiol. 2001;280:L666–L674. doi: 10.1152/ajplung.2001.280.4.L666. [DOI] [PubMed] [Google Scholar]
- Kiesecker C, Zitron E, Scherer D, Lueck S, Bloehs R, Scholz EP, et al. Regulation of cardiac inwardly rectifying potassium current IK1 and Kir2.x channels by endothelin-1. J Mol Med. 2006;84:46–56. doi: 10.1007/s00109-005-0707-8. [DOI] [PubMed] [Google Scholar]
- Lippton HL, Hauth TA, Cohen GA, Hyman AL. Functional evidence for different endothelin receptors in the lung. J Appl Physiol. 1993;75:38–48. doi: 10.1152/jappl.1993.75.1.38. [DOI] [PubMed] [Google Scholar]
- Lopes CM, Gallagher PG, Buck ME, Butler MH, Goldstein SA. Proton block and voltage gating are potassium-dependent in the cardiac leak channel Kcnk3. J Biol Chem. 2000;275:16969–16978. doi: 10.1074/jbc.M001948200. [DOI] [PubMed] [Google Scholar]
- Lopes CM, Rohács T, Czirják G, Balla T, Enyedi P, Logothetis DE. PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J Physiol. 2005;564:117–129. doi: 10.1113/jphysiol.2004.081935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation. 2000;102:2434–2440. doi: 10.1161/01.cir.102.19.2434. [DOI] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lazdunski M, Honoré E. The endocannabinoid anandamide is a direct and selective blocker of the background K+ channel TASK-1. EMBO J. 2001;20:47–54. doi: 10.1093/emboj/20.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurtry IF, Bauer NR, Fagan KA, Nagaoka T, Gebb SA, Oka M. Hypoxia and Rho/Rho-kinase signaling. Lung development versus hypoxic pulmonary hypertension. Adv Exp Med Biol. 2003;543:127–137. [PubMed] [Google Scholar]
- Miyazaki K, Komatsu S, Ikebe M. Dynamics of RhoA and ROKalpha translocation in single living cells. Cell Biochem Biophys. 2006;45:243–254. doi: 10.1385/CBB:45:3:243. [DOI] [PubMed] [Google Scholar]
- Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, et al. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med. 2005;171:494–499. doi: 10.1164/rccm.200405-637OC. [DOI] [PubMed] [Google Scholar]
- Oleksy A, Opaliński L, Derewenda U, Derewenda ZS, Otlewski J. The molecular basis of RhoA specificity in the guanine nucleotide exchange factor PDZ-RhoGEF. J Biol Chem. 2006;281:32891–32897. doi: 10.1074/jbc.M606220200. [DOI] [PubMed] [Google Scholar]
- Olschewski A, Li Y, Tang B, Hanze J, Eul B, Bohle RM, et al. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res. 2006;98:1072–1080. doi: 10.1161/01.RES.0000219677.12988.e9. [DOI] [PubMed] [Google Scholar]
- Priest RM, Robertson TP, Leach RM, Ward JP. Membrane potential-dependent and -independent vasodilation in small pulmonary arteries from chronically hypoxic rats. J Pharmacol Exp Ther. 1998;285:975–982. [PubMed] [Google Scholar]
- Putzke C, Wemhöner K, Sachse FB, Rinné S, Schlichthörl G, Li XT, et al. The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc Res. 2007;75:59–68. doi: 10.1016/j.cardiores.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- Sakurada S, Okamoto H, Takuwa N, Sugimoto N, Takuwa Y. Rho activation in excitatory agonist-stimulated vascular smooth muscle. Am J Physiol Cell Physiol. 2001;281:C571–C578. doi: 10.1152/ajpcell.2001.281.2.C571. [DOI] [PubMed] [Google Scholar]
- Sato J, Iiri T. Rho and Rho-kinase. Nippon Rinsho. 2006;64(Suppl. 5):197–202. [PubMed] [Google Scholar]
- Seo B, Oemar BS, Siebenmann R, von Segesser L, Lüscher TF. Both ETA and ETB receptors mediate contraction to endothelin-1 in human blood vessels. Circulation. 1994;89:1203–1208. doi: 10.1161/01.cir.89.3.1203. [DOI] [PubMed] [Google Scholar]
- Staudacher K, Staudacher I, Ficker E, Seyler C, Gierten J, Kisselbach J, et al. Carvedilol targets human K2P 3.1 (TASK1) K+ leak channels. Br J Pharmacol. 2011;163:1099–1110. doi: 10.1111/j.1476-5381.2011.01319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Lei Q, Sirois JE, Bayliss DA. TASK-1, a two-pore domain K+ channel, is modulated by multiple neurotransmitters in motoneurons. Neuron. 2000;25:399–410. doi: 10.1016/s0896-6273(00)80903-4. [DOI] [PubMed] [Google Scholar]
- Tang B, Li Y, Nagaraj C, Morty RE, Gabor S, Stacher E, et al. Endothelin-1 inhibits background two-pore domain channel TASK-1 in primary human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2009;41:476–483. doi: 10.1165/rcmb.2008-0412OC. [DOI] [PubMed] [Google Scholar]
- Thomas D, Zhang W, Karle CA, Kathöfer S, Schöls W, Kübler W, et al. Deletion of protein kinase A phosphorylation sites in the HERG potassium channel inhibits activation shift by protein kinase A. J Biol Chem. 1999;274:27457–27462. doi: 10.1074/jbc.274.39.27457. [DOI] [PubMed] [Google Scholar]
- Thomas D, Plant LD, Wilkens CM, McCrossan ZA, Goldstein SAN. Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron. 2008;58:859–870. doi: 10.1016/j.neuron.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Vincent S, Settleman J. The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol Cell Biol. 1997;17:2247–2256. doi: 10.1128/mcb.17.4.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jin N, Ganguli S, Swartz DR, Li L, Rhoades RA. Rho-kinase activation is involved in hypoxia-induced pulmonary vasoconstriction. Am J Respir Cell Mol Biol. 2001;25:628–635. doi: 10.1165/ajrcmb.25.5.4461. [DOI] [PubMed] [Google Scholar]