

Abstract
BACKGROUND AND PURPOSE
Gastrointestinal (GI) motility is regulated in part by fatty acid ethanolamides (FAEs), including the endocannabinoid (EC) anandamide (AEA). The actions of FAEs are terminated by fatty acid amide hydrolase (FAAH). We investigated the actions of the novel FAAH inhibitor AM3506 on normal and enhanced GI motility.
EXPERIMENTAL APPROACH
We examined the effect of AM3506 on electrically-evoked contractility in vitro and GI transit and colonic faecal output in vivo, in normal and FAAH-deficient mice treated with saline or LPS (100 µg·kg−1, i.p.), in the presence and absence of cannabinoid (CB) receptor antagonists. mRNA expression was measured by quantitative real time-PCR, EC levels by liquid chromatography-MS and FAAH activity by the conversion of [3H]-AEA to [3H]-ethanolamine in intestinal extracts. FAAH expression was examined by immunohistochemistry.
KEY RESULTS
FAAH was dominantly expressed in the enteric nervous system; its mRNA levels were higher in the ileum than the colon. LPS enhanced ileal contractility in the absence of overt inflammation. AM3506 reversed the enhanced electrically-evoked contractions of the ileum through CB1 and CB2 receptors. LPS increased the rate of upper GI transit and faecal output. AM3506 normalized the enhanced GI transit through CB1 and CB2 receptors and faecal output through CB1 receptors. LPS did not increase GI transit in FAAH-deficient mice.
CONCLUSIONS AND IMPLICATIONS
Inhibiting FAAH normalizes various parameters of GI dysmotility in intestinal pathophysiology. Inhibition of FAAH represents a new approach to the treatment of disordered intestinal motility.
Keywords: endocannabinoids, gastrointestinal motility, lipopolysaccharide, fatty acid amide hydrolase, cannabinoid receptors
Introduction
The endocannabinoid (EC) system consists of the cannabinoids (CBs) receptor type 1 and 2 (CB1 and CB2 receptors), endogenous CB ligands, anandamide (AEA) and 2-arachidonoylglycerol (2-AG), and the enzymes involved in their biosynthesis and degradation (Di Marzo, 2009; Izzo and Sharkey, 2010). This system is involved in regulating gastrointestinal (GI) function (Duncan et al., 2005; Storr and Sharkey, 2007; Izzo and Camilleri, 2008; Izzo and Sharkey, 2010). ECs activate CB1 and CB2 receptors in the enteric nervous system (ENS), smooth muscle and epithelium (Duncan et al., 2005; Wright et al., 2005, 2008; Izzo and Camilleri, 2008; Izzo and Sharkey, 2010). In the ENS, ECs reduce ACh release, thereby inhibiting contractility and peristalsis (Aviello et al., 2008; Izzo and Sharkey, 2010). In the intestinal epithelium, CB1 receptor activation regulates cell proliferation and promotes healing (Wright et al., 2005; Cianchi et al., 2008).
ECs are rapidly degraded (Puffenbarger, 2005; Ahn et al., 2008). Fatty acid amide hydrolase (FAAH) catalyses the hydrolysis of AEA, as well as palmitoylethanolamide (PEA) and oleoylethanolamide (OEA) (Cravatt and Lichtman, 2002; Fegley et al., 2005). Inhibiting FAAH produces behavioural effects consistent with activation of the CB1 receptor, for example, anti-anxiety, analgesia and anti-inflammatory actions (Chang et al., 2006; Scherma et al., 2008; Schlosburg et al., 2009). However, typical adverse behavioural effects of CB agonists (e.g. catalepsy) are not observed in animals treated with FAAH inhibitors or seen in mice deficient in the FAAH gene (Cravatt et al., 2001; Kathuria et al., 2003). These features make FAAH inhibition an attractive therapeutic option for circumstances where CB receptor activation may be beneficial (Cravatt and Lichtman, 2003; Petrosino and Di Marzo, 2010).
AEA, PEA and OEA are able to reduce intestinal motility in mice (Capasso et al., 2001; Pinto et al., 2002; Cluny et al., 2009). While there is compelling evidence in support of the EC system regulating intestinal motility, the role of FAAH remains somewhat enigmatic. FAAH is expressed throughout the wall of the gut; in the ENS, epithelial cells and immune cells (Katayama et al., 1997; Capasso et al., 2005; De Filippis et al., 2008a; Marquez et al., 2009). It is up-regulated in sepsis and colitis, but not in colon cancer (Ligresti et al., 2003; De Filippis et al., 2008a; Marquez et al., 2009). However, FAAH-deficient mice do not display reduced GI motility and may (Capasso et al., 2005) or may not have elevated intestinal levels of AEA despite the increased intestinal PEA and OEA levels (Fegley et al., 2005).
Treatment with the FAAH inhibitor URB597 blocks FAAH activity, but does not elevate levels of fatty acid ethanolamides (FAE) in the small intestine, in contrast to the CNS (Fegley et al., 2005); however, another FAAH inhibitor, AA-5-HT, decreases intestinal motility and increases intestinal AEA and PEA (Capasso et al., 2005). The differences in findings of the studies on FAAH inhibitors and FAAH-deficient mice require further investigation.
Despite the discrepancies that have been seen on the levels of FAEs after treatment with different FAAH inhibitors and in FAAH-deficient mice, recent studies showed a significant association between FAAH genotype and different phenotypes of functional GI disorders (Camilleri et al., 2008). Moreover, patients with Crohn's disease and ulcerative colitis with a FAAH gene polymorphism are more prone to a severe disease phenotype and an earlier disease onset respectively (Storr et al., 2009a).
Given the therapeutic potential of FAAH to regulate intestinal motility, we have investigated a recently developed FAAH inhibitor, AM3506 (Godlewski et al., 2010) in a model of enhanced GI transit induced by LPS. It has been shown that a low dose of LPS increases GI motility without causing inflammation. This model of GI hypermotility mimics some features of functional GI motor disorders (Ceregrzyn et al., 2001; Mathison et al., 2004; Duncan et al., 2008).
We hypothesized that the newly developed potent FAAH inhibitor AM3506 (Godlewski et al., 2010) would normalize intestinal dysmotility, through activation of the EC system. Our goal was to evaluate whether FAAH inhibitors might have therapeutic potential in regulating abnormal GI motility, an approach that could be translated to humans.
Methods
Animals
Male Swiss albino mice (CD1, Charles River, St. Constant, QC, Canada), male CB1-deficient mice (CB1−/−) on a predominant C57BL/6N background, male FAAH-deficient mice on C57BL/6N background and their wild-type pairs at 5–8 weeks of age were used and genotyped as previously described (Cravatt et al., 2001; Marsicano et al., 2002). Mice were housed with free access to food and water, and were allowed 1 week of acclimatization at a constant temperature (22°C) under a 12:12 h light–dark cycle before the study was commenced. All animal care and experimental procedures complied with the Guidelines of the Canadian Council on Animal Care and were approved by the University of Calgary Animal Care Committee.
LPS treatment for in vitro experiments
For the in vitro experiments, LPS (100 µg·kg−1; Escherichia coli 026:B6; Sigma-Aldrich, St. Louis, MO, USA) or saline was injected i.p., and mice were killed 90 min later by cervical dislocation (Mathison et al., 2004; Duncan et al., 2008). The in vivo experiments are discussed separately below.
Determination of tissue myeloperoxidase (MPO) activity
MPO activity was assessed as a marker of neutrophil infiltration (Krawisz et al., 1984; Storr et al., 2009b). Samples of ileum or distal colon of LPS or saline-treated CD1 mice were homogenized in hexadecyltrimethyl ammonium bromide (HTAB) buffer (0.5% HTAB; Sigma-Aldrich). The homogenate was centrifuged, and supernatant was added to potassium phosphate buffer, containing O-dianisidine hydrochloride and H2O2. Absorbance was measured at 460 nm (Thermo Fischer Labsystems Multiskan, Thermo Scientific, Ottawa, ON, Canada), and MPO activity was expressed in U g−1 of wet tissue weight and was calculated from a standard curve performed on purified peroxidase enzyme (Sigma-Aldrich).
Histology
As detailed before (Storr et al., 2009b), segments of distal colon or ileum of LPS or saline-treated CD1 mice were fixed overnight in Zamboni's fixative (2% paraformaldehyde, 15% picric acid; pH 7.4) at 4°C. Tissues were then rinsed in PBS and cross and sagittal sections of the specimens were cryoprotected in PBS containing 20% sucrose for 24 h. Specimens were embedded in optimum cutting temperature (Tissue-Tek, Sakura Finetechnical, Tokyo, Japan), cryostat-sectioned (12 µm) and mounted onto poly-D-lysine-coated slides. Sections were stained with haematoxylin and eosin and examined using a Zeiss Axioplan microscope (Carl Zeiss, Toronto, ON, Canada).
RNA extraction, cDNA generation and quantitative PCR
Distal colon and ileum of LPS- or saline-treated CD1 mice stored at −20°C in RNAlater (Qiagen, Mississauga, ON, Canada) were used for RNA extraction. Total RNA was extracted using the QIAGEN RNeasy Plus Mini Kit (Qiagen, Mississauga, ON, Canada). cDNA was generated from 1 µg of total RNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). In some experiments, reverse transcriptase was withheld from the reaction mixture.
TaqMan Gene Expression assay kits for the CB1 (Mm00432621_s1), CB2 (Mm0438286_m1), FAAH (Mm00515684_m1) and TNF (Mm00443258_m1) genes were purchased from Applied Biosystems (Frederick, MD, USA). The rodent glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probe (VIC) from Applied Biosystems was used as the internal control. GAPDH expression was unchanged after treatment of the animals with LPS. Triplicate samples of each cDNA were amplified by real-time PCR in the ABI Prism 7000 Sequence Detection System (Applied Biosystems; 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min). Results were analysed using the ABI Prism 7000 SDS software (Applied Biosystems). Relative quantification (RQ) values, defined as fold changes in mRNA expressions, were calculated using the ‘δδ Ct’ method. Briefly, a sample of ileum from a saline-treated animal was selected as the calibrator. The Ct values of other samples were calculated and presented as fold decrease or increase relative to the calibrator and presented as mean ± SEM of RQ values in each group.
Immunohistochemistry
Ileum and distal colon of LPS- or saline-treated CD1 mice were removed and immersed in ice-cold PBS (10 min) containing 1 µM nicardipine (Sigma-Aldrich). Tissues were opened along the mesenteric border, and were stretched and pinned mucosal side up in Sylgard (Dow Corning, Midland, MI, USA)-coated Petri dishes. Tissue samples were fixed by overnight immersion in 4% paraformaldehyde. Full-wall thickness cryostat sections or whole mount preparations were washed in PBS (3 × 10 min) containing 0.1% Triton X-100 (Sigma), incubated with a rabbit anti-FAAH primary antibody (1:500, provided by KM), which was raised against the last 103 amino acids of rat FAAH (Helliwell et al., 2004) for 48 h at 4°C. After 48 h, tissues were washed again in PBS (3 × 10 min) and incubated with the donkey anti-rabbit CY3 (Jackson ImmunoResearch, West Grove, PA, USA; 1:100) as a secondary antibody for 90 min at room temperature, mounted in bicarbonate-buffered glycerol, examined using a Zeiss Axioplan fluorescence microscope and photographed with a Sensys digital camera (Photometrics, Tucson, AZ, USA) using V for Windows software (version 3.5; Digital Optics Ltd; Auckland, New Zealand). Intestinal tissues from FAAH-deficient mice were used as a negative control (Supporting Information Figure S1).
Assessment of FAAH activity
Ileal and distal colonic samples of LPS- or saline-treated CD1 mice were incubated at 37°C in Krebs solution with or without AM3506 (100 nM) for 15 min. Tissues were washed three times with Krebs solution and immediately frozen in liquid nitrogen. Frozen tissues were then homogenized in ice-cold 10 mM Tris-HCl buffer pH 7.6 containing 1 mM EDTA and centrifuged to remove cell debris. Supernatants were transferred to other tubes, and protein amounts were adjusted to 20 µg for each reaction. Homogenates were incubated with a mix containing 400 pmol of cold AEA and 500 fmol of [3H]-AEA (specific activity – 60 Ci·mmol−1; American Radiolabeled Chemicals, St. Louis, MO, USA) for 10 min at 37°C. FAAH activity was quantified by the amount of the [3H]-ethanolamine released from [3H]-AEA labelled on the ethanolamine moiety, as described previously (Fowler et al., 2000; Liu et al., 2003). Samples were done in triplicates and the results are presented as pmol AEA hydrolyzed min−1 mg−1 protein.
Identification and quantification of ECs
Quantification of EC levels was performed as described previously (Williams et al., 2006; 2007; Wood et al., 2008). Frozen samples of ileum and distal colon of LPS- or saline-treated CD1 mice pretreated with AM3506 (0.5 mg·kg−1) or its vehicle were weighed before homogenization in ice-cold acetone:PBS (pH 7.4; 3:1) and centrifuged. After the acetone had been evaporated under nitrogen, 100 µL PBS, one volume of methanol and two volumes of chloroform were added to the remaining supernatant,. The chloroform layer was dried under nitrogen stream and reconstituted in methanol before LC/MS/MS analysis. Chromatographic separation was achieved using an Agilent Zorbax SB-CN column (2.1 × 50 mm, 5 mm) on a Finnigan TSQ Quantum Ultra triple quadrupole mass spectrometer (Thermo Electron, San Jose, CA, USA) with an Agilent 1100 HPLC on the front end (Agilent Technologies, Wilmington, DE, USA). Eluted peaks were ionized via atmospheric pressure chemical ionization in positive mode and detected by multiple reaction monitoring. Deuterated internal standards were used for standard curves, and the levels of AEA, PEA, OEA or 2-AG g−1 tissue were determined.
Assessment of intestinal contractility
Segments of ileum and distal colon of LPS- or saline-treated mice were removed and immersed in ice-cold oxygenated Krebs solution. Intestinal segments (1 cm) were tied with a silk thread and suspended in an organ bath filled with oxygenated Krebs solution (95% O2 and 5% CO2) at 37°C. The preparations were attached to an isometric force transducer, were positioned between platinum electrodes and allowed to equilibrate under 500 mg tension (Harvard Apparatus, model 50–7905, Kent, UK). Mechanical activity of the muscle was enhanced by a transducer amplifier (Harvard Apparatus, model 50-7970), relayed to a bioelectric amplifier (Hewlett-Packard, model 8811A, Mississauga, ON, Canada). The data were converted using a CED 1401 PLUS analogue-to-digital converter (Cambridge Electronic Design, Cambridge, UK.) running into Spike 3 software (Cambridge Electronic Design).
We assessed the effect of LPS on tension generated by cumulative addition of bethanechol (1–30 µM) and on electrically-evoked contractions and relaxations of the ileum and colon. Each experiment was done on fresh ileal or colonic tissues, and different tissues were used for each experiment. Electrical field stimulation (EFS) was applied by a Grass Stimulator (S88 stimulator, Grass, Quincy, MA, USA) at 1, 2, 4 and 8 Hz (10 s stimulation duration, 0.5 ms pulse duration, 60 V). EFS relaxations were recorded in the presence of atropine (1 µM) and guanethidine (5 µM). After these studies, the tissues were stimulated at 4 Hz (0.5 ms pulse duration; 60 V) continuously, and the effects of AM3506 or CB receptors agonists and antagonists were investigated. In separate experiments, we assessed if AM3506 altered tension generated by bethanechol (300 nM–10 µM).
AM3506 or CB agonists were added to the organ bath, and 15 min incubation time was allowed for each drug. Before the addition of drugs, the mean of three successive EFS contractions or relaxations were used as an internal control (100%). The amplitude of contractions and relaxations were measured and in the presence of drugs, were reported as the percentage of the internal control. In every set of experiments, vehicle controls were performed.
GI transit and faecal output studies
Mice were transferred from their home cage into individual transparent plastic cages without bedding. After 60 min acclimatization, any faecal pellets were removed from the cage, and mice were administered LPS (100 µg·kg−1, i.p.) or physiological saline. The individual cages were inspected for the presence of faecal pellets for the next 90 min, and the total weight of faecal output was measured. After this, 200 µL of a charcoal (10% charcoal, 5% gum arabic) marker was gavaged into the stomach to investigate the upper GI transit, which is a gross measure of gastric emptying plus small intestinal transit. The mice were killed 15 min later, and the small intestine was removed. The distance travelled by the marker was measured and expressed as a percentage of the total length of the small intestine (pylorus to caecum). To determine whether FAAH inhibition affects intestinal motility, AM3506 (0.5 mg·kg−1, i.p.) or vehicle was injected 20 min before the LPS or saline injection. In separate experiments, vehicle, AM251 (0.5 mg·kg−1, i.p.) or AM630 (1 or 5 mg·kg−1, i.p.) were administered 20 min before AM3506 injection to determine the CB receptor responsible for the effects.
Whole-gut transit was assessed in FAAH-deficient mice and their aged-matched wild-types. Whole-gut transit was performed as described previously (Cluny et al., 2009). Briefly, mice were transferred to individual cages without bedding and were left to acclimatize for 1 h. Then the animals were gavaged with 200 µL of an Evans’ blue (5% Evans’ blue, 5% gum arabic) marker. The latency to the detection of Evans’ blue in the faeces (in min) was recorded and presented as whole-gut transit time.
Assessment of locomotor activity
Ambulatory locomotor activity was measured 90 min after LPS or saline injection using an infrared beam activity monitor (Columbus Instruments, Columbus, OH, USA). AM3506 (0.5 mg·kg−1, i.p.) or vehicle was injected 20 min before the mice were administered an injection of LPS (100 µg·kg−1, i.p.) or physiological saline. Mice were pre-exposed to the recording equipment in the morning and the experiments were done in the afternoon of the same day. Each individual mouse was placed in the apparatus, and the ambulatory count was recorded over a 10 min period. Movement of the mice was recorded as the ambulatory activity count when the infrared beams were sequentially broken. The effect of WIN55,212-2 (5 mg·kg−1) on locomotor activity was used as a positive control.
Drugs
AM3506 was dissolved in ethanol for in vitro experiments. AM3506 is a later generation sulphonyl fluoride analogue with enhanced affinity and selectivity for FAAH (Figure 1A) (Deutsch et al., 1997; Godlewski et al., 2010). Experiments on human recombinant FAAH showed that AM3506 has IC50 of 48 nM for inhibition of FAAH activity, and it is three times more potent compared with its structural analogue phenylmethylsulphonyl fluoride. AM3506 has low affinity for CB1 (5.77 µmol·l−1) and moderate affinity for CB2 receptors (192 nmol·l−1). AM3506 is twice as potent in inhibiting FAAH compared with URB597 (Godlewski et al., 2010). The dose and concentration of AM3506 were selected based on our previous study that showed specific FAAH blockage at 100 nm in vitro and 0.5 mg·kg−1in vivo (Godlewski et al., 2010). The CB1/CB2 receptor agonist WIN55,212-2; CB2 receptor agonist JWH-133; CB1 receptor antagonist AM251; and CB2 receptor antagonist AM630 were purchased from Tocris (Ellisville, MI, USA) and dissolved in 100% ethanol. AM630 was dissolved in 100% dimethyl sulphoxide (DMSO) for in vitro experiments. The maximum concentration of ethanol and DMSO used in the organ bath had no effect on contractility. For the in vivo experiments AM3506, AM251 and AM630 were dissolved in 2% DMSO, 1% Tween 80 in physiological saline (vehicle). Bethanechol, atropine, guanethidine and tetrodotoxin (TTX) were purchased from Sigma-Aldrich and dissolved in water. The concentration of stock solutions for AM3506, WIN55,212-2, JWH133, AM630, AM251, guanethidine and atropine were 0.01 M and for bethanechol 1 M, these were diluted immediately before the experiments.
Figure 1.
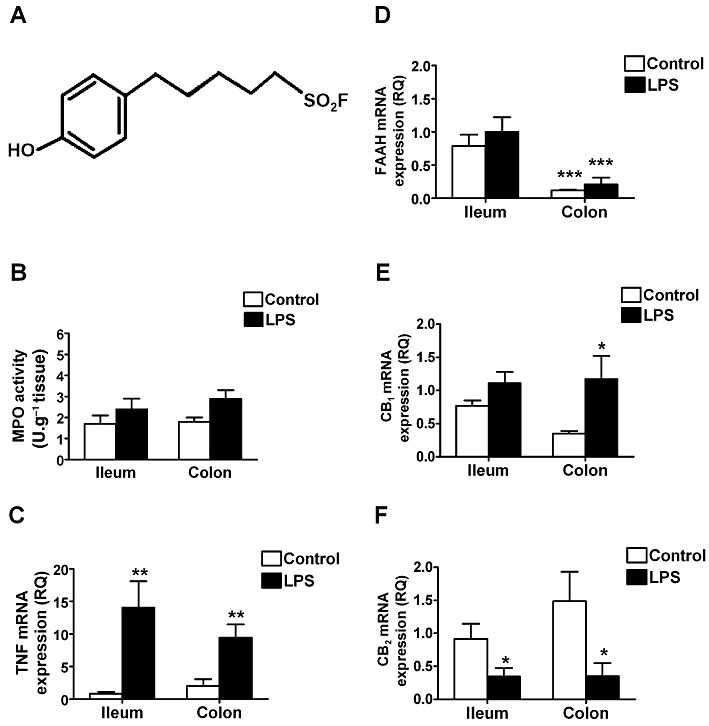
(A) Chemical structure of AM3506. AM3506 is a structural analogue of phenylmethylsulphonyl fluoride. (B) MPO activity; (C) TNF; (D) FAAH mRNA; (E) CB1 receptor; and (F) CB2 receptor expression in the ileum and distal colon of CD1 mice treated with saline (control) or LPS (100 µg·kg−1, i.p.). There were no significant changes in MPO activity after LPS treatment, although TNF mRNA was significantly increased in both ileum and distal colon. LPS treatment had no effect on FAAH mRNA, which was significantly higher in the ileum than the colon. CB1 receptor mRNA was increased in the distal colon, and CB2 receptor mRNA was reduced in both regions of the gut by LPS treatment. *P < 0.05, **P < 0.01, ***P < 0.001 compared with control in (E, C and F) and compared with ileum in (D); n = 4–6 per group.
Statistics
Data are presented as mean ± SEM of n experiments, which indicates the number of individual mice. Student's t-test was used to compare a single treatment mean with control mean, while one-way or two-way anova followed by Bonferroni post hoc testing were used for the analysis of multiple measurements. A value of P < 0.05 was considered statistically significant.
Results
The effects of LPS treatment on inflammation, FAAH, CB1 and CB2 expression
LPS treatment caused no histological evidence of inflammation (not shown) or granulocyte infiltration, as determined by MPO activity (Figure 1B). Nevertheless, LPS gave rise to immune activation as shown by enhanced TNF mRNA expression (Figure 1C). FAAH mRNA levels were significantly higher in the ileum than the colon. LPS treatment had no effect on the levels of FAAH expression in either region of the GI tract (Figure 1D). CB1 receptor mRNA levels were increased in the colon, while CB2 receptor mRNA levels were significantly reduced in both ileum and colon by LPS treatment (Figure 1E, F). Despite the regional variation in FAAH mRNA expression, FAAH immunoreactivity was present in neurons and nerve fibres and/or terminals in the ileal and colonic myenteric plexus (Figure 2), in smooth muscle, and epithelial cells. In the ileum, some neurons displayed higher levels of expression than others, the significance of which is not known. The pattern of FAAH immunoreactivity remained unchanged after LPS treatment (Figure 2). No immunoreactivity was observed in FAAH-deficient mice.
Figure 2.
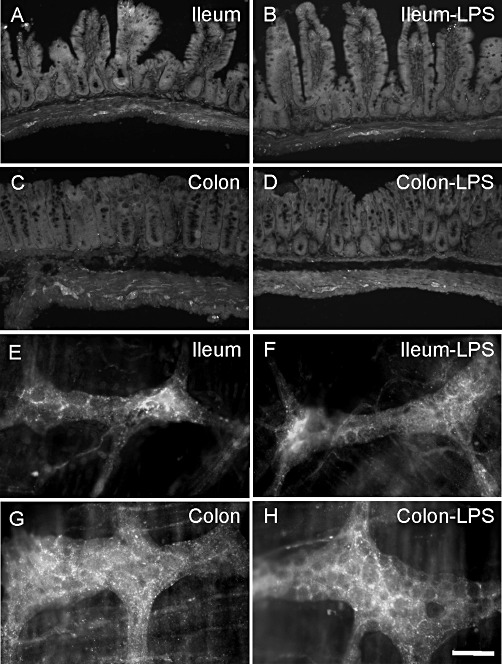
FAAH immunoreactivity in full-wall thickness sections (A–D) and whole mount preparations of the myenteric plexus (E–H) of the ileum and distal colon from animals treated with saline and LPS. LPS (100 µg·kg−1, i.p.) treatment did not alter the distribution of FAAH (B, D, F and H). Scale bars: 100 µm (A–D) and 50 µm (E–H).
Effect of LPS treatment on FAAH activity and EC levels
The activity of FAAH regulates the levels of ethanolamides in the gut. LPS treatment did not change FAAH activity in the ileum or the colon of mice (Figure 3A, B). After incubation with AM3506 (100 nM), we observed a significant reduction in FAAH activity in the ileum of LPS-treated mice (Figure 3A). LPS treatment had no effect on the levels of AEA, 2-AG, PEA and OEA (Figure 4). In LPS-treated mice, which were pretreated with AM3506 (0.5 mg·kg−1), there was a significant increase in AEA levels the ileum and the tended to be increased in the colon (Figure 4A, B). As previously reported (Pinto et al., 2002), levels of 2-AG were over a thousand times higher than AEA in both regions of the gut. 2-AG tended to be decreased in the ileum and colon of LPS-treated mice that were pretreated with AM3506, although this did not reach significance with a multiple group comparison.
Figure 3.
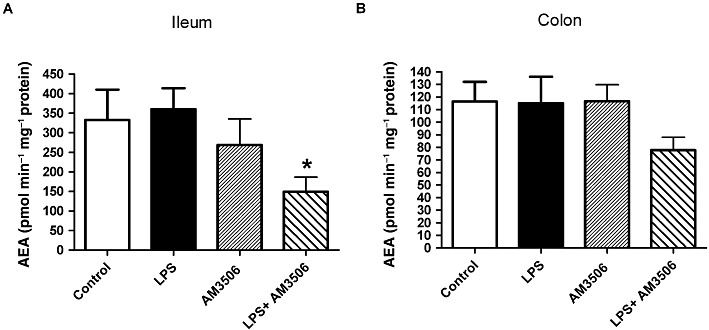
FAAH activity in the ileum (A) and the distal colon (B) of CD1 mice. Note that FAAH activity in the colon of control group was significantly lower than in the ileum (P < 0.05). LPS (100 µg·kg−1, i.p.) treatment or incubation with AM3506 (100 nM) did not inhibit FAAH activity in the ileum and the distal colon. After incubation with AM3506 (100 nM), a significant reduction in the FAAH activity of the ileum of LPS-treated mice was observed (A). *P < 0.01 compared with control; n = 6–10 per group.
Figure 4.
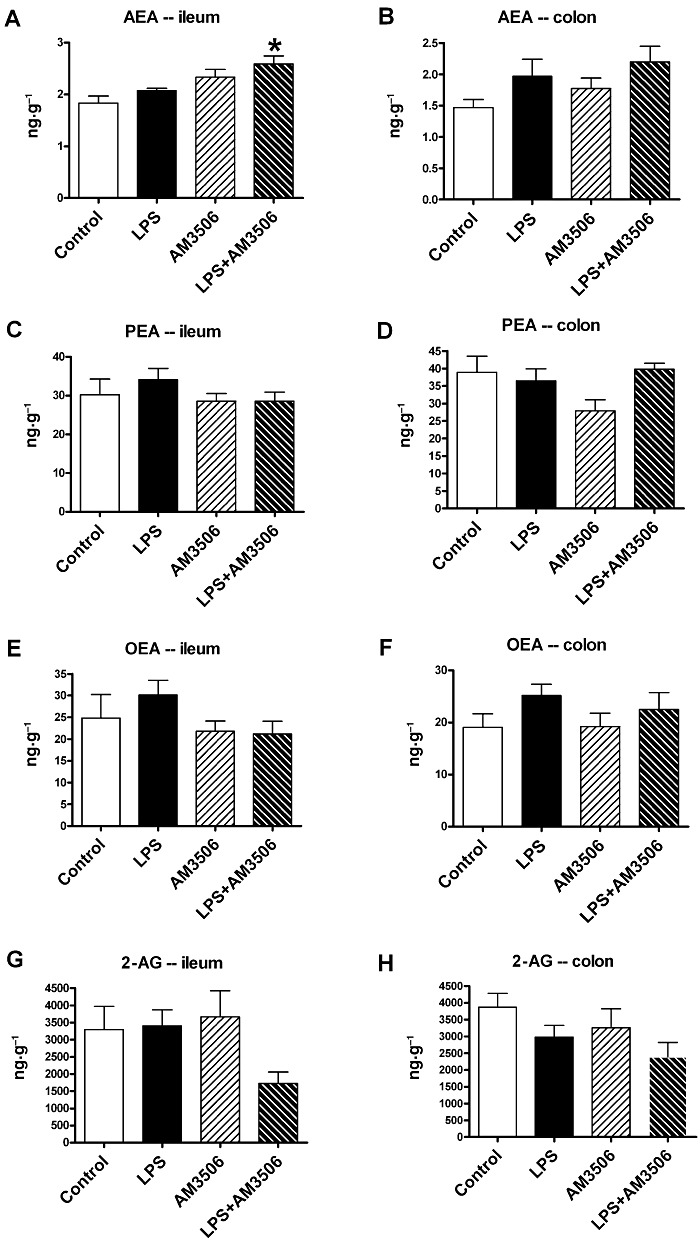
FAE and 2-AG levels in the ileum and distal colon of saline-treated (control) or LPS (100 µg·kg−1, i.p.)-treated mice pretreated with AM3506 (0.5 mg·kg−1, i.p.) or its vehicle. AM3506 treatment enhanced AEA levels in the ileum (A), but not the colon (B) of LPS-treated mice. There were no significant differences in the levels of PEA (C and D), OEA (E and F) or 2-AG (G and H) in the ileum (C, E and G) or colon (D, F and H). *P < 0.05 compared with control; n = 3–10 per group.
The effects of LPS treatment on GI contractility
Electrical stimulation of the mouse ileum produced a largely monophasic contraction, which was converted to a relaxation under NANC conditions after adrenergic and cholinergic blockade with guanethidine and atropine. The mouse colon typically gave a relaxation followed by a contraction after electrical stimulation, and this was converted to a larger relaxation and a smaller contraction in NANC conditions. We assessed the effect of LPS on ileal contractility in the mouse ileum and colon (Figure 5A and B). We observed a frequency-dependent contraction of the ileum, which was significantly enhanced at 4 and 8 Hz stimulations in tissues taken from LPS-treated mice (Figure 5A). Similarly, we observed a frequency-dependent contraction in the mouse colon, but by contrast, this was not enhanced in animals treated with LPS (Figure 5B). In control or LPS-treated mice, the contractions evoked by EFS in the ileum were completely abolished by atropine (1 µM) and TTX (100 nM) (data not shown). In contrast, atropine reduced colonic contractions by only 31.7 ± 5.5% (P < 0.01, n = 4) and TTX abolished them, albeit at a higher concentration (2 µM). LPS did not change the bethanechol-induced contractions (1–30 µM) or EFS-induced relaxations in either the ileum or colon (data not shown). The effects of bethanechol (10 µM) were not altered by TTX (100 nM), but they were completely blocked by atropine (1 µM).
Figure 5.
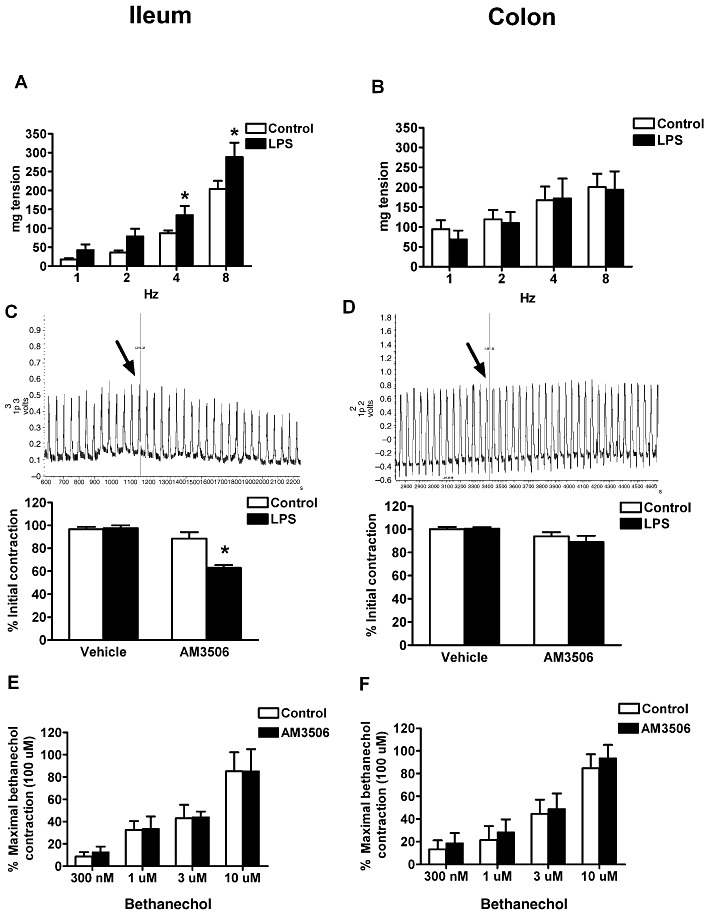
Effect of LPS (100 µg·kg−1, i.p.) and AM3506 (100 nM) on EFS-evoked contractions (4 Hz in C and D) of mouse ileum (A and C) and distal colon (B and D) and bethanechol(300 nM–10 µM)-induced contractions of mouse ileum and distal colon (E and F). Note that LPS enhanced contractility in the ileum (A) but not the distal colon (B) of the CD1 mice at all frequencies. (C) AM3506 reduced ileal contractions in LPS-treated CD1 mice but not in control mice. The trace represents the effect of AM3506 on ileal EFS contractility in a LPS-treated mouse. The arrow shows the time that tissue was exposed to AM3506 (100 nM). (D) AM3506 had no effect on colonic electrical contractions in control and LPS-treated animals. The trace represents the effect of AM3506 on colonic EFS contractility in a LPS-treated mouse. The arrow shows the time that tissue was exposed to AM3506 (100 nM). (E and F) Contractilities induced by bethanechol were not changed after LPS treatment. *P < 0.05 compared with control; n = 4–8 per group in all cases.
Effect of FAAH inhibition on intestinal contractility
Addition of AM3506 (100 nM) to the organ bath caused a reduction of electrically evoked contractions in the ileum of LPS-treated mice, but not that of normal animals (Figure 5C). The magnitude of the reduction (37 ± 6%) seen in LPS-treated mice was similar to the extent of the enhancement seen after LPS treatment (48 ± 8%), suggesting that FAAH inhibition normalizes contractility. In the same manner that LPS had no effect on contractility of the colon, AM3506 also had no effect on either normal or LPS-treated colonic contractility (Figure 5D). AM3506 did not alter bethanechol contractility in the ileum and colon (Figure 5E and F).
The effect of AM3506 on the ileum of LPS-treated mice was completely abolished in FAAH−/− mice (Figure 6A). To test whether the effects of FAAH inhibition by AM3506 on GI contractility are mediated via CB1 and/or CB2 receptors, we used CB1 and CB2 receptor antagonists, as well as CB1−/− mice. The CB1 receptor antagonist AM251 (100 nM) completely abolished the effect of AM3506 (100 nM) in the ileum of LPS-treated mice, as did the CB2 receptor antagonist AM630 (300 nM) (Figure 6B). A combination of both antagonists also completely abolished the effects of AM3506. The actions of AM3506 were absent in the ileum of CB1−/− mice (Figure 6C). AM251 and AM630 at the concentrations used in this study did not change EFS- or bethanechol-induced contractilities.
Figure 6.
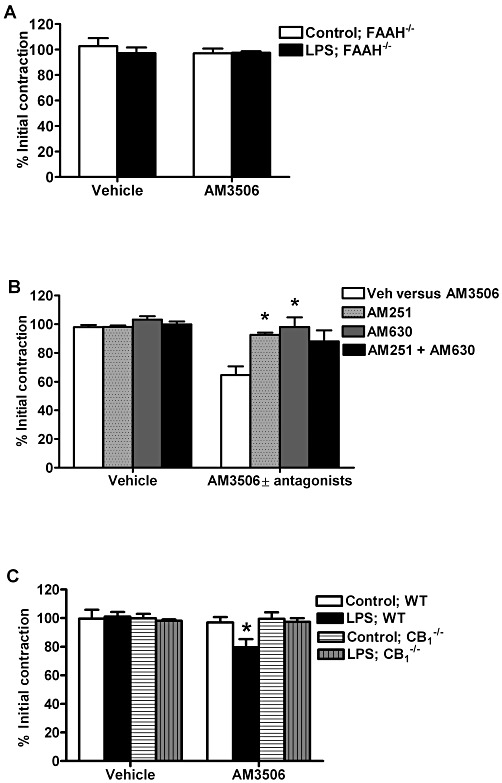
Effect of AM3506 (100 nM) on ileal contractions in gene-deficient mice and in the presence of CB receptor antagonists. (A) AM3506 did not change EFS contractility in control or LPS(100 µg·kg−1, i.p.)-treated FAAH−/− mice. (B) AM251 (100 nM) and AM630 (300 nM) blocked the effect of AM3506 on ileal contractility in LPS treated CD1 mice. (C) AM3506 had no effect on ileal contractions of vehicle or LPS-treated CB1−/− mice; however, it decreased EFS contractility in C57BL/6N wild-type mice treated with LPS (100 µg·kg−1, i.p.). *P < 0.05 compared with AM3506 in (B) and compared with vehicle in (C); n = 4–8 per group in all cases.
Effect of CB1 and CB2 receptor agonists on EFS-induced contractions after LPS treatment
Because LPS treatment could alter the expression and/or function of CB receptors, we next examined the effects of CB agonists in LPS-treated animals.
WIN55,212-2 (10–100 nM), a prototypical CB agonist, reduced ileal contractility evoked by EFS in a concentration-dependent manner (Figure 7A), but did not significantly reduce colonic contractility at these concentrations (Figure 7B). After LPS treatment, the effects of WIN55,212-2 were virtually identical in the ileum; however, it significantly decreased electrical contractility in the colon (Figure 7B), consistent with the observed increase in CB1 receptor mRNA expression. The effect of WIN55,212-2 in the ileum of saline or LPS-treated mice was abolished by the CB1 receptor antagonist AM251 (100 nM) but not by the CB2 receptor antagonist AM630 (300 nM) (Figure 7A). After LPS treatment, the effects of WIN55,212-2 (100 nM) in the colon was partially reversed by 100 nM AM251 (Figure 7B). AM630 (300 nM) did not change the response to WIN55,212-2 in the colon (Figure 7B).
Figure 7.
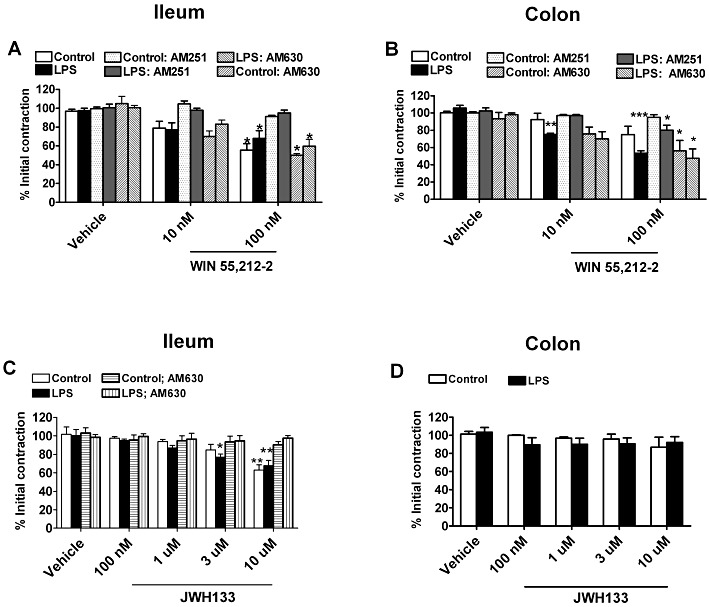
The effect of WIN55,212-2 (10–100 nM) and JWH133 (100 nM–10 µM) in the ileum and distal colon of CD1 mice after treatment with saline (control) or LPS (100 µg·kg−1, i.p.). (A) WIN55,212-2 significantly reduced contractility of the ileum in control and LPS-treated mice to about the same extent and this effect was fully reversed by the CB1 receptor antagonist AM251 (100 nM) but not the CB2 receptor antagonist AM630 (300 nM). (B) WIN55,212-2 reduced contractility of the colon in control mice, but this was only significant after LPS treatment. AM251 did not fully reverse the effects of WIN55,212-2 (100 nM) after LPS treatment in the distal colon. (C) JWH133 concentration-dependently reduced contractions evoked by EFS in the ileum. This effect was abolished by AM630 (300 nM). (D) JWH133 had no effect on evoked contractions in the distal colon. *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle; n = 3–6 per group for all panels.
To assess if there is a role for CB2 receptors in GI contractility in the mouse, we tested the selective CB2 receptor agonist JWH-133. JWH-133 reduced electrically-evoked contractions at the highest concentration (10 µM) in the ileum, but was without effect in the colon (Figure 7C and D). The effect of JWH-133 in the ileum was completely blocked by the CB2 receptor antagonist AM630. After LPS treatment, JWH-133 was slightly more potent in the ileum, completely reversed by AM630 and ineffective in the colon (Figure 7C and D).
Effect of LPS treatment and FAAH inhibition on intestinal transit
Upper GI transit was increased in LPS-treated mice (Figure 8A), as it is in rats (Mathison et al., 2004). AM3506 (0.5 mg·kg−1) had no effect on transit in vehicle-treated mice, but slowed the LPS-enhanced upper GI transit back to control levels (Figure 8A). The effect of AM3506 was abolished by AM251 (0.5 mg·kg−1) and the highest dose of AM630 (5 mg·kg−1) (Figure 8A). AM251 and AM630 at these doses did not alter upper GI transit when administered alone in both control and LPS-treated animals (data not shown).
Figure 8.
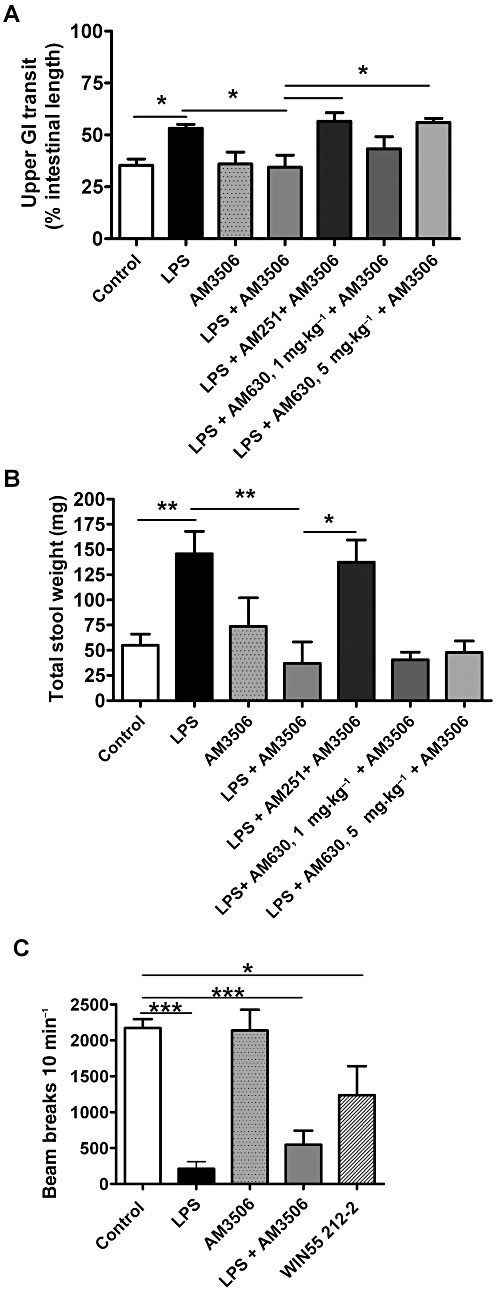
Effect of AM3506 (0.5 mg·kg−1, i.p.) on upper GI transit of a charcoal marker (A) and total stool weight (B) of control and LPS (100 µg·kg−1, i.p.)-treated mice. (A) AM3506 reversed the enhanced transit produced by LPS while having no effect alone. The effect of AM3506 was reversed by AM251 and the higher dose of AM630. (B) AM3506 reversed the enhanced stool output produced by LPS while having no effect alone. The effect of AM3506 was reversed by AM251, but not by AM630. (C) Effect of LPS (100 µg·kg−1, i.p.) and AM3506 (0.5 mg·kg−1, i.p.) on ambulatory motor activity of mice; AM3506 did not decrease ambulatory motor activity. LPS decreased ambulatory activity to the same extent in AM3506- or vehicle-pretreated mice. WIN55,212-2 (5 mg·kg−1) significantly decreased ambulatory activity. *P < 0.05, **P < 0.01, ***P < 0.001, bar indicates significant differences between the groups. n = 4–14 per group.
Effect of LPS treatment and FAAH inhibition on faecal output
In order to assess the effects of LPS and FAAH inhibition on faecal output, we measured the mass of faecal output over 90 min. The total weight of faecal output significantly increased in LPS-treated mice (Figure 8B). This enhanced faecal output was completely reversed by AM3506 (0.5 mg·kg−1), which alone had no significant effect on control stool output (Figure 8B). AM251 abolished the effect of AM3506 on total stool output; indicating that the effect of AM3506 is mediated by CB1 receptors. AM630 had no effect on AM3506 at either dose used (Figure 8B). AM251 and AM630 alone at the doses used did not change total faecal output in either control or LPS-treated animals (data not shown).
Effect of AM3506 on locomotor activity
Finally, in order to examine whether AM3506 produces any cataleptic effects, we examined the motor activity of mice treated with AM3506. AM3506 (0.5 mg·kg−1 i.p.), compared with WIN55,212-2, did not change the ambulatory score in saline-treated mice; however, LPS-treated animals that were/were not pretreated with AM3506 (0.5 mg·kg−1) showed a similar decrease in ambulatory score (Figure 8C).
GI motility in FAAH-deficient mice
Whole gut and upper GI transit were identical in FAAH-deficient mice compared with the wild-type mice (Figure 9A and B). LPS increased upper GI transit in the wild-type but not in the FAAH-deficient mice (Figure 9B).
Figure 9.
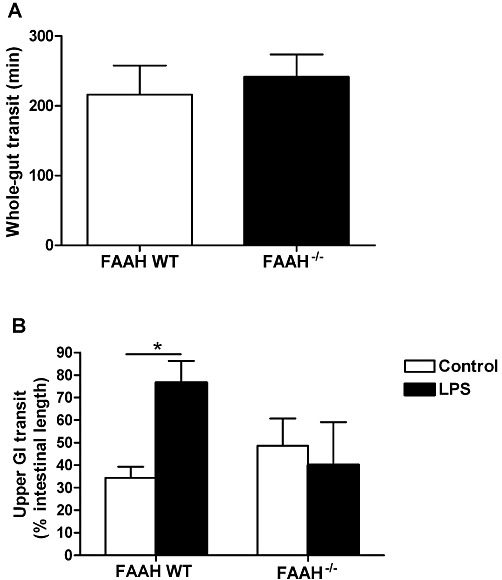
GI transit in FAAH-deficient (FAAH−/−) and wild-type (WT) mice. (A) Whole-gut transit was identical in both groups. (B) Upper GI transit was identical in both groups under control conditions; however, LPS (100 µg·kg−1, i.p.) increased transit in wild-type but not the FAAH−/− mice. *P < 0.05; n = 4–8 per group.
Discussion and conclusions
FAAH is an important enzyme whose activity is involved in the regulation of the levels of ECs and their related FAEs (Puffenbarger, 2005; Ahn et al., 2008; Fezza et al., 2008). FAAH-deficient mice display a behavioural and functional phenotype; they have heightened pain thresholds, reduced anxiety and, among other things, are protected from the development of experimental colitis (Cravatt et al., 2001; Massa et al., 2004; Moreira et al., 2008; Wise et al., 2008). FAAH inhibitors produce a very similar spectrum of actions to that of genetic ablation of FAAH (Gaetani et al., 2003; Schlosburg et al., 2009). In the GI tract, FAAH inhibitors protect animals from the development of colitis and against the development of aberrant crypt foci, considered to be precancerous lesions of the colon (Izzo et al., 2008; Storr et al., 2008). These compounds offer significant promise as therapeutic agents because they do not appear to cause the unwanted central actions of CB agonists, even though their effects can largely be blocked by CB1 receptor antagonists and are not observed in CB1 receptor gene-deficient mice (Petrosino and Di Marzo, 2010). We have investigated a novel FAAH inhibitor AM3506 (Godlewski et al., 2010) in a model of intestinal dysmotility and shown that this compound normalizes LPS-enhanced small intestinal motility in vitro and in vivo, without obvious behavioural side effects or actions in normal animals.
Previously, we demonstrated in rats that the EC system is involved in enhanced motility in the LPS model, with the notable finding that the enhanced intestinal contractility and motility were regulated by CB2 receptors (Mathison et al., 2004; Duncan et al., 2008). Here we have extended these findings to a mouse model and have focused on the role of FAAH. Examination of the gut from LPS-treated mice revealed no obvious structural changes or evidence of leucocyte infiltration. Nevertheless, immune activation was demonstrated by the increased TNF mRNA levels in the ileum and colon of LPS-treated mice, and this mechanism probably underlies the functional alterations observed (Ceregrzyn et al., 2001). Ileus, histological changes and increased macrophage activation in the mouse intestine have been reported in mouse models of sepsis induced with much higher doses of LPS than we used (de Winter et al., 2005; De Filippis et al., 2008b). Both increased and decreased GI motility can be seen after LPS, depending on the dose, timing between injection and assessments of GI motility, the region of the GI system that is investigated and the type of LPS.
In this study we used a model of intestinal dysmotility in the absence of overt intestinal inflammation. Our model has features of acute gastroenteritis and irritable bowel syndrome (IBS). The enteric nervous system has been implicated in dysmotility in IBS (Gershon, 2005). In this regard it is worth noting that the ENS is activated in the LPS model (Mathison et al., 2004; Duncan et al., 2008). A second feature of this LPS model is its association with increased excitability of colonic nociceptive dorsal root ganglion neurons (Ochoa-Cortes et al., 2010). This is consistent with the enhanced visceral sensitivity in IBS patients (Ohman and Simren, 2010).
Interestingly, in the colon, LPS did not have any effect on EFS-evoked contractility, although faecal output in vivo was increased. This is possibly due to increased secretomotor activity, central actions or the enhanced transit of small intestinal contents into the large intestine. The latter is less likely because of the lack of sensitivity to CB2 receptor antagonists. In the ileum, contractility is completely cholinergic whereas in the colon, it is partially cholinergic (Mule et al., 2007a,b) as confirmed in this study. The differences in the motor responses in the ileum versus the colon may be because of the balance of cholinergic versus non-cholinergic excitation. LPS did not appear to have a direct effect on intestinal smooth muscle motor activity or enteric inhibitory motor neurons, as the response did not change for bethanechol- or EFS-induced relaxation. Thus, it appears that our model is largely one of enhanced ACh release in the ENS. Since CB1 and CB2 receptors are localized to enteric cholinergic nerves and CB receptors regulate the release of ACh, our focus on modulation of the EC system offers the possibility to target these nerves with a degree of specificity.
The EC system is activated in the LPS model as previously demonstrated (Mathison et al., 2004; Duncan et al., 2008; Li et al., 2010). This state of activation does not seem to depend on changes to CB receptor expression or on enhanced EC levels. CB1 receptor mRNA in the ileum was not affected by LPS treatment to any great extent, although LPS significantly elevated CB1 receptor mRNA levels in the colon. This finding helps to explain the slightly greater potency of the CB agonist WIN55,212-2 after LPS treatment in the colon. In contrast, CB2 receptor mRNA expression was reduced by LPS treatment in the ileum and colon. A similar reduction in CB2 receptor expression in response to LPS has previously been reported in immune cells (Lee et al., 2001). However, it is apparent from our findings, and previous reports, that despite a reduced expression, CB2 receptor function in the small intestine is increased by LPS treatment (Mathison et al., 2004; Duncan et al., 2008). In fact, we observed what appears to be a fully functional CB2 receptor in our model, since AM3506 inhibited both small intestinal contractility in vitro and upper GI motility in vivo in a manner that was reversed by the CB2 receptor antagonist AM630 and, as well, the CB2 receptor agonist JWH-133 reduced electrically evoked contractions in vitro. While CB2 receptor mRNA is present in both the ileum and colon, no functional CB2 receptor actions were found in the colon. The reasons for these regional differences is not clear, but these findings suggest that targeting the CB2 receptor in small bowel pathophysiology with endogenous ligands represents an attractive therapeutic option because it has no psychotropic central actions and is apparently inactive in the healthy gut.
The main goal of the study was to examine whether inhibition of FAAH represents a mechanism to reverse dysmotility in animals treated with LPS. Here we showed that AM3506 was able to inhibit enhanced contractility evoked by EFS in vitro in tissues from animals treated with LPS, but not under control conditions. Our immunohistochemical localization of FAAH in the enteric nervous system suggests that AM3506 is acting there, which is also consistent with the functional activity of AM3506 in vitro. Perhaps more importantly, AM3506 was effective in vivo at reducing the enhanced intestinal motility and faecal output in LPS-treated mice.
AM3506 had virtually no effect on the GI motility of healthy animals and GI transit was identical in FAAH-deficient and wild-type mice. Moreover, we have shown that AM3506 does not inhibit FAAH activity in the ileum of the healthy animals and does not alter intestinal AEA, 2-AG, PEA and OEA levels. Capasso et al. (2005) showed that FAAH-deficient mice have similar intestinal transit compared with the wild-type mice; however, in their hands, the FAAH inhibitor AA-5-HT reduced intestinal transit, partially through a CB1 receptor-mediated mechanism of action. The reason for the differences between Capasso's findings and our results may relate to the different specificities between the FAAH inhibitors or other factors. AA-5-HT antagonizes transient receptor potential vanilloid 1 with an IC50 of 36.8–39.9 nM, which is far lower compared than its IC50 for FAAH (1–12 µM). Moreover, AA-5-HT is known to increase not only the intestinal levels of AEA, but also those of 2-AG and PEA, which may reduce intestinal motility. On the other hand, AM3506 has affinity for CB1 and CB2 at 5.77 µM and 192 nM, respectively; however, the Ki of AA-5-HT for CB1 and CB2 receptors is more than 50 and 10 µM, respectively (Maione et al., 2007; Godlewski et al., 2010). In agreement with our findings, in a recent study that examined three different FAAH inhibitors, that is, AA-5-HT, PMSF and URB597, it was shown that these inhibitors caused no changes in electrically-evoked contractility of the small intestine in rats and guinea-pigs (Makwana et al., 2010). These data support the idea that under physiological conditions, inhibition of FAAH produces little effect.
In the presence of LPS, AM3506 significantly decreased FAAH activity in the ileum. Moreover, AM3506 increased AEA levels in the ileum in LPS-treated mice. In another study, Capasso et al. (2008) showed that cannabidiol, which is a cannabis-derived non-psychotropic compound, normalizes croton oil-induced hypermotility in the small intestine of mouse without affecting motility in the control mice, and AA-5-HT abolished the effect of cannabidiol. These findings, as well as the recent publication by De Petrocellis et al. (2011) about the role of cannabidiol in the inhibition of FAAH activity, emphasize the role of FAAH in the regulation of GI dysmotility in pathological conditions.
Another interesting finding of the current study was the lack of enhancement of the upper GI transit in LPS-treated FAAH-deficient mice. This suggests a role of FAAH in LPS-enhanced motility in the GI tract and supports our pharmacological experiments, which showed that inhibiting FAAH normalizes upper GI motility in LPS-treated wild-type mice. Whether this is due to an anti-inflammatory role of FAEs or other mechanisms needs further investigation.
We did not observe any change in the colonic contractility after LPS and/or AM3506 treatments. Moreover, we observed a far lower expression of FAAH mRNA and FAAH activity in the colon, which is in agreement with the lack of functional effects of AM3506 in this region. Given the similar baseline levels of ECs in the ileum and colon, there might be at least two enzymes capable of hydrolyzing AEA in different regions of the GI tract. One is FAAH, the other is like FAAH in some respects but not in terms of its sensitivity to AM3506 or LPS, explaining how AM3506 could inhibit FAAH and also reverse the actions of LPS in vivo. A similar conclusion regarding AEA hydrolase activity was drawn by Fegley et al. (2005) who examined another FAAH inhibitor, URB597, in the GI tract. Although URB597 clearly inhibited AEA hydrolysis in the duodenum and was inactive in FAAH gene-deficient mice, the levels of AEA, OEA and PEA were unaffected by this compound (Fegley et al., 2005). One potential candidate is the acid amidase enzyme discovered by Ueda et al. (2001), found in the gut, albeit at higher levels in the small intestine. Other possibilities include the ability of colonic bacteria to metabolize ECs or differences in EC biosynthesis between the regions of the gut. These alternative possibilities require further investigation.
Interestingly, in vivo, we observed that the actions of AM3506 on stool output from the colon were entirely CB1 receptor-sensitive, whereas in the ileum, it appears that there is a CB1 and CB2 receptor component to the enhanced motility. These results contrast with those from an earlier study in rats that found a purely CB2-mediated response (Mathison et al., 2004; Duncan et al., 2008). AM3506 did not reduce locomotor behaviours in mice suggesting that it was not acting as an exogenous CB agonist, which would be expected to reduce locomotion. These findings support the notion that FAAH inhibitors represent a new class of potential therapeutic agents in the treatment of disorders of GI motility and inflammation (Capasso et al., 2005; Storr et al., 2008).
The expression of FAAH may be regulated up or down by LPS according to the conditions. In a report by De Filippis et al. (2008a) FAAH protein expression in the small intestine during sepsis was increased, but in isolated lymphocytes, LPS reduced FAAH gene and protein expression (Maccarrone et al., 2001). Therefore, it seems that the regulation of ECs and fatty acid amide levels are under complex control, and their levels vary in the gut dependent upon several conditions (Gomez et al., 2002; Darmani et al., 2005). This is likely to be a reflection of both synthesis and degradation through a variety of enzymatic pathways. This idea is supported by the findings of Pinto et al. (2002), who reported 100-fold greater activity of an AEA hydrolase in the colon than in the ileum. Further characterization of the enzymes of EC synthesis and degradation in the GI tract are required.
In conclusion, we have shown that inhibition of FAAH normalizes enhanced motility in vitro and in vivo in the mouse GI tract through the EC system. Inhibition of FAAH with compounds, which appear to be devoid of the side effects of exogenous CBs, may represent a novel treatment for abnormal GI motility in conditions such as IBS.
Acknowledgments
Grant Support: This work was supported by grants from the Canadian Institutes of Health Research (to KAS, KDP and JSD) and National Institutes of Health (NIH; grants DA09158, DA7215, DA3801 and DA023142 to AM). MAS was supported by the University of Calgary Research Grant Committee. KM was supported by NIH grants DA11322 and DA21696. KAS and KDP are Alberta Heritage Foundation for Medical Research Medical Scientists. KDP holds a Canada Research Chair. KAS holds the CCFC Chair in IBD Research at University of Calgary.
Glossary
- 2-AG
2-arachidonoylglycerol
- AEA
anandamide
- AM251
N-(piperidin-1-yl)-5-(4-iodophenyl) -1-(2,4-dichlorophenyl)-4-methyl-1H- pyrazole-3-carboxamide
- AM630
6-iodo-2-methyl-1-[2-(4-morpholinyl) ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone
- CB
cannabinoid
- CB1
receptor, cannabinoid receptor type 1
- CB2
receptor, cannabinoid receptor type 2
- EC
endocannabinoid
- EFS
electrical field stimulation
- ENS
enteric nervous system
- FAAH
fatty acid amide hydrolase
- FAE
fatty acid ethanolamides
- GI
gastrointestinal
- JWH133
(6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b, d] pyran
- OEA
oleoylethanolamide
- PEA
palmitoylethanolamide
- TTX
tetrodotoxin
- WIN55
212-2, (R)-(+)-[2,3-dihydro-5-methyl-3-(4- morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate
Conflict of interests
GK and AM are co-inventors on patent application 200100261674 published 14 October 2010 covering AM3506. The other authors declare they have no competing interests.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 FAAH immunoreactivity in whole mountpreparations of the myenteric plexus of the distal colon from awild-type (A, WT) and FAAH-deficient (KO) mouse (B). Note that inFAAH −/− mice, there was no immunoreactivityobserved. Scale bar: 50 μm.
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviello G, Romano B, Izzo AA. Cannabinoids and gastrointestinal motility: animal and human studies. Eur Rev Med Pharmacol Sci. 2008;12(Suppl. 1):81–93. [PubMed] [Google Scholar]
- Camilleri M, Carlson P, McKinzie S, Grudell A, Busciglio I, Burton D, et al. Genetic variation in endocannabinoid metabolism, gastrointestinal motility, and sensation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G13–G19. doi: 10.1152/ajpgi.00371.2007. [DOI] [PubMed] [Google Scholar]
- Capasso R, Izzo AA, Fezza F, Pinto A, Capasso F, Mascolo N, et al. Inhibitory effect of palmitoylethanolamide on gastrointestinal motility in mice. Br J Pharmacol. 2001;134:945–950. doi: 10.1038/sj.bjp.0704339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso R, Matias I, Lutz B, Borrelli F, Capasso F, Marsicano G, et al. Fatty acid amide hydrolase controls mouse intestinal motility in vivo. Gastroenterology. 2005;129:941–951. doi: 10.1053/j.gastro.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Capasso R, Borrelli F, Aviello G, Romano B, Scalisi C, Capasso F, et al. Cannabidiol, extracted from Cannabis sativa, selectively inhibits inflammatory hypermotility in mice. Br J Pharmacol. 2008;154:1001–1008. doi: 10.1038/bjp.2008.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceregrzyn M, Kamata T, Yajima T, Kuwahara A. Biphasic alterations in gastrointestinal transit following endotoxaemia in mice. Neurogastroenterol Motil. 2001;13:605–613. doi: 10.1046/j.1365-2982.2001.00291.x. [DOI] [PubMed] [Google Scholar]
- Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, et al. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148:102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianchi F, Papucci L, Schiavone N, Lulli M, Magnelli L, Vinci MC, et al. Cannabinoid receptor activation induces apoptosis through tumor necrosis factor alpha-mediated ceramide de novo synthesis in colon cancer cells. Clin Cancer Res. 2008;14:7691–7700. doi: 10.1158/1078-0432.CCR-08-0799. [DOI] [PubMed] [Google Scholar]
- Cluny NL, Keenan CM, Lutz B, Piomelli D, Sharkey KA. The identification of peroxisome proliferator-activated receptor alpha-independent effects of oleoylethanolamide on intestinal transit in mice. Neurogastroenterol Motil. 2009;21:420–429. doi: 10.1111/j.1365-2982.2008.01248.x. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Lichtman AH. The enzymatic inactivation of the fatty acid amide class of signaling lipids. Chem Phys Lipids. 2002;121:135–148. doi: 10.1016/s0009-3084(02)00147-0. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Lichtman AH. Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system. Curr Opin Chem Biol. 2003;7:469–475. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmani NA, McClanahan BA, Trinh C, Petrosino S, Valenti M, Di Marzo. Cisplatin increases brain 2-arachidonoylglycerol (2-AG) and concomitantly reduces intestinal 2-AG and anandamide levels in the least shrew. Neuropharmacology. 2005;49:502–513. doi: 10.1016/j.neuropharm.2005.04.007. [DOI] [PubMed] [Google Scholar]
- De Filippis D, Iuvone T, D'Amico A, Esposito G, Steardo L, Herman AG, et al. Effect of cannabidiol on sepsis-induced motility disturbances in mice: involvement of CB receptors and fatty acid amide hydrolase. Neurogastroenterol Motil. 2008a;20:919–927. doi: 10.1111/j.1365-2982.2008.01114.x. [DOI] [PubMed] [Google Scholar]
- De Filippis D, Iuvone T, Esposito G, Steardo L, Arnold GH, Paul AP, et al. Melatonin reverses lipopolysaccharide-induced gastro-intestinal motility disturbances through the inhibition of oxidative stress. J Pineal Res. 2008b;44:45–51. doi: 10.1111/j.1600-079X.2007.00526.x. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Ligresti A, Moriello AS, Allara M, Bisogno T, Petrosino S, et al. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol. 2011;163:1479–1494. doi: 10.1111/j.1476-5381.2010.01166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch DG, Lin S, Hill WA, Morse KL, Salehani D, Arreaza G, et al. Fatty acid sulfonyl fluorides inhibit anandamide metabolism and bind to the cannabinoid receptor. Biochem Biophys Res Commun. 1997;231:217–221. doi: 10.1006/bbrc.1997.6072. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. The endocannabinoid system: its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol Res. 2009;60:77–84. doi: 10.1016/j.phrs.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Duncan M, Davison JS, Sharkey KA. Review article: endocannabinoids and their receptors in the enteric nervous system. Aliment Pharmacol Ther. 2005;22:667–683. doi: 10.1111/j.1365-2036.2005.02648.x. [DOI] [PubMed] [Google Scholar]
- Duncan M, Mouihate A, Mackie K, Keenan CM, Buckley NE, Davison JS, et al. Cannabinoid CB2 receptors in the enteric nervous system modulate gastrointestinal contractility in lipopolysaccharide-treated rats. Am J Physiol Gastrointest Liver Physiol. 2008;295:G78–G87. doi: 10.1152/ajpgi.90285.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, et al. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther. 2005;313:352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Fezza F, De Simone C, Amadio D, Maccarrone M. Fatty acid amide hydrolase: a gate-keeper of the endocannabinoid system. Subcell Biochem. 2008;49:101–132. doi: 10.1007/978-1-4020-8831-5_4. [DOI] [PubMed] [Google Scholar]
- Fowler CJ, Borjesson M, Tiger G. Differences in the pharmacological properties of rat and chicken brain fatty acid amidohydrolase. Br J Pharmacol. 2000;131:498–504. doi: 10.1038/sj.bjp.0703569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaetani S, Cuomo V, Piomelli D. Anandamide hydrolysis: a new target for anti-anxiety drugs? Trends Mol Med. 2003;9:474–478. doi: 10.1016/j.molmed.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Gershon MD. Nerves, reflexes, and the enteric nervous system: pathogenesis of the irritable bowel syndrome. J Clin Gastroenterol. 2005;39:S184–S193. doi: 10.1097/01.mcg.0000156403.37240.30. [DOI] [PubMed] [Google Scholar]
- Godlewski G, Alapafuja SO, Batkai S, Nikas SP, Cinar R, Offertaler L, et al. Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects. Chem Biol. 2010;17:1256–1266. doi: 10.1016/j.chembiol.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez R, Navarro M, Ferrer B, Trigo JM, Bilbao A, Del Arco I, et al. A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J Neurosci. 2002;22:9612–9617. doi: 10.1523/JNEUROSCI.22-21-09612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helliwell RJ, Chamley LW, Blake-Palmer K, Mitchell MD, Wu J, Kearn CS, et al. Characterization of the endocannabinoid system in early human pregnancy. J Clin Endocrinol Metab. 2004;89:5168–5174. doi: 10.1210/jc.2004-0388. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Camilleri M. Emerging role of cannabinoids in gastrointestinal and liver diseases: basic and clinical aspects. Gut. 2008;57:1140–1155. doi: 10.1136/gut.2008.148791. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Sharkey KA. Cannabinoids and the gut: new developments and emerging concepts. Pharmacol Ther. 2010;126:21–38. doi: 10.1016/j.pharmthera.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Aviello G, Petrosino S, Orlando P, Marsicano G, Lutz B, et al. Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J Mol Med. 2008;86:89–98. doi: 10.1007/s00109-007-0248-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama K, Ueda N, Kurahashi Y, Suzuki H, Yamamoto S, Kato I. Distribution of anandamide amidohydrolase in rat tissues with special reference to small intestine. Biochim Biophys Acta. 1997;1347:212–218. doi: 10.1016/s0005-2760(97)00078-7. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Krawisz JE, Sharon P, Stenson WF. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–1350. [PubMed] [Google Scholar]
- Lee SF, Newton C, Widen R, Friedman H, Klein TW. Downregulation of cannabinoid receptor 2 (CB2) messenger RNA expression during in vitro stimulation of murine splenocytes with lipopolysaccharide. Adv Exp Med Biol. 2001;493:223–228. doi: 10.1007/0-306-47611-8_26. [DOI] [PubMed] [Google Scholar]
- Li YY, Li YN, Ni JB, Chen CJ, Lv S, Chai SY, et al. Involvement of cannabinoid-1 and cannabinoid-2 receptors in septic ileus. Neurogastroenterol Motil. 2010;22:350–e88. doi: 10.1111/j.1365-2982.2009.01419.x. [DOI] [PubMed] [Google Scholar]
- Ligresti A, Bisogno T, Matias I, De Petrocellis L, Cascio MG, Cosenza V, et al. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology. 2003;125:677–687. doi: 10.1016/s0016-5085(03)00881-3. [DOI] [PubMed] [Google Scholar]
- Liu J, Batkai S, Pacher P, Harvey-White J, Wagner JA, Cravatt BF, et al. Lipopolysaccharide induces anandamide synthesis in macrophages via CD14/MAPK/phosphoinositide 3-kinase/NF-kappaB independently of platelet-activating factor. J Biol Chem. 2003;278:45034–45039. doi: 10.1074/jbc.M306062200. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, De Petrocellis L, Bari M, Fezza F, Salvati S, Di Marzo V, et al. Lipopolysaccharide downregulates fatty acid amide hydrolase expression and increases anandamide levels in human peripheral lymphocytes. Arch Biochem Biophys. 2001;393:321–328. doi: 10.1006/abbi.2001.2500. [DOI] [PubMed] [Google Scholar]
- Maione S, De Petrocellis L, de Novellis V, Moriello AS, Petrosino S, Palazzo E, et al. Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol. 2007;150:766–781. doi: 10.1038/sj.bjp.0707145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makwana R, Molleman A, Parsons ME. Evidence for both inverse agonism at the cannabinoid CB1 receptor and the lack of an endogenous cannabinoid tone in the rat and guinea-pig isolated ileum myenteric plexus-longitudinal muscle preparation. Br J Pharmacol. 2010;160:615–626. doi: 10.1111/j.1476-5381.2010.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez L, Suarez J, Iglesias M, Bermudez-Silva FJ, Rodriguez FF, Andreu M. Ulcerative colitis induces changes on the expression of the endocannabinoid system in the human colonic tissue. PLoS ONE. 2009;4:e6893. doi: 10.1371/journal.pone.0006893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Massa F, Marsicano G, Hermann H, Cannich A, Monory K, Cravatt BF, et al. The endogenous cannabinoid system protects against colonic inflammation. J Clin Invest. 2004;113:1202–1209. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathison R, Ho W, Pittman QJ, Davison JS, Sharkey KA. Effects of cannabinoid receptor-2 activation on accelerated gastrointestinal transit in lipopolysaccharide-treated rats. Br J Pharmacol. 2004;142:1247–1254. doi: 10.1038/sj.bjp.0705889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira FA, Kaiser N, Monory K, Lutz B. Reduced anxiety-like behaviour induced by genetic and pharmacological inhibition of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) is mediated by CB1 receptors. Neuropharmacology. 2008;54:141–150. doi: 10.1016/j.neuropharm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Mule F, Amato A, Baldassano S, Serio R. Evidence for a modulatory role of cannabinoids on the excitatory NANC neurotransmission in mouse colon. Pharmacol Res. 2007a;56:132–139. doi: 10.1016/j.phrs.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Mule F, Amato A, Serio R. Role for NK(1) and NK(2) receptors in the motor activity in mouse colon. Eur J Pharmacol. 2007b;570:196–202. doi: 10.1016/j.ejphar.2007.05.036. [DOI] [PubMed] [Google Scholar]
- Ochoa-Cortes F, Ramos-Lomas T, Miranda-Morales M, Spreadbury I, Ibeakanma C, Barajas-Lopez C, et al. Bacterial cell products signal to mouse colonic nociceptive dorsal root ganglia neurons. Am J Physiol Gastrointest Liver Physiol. 2010;299:G723–G732. doi: 10.1152/ajpgi.00494.2009. [DOI] [PubMed] [Google Scholar]
- Ohman L, Simren M. Pathogenesis of IBS: role of inflammation, immunity and neuroimmune interactions. Nat Rev Gastroenterol Hepatol. 2010;7:163–173. doi: 10.1038/nrgastro.2010.4. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Di Marzo V. FAAH and MAGL inhibitors: therapeutic opportunities from regulating endocannabinoid levels. Curr Opin Investig Drugs. 2010;11:51–62. [PubMed] [Google Scholar]
- Pinto L, Izzo AA, Cascio MG, Bisogno T, Hospodar-Scott K, Brown DR, et al. Endocannabinoids as physiological regulators of colonic propulsion in mice. Gastroenterology. 2002;123:227–234. doi: 10.1053/gast.2002.34242. [DOI] [PubMed] [Google Scholar]
- Puffenbarger RA. Molecular biology of the enzymes that degrade endocannabinoids. Curr Drug Targets CNS Neurol Disord. 2005;4:625–631. doi: 10.2174/156800705774933050. [DOI] [PubMed] [Google Scholar]
- Scherma M, Medalie J, Fratta W, Vadivel SK, Makriyannis A, Piomelli D, et al. The endogenous cannabinoid anandamide has effects on motivation and anxiety that are revealed by fatty acid amide hydrolase (FAAH) inhibition. Neuropharmacology. 2008;54:129–140. doi: 10.1016/j.neuropharm.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Kinsey SG, Lichtman AH. Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J. 2009;11:39–44. doi: 10.1208/s12248-008-9075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storr MA, Sharkey KA. The endocannabinoid system and gut-brain signalling. Curr Opin Pharmacol. 2007;7:575–582. doi: 10.1016/j.coph.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Storr MA, Keenan CM, Emmerdinger D, Zhang H, Yuce B, Sibaev A, et al. Targeting endocannabinoid degradation protects against experimental colitis in mice: involvement of CB1 and CB2 receptors. J Mol Med. 2008;86:925–936. doi: 10.1007/s00109-008-0359-6. [DOI] [PubMed] [Google Scholar]
- Storr M, Emmerdinger D, Diegelmann J, Yuce B, Pfennig S, Ochsenkuhn T, et al. The role of fatty acid hydrolase gene variants in inflammatory bowel disease. Aliment Pharmacol Ther. 2009a;29:542–551. doi: 10.1111/j.1365-2036.2008.03910.x. [DOI] [PubMed] [Google Scholar]
- Storr MA, Keenan CM, Zhang H, Patel KD, Makriyannis A, Sharkey KA. Activation of the cannabinoid 2 receptor (CB(2)) protects against experimental colitis. Inflamm Bowel Dis. 2009b;15:1678–1685. doi: 10.1002/ibd.20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda N, Yamanaka K, Yamamoto S. Purification and characterization of an acid amidase selective for N-palmitoylethanolamine, a putative endogenous anti-inflammatory substance. J Biol Chem. 2001;276:35552–35557. doi: 10.1074/jbc.M106261200. [DOI] [PubMed] [Google Scholar]
- Williams J, Pandarinathan L, Wood J, Vouros P, Makriyannis A. Endocannabinoid metabolomics: a novel liquid chromatography-mass spectrometry reagent for fatty acid analysis. AAPS J. 2006;8:E655–E660. doi: 10.1208/aapsj080474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J, Wood J, Pandarinathan L, Karanian DA, Bahr BA, Vouros P, et al. Quantitative method for the profiling of the endocannabinoid metabolome by LC-atmospheric pressure chemical ionization-MS. Anal Chem. 2007;79:5582–5593. doi: 10.1021/ac0624086. [DOI] [PubMed] [Google Scholar]
- de Winter BY, van Nassauw L, de Man JG, de Jonge F, Bredenoord AJ, Seerden TC, et al. Role of oxidative stress in the pathogenesis of septic ileus in mice. Neurogastroenterol Motil. 2005;17:251–261. doi: 10.1111/j.1365-2982.2004.00618.x. [DOI] [PubMed] [Google Scholar]
- Wise LE, Cannavacciulo R, Cravatt BF, Martin BF, Lichtman AH. Evaluation of fatty acid amides in the carrageenan-induced paw edema model. Neuropharmacology. 2008;54:181–188. doi: 10.1016/j.neuropharm.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JT, Williams JS, Pandarinathan L, Courville A, Keplinger MR, Janero DR, et al. Comprehensive profiling of the human circulating endocannabinoid metabolome: clinical sampling and sample storage parameters. Clin Chem Lab Med. 2008;46:1289–1295. doi: 10.1515/CCLM.2008.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright K, Rooney N, Feeney M, Tate J, Robertson D, Welham M, et al. Differential expression of cannabinoid receptors in the human colon: cannabinoids promote epithelial wound healing. Gastroenterology. 2005;129:437–453. doi: 10.1016/j.gastro.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Wright KL, Duncan M, Sharkey KA. Cannabinoid CB2 receptors in the gastrointestinal tract: a regulatory system in states of inflammation. Br J Pharmacol. 2008;153:263–270. doi: 10.1038/sj.bjp.0707486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.