

Abstract
BACKGROUND AND PURPOSE
Atorvastatin metabolites differ in their potential for drug interaction because of differential inhibition of drug-metabolizing enzymes and transporters. We here investigate whether they exert differential effects on the induction of these genes via activation of pregnane X receptor (PXR) and constitutive androstane receptor (CAR).
EXPERIMENTAL APPROACH
Ligand binding to PXR or CAR was analysed by mammalian two-hybrid assembly and promoter/reporter gene assays. Additionally, surface plasmon resonance was used to analyse ligand binding to CAR. Primary human hepatocytes were treated with atorvastatin metabolites, and mRNA and protein expression of PXR-regulated genes was measured. Two-hybrid co-activator interaction and co-repressor release assays were utilized to elucidate the molecular mechanism of PXR activation.
KEY RESULTS
All atorvastatin metabolites induced the assembly of PXR and activated CYP3A4 promoter activity. Ligand binding to CAR could not be proven. In primary human hepatocytes, the para-hydroxy metabolite markedly reduced or abolished induction of cytochrome P450 and transporter genes. While significant differences in co-activator recruitment were not observed, para-hydroxy atorvastatin demonstrated only 50% release of co-repressors.
CONCLUSIONS AND IMPLICATIONS
Atorvastatin metabolites are ligands of PXR but not of CAR. Atorvastatin metabolites demonstrate differential induction of PXR target genes, which results from impaired release of co-repressors. Consequently, the properties of drug metabolites have to be taken into account when analysing PXR-dependent induction of drug metabolism and transport. The drug interaction potential of the active metabolite, para-hydroxy atorvastatin, might be lower than that of the parent compound.
Keywords: atorvastatin, metabolites, para-hydroxy atorvastatin, PXR, CAR, induction, drug interaction
Introduction
Statins are widely used in the treatment of hypercholesterolaemia, which is a key feature of the metabolic syndrome in humans and an important risk factor for the development of cardiovascular diseases, such as myocardial infarction. By competitive inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), the rate-limiting enzyme in cholesterol synthesis, statins cause the intracellular depletion of cholesterol, thereby inducing the proteolytic activation of sterol response element-binding proteins, which stimulates the expression of the low-density lipoprotein (LDL) receptor (Horton et al., 2002). As one consequence, the plasma concentration of LDL cholesterol is lowered by increased hepatocellular uptake (Poli, 2007).
Atorvastatin has been prescribed for many years and is considered as one of the most potent agents within the statin drug class, in terms of the LDL cholesterol-lowering effect (Poli, 2007). However, the therapeutic response at a given dose is highly variable between individuals (Pedro-Botet et al., 2001), and the correlation between plasma concentrations and response to atorvastatin is poor (Lennernäs, 2003). One reason may be the extensive first-pass metabolism and the consequent metabolism-dependent interconversion of active and inactive metabolites of atorvastatin (Figure 1). Conversion of the active acid form to the inactive lactone mainly requires an acylglucuronide intermediate (Prueksaritanont et al., 2002), which is predominantly catalysed by UDP glucuronosyltransferase (UGT) 1A3 (Riedmaier et al., 2010). Metabolism of the drug by cytochrome P450 (CYP) 3A4 results in the formation of the two active metabolites ortho-hydroxy and para-hydroxy atorvastatin (Jacobsen et al., 2000). These two active metabolites are said to be responsible for the atorvastatin-related prolonged inhibition of HMGCR and the higher efficacy in lowering LDL cholesterol, as compared with other statins (Poli, 2007).
Figure 1.
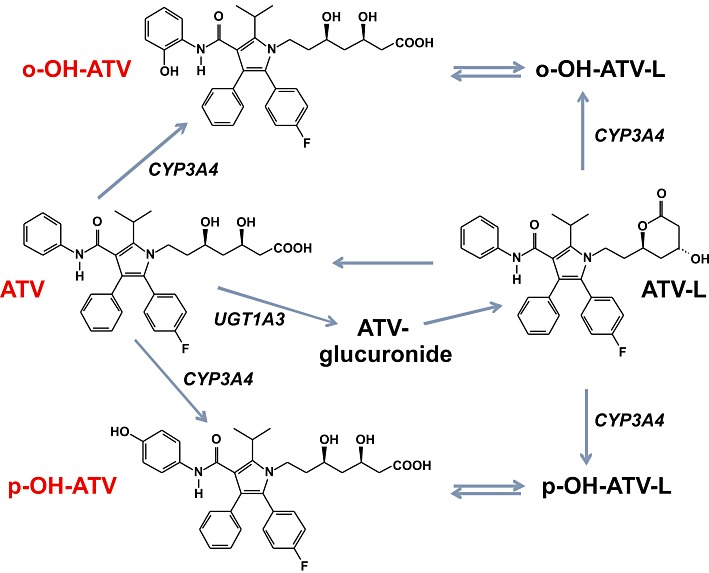
Chemical structures and metabolic pathways of atorvastatin and metabolites. Only the chemical structures of metabolites used in this study are shown. Metabolic pathways are displayed according to Jacobsen et al. (2000) and Prueksaritanont et al. (2002). ATV, atorvastatin; ATV-L, atorvastatin lactone, o-OH-ATV, ortho-hydroxy atorvastatin; p-OH-ATV, para-hydroxy atorvastatin.
Disposition of a drug is also strongly influenced by transport processes. Hepatocellular uptake of atorvastatin, which is a substrate of the transporter SLCO1B1 (OATP1B1) (Kameyama et al., 2005), is largely mediated by this membrane transport protein. For instance, atorvastatin pharmacokinetics is significantly altered by OATP1B1 inhibitors in humans (Lau et al., 2007), as well as by SLCO1B1 genetic variants as shown by two independent studies (Pasanen et al., 2007; Lee et al., 2010). Moreover, atorvastatin is also a substrate of the efflux transporter ABCB1 (MDR1/P-glycoprotein) (Wu et al., 2000; Hochman et al., 2004), which influences pharmacokinetics of atorvastatin (Keskitalo et al., 2008). Thus, multiple drug interactions because of the competitive inhibition of enzymes and/or transporters are conceivable and have been reported in the literature (see Lennernäs, 2003).
Additionally, atorvastatin, in common with many statins, induces the hepatic expression of CYP3A4 and CYP2B6 (Kocarek et al., 2002), which suggests activation of the pregnane X receptor (PXR, NR1I2) and/or the constitutive androstane receptor (CAR, NR1I3; receptor nomenclature follows Alexander et al., 2011). These two receptors are the main xenosensors in the body and are responsible for the adaptive response to xenobiotics, including drugs, by inducing the expression of drug-metabolizing enzymes and transporters. Especially, PXR is activated by a large spectrum of structurally diverse chemicals, which bind to the receptor as ligands (di Masi et al., 2009). In contrast, the majority of CAR activators act by promoting the translocation of the receptor from the cytosol to the nucleus by an indirect mechanism, which is only partially understood and not involving ligand binding (Li and Wang, 2010). Thus, induction via activation of PXR and/or CAR may further contribute to atorvastatin-dependent drug interaction.
Given the extensive metabolism of atorvastatin and the contribution of the active metabolites, ortho-hydroxy and para-hydroxy atorvastatin, to the drug's therapeutic effect, it is important to consider their respective effects on drug interaction. Whereas the inhibition of drug-metabolizing enzymes and transporters by atorvastatin metabolites has been already studied (Chen et al., 2005; Sakaeda et al., 2006), an analysis regarding the induction of absorption distribution metabolism excretion (ADME) genes via PXR and/or CAR activation is still missing. Thus, the objective of the present study was to characterize atorvastatin, its lactone form, as well as both active hydroxy metabolites with respect to activation of PXR and/or CAR and subsequent induction of the corresponding target genes.
Here, we show that atorvastatin and its metabolites are ligands of PXR, but not of CAR. Interestingly, only the para-hydroxy metabolite showed significantly impaired induction of PXR-regulated genes, which may be explained by the impaired release of co-repressors from PXR by the metabolite. We conclude that the active metabolite, para-hydroxy atorvastatin, might have a reduced capacity to interact with other drugs.
Methods
Cell culture and generation of stable transfectants
The human hepatoblastoma cell line HepG2 was obtained from ATCC (Manassas, VA, USA) and grown in minimal essential medium, supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U·mL−1 penicillin and 100 U·mL−1 streptomycin. A pool of cells stably expressing human PXR (HepG2-PXR) was generated by selection with 800 µg·mL−1 G-418 sulphate of HepG2 cells, which were transfected with the expression plasmid pNTAP-huPXR by calcium phosphate co-precipitation as described by Geick et al. (2001), and further propagated in the presence of 200 µg·mL−1 G-418. Expression of human PXR protein in HepG2-PXR cells was confirmed by Western blotting using the PXR-specific antibody N16. COS1 cells were cultivated in Dulbecco's modified Eagle's medium buffered with 25 mM HEPES, supplemented with 100 U·mL−1 penicillin, 100 U·mL−1 streptomycin, 2 mM L-glutamine, and 10% fetal calf serum.
Transient transfections, mammalian two-hybrid and reporter gene assays
One day before transfection, HepG2 or HepG2-PXR cells were plated in 24-well plates at a density of 1.5 × 105 cells per well, whereas COS1 cells were plated at a density of 3 × 104 cells per well. Transfections were performed using Effectene™ transfection reagent (QIAGEN, Hilden, Germany), according to the manufacturer's recommendations. For two-hybrid PXR assembly assays HepG2 cells were transfected with 150 ng of the reporter gene plasmid pGL3-G5, 20 ng of the expression plasmid encoding GAL4-DBD/PXR(132–188) and 10 ng of the expression plasmid encoding VP16-AD/PXR(189–434) per well. Two-hybrid CAR assembly assays were performed in COS1 cells as described previously (Burk et al., 2005). For CYP3A4 promoter reporter gene assays, HepG2-PXR cells were transfected with 150 ng of the reporter gene plasmid pGL3-CYP3A4(-7830/Δ7208–364). Two-hybrid assays for the analysis of PXR interactions with co-activator or co-repressor were performed as described previously (Burk et al., 2005). The β-galactosidase reference plasmid pCMVβ (20ng) was always co-transfected. If necessary, respective empty expression vectors or pUC18 were used to fill up to a total amount of 200 ng of plasmid DNA per well. Subsequently, cells were treated for 24 h or 40 h with the indicated drugs dissolved in dimethyl sulphoxide (DMSO) or with an equivalent amount of DMSO (final 0.1%). In case of treatment with statins for more than 24 h, 400 µM mevalonolactone was added to preserve normal cell growth. Luciferase and β-galactosidase activities were analysed as described previously (Burk et al., 2002). Luciferase activity was normalized with respect to transfection efficiency using the corresponding β-galactosidase activity.
Bacterial protein expression and purification
Expression of soluble CAR-LBD and steroid receptor co-activator 1 (SRC-1)-RID His-tag fusion proteins was performed in Escherichia coli BL21(DE3), which were transformed with the respective bacterial expression plasmids and grown at 16°C for 20 h (CAR-LBD) or at 25°C for 4 h (SRC-1-RID) after induction of recombinant protein expression by 0.2 mM isopropyl β-D-1-thiogalactopyranoside. Recombinant proteins were affinity purified using Talon His-tag purification resin (Clontech, Mountain View, CA, USA) and immediately diluted into the running buffer (see below). The purity of the eluted proteins was controlled by SDS-PAGE and silver staining of the protein gel.
Surface plasmon resonance
Measurement of protein–protein interaction was performed by surface plasmon resonance using the Biacore 3000 instrument (Biacore/GE Healthcare, Freiburg, Germany). All buffers were degassed before use. The receptor interaction domain of the steroid receptor co-activator 1 (SRC-1-RID) protein was diluted into 10 mM sodium acetate pH 5 and immobilized on the dextrane matrix of a CM5 sensor chip using amine coupling. A reference cell was used to subtract possible non-specific binding of CAR-LBD protein to the chip surface. Running buffer consisted of 50 mM sodium phosphate pH 7.5, 150 mM sodium chloride, 0.1% Tween 20, 1 mM β-mercaptoethanol and 1% DMSO. Assays were performed at 25°C using a flow rate of 50 µL·min−1 to minimize mass transfer. Atorvastatin metabolites and 6-(4-chlorophenyl)imidazo[2,1-b]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl) oxime (CITCO) were prepared in DMSO as 30 mM and 100 mM stock solutions respectively. Compounds were diluted into running buffer to yield a final concentration of 100 µM. Before each assay, the system was primed at least three times, and three injections of running buffer and regeneration solution, respectively, were performed to optimize baseline stability. Each assay contained at least three injections of running buffer, ligands only and a 1% DMSO solvent control. Before injection, each cycle was able to establish a stable baseline for at least 90 s. CAR-LBD protein (0.21 µM) was injected either alone to define the constitutive binding to the immobilized co-activator or after an incubation of at least 30 min with the compounds at room temperature. Both association and dissociation were measured for 1 min. The post-dissociation phase comprised two-step regeneration and an extra clean washing step with running buffer. The first regeneration solution consisted of 10 mM sodium hydroxide, 150 mM sodium chloride and 0.03% SDS, whereas the second was made up by normal running buffer.
Primary human hepatocytes
Experimental procedures complied with the Guidelines of the charitable state-controlled Foundation for Human Tissue and Cell Research (Thasler et al., 2003), the respective institutional guidelines and were approved by the local ethical committees of the Ludwig-Maximilians-University of Munich, the University Medical Centre Regensburg and the Charité, Humboldt University Berlin, Germany. Tissue samples from human liver resections were obtained, with full consent, from patients undergoing partial hepatectomy because of primary or secondary liver tumours. Detail of the donors are given in Supporting Information Table S1. Human hepatocytes were isolated from the samples using a modified two-step EGTA/collagenase perfusion procedure as described previously (Weiss et al., 2003; Nussler et al., 2009). Viability of isolated hepatocytes was determined by Trypan blue exclusion. Only cell preparations with a viability >80% were used for experiments. The isolated cells were seeded at a density of 1.5 × 106 cells per well into collagen type I-coated six-well plates and cultivated at 37°C in a humidified incubator with 5% CO2 in Williams’ E medium, supplemented with 10% fetal calf serum, 15 mM HEPES, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 2 mM L-glutamine, 100 U·mL−1 each of penicillin and streptomycin, 0.032 IU·mL−1 insulin and 100 nM dexamethasone. After 2 days, cultures were maintained for 24 h in serum-free Williams’ E medium supplemented with 2 mM L-glutamine, 100 nM dexamethasone, 1% insulin-transferrin-selenium-G (ITS-G), and 100 U·mL−1 each of penicillin and streptomycin. Thereafter, cells were treated for 48 h with the indicated chemicals or with an equivalent amount (final 0.1%) of DMSO only. As shown in Supporting Information Figure S1, these cultures of human hepatocytes, as described above, generally maintained the expression of xenosensor PXR and co-activator SRC-1, whereas expression of co-repressor silencing mediator for retinoid and thyroid receptors (SMRT) was increased compared with matched, freshly isolated cells. CYP3A4 and CYP2B6 expression were down-regulated to at most 25% and <10% respectively. Thus, these culture conditions were suitable for induction studies.
Quantitative real-time PCR analysis
Total RNA and first-strand cDNA were prepared as previously described (Burk et al., 2002). The integrity of RNA samples was confirmed by formaldehyde agarose gel electrophoresis. Only the samples that showed sharp ribosomal RNA bands (28S twice as intense as 18S) and did not show any visible signs of degradation, were further processed. PCR reactions were set up with cDNA corresponding to 25 pg (18S rRNA) or 25 ng (all other assays) of total RNA and the qPCR MasterMix Plus Low ROX (Eurogentec, Seraing, Belgium). Gene expression levels were quantified by TaqMan real-time quantitative PCR using the 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The experiments were performed in a final volume of 25 µL using the default settings of the 7500 Real-Time PCR system. Assays were done in triplicate. Oligonucleotide primers and probes, designed with PrimerExpress software version 2.0 (Applied Biosystems), are shown in Supporting Information Table S2. The TaqMan gene expression assays Hs00168352_m1 and Hs00196955_m1 (Applied Biosystems) were used for the quantification of HMGCR and SMRT mRNA expression respectively. Serial dilutions of respective linearized cDNA plasmids were used to create the calibration curves, ranging from 30 to 3 × 107 copies. The respective gene expression levels were normalized to the corresponding 18S rRNA level and calculated as copies per 108 copies of 18S rRNA.
Protein analysis
Hepatocytes were scraped off the plate using ice-cold PBS. Cells were lysed by 30 min incubation on ice in 50 mM Tris-Cl pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% (v/v) Nonidet P-40, supplemented with 1× Halt protease inhibitor cocktail. Total protein lysates were quantified using the bicinchoninic acid method and a calibration curve generated with serial dilutions of BSA. Samples (30 µg protein) of each lysate were separated on a 10% SDS-polyacrylamide gel and transferred to nitrocellulose membrane by electroblotting. Transfer was controlled by Ponceau S staining. Blots were sequentially incubated with primary antibodies specific for CYP2B6, CYP3A4 and β-actin, each followed by incubation with the respective secondary peroxidase-conjugated antibody and detection by means of chemiluminescence using substrate SuperSignal West Dura. Protein bands were quantified with Fuji LAS-1000 CCD camera (Raytest, Straubenhardt, Germany) and AIDA software (Raytest).
Data analysis and statistical procedures
For transient transfections, the mean values of at least three independent experiments, each performed in triplicate, were used for statistical analysis. Multiple comparisons were generally performed using one-way anova with post tests, as indicated in the respective figure legends. To concurrently analyse the effect of treatment with chemicals and co-transfections of co-factors, two-way anova with Bonferroni's post-test was used (see Figure 6). Comparisons to a hypothetical mean were performed using one sample t-test, and the resulting P-values were corrected for multiple testing by the method of Bonferroni.
Figure 6.
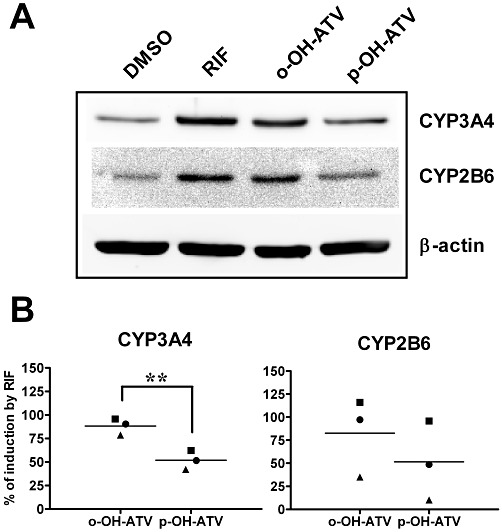
para-Hydroxy atorvastatin shows reduced induction of CYP protein expression. Primary human hepatocyte cultures (n = 3) were treated with 30 µM of the indicated chemicals or 0.1% DMSO only. After 48 h of treatment, total cellular protein lysates were prepared, followed by Western blot analysis of the indicated proteins. (A) Representative Western blot results from one individual culture. (B) Mean induction of CYP3A4 and CYP2B6 protein by ortho-hydroxy (o-OH-ATV) and para-hydroxy atorvastatin (p-OH-ATV) was calculated by normalizing CYP expression to β-actin and further comparing it with the extent of induction by rifampin (RIF), which was designated as 100%. Data were analysed by paired t-test. **P < 0.01, significant differences between groups as shown.
As hepatocyte mRNA expression data were not normally distributed, multiple comparisons were performed using Kruskal–Wallis test or Friedman test with Dunn's multiple comparisons post-test, whereas comparisons to a hypothetical mean were performed using Wilcoxon signed rank test. Pairwise comparisons of CYP protein induction data in hepatocytes were performed using the paired t-test.
All calculations were done using GraphPad Prism 5.04 or InStat 3.10 (GraphPad Software, La Jolla, CA, USA). Statistical significance was defined as P < 0.05.
Plasmids
All plasmids used in transient transfection analyses have been described previously: human CYP3A4 enhancer/promoter reporter gene plasmid pGL3-CYP3A4(-7830/Δ7208–364) (Hustert et al., 2001); the Gal4-dependent reporter gene construct pGL3-G5 and the expression plasmids encoding fusion proteins of the GAL4 DNA-binding domain (DBD) and receptor interaction domains (RID) of human co-activators SRC-1 (NCOA1, amino acids 583–783), transcriptional intermediary factor (TIF-2; NCOA2, amino acids 583–779) and vitamin D receptor interacting protein 205 (DRIP205; MED1, amino acids 527–774) (Arnold et al., 2004); expression plasmids encoding fusion proteins of the VP16-activation domain (AD) and human PXR or CAR ligand-binding domain (LBD) or parts of it (PXR amino acids 108–434, 189–434; CAR amino acids 151–348), expression plasmids encoding fusion proteins of the GAL4-DBD and the helix 1 parts of human PXR or CAR-LBD (PXR amino acids 132–188; CAR amino acids 105–150) and the expression plasmid encoding a fusion protein of the GAL4-DBD and RID of human co-repressor SMRT (NCOR2, amino acids 1109–1330) (Burk et al., 2005). The β-galactosidase expression plasmid pCMVβ was purchased from Clontech.
The expression plasmid pNTAP-huPXR was constructed by cloning the open reading frame of human PXR, amplified by PCR out of pcDhPXR (Geick et al., 2001) using primers 5′-AGT GGA TCC ATG GAG GTG AGA CCC AAA GAA AGC −3′ and 5′- GAT GAA TTC TCA GCT ACC TGT GAT GCC GAA CAA −3′. The oligonucleotides introduced BamHI and EcoRI restriction sites at the 5′- and 3′-ends of the amplified fragment respectively. The BamHI/EcoRI digested PCR fragment was cloned into appropriately digested vector pNTAP-B (Stratagene, La Jolla, CA, USA).
Bacterial expression plasmids encoding the LBD (amino acids 105–348) of human CAR or the RID (amino acids 583–783) of human SRC-1 were constructed by cloning PCR-amplified appropriate DNA fragments into pET-28a(+) or pET-22b(+) vector (Novagen/Merck Biosciences, Darmstadt, Germany) respectively. The PCR-amplified SRC-1 fragment additionally contains a sequence encoding N-terminal 10xHis-tag in frame with the SRC-1 part and a downstream stop codon. Thus, both bacterial expression plasmids encode the respective proteins as N-terminal His-tag fusions. The identities of all PCR-amplified DNA fragments were verified by sequencing.
Materials
Atorvastatin, atorvastatin lactone, ortho-hydroxy atorvastatin, and para-hydroxy atorvastatin were purchased from Toronto Research Chemicals (North York, Canada). DMSO (+/−)-mevalonolactone, rifampin, phenobarbital, clofibrate, triphenylphosphate, clotrimazole, PK11195, pravastatin and dexamethasone were purchased from Sigma-Aldrich (Taufkirchen, Germany). CITCO and G418-sulphate were purchased from BIOMOL/Enzo Life Sciences (Plymouth Meeting, PA, USA) and Calbiochem/Merck Biosciences (Darmstadt, Germany) respectively.
The following cell culture reagents were purchased from Invitrogen (Carlsbad, CA, USA): minimal essential medium, Williams’ E medium, non-essential amino acids, sodium pyruvate and ITS-G supplement. Fetal calf serum was obtained from Sigma-Aldrich, whereas Dulbecco's modified Eagle's medium buffered with 25 mM HEPES, L-glutamine and penicillin–streptomycin solution were purchased from BioWhittaker/Lonza (Verviers, Belgium). Twenty-four-well cell culture plates were obtained from BD Biosciences Discovery Labware (Bedford, MA, USA).
Antibodies used in immunoblotting were obtained as follows: PXR-specific antibody N16 (sc-9690) from Santa Cruz (Santa Cruz, CA, USA), antibodies WB-3A4 and INHIBITION MAB-2B6, specific for CYP3A4 and CYP2B6, respectively, from BD Biosciences (Heidelberg, Germany), the β-actin antibody (clone AC-15) from Sigma-Aldrich and the respective secondary peroxidase-conjugated antibodies from Dako (Glostrup, Denmark).
Further reagents were purchased as indicated: Effectene transfection reagent from QIAGEN, Talon His-tag purification resin from Clontech, PageSilver silver staining kit from Fermentas (Sankt Leon-Rot, Germany), sensor chip CM5 and amine coupling kit from Biacore/GE Healthcare, qPCR MasterMix Plus Low ROX from Eurogentec, Halt protease inhibitor cocktail and SuperSignal West Dura chemiluminescent substrate from Thermo Scientific/Pierce (Rockford, IL, USA). Oligonucleotide primers and TaqMan probes were custom-synthesized by Biomers (Ulm, Germany) and Applied Biosystems respectively.
Results
Atorvastatin and its metabolites are ligands of human PXR and induce PXR-dependent CYP3A4 promoter activity
Atorvastatin has been shown to induce CYP3A4 and CYP2B6 gene expression in primary human hepatocytes (Kocarek et al., 2002), thereby suggesting activation of PXR and/or CAR. However, the respective molecular mechanism has not yet been fully elucidated, as much as the role of atorvastatin lactone and hydroxy metabolites in the induction of CYP expression has not been addressed. To analyse whether atorvastatin and/or its metabolites are ligands of PXR, we applied a previously developed assembly assay, relying on the ligand-dependent interaction of fragments of the PXR-LBD (Burk et al., 2005). Figure 2A shows that rifampin, a well-known prototypical ligand of human PXR, efficiently induced the interaction of PXR helix 1 with the remainder of the LBD. Similarly, atorvastatin lactone and both hydroxy metabolites also induced PXR-LBD assembly, however, weaker than rifampin, whereas the parent compound showed only a marginal effect. Among all the metabolites, atorvastatin lactone was the most potent inducer of PXR-LBD assembly.
Figure 2.
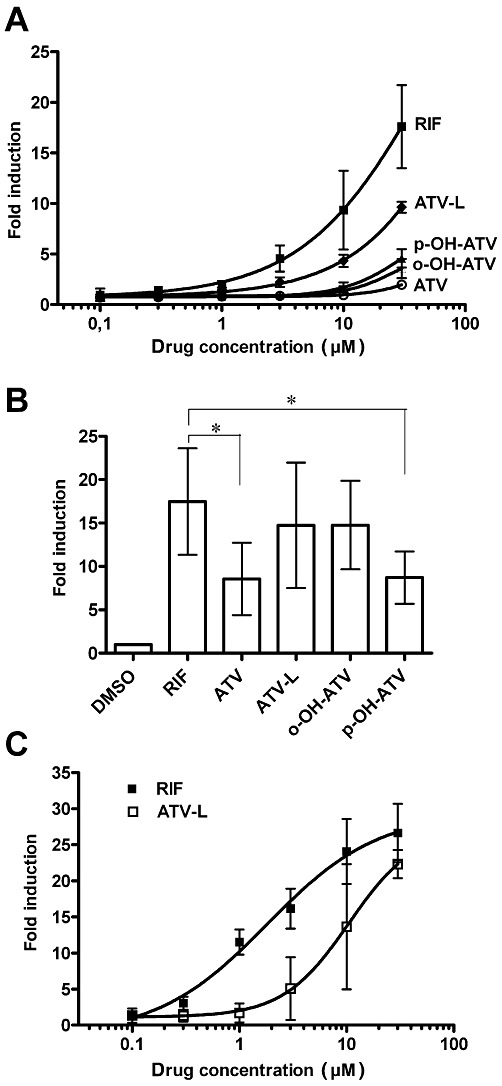
Atorvastatin and its metabolites induce assembly of the human PXR-LBD and CYP3A4 promoter activity. (A) PXR-assembly assay in HepG2 cells, co-transfected with expression plasmids encoding GAL4-DBD/hPXR-LBD (132–188) and VP16-AD/hPXR-LBD (189–434) fusion proteins. Cells were treated for 40 h with 0.1% DMSO or increasing concentrations of the indicated compounds. Mean fold induction (±SD) of the respective normalized activity of co-transfected reporter plasmid pGL3-G5 by treatment with the indicated concentrations of compounds is shown. (B) and (C) HepG2-PXR cells were transfected with pGL3-CYP3A4(-7830/Δ7208–364) and treated with increasing concentrations (C) or with 30 µM of the indicated compounds (B) for 24 h. The columns (B) and graphs (C) show mean fold induction (±SD) of normalized CYP3A4 promoter reporter gene activity by treatment with compounds. The respective reporter activity of transfected cells, which were treated with DMSO only, was designated as 1. Data were analysed by repeated measures one-way anova with Bonferroni's multiple comparisons post–test. *P < 0.05, significant differences between groups as shown. RIF, rifampin; ATV, atorvastatin; ATV-L, atorvastatin lactone, o-OH-ATV, ortho-hydroxy atorvastatin; p-OH-ATV, para-hydroxy atorvastatin.
Next, we asked whether atorvastatin and its metabolites may act as PXR agonists because antagonists also induce the assembly of nuclear receptor helix 1 fragments with the remainder of the LBD (Pissios et al., 2000 and data not shown). Figure 2B clearly shows that all of them induced the PXR-dependent transactivation of the CYP3A4 enhancer/promoter reporter gene in transiently transfected HepG2-PXR cells. The parent compound and the para-hydroxy metabolite showed significantly reduced activity, whereas the lactone and the ortho-hydroxy metabolite induced PXR-activity similar to rifampin. Dose-response analysis further revealed that rifampin activated PXR with an EC50 value of 1.8 µM (95% CI 1–4 µM), whereas atorvastatin lactone exhibited an EC50 value of >8 µM, which could not be determined more precisely as the activation plateau was not reached by 30 µM (Figure 2C).
Atorvastatin and its metabolites do not bind to human CAR as ligands
By applying our previously described cellular assembly assay for human CAR (Burk et al., 2005), we analysed whether atorvastatin and its metabolites may bind to CAR as ligands. Figure 3A shows that they did not induce the assembly of the human CAR-LBD, in clear contrast to CITCO, a well-known ligand for human CAR. The suitability of this assay for the identification of CAR ligands, whether agonists or inverse agonists, is shown in Supporting Information Figure S2A. Further cellular two-hybrid co-activator interaction assays using co-activators SRC-1 or TIF-2 together with reference variant CAR (Supporting Information Figure S3A,B) or using co-activator DRIP205 with the ligand-dependent CAR-SV2 splice variant (Arnold et al., 2004) (Supporting Information Figure S3C) confirmed that atorvastatin and its metabolites did not change human CAR activity by ligand binding. These compounds also did not change the interaction of CAR with co-repressor SMRT in a cellular two-hybrid assay (Supporting Information Figure S3D). Additionally, we established an in vitro Biacore protein–protein interaction assay using bacterially expressed human CAR-LBD and co-activator SRC-1-RID proteins. Treatment with CITCO clearly increased the binding of CAR to SRC-1, whereas atorvastatin and its metabolites did not show any significant effect (Figure 3B). Further negative and positive controls for the in vitro Biacore interaction assay are shown in Supporting Information Figure S2B.
Figure 3.
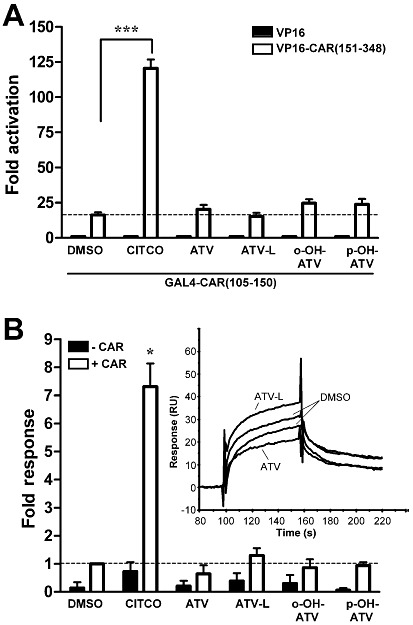
Atorvastatin and its metabolites neither induce the assembly of CAR-LBD nor the in vitro interaction with co-activator SRC-1. (A) CAR assembly assay in COS1 cells, which were co-transfected with expression plasmids encoding GAL4-DBD/hCAR-LBD (105–150) and VP16-AD/hCAR-LBD (151–348) fusion proteins (open columns) or empty expression vector pVP16-AD (filled columns). Cells were treated for 40 h with 1 µM CITCO, 30 µM of atorvastatin or metabolites or 0.1% DMSO only. Mean fold activation (±SD) of the normalized activity of co-transfected reporter plasmid pGL3-G5 by treatment with the indicated compounds is shown. The respective activity of cells transfected with GAL4-DBD/hCAR-LBD(105–150) expression plasmid and pVP16-AD, treated with DMSO only, was designated as 1. Data were analysed by one-way anova with the Tukey-Kramer post-test. ***P < 0.001, significant effect of CITCO. (B) Biacore analysis of ligand-induced interaction of co-activator SRC-1 with CAR in vitro. CAR-LBD protein, pre-incubated with 10 µM CITCO or 100 µM of the indicated atorvastatin metabolites or 1% DMSO only, was injected onto immobilized SRC-1-RID protein (+CAR). As a control, compounds were also injected alone (-CAR). Data are shown as means ± SD of four to six independent experiments. Open columns show increase in binding by pre-incubation of CAR with compounds. Binding of CAR to SRC-1 in the presence of DMSO only, was designated as 1. Data were analysed by one sample t-test and P-values corrected for multiple testing by the method of Bonferroni, *P < 0.05, significant effect of CITCO. The inset shows representative individual sensorgrams of CAR pre-incubated with ATV, ATV-L or vehicle DMSO only. ATV, atorvastatin; ATV-L, atorvastatin lactone, o-OH-ATV, ortho-hydroxy atorvastatin; p-OH-ATV, para-hydroxy atorvastatin.
Atorvastatin and its metabolites differentially induce the hepatic expression of PXR-regulated genes
Having shown that atorvastatin and its metabolites are ligands only for human PXR, we assessed their respective induction capacity using primary human hepatocytes and the induction of endogenous CYP3A4 and UGT1A3 expression, as measured by quantitative real-time PCR. Treatment with the prototypical PXR ligand rifampin induced CYP3A4 mRNA expression with median 8.6-fold (range: 4.7–16.3). Atorvastatin, atorvastatin lactone and ortho-hydroxy atorvastatin all induced CYP3A4 expression to a similar extent, with medians (range) of 6.5- (3.1–12.7), 7.1- (3.9–12.3) and 7.9- (1.8–12.7) fold respectively. In contrast, induction of CYP3A4 by para-hydroxy atorvastatin was significantly reduced, with median 2.5-fold, ranging from 0.4- to 4.4-fold in individual cultures. As expected (Kocarek et al., 2002) and thus used as a negative control, pravastatin did not induce CYP3A4 expression at all (Figure 4A). Regarding UGT1A3 expression, Figure 4B shows that this gene was induced with similar median fold changes by rifampin, atorvastatin and its metabolites. To prove the successful inhibition of cholesterol biosynthesis, which has to result in the compensatory increased expression of HMGCR (Horton et al., 2002), we further analysed HMGCR mRNA expression in the drug-treated hepatocytes. HMGCR expression showed the expected increase in cultures, which were treated with the pharmacologically active atorvastatin (median 10.0-fold, range 6.5–12.3), ortho-hydroxy atorvastatin (median 8.0-fold, range 4.2–9.8), and para-hydroxy atorvastatin (median 8.4-fold, range 5.2–15.3), but also demonstrated an increase in cultures treated with the inactive atorvastatin lactone (median 8.6-fold, range 4.4–9.5) (Figure 4C), which can only be explained by substantial conversion of the lactone to the acid form in human hepatocytes. As expected, HMGCR gene expression was not induced by rifampin (Figure 4C).
Figure 4.
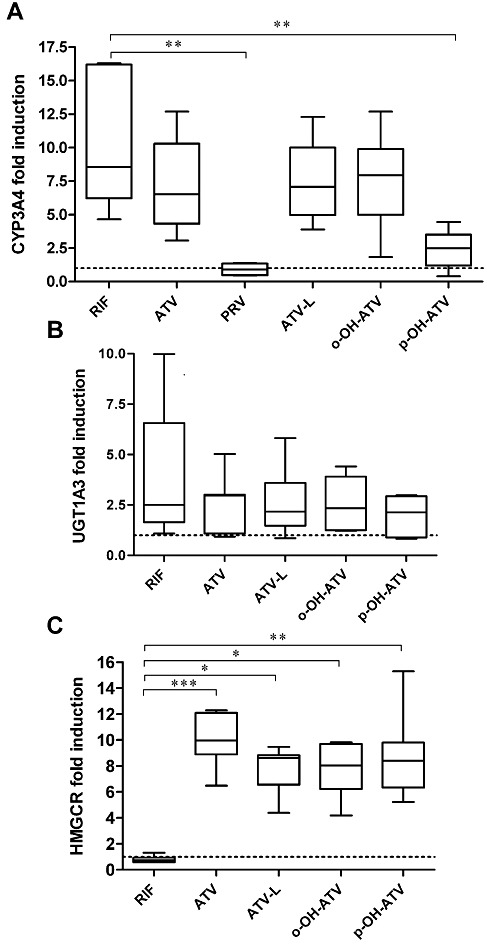
Atorvastatin and its metabolites differentially induce hepatic gene expression in primary human hepatocytes. Primary human hepatocyte cultures (n = 7, PRV n = 4) were treated with 30 µM of the indicated chemicals or 0.1% DMSO only. After 48 h of treatment total RNA was prepared, followed by analysis of CYP3A4 (A), UGT1A3 (B) and HMGCR (C) mRNA expression by TaqMan real-time RT-PCR. Expression levels were normalized with respect to the corresponding expression of 18S rRNA. Data are shown as fold induction by treatment with chemicals as compared with the treatment with DMSO only, which was designated as 1, and are presented as box and whisker plots, with boxes representing the 25th–75th percentiles, medians indicated by horizontal lines and whiskers showing minimum and maximum values. Data were analysed by Kruskal–Wallis test with Dunn's multiple comparisons post–test. ***P < 0.001, **P < 0.01, *P < 0.05, significant differences between groups as shown. RIF, rifampin; ATV, atorvastatin; ATV-L, atorvastatin lactone, o-OH-ATV, ortho-hydroxy atorvastatin; p-OH-ATV, para-hydroxy atorvastatin; PRV, pravastatin.
Having shown the differential impact of the two hydroxy metabolites of atorvastatin on the expression of CYP3A4 and UGT1A3, we investigated their effect on the expression of other genes, known to be regulated by PXR. Figure 5A–D shows that rifampin and the ortho-hydroxy metabolite both significantly induced the expression of CYP2B6, EPHX1, ABCB1 and SLCO1B1, compared with treatment with DMSO only (P < 0.05, Wilcoxon signed rank test). In contrast, the para-hydroxy metabolite only weakly induced ABCB1 (P < 0.05, Wilcoxon signed rank test).
Figure 5.
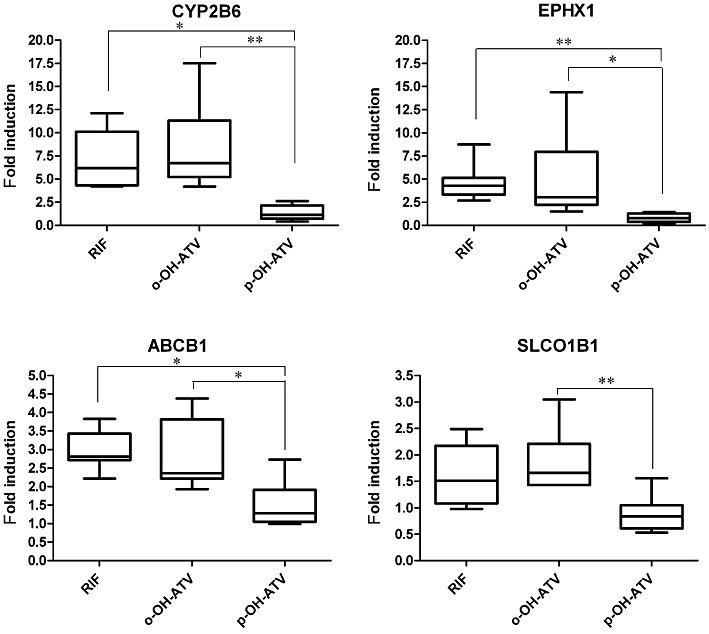
The hydroxy metabolites of atorvastatin differentially induce the expression of PXR target genes. Expression of CYP2B6, EPHX1, ABCB1 and SLCO1B1 was analysed by TaqMan real-time RT-PCR of the mRNA samples of primary human hepatocyte cultures, which were treated with rifampin (RIF), ortho-hydroxy atorvastatin (o-OH-ATV), para-hydroxy atorvastatin (p-OH-ATV) or DMSO (see legend of Figure 4). Fold induction by treatment with rifampin and the atorvastatin hydroxy metabolites was calculated with respect to expression levels by treatment with DMSO only, which was designated as 1. The data are presented as box and whisker plots as described in Figure 4. Data were analysed by Friedman's test with Dunn's multiple comparisons post–test. *P < 0.05, **P < 0.01, significant differences between groups as shown.
Impaired induction of CYP3A4 and CYP2B6 genes by para-hydroxy atorvastatin is manifested on the protein level
Induction of CYP and ABCB1 genes is functionally relevant, as it results in elevated protein levels, which influence the pharmacokinetics of drug substrates. Using hepatocytes from three additional donors, we assayed the protein levels of CYP3A4, CYP2B6 and ABCB1, after treatment with the hydroxy atorvastatin metabolites. Figure 6 shows that CYP3A4 and CYP2B6 proteins were induced by ortho-hydroxy atorvastatin and rifampin, to a similar extent. Mean induction of CYP3A4 protein by para-hydroxy atorvastatin was significantly reduced by 40% (P = 0.0017, paired t-test), compared with the ortho-hydroxy metabolite. A similar trend, however, not reaching significance, was observed for CYP2B6 protein (P = 0.0712, paired t-test). The MDR1/P-glycoprotein, which is encoded by the ABCB1 gene, was induced by rifampin but there was no consistent induction by the two hydroxy metabolites of atorvastatin (data not shown). The difference in induction of CYP3A4 and CYP2B6 between the two metabolites is smaller, in terms of the protein than the corresponding mRNA (see Figures 4 and 5), which we also confirmed for these three hepatocyte cultures, by analysing mRNA induction, showing that expression of both CYP genes was approximately 3.5-fold higher by ortho-hydroxy atorvastatin than by the para-hydroxy metabolite.
Atorvastatin and its metabolites differentially modulate the interaction of co-regulatory proteins with human PXR
Having shown that the induction of some PXR target genes was strongly impaired in para-hydroxy atorvastatin-treated primary human hepatocytes, we attempted to elucidate the corresponding molecular mechanisms. In general, activation of nuclear receptors involves the ligand-dependent release of co-repressors and subsequent ligand-dependent interaction with co-activators. Thus, we performed mammalian two-hybrid assays that rely on the interaction of the PXR-LBD with RIDs of co-regulatory proteins. First, we analysed the ligand-dependent interaction with co-activators SRC-1 (NCOA1), TIF-2 (NCOA2) and DRIP205 (MED1), which are known to be recruited to PXR in a ligand-dependent manner. Atorvastatin only induced the interaction of PXR with SRC-1, whereas the lactone and both hydroxy metabolites stimulated the interaction of PXR with all three co-activators tested. However, the effect was not as pronounced as with the prototypical ligand rifampin (Figure 7A–C). Most importantly, significant differences in the interaction of PXR and co-activators were not observed between ortho-hydroxy and para-hydroxy atorvastatin treatment. Next, we analysed the ligand-dependent release of co-repressor SMRT (NCOR2). In the absence of ligands, the PXR-LBD constitutively interacted with this co-repressor. Rifampin, atorvastatin, atorvastatin lactone and ortho-hydroxy atorvastatin abolished this interaction. In contrast, para-hydroxy atorvastatin only produced a limited (50%) release of co-repressor SMRT (Figure 7D).
Figure 7.
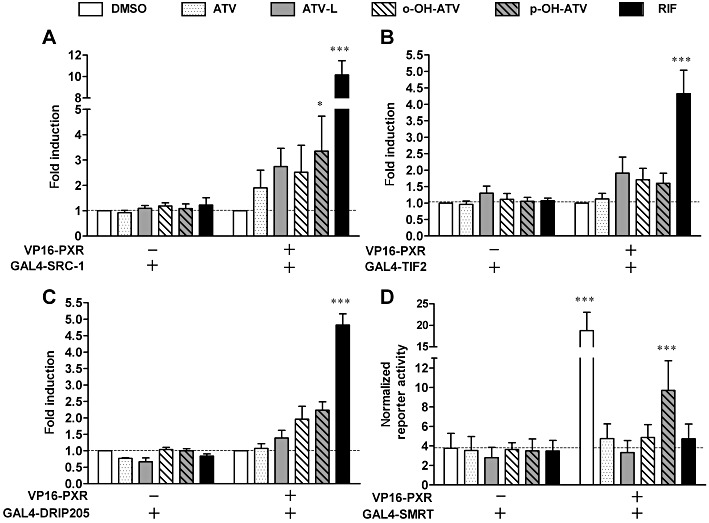
Atorvastatin and its metabolites differentially modulate the interaction of PXR with co-regulators. (A–C) HepG2 cells were co-transfected with expression plasmids encoding GAL4-DBD/co-activator-RID fusion proteins, as indicated, and an expression plasmid encoding VP16-AD/PXR-LBD (108–434) fusion protein (+) or empty expression vector pVP16-AD (–). Columns show the mean fold induction (±SD) of the normalized activity of co-transfected pGL3-G5 after 40 h of treatment with 0.1% DMSO, 30 µM atorvastatin (ATV), 30 µM atorvastatin lactone, (ATV-L), 30 µM ortho-hydroxy atorvastatin(o-OH-ATV), 30 µM para-hydroxy atorvastatin (p-OH-ATV) and 10 µM rifampin (RIF). The activity of cells transfected with the respective expression plasmid combination and treated with DMSO only, was designated as 1. (D) HepG2-PXR cells were co-transfected with plasmids encoding GAL4-DBD/SMRT-RID fusion protein and VP16-AD/PXR-LBD (108–434) fusion protein (+) or empty expression vector pVP16-AD (–). Columns show the means (±SD) of normalized pGL3-G5 reporter activity after 24 h of treatment with compounds, as described above. Data were analysed by two-way anova with Bonferroni's post-test. ***P < 0.001, *P < 0.05, significantly different from corresponding value with empty expression vector (–).
Discussion and conclusions
Atorvastatin induces the expression of CYP2B6, CYP2C8, CYP2C9 and CYP3A4 mRNA, protein and enzymic activity in hepatocytes (Kocarek et al., 2002; Feidt et al., 2010) and activates human PXR and CAR in reporter gene assays (Kobayashi et al., 2005; Monostory et al., 2009; Yamasaki et al., 2009), thereby suggesting ligand binding to these xenosensing nuclear receptors as the molecular mechanism of induction. It was the aim of this study to elucidate this hypothesis and to analyse any potential role of atorvastatin metabolites in induction.
Atorvastatin and its primary metabolites are ligands for human PXR but not for CAR. In the PXR assembly assay (Burk et al., 2005), atorvastatin, both in its acid and lactone forms, and atorvastatin ortho- and para-hydroxy metabolites all were ligands of PXR. Nuclear receptor assembly assays, which rely on the ligand-dependent interaction of the respective LBD helix 1 fragment with the remainder of its LBD, reflect the intramolecular conformational change of helix 1 and subsequent stabilization of the LBD structure upon ligand binding (Pissios et al., 2000). The usefulness of the PXR assembly assay for the identification of ligands has recently been confirmed (Kobayashi et al., 2010). In this assay, the lactone form of atorvastatin showed the highest potency; however, this does not necessarily reflect the highest binding affinity to PXR. Atorvastatin lactone is more lipophilic than the acid form and may simply enter cells more readily (Yamasaki et al., 2009).
It was claimed that statins, including atorvastatin, are ligands of CAR (Willrich et al., 2009), suggested by the activation of co-transfected CAR in hepatoma cell lines by statins, using CYP2B6 and CYP3A4 promoter reporter gene assays (Kobayashi et al., 2005; Monostory et al., 2009). However, very recent studies also using promoter reporter gene assays have questioned the activation of CAR by statins (Howe et al., 2011; Omiecinski et al., 2011). Using different cellular and in vitro assays, all relying on ligand-dependent protein–protein interactions as a measure of ligand binding, it is unequivocally shown that neither atorvastatin nor any of its metabolites act as ligands of reference variant human CAR or of the ligand-dependent splice variant CAR-SV2 (Arnold et al., 2004). CAR-SV2 is said to account for approximately 40% of human CAR transcripts (Jinno et al., 2004) and exhibits specific ligand binding (Dring et al., 2010). Treatment with atorvastatin activated AMP-activated kinase (Sun et al., 2006), which was further demonstrated to be involved in the activation of CAR by phenobarbital-type activators and subsequent translocation of the receptor into the nucleus (Rencurel et al., 2005; Shindo et al., 2007). Thus, the discrepancy between the findings reported here and the previously published results (Kobayashi et al., 2005; Monostory et al., 2009), with respect to the activation of CAR by atorvastatin, may be explained by ligand-independent, phenobarbital-type activation of CAR in the cells used in these studies. Studies directly addressing nuclear translocation of CAR in hepatocytes by treatment with atorvastatin are clearly required to resolve the issue.
Atorvastatin induces a broader spectrum of ADME genes in hepatocytes than previously thought, now including phase II drug-metabolizing enzyme and drug transporter genes, besides the already known CYP genes. As SLCO1B1/OATP1B1 and ABCB1/P-glycoprotein mediate hepatic uptake and efflux of atorvastatin, respectively, and CYP3A4 and UGT1A3 are the main detoxifying enzymes, atorvastatin may induce all the steps of its own elimination. Autoinduction of elimination may thus further contribute to the inter-individual variability in disposition and therapeutic efficacy of atorvastatin. However, induction of drug metabolism and transport by atorvastatin has, up to now, been demonstrated only in cell-based promoter reporter gene assays and in hepatocytes ex vivo (Marino et al., 2011). Evidence for the mechanism to be active in liver in vivo is still missing, to the best of our knowledge, clearly asking for appropriately designed clinical studies, which may also address the autoinduction of elimination suggested here, by analysing the time dependency of the drug's pharmacokinetics with repeated dosing regimens.
Apart from the parent drug, the metabolites of atorvastatin also participate in the induction by PXR but not all are equally efficient as inducers. The para-hydroxy metabolite showed a significantly reduced induction of PXR target genes, except for UGT1A3, compared with the other metabolites. Because UGT1A3 was as well induced by both hydroxy metabolites, the difference seen in induction of the other genes is most likely not to be the consequence of altered metabolism or transport of para-hydroxy atorvastatin in hepatocytes. para-Hydroxy atorvastatin also induced HMGCR expression as strong as the other compounds, indicating that it is efficiently taken up into cells. This conclusion is further supported by studies reporting that both hydroxy metabolites are equally well transported by OATP1B1 (Lau et al., 2007). Similarly, as the PXR assembly and co-activator interaction assays did not show weaker activity of the para-hydroxy as compared with the ortho-hydroxy metabolite, differences in ligand binding affinity and co-activator recruitment most likely do not account for the impaired induction capacity of para-hydroxy atorvastatin. However, this metabolite showed less co-repressor release, which suggests the molecular mechanism of its impaired induction capacity in hepatocytes (Figure 8). In the absence of an agonist, PXR is bound to co-repressors, as, for example, SMRT, resulting in inactive receptor complexes. Upon agonist binding, conformation of the receptor is changed, the co-repressors dissociate and co-activators, as, for example, SRC-1, are recruited to the receptor, resulting in active receptor complexes and subsequently in transcriptional activation of PXR target genes (di Masi et al., 2009). Thus, failure or decrease in the release of co-repressors upon ligand binding will preclude or diminish transcriptional activation by PXR, even if a ligand is, in principle, able to fully induce a receptor conformation allowing co-activator binding in vitro, as it is the case with para-hydroxy atorvastatin. This is because the balance of PXR complexes remains with the inactive co-repressor complexes, even after ligand binding (Figure 8).
Figure 8.
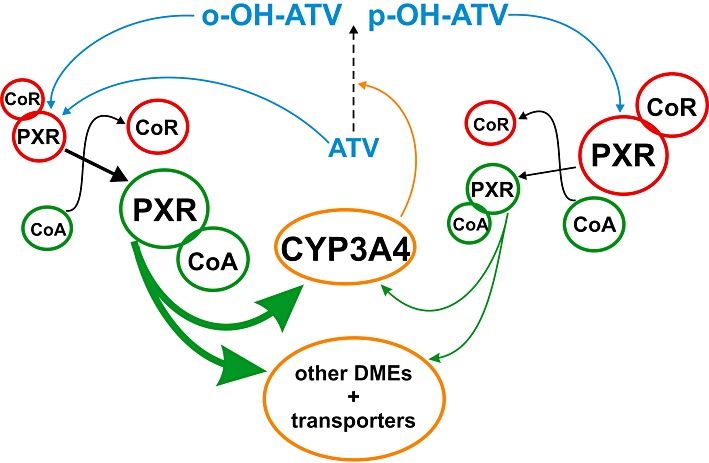
Proposed molecular mechanism of the impaired induction of PXR activation by para-hydroxy atorvastatin. Because of the reduced release of co-repressors (CoR), para-hydroxy atorvastatin (p-OH-ATV) may result in fewer transcriptionally active PXR-co-activator (CoA) complexes than ortho-hydroxy atorvastatin (o-OH-ATV), thereby keeping more PXR in the inactive co-repressor complexes. Consequently, p-OH-ATV results in reduced induction of drug-metabolizing enzymes and transporters.
In this respect, para-hydroxy atorvastatin resembles docetaxel, which was shown to recruit co-activators while failing to release co-repressors in vitro. However in contrast to the atorvastatin metabolite, docetaxel did not induce PXR target gene expression at all and was thus called a ‘PXR-transparent’ drug (Synold et al., 2001). para-Hydroxy atorvastatin is clearly not such a ‘PXR-transparent’ compound but it may be regarded as a selective receptor modulator of PXR, as it demonstrates gene and tissue specificity of induction. It induces some PXR target genes (e.g. UGT1A3) as effectively as the parent drug and the other metabolites, while not inducing (e.g. SLCO1B1) or only weakly inducing (e.g. CYP3A4) others. Furthermore, its inductive capacity also depends on cell type; in contrast to HepG2-PXR cells, para-hydroxy atorvastatin proved to be as efficient as the ortho-hydroxy metabolite for inducing CYP3A4 promoter activity in the intestinal LS174T cells (data not shown).
Given its impaired activation of PXR in hepatic cell systems, especially regarding the induction of CYP enzymes, para-hydroxy atorvastatin might also have a reduced capacity for drug interactions in vivo. Induction of CYP3A4 protein expression by para-hydroxy atorvastatin is, on average, only 60% of that observed with the ortho-hydroxy metabolite. Thus, drug–drug interactions because of CYP3A4 induction may be accordingly diminished if para-hydroxy atorvastatin were to be used therapeutically instead of the parent drug. Moreover, the clinical use of an active metabolite, which is no longer subject to metabolism by CYP3A4 itself, would also help to avoid the serious drug interactions, which have been reported for the parent drug atorvastatin, when co-administered with inhibitors or inducers of CYP3A4 (see Neuvonen et al., 2006). The potent selective CYP3A4 enzyme inhibitor itraconazole was reported to increase the area under the plasma concentration time curve (AUC) and elimination half-life of atorvastatin by threefold (Kantola et al., 1998). Increasing plasma levels of atorvastatin may result in life-threatening muscle toxicity, as reported for the combination of atorvastatin and fluconazole, another inhibitor of CYP3A enzymes (Kahri et al., 2005). On the other hand, the PXR ligand rifampin, a potent inducer of CYP3A expression in vivo, decreased the AUC of atorvastatin by 80% (Backman et al., 2005). Additional clinical benefit may result from the potent antioxidant properties of para-hydroxy atorvastatin because in contrast to the parent drug, the hydroxy metabolites of atorvastatin protect lipoproteins from oxidation (Aviram et al., 1998).
In conclusion, evidence is provided for the activation of PXR by atorvastatin and its metabolites by demonstrating ligand binding to the receptor. The differential activity of the para-hydroxy metabolite in induction of PXR-dependent gene expression, which is most likely caused by impaired co-repressor release, may suggest this active metabolite as a selective PXR modulator and as a promising candidate for future drug development due to its expected lower capacity of drug interactions. This study further demonstrates that it is important to take into account the properties of metabolites when analysing the PXR-dependent induction of drug metabolism and transport.
Acknowledgments
We greatly appreciate the expert technical assistance of K. Abuazi Rincones. We also would like to thank U. Hofmann for drawing the atorvastatin chemical formulas.
This work was supported by the German Federal Ministry of Education and Research (BMBF) HepatosSys network grants 0313081B (A.K.N.), 0313081D (T.S.W and W.E.T.), 0313080F to Rolf Schmid, Institute of Technical Biochemistry, University of Stuttgart, Germany (L.G.) and 0313080I (O.B)., the BMBF grant 03 IS 2061C (M.S.) and the FP7 grant PITN-GA-2009–238132 (M.S.), and by the Robert Bosch Foundation, Stuttgart, Germany.
This work contains parts of the doctoral theses of E.H and L.G.
Glossary
- AD
activation domain
- ADME
absorption distribution metabolism excretion
- CAR
constitutive androstane receptor
- CITCO
6-(4-chlorophenyl)imidazo[2,1-b]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl) oxime
- CYP
cytochrome P450
- DBD
DNA-binding domain
- DMSO
dimethyl sulphoxide
- DRIP205
vitamin D receptor interacting protein 205
- HMGCR
3-hydroxy-3-methylglutaryl-coenzyme A reductase
- kb
kilobase pair(s)
- LBD
ligand-binding domain
- LDL
low-density lipoprotein
- PXR
pregnane X receptor
- RID
receptor interaction domain
- SMRT
silencing mediator for retinoid and thyroid receptors
- SRC-1
steroid receptor co-activator 1
- TIF-2
transcriptional intermediary factor 2
- UGT
UDP-glucuronosyltransferase
Conflicts of interest
The authors state no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Expression of PXR and co-factors ismaintained in plated primary human hepatocytes. Expression levelsof the indicated genes, as determined by TaqMan real-time RT-PCR,were compared between human hepatocytes grown for 5 days asmonolayer on collagen I-coated plates and matched freshly isolatedcells (n = 4). During the last 2 days of culture, hepatocytes were treated with 0.1% DMSO. Data are shown as relative to the expression level in freshly isolated cells, which was designated as 1. Lines indicate means.
Figure S2 CAR ligands induce the assembly ofCAR-LBD and in vitro interaction with co-activator SRC-1.(A) CAR assembly assay in COS1 cells, which were co-transfectedwith expression plasmids encoding GAL4-DBD/hCAR-LBD (105–150) and VP16-AD/hCAR-LBD (151–348) fusion proteins. Cells weretreated for 40 h with 10 μM rifampin (RIF), 1 mM phenobarbital(PB), 1 μM CITCO, 100 μM clofibrate (CLO), 30 μMtriphenyphosphate (TPP), 10 μM clotrimazole (CLOT), 10 μMPK11195 (PK) or 0.1% DMSO only. Mean fold induction (±SD) ofthe normalized activity of co-transfected reporter plasmid pGL3-G5by treatment with the indicated chemicals is shown. Hatched andcross-hatched columns indicate treatment with negative controls.Columns in black or grey indicate treatment with agonists orinverse agonists respectively. The respective activity of cellstreated with DMSO only, was designated as 1. (B) Biacore analysisof ligand-induced interaction of co-activator SRC-1 with CAR invitro. CAR-LBD protein, pre-incubated with 100 μM of theindicated chemicals or 1% DMSO only, was injected onto immobilizedSRC-1-RID protein (open columns). As a control, chemicals were alsoinjected alone (filled columns). Data are shown as means ±SD of three to six independent experiments. Open columns showincrease in binding by pre-incubation of CAR with chemicals.Binding of CAR to SRC-1 in the presence of DMSO only, wasdesignated as 1. (A) and (B) Statistically significant differenceswere analysed by one sample t-test, resultingP-values were corrected for multiple testing by the methodof Bonferroni and indicated by asterisks (*P < 0.05;**P < 0.01; ***P < 0.001).
Figure S3 Atorvastatin and its metabolites donot change the interaction of CAR with co-regulators. (A–C) COS1 cells were co-transfected with expression plasmids encodingGAL4-DBD/co-activator-RID fusion proteins, as indicated, andexpression plasmids encoding VP16-AD/CAR-LBD (105–348) (A) and (B) or VP16-AD/CAR-SV2-LBD (105–353) (C) fusion proteins(open columns) or empty expression vector pVP16-AD (filledcolumns). Cells were treated for 40 h with 1 μM CITCO, 30 μMof atorvastatin metabolites or 0.1% DMSO only. Mean fold activation(±SD) of the normalized activity of co-transfected reporterplasmid pGL3-G5 by treatment with the indicated chemicals is shown.The respective activity of cells transfected with the particularGAL4-DBD/co-activator-RID fusion protein expression plasmids andpVP16-AD, treated with DMSO only, was designated as 1. (D) COS1cells were co-transfected with plasmids encoding GAL4-DBD/SMRT-RIDfusion protein and VP16-AD/CAR-LBD (105–348) fusion protein.An expression plasmid encoding human RXRα was additionallyco-transfected. Columns show the mean fold activation (±SD) of normalized pGL3-G5 reporter activity after 40 h of treatmentwith chemicals (AND, 10 μM androstenol), as compared with therespective activity of cells transfected with GAL4-DBD/SMRT-RIDplasmid and pVP16-AD, treated with DMSO only, which was designatedas 1. (A–D) Statistical significant differences to DMSOvehicle treatments were analysed by repeated measures one-way ANOVAwith Dunett's multiple comparisons test and indicated by asterisks(**P < 0.01).
Table S1 Hepatocyte donor data
Table S2 Oligonucleotides for quantitative real-time RT-PCR
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold KA, Eichelbaum M, Burk O. Alternative splicing affects the function and tissue-specific expression of the human constitutive androstane receptor. Nucl Recept. 2004;2:1. doi: 10.1186/1478-1336-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviram M, Rosenblat M, Bisgaier CL, Newton RS. Atorvastatin and gemfibrozil metabolites, but not the parent drugs, are potent antioxidants against lipoprotein oxidation. Artherosclerosis. 1998;138:271–280. doi: 10.1016/s0021-9150(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Backman JT, Luurila H, Neuvonen M, Neuvonen PJ. Rifampin markedly decreases and gemfibrozil increases the plasma concentrations of atorvastatin and its metabolites. Clin Pharmacol Ther. 2005;78:154–167. doi: 10.1016/j.clpt.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Burk O, Tegude H, Koch I, Hustert E, Wolbold R, Glaeser H, et al. Molecular mechanisms of polymorphic CYP3A7 expression in adult human liver and intestine. J Biol Chem. 2002;277:24280–24288. doi: 10.1074/jbc.M202345200. [DOI] [PubMed] [Google Scholar]
- Burk O, Arnold KA, Nussler AK, Schaeffeler E, Efimova E, Avery BA, et al. Antimalarial artemisinin drugs induce cytochrome P450 and MDR1 expression by activation of xenosensors pregnane X receptor and constitutive androstane receptor. Mol Pharmacol. 2005;67:1954–1965. doi: 10.1124/mol.104.009019. [DOI] [PubMed] [Google Scholar]
- Chen C, Mireles RJ, Campbell SD, Lin J, Mills JB, Xu JJ, et al. Differential interaction of 3 hydroxy-3-methylglutaryl-CoA reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab Dispos. 2005;33:537–546. doi: 10.1124/dmd.104.002477. [DOI] [PubMed] [Google Scholar]
- Dring AM, Anderson LE, Qamar S, Stoner MA. Rational quantitative structure-activity relationship (RQSAR) screen for PXR and CAR isoform-specific nuclear receptor ligands. Chem Biol Interact. 2010;188:512–525. doi: 10.1016/j.cbi.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feidt D, Klein K, Hofmann U, Riedmaier S, Knobeloch D, Thasler WE, et al. Profiling induction of cytochrome P450 enzyme activity by statins using a new liquid chromatography-tandem mass spectrometry cocktail assay in human hepatocytes. Drug Metab Dispos. 2010;38:1589–1597. doi: 10.1124/dmd.110.033886. [DOI] [PubMed] [Google Scholar]
- Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276:14581–14587. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- Hochman JH, Pudvah N, Qiu J, Yamazaki M, Tang C, Lin JH, et al. Interactions of human P-glycoprotein with simvastatin, simvastatin acid, and atorvastatin. Pharm Res. 2004;21:1686–1691. doi: 10.1023/b:pham.0000041466.84653.8c. [DOI] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, Sanat F, Thumser AE, Coleman T, Plant N. The statin class of HMG-CoA reductase inhibitors demonstrate differential activation of the nuclear receptors PXR, CAR and FXR, as well as their downstream target genes. Xenobiotica. 2011;41:519–529. doi: 10.3109/00498254.2011.569773. [DOI] [PubMed] [Google Scholar]
- Hustert E, Zibat A, Presecan-Siedel E, Eiselt R, Mueller R, Fuss C, et al. Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug Metab Dispos. 2001;29:1454–1459. [PubMed] [Google Scholar]
- Jacobsen W, Kuhn B, Soldner A, Kirchner G, Sewing KF, Kollman PA, et al. Lactonization is the critical first step in the disposition of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor atorvastatin. Drug Metab Dispos. 2000;28:1369–1378. [PubMed] [Google Scholar]
- Jinno H, Tanaka-Kagawa T, Hanioka N, Ishida S, Saeki M, Soyama A, et al. Identification of novel alternative splice variants of human constitutive androstane receptor and characterization of their expression in the liver. Mol Pharmacol. 2004;65:496–502. doi: 10.1124/mol.65.3.496. [DOI] [PubMed] [Google Scholar]
- Kahri J, Valkonen M, Bäcklund T, Vuoristo M, Kivistö KT. Rhabdomyolysis in a patient receiving atorvastatin and fluconazole. Eur J Clin Pharmacol. 2005;60:905–907. doi: 10.1007/s00228-004-0858-5. [DOI] [PubMed] [Google Scholar]
- Kameyama Y, Yamashita K, Kobayashi K, Hosakawa M, Chiba K. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics. 2005;15:513–522. doi: 10.1097/01.fpc.0000170913.73780.5f. [DOI] [PubMed] [Google Scholar]
- Kantola T, Kivistö KT, Neuvonen PJ. Effect of itraconazole on the pharmacokinetics of atorvastatin. Clin Pharmacol Ther. 1998;64:58–65. doi: 10.1016/S0009-9236(98)90023-6. [DOI] [PubMed] [Google Scholar]
- Keskitalo JE, Kurkinen KJ, Neuvoneni PJ, Niemi M. ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin Pharmacol Ther. 2008;84:457–461. doi: 10.1038/clpt.2008.25. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Yamanaka Y, Iwazaki N, Nakajo I, Hosokawa M, Negishi M, et al. Identification of HMG-CoA reductase inhibitors as activators for human, mouse and rat constitutive androstane receptor. Drug Metab Dispos. 2005;33:924–929. doi: 10.1124/dmd.104.002741. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Saito K, Takagi S, Chiba K. Ligand-dependent assembly of Pregnane X Receptor, constitutive androstane receptor and liver X receptor is applicable to identify ligands. Drug Metab Lett. 2010;4:88–94. doi: 10.2174/187231210791292744. [DOI] [PubMed] [Google Scholar]
- Kocarek TA, Dahn MS, Cai H, Strom SC, Mercer-Haines NA. Regulation of CYP2B6 and CYP3A expression by hydroxymethylglutaryl coenzyme A inhibitors in primary cultured human hepatocytes. Drug Metab Dispos. 2002;30:1400–1405. doi: 10.1124/dmd.30.12.1400. [DOI] [PubMed] [Google Scholar]
- Lau YY, Huang Y, Frassetto L, Benet L. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther. 2007;81:194–204. doi: 10.1038/sj.clpt.6100038. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Lee MG, Lim LA, Jang SB, Chung JY. Effects of SLCO1B1 and ABCB1 genotypes on the pharmacokinetics of atorvastatin and 2-hydroxyatorvastatin in healthy Korean subjects. Int J Clin Pharmacol Ther. 2010;48:36–45. doi: 10.5414/cpp48036. [DOI] [PubMed] [Google Scholar]
- Lennernäs H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42:1141–1160. doi: 10.2165/00003088-200342130-00005. [DOI] [PubMed] [Google Scholar]
- Li H, Wang H. Activation of xenobiotic receptors: driving into the nucleus. Expert Opin Drug Metab Toxicol. 2010;6:409–426. doi: 10.1517/17425251003598886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M, di Masi A, Trezza V, Pallottini V, Polticelli F, Ascenzi P. Xenosensors CAR and PXR at work: impact on statin metabolism. Curr Drug Metab. 2011;12:300–311. doi: 10.2174/138920011795101859. [DOI] [PubMed] [Google Scholar]
- di Masi A, De Marinis E, Ascenzi P, Marino M. Nuclear receptors CAR and PXR: molecular, functional, and biomedical aspects. Mol Aspects Med. 2009;30:297–343. doi: 10.1016/j.mam.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Monostory K, Pascussi JM, Szabó P, Temesvári M, Köhalmy K, Acimovic J, et al. Drug interaction potential of 2-((3,4-dichlorophenethyl)(propyl)amino)-1-(pyridin-3-yl)ethanol (LK-935), the novel nonstatin-type cholesterol-lowering agent. Drug Metab Dispos. 2009;37:375–385. doi: 10.1124/dmd.108.023887. [DOI] [PubMed] [Google Scholar]
- Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006;80:565–581. doi: 10.1016/j.clpt.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Nussler AK, Nussler NC, Merk V, Brulport M, Schormann W, Yao P. The holy grail of hepatocyte culturing and therapeutic use. In: Santin M, et al., editors. Strategies in Regenerative Medicine. New York: Springer; 2009. pp. 283–320. [Google Scholar]
- Omiecinski CJ, Coslo DM, Chen T, Laurenzana EM, Peffer RC. Multi-species analyses of direct activators of the constitutive androstane receptor. Toxicol Sci. 2011;123:550–562. doi: 10.1093/toxsci/kfr191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2007;82:726–733. doi: 10.1038/sj.clpt.6100220. [DOI] [PubMed] [Google Scholar]
- Pedro-Botet J, Schaefer EJ, Bakker-Arkema RG, Black DM, Stein EM, Corella D, et al. Apolipoprotein E genotype affects plasma lipid response to atorvastatin in a gender specific manner. Atherosclerosis. 2001;158:183–193. doi: 10.1016/s0021-9150(01)00410-5. [DOI] [PubMed] [Google Scholar]
- Pissios P, Tzameli I, Kushner P, Moore DD. Dynamic stabilization of nuclear receptor ligand binding domains by hormone or corepressor binding. Mol Cell. 2000;6:245–253. doi: 10.1016/s1097-2765(00)00026-5. [DOI] [PubMed] [Google Scholar]
- Poli A. Atorvastatin: pharmacological characteristics and lipid-lowering effects. Drugs. 2007;67(Suppl. 1):3–15. doi: 10.2165/00003495-200767001-00002. [DOI] [PubMed] [Google Scholar]
- Prueksaritanont T, Subramanian R, Fang X, Ma B, Qiu Y, Lin JH, et al. Glucuronidation of statins in animals and humans: a novel mechanism of statin lactonization. Drug Metab Dispos. 2002;30:505–512. doi: 10.1124/dmd.30.5.505. [DOI] [PubMed] [Google Scholar]
- Rencurel F, Stenhouse A, Hawley SA, Friedberg T, Hardie DG, Sutherland C, et al. AMP-activated protein kinase mediates phenobarbital induction of CYP2B gene expression in hepatocytes and a newly derived human hepatoma cell line. J Biol Chem. 2005;280:4367–4373. doi: 10.1074/jbc.M412711200. [DOI] [PubMed] [Google Scholar]
- Riedmaier S, Klein K, Hofmann U, Keskitalo JE, Neuvonen PJ, Schwab M, et al. UDP-glucuronosyltransferase (UGT) polymorphisms affect atorvastatin lactonization in vitro and in vivo. Clin Pharmacol Ther. 2010;87:65–73. doi: 10.1038/clpt.2009.181. [DOI] [PubMed] [Google Scholar]
- Sakaeda T, Fujino H, Komoto C, Kakumoto M, Jin JS, Iwaki K, et al. Effects of acid and lactone forms of eight HMG-CoA reductase inhibitors on CYP-mediated metabolism and MDR1-mediated transport. Pharm Res. 2006;23:506–512. doi: 10.1007/s11095-005-9371-5. [DOI] [PubMed] [Google Scholar]
- Shindo S, Numazawa S, Yoshida T. A physiological role of AMP-activated protein kinase in phenobarbital-mediated constitutive androstane receptor activation and CYP2B induction. Biochem J. 2007;401:735–741. doi: 10.1042/BJ20061238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Lee TS, Zhu M, Gu C, Wang Y, Zhu Y, et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006;114:2655–2662. doi: 10.1161/CIRCULATIONAHA.106.630194. [DOI] [PubMed] [Google Scholar]
- Synold TW, Dussault I, Forman BM. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med. 2001;7:584–590. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- Thasler WE, Weiss TS, Schillhorn K, Stoll PT, Irrgang B, Jauch KW. Charitable state-controlled foundation Human Tissue and Cell Research: ethic and legal aspects in the supply of surgically removed human tissue for research in the academic and commercial sector in Germany. Cell Tissue Bank. 2003;4:49–56. doi: 10.1023/A:1026392429112. [DOI] [PubMed] [Google Scholar]
- Weiss TS, Pahernik S, Scheruebl I, Jauch KW, Thasler WE. Cellular damage to human hepatocytes through repeated application of 5-aminolevulinic acid. J Hepatol. 2003;38:476–482. doi: 10.1016/s0168-8278(02)00454-3. [DOI] [PubMed] [Google Scholar]
- Willrich MA, Hirata MH, Hirata RD. Statin regulation of CYP3A4 and CYP3A5 expression. Pharmacogenomics. 2009;10:1017–1024. doi: 10.2217/pgs.09.42. [DOI] [PubMed] [Google Scholar]
- Wu X, Whitfield LR, Stewart BH. Atorvastatin transport in the Caco-2 cell model: contribution of P-glycoprotein and the proton-monocarboxylic acid co-transporter. Pharm Res. 2000;17:209–215. doi: 10.1023/a:1007525616017. [DOI] [PubMed] [Google Scholar]
- Yamasaki D, Nakamura T, Okamura N, Kokudai M, Inui N, Takeuchi K, et al. Effects of acid and lactone forms of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors on the induction of MDR1 expression and function in LS180 cells. Eur J Pharm Sci. 2009;37:126–132. doi: 10.1016/j.ejps.2009.01.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.