

Abstract
Oxidized low-density lipoprotein (oxLDL) and oxLDL-containing immune complexes (oxLDL-IC) contribute to the formation of lipid-laden macrophages (foam cells). Fcγ receptors mediate uptake of oxLDL-IC, whereas scavenger receptors internalize oxLDL. We have previously reported that oxLDL-IC, but not free oxLDL, activate macrophages and prolong their survival. Sphingomyelin is a major constituent of cell membranes and lipoprotein particles and acid sphingomyelinase (ASMase) hydrolyses sphingomyelin to generate the bioactive lipid ceramide. ASMase exists in two forms: lysosomal (L-ASMase) and secretory (S-ASMase). In this study we examined whether oxLDL and oxLDL-IC regulate ASMase differently, and whether ASMase mediates monocyte/macrophage activation and cytokine release. The oxLDL-IC, but not oxLDL, induced early and consistent release of catalytically active S-ASMase. The oxLDL-IC also consistently stimulated L-ASMase activity, whereas oxLDL induced a rapid transient increase in L-ASMase activity before it steadily declined below baseline. Prolonged exposure to oxLDL increased L-ASMase activity; however, activity remained significantly lower than that induced by oxLDL-IC. Further studies were aimed at defining the function of the activated ASMase. In response to oxLDL-IC, heat-shock protein 70B’ (HSP70B’) was up-regulated and localized with redistributed ASMase in the endosomal compartment outside the lysosome. Treatment with oxLDL-IC induced the formation and release of HSP70-containing and IL-1β-containing exosomes via an ASMase-dependent mechanism. Taken together, the results suggest that oxLDL and oxLDL-IC differentially regulate ASMase activity, and the pro-inflammatory responses to oxLDL-IC are mediated by prolonged activation of ASMase. These findings may contribute to increased understanding of mechanisms mediating macrophage involvement in atherosclerosis.
Keywords: acid sphingomyelinase, HSP70, interleukin-1, oxidized low-density lipoprotein immune complexes
Introduction
Oxidatively modified low-density lipoprotein (oxLDL) plays an important role in the development of atherosclerosis. Macrophages internalize oxLDL through several membrane scavenger receptors,1,2 and once internalized, oxLDL is transported to lysosomes for processing and degradation.3,4 In addition, oxLDL is immunogenic and induces the generation of auto-antibodies that form circulating immune complexes (oxLDL-IC).5,6 The uptake of oxLDL-IC by macrophages is mediated primarily through FcγR1 on the cell surface and results in the release of pro-inflammatory cytokines, such as interleukin-1 β (IL-1β) and tumour necrosis factor, and the formation of lipid-laden foam cells.7–11 We have previously shown that oxLDL-IC, as opposed to oxLDL, up-regulate the genes involved in both the inflammatory response and in cell survival.12 Whereas oxLDL can be toxic to monocytes, it has been shown that the engagement of FcγR1 by oxLDL-IC promotes macrophage survival through a sphingosine 1-phosphate-mediated pathway and through Akt phosphorylation.12,13 We have also shown that cross-linking of the FcγR1 alters how macrophages process internalized oxLDL.4 The lipid moiety from internalized oxLDL-IC is ‘trapped’ in the endosomal compartment, whereas the apolipoprotein moiety is directed to the lysosomal compartment.4 This difference in trafficking may determine macrophage activation and survival in response to oxLDL.
Sphingomyelin (SM) is an abundant cellular sphingolipid that has a tendency to self-associate into cell membrane rafts and can account for up to 25% of the total phospholipids in LDL particles.14,15 Acid sphingomyelinase (ASMase), located in the luminal leaflet of endosomes, lysosomes and phagosomes, can hydrolyse SM to form ceramide.16,17 The ASMase has also been reported at the outer leaflet of the plasma membrane upon the activation of certain transmembrane receptors, and has been implicated in the hydrolysis of SM in the plasma membrane.18,19 It is believed that the generation of ceramide at the cell surface might promote enhanced receptor cross-linking and increased initiation of receptor-mediated signal transduction.20,21
The functionality of ASMase is critical for the macrophage response against both bacterial infections and in the processing and trafficking of internalized lipids.22–26 Defects in lysosomal ASMase (L-ASMase) can lead to the development of Niemann–Pick disease, a lysosomal storage disorder characterized by the accumulation of SM.27,28 A secreted form of ASMase (S-ASMase) has also been described29 and is generated through differential processing and trafficking of the same precursor as L-ASMase.30,31 It has been reported that S-ASMase in vitro hydrolyses SM in LDL particles resulting in their aggregation into larger units.32,33 In addition, increased S-ASMase activity has been reported in the arterial intima and correlated with atherosclerotic plaque development.33 Interestingly, S-ASMase activity was shown to be higher with oxLDL than native LDL particles, suggesting that the oxidation of lipids favours SM hydrolysis.31 It has also been suggested that arterial wall factors such as collagen and lipases may enhance ceramide-mediated aggregation of LDL.31 Moreover, LDL receptor/ASMase double knockout mice (ldlr−/−asm−/−) exhibit reduced arterial lipoprotein retention and reduced development of the atheromata.34 However, little is known about the role of macrophage-derived ASMase isoforms in the functionality of lipoprotein-stimulated macrophages.
We now describe differential activation profiles of both L-ASMase and S-ASMase in response to oxLDL and oxLDL-IC in U937 monocytic cells and in monocytes isolated from ASMase knockout (KO) mice. We also show that the uptake of oxLDL-IC promotes the redistribution of intracellular ASMase and its association with HSP70B’ in the endosomal compartment outside the lysosomes. We further demonstrate that prolonged activity of ASMase could be responsible for macrophage IL-1β release in response to oxLDL-IC through the generation of exosomes. We propose a potential novel role of macrophage-derived ASMase in the development of atherosclerosis under conditions of inflammation and immune complex formation.
Materials and methods
Cells
Adherent mouse macrophage-like RAW 264.7 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and grown in RPMI-1640 (Gibco, Grand Island, NY) supplemented with 100 U/ml penicillin and 50 μg/ml streptomycin, and 10% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA). U937 cells were obtained from ATCC and were grown in Iscove's modified Dulbecco's medium (Gibco) supplemented with 100 U/ml penicillin and 50 μg/ml streptomycin, and 10% FBS. Mouse monocytes were obtained from ASMase−/− and ASMase+/+ C57BL/6 mice. Animals were maintained under standard laboratory conditions. All animal procedures were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee and followed the guidelines of the American Veterinary Medical Association. Mouse peripheral blood was collected via cardiac puncture and monocytes were purified using a two-step negative selection method as described by Swirski et al.35 Monocyte purity was verified using flow cytometry to be at least 90%. Unless otherwise stated, cells were seeded at 106/ml before priming with 200 ng/ml interferon-γ (IFN-γ) and cultured overnight in Iscove's modified Dulbecco's medium containing 1% FBS. Cells were treated with either Dulbecco's phosphate-buffered saline (DPBS) vehicle, 75 μg/ml oxLDL, or 100 μg/ml oxLDL-IC or keyhole limpet haemocyanin immune complexes (KLH-IC) for various times. The concentrations of oxLDL and oxLDL-IC were selected to account for the increased protein content of immune complexes, and therefore contain similar levels of oxLDL. The KLH-IC was used as a control for immune complexes because KLH has a molecular weight comparable to LDL and is able to engage Fcγ receptors similar to oxLDL-IC without containing lipoproteins. Exogenously added bacterial sphingomyelinase (bSMase) (Sigma, Saint Louis, MO) was used at 200 mU/ml. Desipramine (Sigma) was used at 20 μm and added 2 hr before stimulation to inhibit ASMase activity.
Lipoprotein isolation and oxidation
The LDL (d = 1·019–1·063 g/ml) was isolated from the plasma of donors who were free from clinically apparent disease, and oxidatively modified using Cu2+ as described previously.4,12,36 The oxidative modification of LDL was evaluated by quantification of conjugated dienes as previously described.37
Preparation of immune complexes
Immune complexes containing oxLDL were prepared with human oxLDL and human anti-oxLDL antibodies as described previously.5,12 After precipitation, immune complexes were re-suspended in DPBS and the concentrations of total protein were determined using the bicinchoninic acid (BCA) protein assay (Pierce). KLH-IC was prepared as described previously.10,12
Labelling of oxLDL with lipophilic fluorescent dyes
Fluorescent labelling of the lipid moiety of oxLDL or oxLDL-IC with 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) or 1,1′-dioctadecyl-3,3,3′,3-tetramethy lindocarbocyanineperchlorate (DiI) (Invitrogen, Carlsbad, CA) was performed as described previously38 with modifications as previously detailed.4
Transfection of RAW 264.7 cells
We have previously shown that U937 and RAW 264.7 cell lines respond similarly with regard to internalization of fluorescently labelled oxLDL and oxLDL-IC.4,11 We used RAW 264.7 cells for ASMase-dsRed expression experiments because they are more amenable to plasmid transfection. Construction and characterization of the plasmid encoding the green fluorescent protein-tagged HSP70B’ (HSP70B’-GFP) protein has been previously described,11 as has the plasmid encoding dsRed-tagged ASMase protein (ASMase-dsRed).39 Cells were transfected with 1 μg/ml of each plasmid using FuGENE HD Transfection reagent (Promega, San Luis Obispo, CA) according to the manufacturer's instructions. Cells were cultured for 24 hr before replacing the culture medium with serum-free RPMI.
Confocal microscopy
After priming with IFN-γ overnight, cells were re-suspended in fresh medium containing IFN-γ and FBS 1 hr. Cells were treated with either DiO-labelled oxLDL (DiO-oxLDL; 75 μg/ml) or DiO-oxLDL-IC (100 μg/ml) for 5 hr before treatment with human plasma for 30 min at 4° to block Fc receptors. Cells were fixed in 4% formaldehyde and non-specific binding was blocked by incubating the cells for 1 hr with 10% human plasma and 10% BSA. The cells were then incubated with anti-ASMase antibody (rabbit polyclonal antibody #1599, 1 : 50 dilution volume/volume, kindly provided by Dr Richard Kolesnick, Memorial Sloan-Kettering Cancer Center, New York, NY) in DPBS containing 10% human serum and 10% BSA. Alexa-Fluor 488-conjugated goat anti-mouse F(ab’)2 antibody (Invitrogen) was used as a secondary antibody. Cells were suspended in 100 μl DPBS and 10-μl aliquots were loaded into 10-μl glass capillaries (Idaho Technology, Salt Lake City, UT). The capillaries were sealed and cells were visualized using confocal microscopy (Zeiss LSM 510 Meta Laser Scanning Confocal Microscope, Thornwood, NY). Adherent RAW 264.7 cells were grown on Lab-Tek II 8 well chamber slides (Nalge Nunc International, Vernon Hill, IL). After treatment, the cells were fixed in 4% paraformaldehyde before mounting with Mowiol (Sigma). Lysosomal staining was performed in saponin-permeabilized cells using antibody against lysosomal-associated membrane protein 1 (LAMP-1) conjugated to Alexa-Fluor 647 (Santa Cruz Biotechnology, Santa Cruz, CA). Alternatively, lysosomes were visualized by incubating the cells with LysoTracker Green or Blue (Invitrogen) for 30 min before live imaging.
Small interfering RNA disruption of ASMase
U937 cells were transfected with non-targeting or ASMase gene40 small interfering RNA (siRNA) using the Nucleofector™ device (Amaxa Inc., Walkersville, MD) according to the manufacturer's instructions. Knockdown was verified using quantitative PCR analysis. Twenty-four hours after transfection, cells were primed with IFN-γ and incubated with treatments as described above. The non-specific ASMase inhibitor desipramine was also used to reduce ASMase expression and displayed similar ASMase inhibition when compared with ASMase siRNA (data not shown).
Assay of ASMase activity
Activity of ASMase was determined using a minor modification of the method of Jenkins et al.39 For L-ASMase activity, cells were pelleted and re-suspended in L-ASMase lysis buffer (0·2% Triton X-100, 50 mm Tris–HCl, pH 7·4, 1 mm EDTA, with phosphatase and protease inhibitors). Lysates were sonicated (one or two pulses, 10 s), and cellular debris and unbroken cells were pelleted by centrifugation at 1000 g for 5 min at 4°. After determination of protein concentration (BCA assay, Pierce), 100 μg protein (in a total volume of 100 μl) was added to 100 μl reaction mixture containing 100 μm porcine brain SM, 1 × 105 counts/min of choline-[methyl-14C] in micelles containing 0·2% Triton X-100 in sodium acetate buffer (250 mm, pH 5·0) with 1 mm EDTA. S-ASMase activity was performed with 100 μl conditioned medium and assayed with a similar reaction buffer, differing only in the substitution of 0·1 mm ZnCl2 for 1 mm EDTA. The S-ASMase activity (nmol/ml/hr) was normalized to total cellular protein to correct for variations in cell number (nmol/ml of medium/mg of cellular protein/hr). For both S-ASMase and L-ASMase assays, the reaction was run for 60 min at 37°. The reaction was terminated by the addition of 800 μl chloroform/methanol (2 : 1, volume/volume) followed by the addition of 0·2 ml Milli-Q water. After mixing and centrifugation at 2000 g for 5 min, the upper (aqueous) phase was removed and used for liquid scintillation counting.
Isolation of immune complexes after incubation with U937 cells
U937 cells (2 × 106 cells/ml) were treated with DPBS, oxLDL, oxLDL-IC or KLH-IC for 15 min, 30 min or 5 hr. Cells were then washed with ice-cold DPBS and incubated with magnetically labelled Protein G (Dynal, Grand Island, NY) pre-conjugated to rabbit anti-oxLDL antibody for 30 min at 4°. The beads were then recovered using a magnet. After three washes the beads were suspended in elution buffer (Dynal) containing protease inhibitors (Roche, Indianapolis, IL). The suspension was sonicated twice on ice, 10 s each time and then the ASMase activity assay was performed. In these experiments oxLDL-IC formed with rabbit anti-oxLDL were also used,41 because the affinity of antibodies is much greater when antibodies from immunized rabbits are used than when we use purified spontaneously formed human antibodies.36
Isolation of exosomes from conditioned media
Exosomes were isolated according to the previously published protocol with a slight modification.42 Briefly, 2 × 107 cells were incubated with oxLDL or oxLDL-IC in RPMI-1640 containing 1% FBS that had been previously spun at 100 000 g overnight to remove bovine exosomes. After separation of cells, conditioned media were centrifuged using Beckman SW41 rotor at 10 000 g for 30 min to remove high-density debris, then at 100 000 g for 4 hr to pellet exosomes. Purification of exosomes using a discontinuous sucrose gradient was performed as previously described.43 The exosome-containing layer was washed by re-suspension in PBS then centrifugation at 100 000 g for 1 hr. The washed pellets were re-suspended in radioimmunoprecipitation assay buffer containing 0·5% Triton X-100 in preparation for Western blotting. Samples were run in NuPAGE 4-12% Bis-Tris Gels (Invitrogen) and probed using antibodies against ASMase (Abcam, Cambridge, MA), HSP70, HSP70B’ (Pharmingen, Chicago, IL), and IL-1β (Santa Cruz). Proteins were visualized using enhanced chemiluminescence (Pierce).
Quantification of IL-1β secretion
The extracellular concentration of IL-1β from treated human U937 cells was measured using the human IL-1β ELISA kit (Abcam) and from mouse monocytes using the mouse IL-1β ELISA (Invitrogen) kit following the manufacturer's instructions.
Statistical analysis
Significant differences between two groups were evaluated by Student's t-test and one way analysis of variance followed by Tukey's post hoc test for mean separation (P < 0·05). All data are expressed as mean ± SE.
Results
oxLDL-IC but not oxLDL induce ASMase release
To assess whether oxLDL or oxLDL-IC could activate and cause translocation of ASMase, U937 cells were incubated with DiO-labelled oxLDL or DiO-labelled oxLDL-IC for either 30 min or 5 hr, and ASMase was detected using fluorescent labelling. At 30 min, oxLDL could already be detected on the cell surface, and after 5 hr, oxLDL was internalized (Fig. 1a). However, ASMase was not translocated in response to oxLDL. In contrast, at 30 min, the extracellular insoluble oxLDL-IC had not enough time to fully engage the Fcγ receptors on the cell surface (Fig. 1b). Intriguingly, ASMase was detected on the exterior of the cell co-localizing with oxLDL-IC (Fig. 1b), whereas intracellular ASMase in oxLDL-IC-treated cells was decreased compared with oxLDL-treated cells. As cells were washed before the addition of oxLDL-IC, the presence of co-localizing S-ASMase is probably a result of de novo secretion. At 5 hr post-treatment, oxLDL-IC were evenly distributed on the cell surface in organized macrodomains, possibly cross-linked to Fcγ receptors, and continued to be co-localized with ASMase. Expression patterns of ASMase in oxLDL-treated (Fig. 1a) and non-stimulated cells after 5 hr (Fig. 1c) did not exhibit any significant differences.
Figure 1.
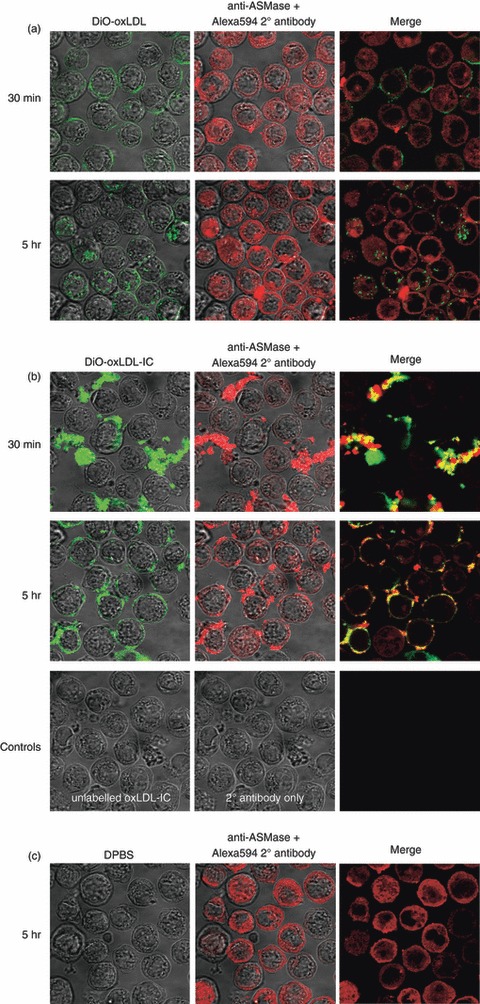
Oxidized low-density lipoprotein-containing immune complexes (oxLDL-IC) but not oxLDL induce acid sphingomyelinase (ASMase) release. After priming with interferon-γ (IFN-γ) overnight, U937 cells were re-suspended in fresh medium containing IFN-γ and 1% fetal bovine serum for 1 hr before treatment with either 3,3′-dioctadecyloxacarbocyanine perchlorate-labelled (DiO-) oxLDL (75 μg/ml) (a), DiO-oxLDL-IC (100 μg/ml) (b), or Dulbecco's PBS-treated for 5 hr (c) then fixed at 30 min or 5 hr post-treatment; ASMase was visualized using anti-ASMase antibody and secondary Alexa-Fluor 594-labelled F(ab)’2 anti-rabbit antibody. Cells were treated with unlabelled oxLDL-IC, and secondary antibody only as a control (lower panel). Yellow denotes areas of ASMase and oxLDL-IC co-localization. Results are representative of two independent experiments.
ASMase activity is regulated in a time- and dose-dependent manner
The activity of the extracellular oxLDL-IC-associated S-ASMase was then measured to determine whether the released S-ASMase was catalytically active. At 30 min post-incubation an increase in S-ASMase activity of 50% and 100% was detected in response to oxLDL-IC and rabbit oxLDL-IC, respectively, compared with corresponding baseline controls (Fig. 2a). These results suggest that cross-linking of the Fcγ receptors can induce the release of activated S-ASMase, which specifically binds to the oxLDL-IC. It has been previously shown that LDL modified by oxidation is hydrolysed by S-ASMase forming aggregates at pH 7·4.32 Extracellular aggregated oxLDL has not been found in our studies, indicating that free oxLDL did not stimulate S-ASMase release (Fig. 1a).
Figure 2.
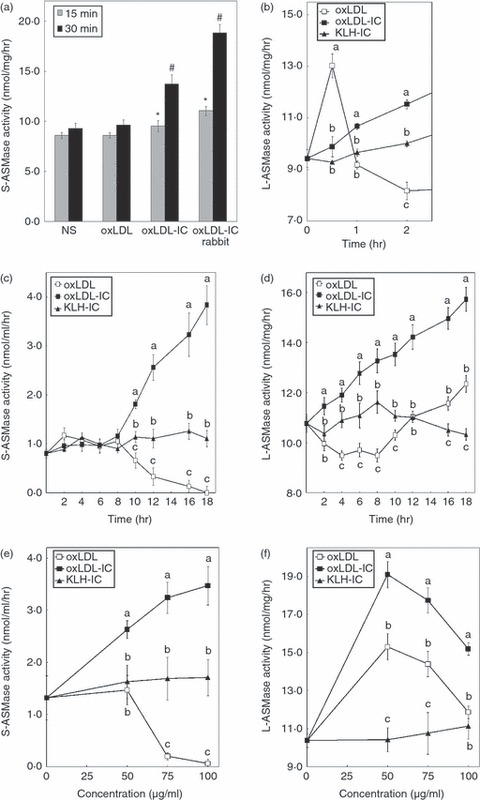
Acid sphingomyelinase (ASMase) activity is regulated in a time- and dose-dependent manner. U937 cells were incubated with oxidized low-density lipoprotein (oxLDL; 75 μg/ml), oxLDL-containing immune complexes (oxLDL-IC), or keyhole limpet haemocyanin-containing immune complexes (KLH-IC; 100 μg/ml) and ASMase activity was measured in conditioned media [secretory (S) –ASMase] (a, c) or in cell lysates [lysosomal (L) -ASMase] (b, d). (a) Extracellular S-ASMase activity associated with oxLDL and oxLDL-IC was measured after immunoprecipitation of oxLDL moiety using magnetically labelled Protein G conjugated to anti-oxLDL antibody. (b) A short time–course of L-ASMase activity was measured in cell lysates. ASMase activity was measured in the conditioned media (S-ASMase) (c) or in cell lysates (L-ASMase) (d) from 0 to 18 hr. Different letters denote significant differences among means at each time-point (P < 0·05). S-ASMase (e) and L-ASMase (f) activity was measured after 18 hr-treatment of U937 cells incubated with various doses of oxLDL, oxLDL-IC and KLH-IC. Different letters denote significant difference among means at each concentration (P < 0·05). Data shown are mean ± SE from duplicate samples from two independent experiments (n = 4).
Early time-points were examined to determine whether there were any transient increases in L-ASMase activity. Interestingly, after 30 min incubation with oxLDL, but not oxLDL-IC or KLH-IC, L-ASMase activity increased to 1·4-fold compared with baseline levels, then dropped to baseline levels by 1 hr before it decreased further (Fig. 2b). To further characterize the kinetics of S-ASMase and L-ASMase activation by oxLDL-IC, U937 cells were incubated with oxLDL, oxLDL-IC or KLH-IC for various times. Treatment with oxLDL (75 μg/ml) gradually reduced S-ASMase activity after 8 hr of incubation, and almost completely abolished activity by 18 hr (Fig. 2c). In contrast, oxLDL-IC treatment (with an equal amount of oxLDL in the immune complex) gradually increased S-ASMase activity after 8 hr of incubation, reaching fourfold increase at 18 hr compared with base line (Fig. 2c). The control KLH-IC-treated cells did not demonstrate any change in S-ASMase activity.
Incubation with oxLDL significantly reduced L-ASMase activity below baseline levels for the first 8 hr, but started to rise above baseline constantly for the duration of the experiment (Fig. 2d). In contrast, oxLDL-IC treatment steadily and significantly increased L-ASMase activity during the entire time–course, reaching a 1·5-fold increase at 18 hr (Fig. 2d). Interestingly, in cells treated with KLH-IC control immune complexes there was a gradual increase in L-ASMase activity from basal levels for up to 8 hr, but this then dropped back to around basal levels by 18 hr (Fig. 2d). The L-ASMase activity in response to oxLDL-IC was significantly higher than KLH-IC at each time after engagement with cell surface.
To determine the dose effect of oxLDL, oxLDL-IC and KLH-IC on S-ASMase and L-ASMase activity, U937 cell were incubated with different concentrations of the treatments for 18 hr. Treatment with lower doses of oxLDL (50 μg/ml) did not affect S-ASMase activity; however, higher doses of oxLDL (above 50 μg/ml) were inhibitory to S-ASMase activity (Fig. 2e). In contrast, increasing concentrations of oxLDL-IC increased S-ASMase activity (Fig. 2e). Intriguingly, L-ASMase activity was significantly elevated in response to both oxLDL and oxLDL-IC at low doses (50 μg/ml) (Fig. 2f); however, concentrations above 50 μg/ml of both oxLDL and oxLDL-IC were inhibitory to L-ASMase activity (Fig. 2f). Increasing concentrations of KLH-IC did not significantly affect either S-ASMase or L-ASMase activity. Studies on ASMase activity in the conditioned media have shown that the majority is Zn2+-dependent, indicating that S-ASMase is primarily the secreted form (see supplementary material, Fig. S1).
We then evaluated levels of ASMase protein using Western blot analysis in both U937 and RAW 264.7 cells. There were no significant changes detected in protein levels at time-points up to 24 hr in U937 cells (see supplementary material, Fig. S2A) or up to 5 hr in RAW 264.7 cells (see supplementary material, Fig. S2B). Therefore, we conclude that the increase in ASMase activity could be mainly the result of a direct effect on the enzyme function.
ASMase co-localizes with the lipid moiety of oxLDL-IC but not with oxLDL
To determine whether ASMase is involved with the uptake of oxLDL and oxLDL-IC, RAW 264.7 cells were transfected with ASMase-dsRed and incubated with either DiO-oxLDL or DiO-oxLDL-IC for 3 hr. As shown in Fig. 3(a) (top panel) ASMase-dsRed co-localized only slightly with internalized oxLDL, whereas it completely co-localized with internalized oxLDL-IC (Fig. 3a, bottom panel). We have previously reported that both the protein and lipid moieties of free oxLDL are internalized into lysosomes whereas the lipid moiety oxLDL-IC is ‘trapped’ in endosomes outside the lysosomal compartment.4 The data in Fig. 3(a) suggest that oxLDL-IC-associated L-ASMase could be in the endosomal compartment, whereas in oxLDL-treated cells ASMase remained lysosomal. Hence, the uptake of oxLDL and oxLDL-IC may have induced differential redistribution of L-ASMase. Additionally, ASMase-dsRed was detected associated with DiO-oxLDL-IC around the periphery of the phagosome (Fig. 3b). A video showing ASMase-dsRed associated with the uptake of DiO-oxLDL-IC is presented in Video S1 (see supplementary material). These data suggest that phagocytosis of oxLDL-IC may require the presence of L-ASMase.
Figure 3.
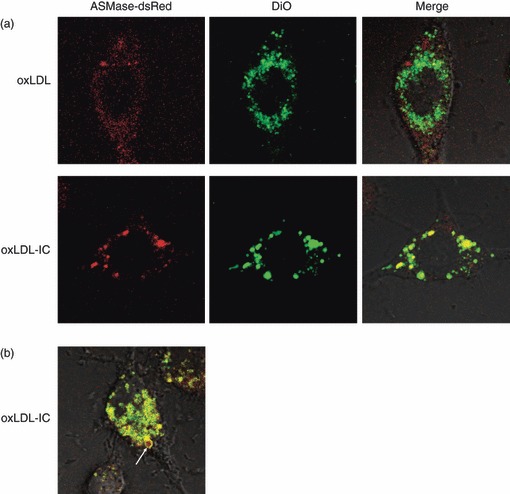
Acid sphingomyelinase (ASMase) co-localizes with the lipid moiety oxidized low-density lipoprotein-containing immune complexes (oxLDL-IC) but not with oxLDL. (a) RAW 264·7 cells were transfected with ASMase-dsRed and treated with either 3,3′-dioctadecyloxacarbocyanine perchlorate-labelled (DiO-) oxLDL (top panel) or DiO-oxLDL-IC (bottom panel) (24 and 32 μg/ml, respectively) for 4 hr before live visualization by confocal microscopy. (b) ASMase is involved with the phagocytosis of DiO-oxLDL-IC. Arrow denotes an area of ASMase-dsRed and DiO-oxLDL-IC co-localization (yellow) in a phagosome.
Exogenous sphingomyelinase enhances the uptake of oxLDL-IC
To ascertain the role of the early S-ASMase secretion observed with oxLDL-IC treatment, cells were incubated with DiI-oxLDL or DiI-oxLDL-IC and LysoTracker Green for 5 hr in the presence and absence of bSMase. Eight to ten microscope fields containing approximately 20 cells each were examined per treatment. Results show that bSMase had no apparent effect on the magnitude of uptake and trafficking of oxLDL to lysosomes (Fig. 4a); however, bSMase treatment increased oxLDL-IC internalization (Fig. 4a). These data suggest that exogenous SMase activity enhances the uptake of oxLDL-IC. Furthermore, the presence of bSMase at the concentrations used in our study did not affect cell proliferation or cell survival of U937 cells (see supplementary material, Fig. S3).
Figure 4.
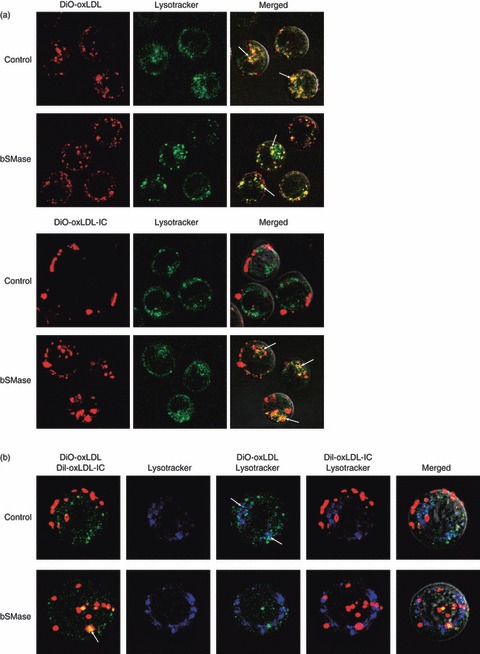
Exogenous sphingomyelinase enhances the uptake of oxidized low-density lipoprotein-containing immune complexes (oxLDL-IC). U937 cells were incubated with and without 200 mU bacterial sphingomyelinase (bSMase) and with (a) 1,1′-dioctadecyl-3,3,3′,3-tetramethy lindocarbocyanineperchlorate-labelled (DiI-) oxLDL (24 μg/ml, top panels) or DiI-oxLDL-IC (32 μg/ml); or (b) with both 3,3′-dioctadecyloxacarbocyanine perchlorate-labelled (DiO-) oxLDL and DiI-oxLDL-IC simultaneously for 5 hr. Thirty minutes before live visualization by confocal microscopy, the cells were treated with LysoTracker Green (a) or LysoTracker Blue (b). Arrows in (a) denote co-localization (yellow) of DiI-oxLDL (red) with LysoTracker Green. Arrows in (b) top panel denote co-localization (turquoise blue) of DiO-oxLDL (green) with LysoTracker Blue; arrow in lower panel denotes co-localization (yellow) of DiO-oxLDL (green) and DiI-oxLDL-IC (red).
We have previously shown that simultaneous addition of oxLDL and oxLDL-IC resulted in their separate intracellular localization, with the lipid moiety of oxLDL located in the lysosomes and that of oxLDL-IC in the endosomes.4 However, when oxLDL-IC was added 2 hr before the addition of oxLDL, the lipid moiety of both was internalized to the endosomal compartment.4 To determine if SMase activity could influence the intracellular trafficking of oxLDL when introduced simultaneously with oxLDL-IC, bSMase was added along with the simultaneous addition of DiO-oxLDL and DiI-oxLDL-IC (Fig. 4b). The yellow colour in the cells shown in the presence of bSMase indicates that oxLDL and oxLDL-IC were internalized into the same non-lysosomal compartment (Fig. 4b). When DiO-oxLDL was mixed with DiO-oxLDL-IC in a cell-free system in the presence of either bSMase or supernatant from 18 hr-oxLDL-IC-treated U937 co-aggregation of the lipid moieties was observed (see supplementary material, Fig. S4). Therefore, the intracellular co-localization of the lipid moieties of oxLDL and oxLDL-IC shown in Fig. 4(b) is possibly the result of internalization of the co-aggregates via the Fc receptors.
ASMase co-localizes with HSP70B’ in the endosomal compartment outside lysosomes
It has been previously reported that HSP70 can stabilize lysosomes by binding to an endolysosomal anionic phospholipid bis(monoacylglycero)phosphate, an essential co-factor for ASMase activity.44 We have previously shown that the expression of the HSP70 family member HSP70B’ is up-regulated in response to oxLDL-IC.11 We also have shown that the lipid moiety from internalized oxLDL-IC is ‘trapped’ in the endosomal compartment, whereas the apolipoprotein moiety is directed to the lysosomal compartment.4 In the current study we examined whether ASMase is associated with HSP70B’. We co-transfected RAW 264.7 cells with both ASMase-dsRed and HSP70B’-GFP, then added either oxLDL or oxLDL-IC for 5 hr. There was significant co-localization of both ASMase and HSP70B’ in oxLDL-IC-treated cells (Fig. 5, bottom panel). Interestingly, ASMase did not co-localize with LAMP-1 in cells treated with oxLDL-IC (Fig. 5, bottom panel). This suggests that oxLDL-IC up-regulates HSP70B’ expression and causes the redistribution of ASMase outside the lysosome. On the other hand, ASMase co-localized with the lysosomal marker LAMP-1 in both the control and oxLDL-treated cells (Fig. 5, top and middle panel).
Figure 5.
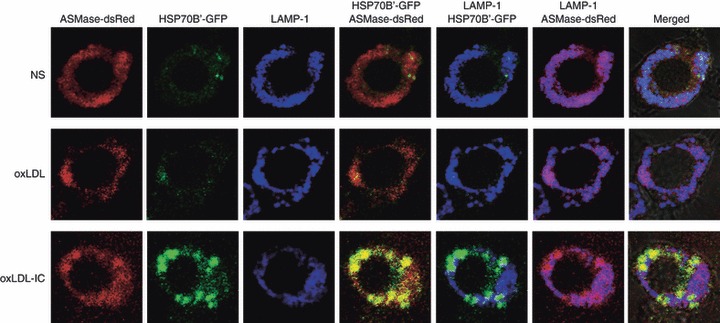
Acid sphingomyelinase (ASMase) co-localizes with heat-shock protein 70B’ (HSP70B’) in the endosomal compartment outside lysosomes. RAW 264·7 cells were co-transfected with both HSP70B’-GFP and ASMase-dsRed before treatment with either unlabelled oxidized low-density lipoprotein (oxLDL; 75 μg/ml) or oxLDL-containing immune complexes (oxLDL-IC; 100 μg/ml) for 5 hr. The cells were fixed in 4% paraformaldehyde and permeabilized using 0·25% saponin and 10% fetal bovine serum in PBS. Lysosomes were probed with anti-LAMP-1 conjugated to Alexa-fluor 647 (blue). Yellow, co-localization of HSP70B’-GFP with ASMase-dsRed; purple, co-localization of ASMase-dsRed with LAMP-1; NS, non-stimulated.
Disruption of L-ASMase inhibits internalization of extracellular oxLDL-IC aggregated with HSP70B’ and ASMase
To determine whether L-ASMase could affect HSP70B’ localization, we transfected RAW 264.7 cells with both ASMase-dsRed and HSP70B’-GFP, then incubated the cells with oxLDL-IC in the presence and absence of 20 μm desipramine and visualized live cells. Treatment of the doubly transfected cells with oxLDL-IC induced HSP70B’-GFP expression and intracellular co-localization with ASMase-dsRed (Fig. 6a), as shown also in Fig. 5 using fixed cells (the effects of oxLDL-IC on total expression of HSP70B’-GFP could be the result of activation of the cytomegalovirus early immediate promoter in stimulated RAW 264.7 cells).11,45 Treatment with tricyclic antidepressants including desipramine has been shown to accumulate in the lysosomes, where it uncouples ASMase, which is subsequently degraded. S-ASMase, by virtue of its different processing in the Golgi, never enters the lysosome and therefore is secreted unaffected by these drugs. Interestingly, desipramine treatment resulted in significant co-localization of ASMase with HSP70B’ extracellularly associated with oxLDL-IC aggregates (Fig. 6b). However, the ASMase–HSP70B’ complexes could not be re-internalized because of the dysfunctional lysosomes that were devoid of ASMase activity.
Figure 6.
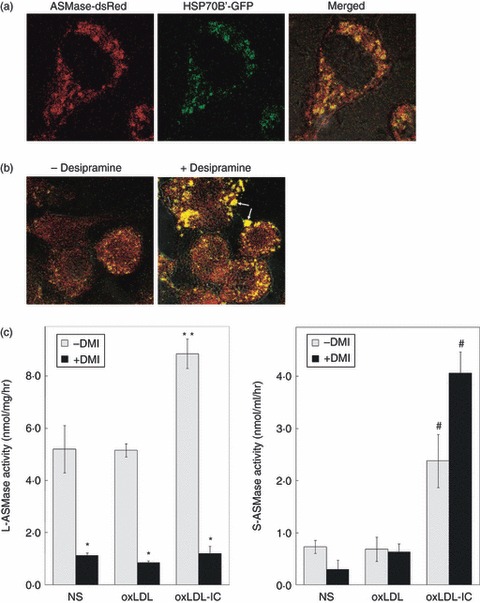
Disruption of lysosomal acid sphingomyelinase (L-ASMase) inhibits internalization of extracellular oxidized low-density lipoprotein-containing immune complexes (oxLDL-IC) aggregated with heat-shock protein 70B’ (HSP70B’) and ASMase. RAW 264.7 cells were transfected with both ASMase-dsRed and HSP70B’-GFP and treated with oxLDL-IC (100 μg/ml) (a) in the presence and absence of desipramine for 4 hr (b). Pretreatment with desipramine (20 μm) was for 2 hr. Cells were visualized live by confocal microscopy. Yellow, co-localization of HSP70B’-GFP with ASMase-dsRed; arrows denote co-localization of HSP70B’-GFP with ASMase-DsRed extracellularly associated with oxLDL-IC aggregates. (c) ASMase activity was measured after 5 hr of incubation with oxLDL (75 μg/ml) or oxLDL-IC (100 μg/ml) in cell lysates (left panel, L-ASMase) or conditioned medium (right panel, S-ASMase). * denotes significant difference between desipramine and no-desipramine treatments (P < 0·05). ** denotes significant difference between oxLDL-IC and each of oxLDL and non-stimulated (NS) treatments. # denotes significant difference between oxLDL-IC and each of the corresponding oxLDL and NS treatments; there was no significant difference in S-ASMase activity in response to desipramine treatment. Data shown are mean ± SE from duplicate samples from two independent experiments (n = 4).
To determine whether the S-ASMase complexed with HSP70B’ was active, RAW 264.7 cells were treated for 2 hr with desipramine before stimulation with oxLDL or oxLDL-IC. The data show that Zn2+-independent ASMase (L-ASMase) activity was higher in oxLDL-IC-stimulated cells than in either oxLDL-stimulated or non-stimulated cells, and desipramine robustly inhibited L-ASMase activity (Fig. 6c, left panel). Zn2+-dependent ASMase (S-ASMase) activity was also higher in oxLDL-IC-stimulated cells compared with oxLDL-stimulated or non-stimulated cells, but was not inhibited by desipramine (Fig. 6c, right panel). These results suggest that in response to stimulation with oxLDL-IC, S-ASMase is secreted along with HSP70B’, and that L-ASMase activity is required for the uptake of S-ASMase/HSP70B’/oxLDL-IC complexes.
Treatment with oxLDL-IC induces the formation of exosomes
Extracellular HSP70B’ and S-ASMase were closely associated, so we investigated whether the two proteins may have also been part of exosomes secreted in the media. Exosomes are approximately 100-nm diameter membrane-bound vesicles implicated in a wide variety of biological processes from cell-to-cell communication to cytokine release.46,47 U937 cells were treated with either oxLDL or oxLDL-IC in the presence and absence of desipramine. Western blot analysis of isolated exosomes showed that they contained ASMase and the known exosome marker, HSP70, but not HSP70B’ (Fig. 7). Interestingly, oxLDL-IC produced more exosomes that contained higher levels of ASMase protein when compared with oxLDL. In addition, desipramine treatment abolished the formation of exosomes, so indicating that ASMase activity is required for the formation of oxLDL-IC-induced exosomes.
Figure 7.
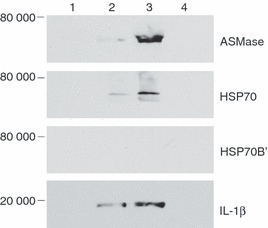
Treatment with oxidized low-density lipoprotein-containing immune complexes (oxLDL-IC) induces the formation of exosomes. U937 cells were either not stimulated (lane 1), or were treated for 5 hr with oxLDL (75 μg/ml) (lane 2), oxLDL-IC (100 μg/ml) (lane 3), or oxLDL-IC (100 μg/ml) in the presence of desipramine (20 μm) (lane 4). Exosomes were isolated from conditioned media, purified using a discontinuous sucrose gradient, and prepared for Western blotting as described in Materials and Methods. Data shown are representative of three independent experiments.
ASMase activity is required for IL-1β secretion in exosomes
We have previously reported that oxLDL-IC-treatment of U937 cells promotes the release of IL-1β.11,12 In this study we examined whether ASMase mediates IL-1β release and whether IL-1β is secreted in the ASMase-containing exosomes. Release of IL-1β in response to oxLDL-IC was consistently higher than in response to oxLDL at time-points measured starting 2 hr after stimulation (Fig. 8a), and importantly was inhibited with desipramine. Using quantitative reverse transcription-PCR we detected constitutive expression of IL-1β mRNA in U937 cells (data not shown). U937 cells transfected with ASMase siRNA exhibited significantly reduced IL-1β levels in response to both oxLDL and oxLDL-IC treatments (Fig. 8b). Knockdown of ASMase protein expression was verified by Western blotting (see supplementary material, Fig. S5).
Figure 8.
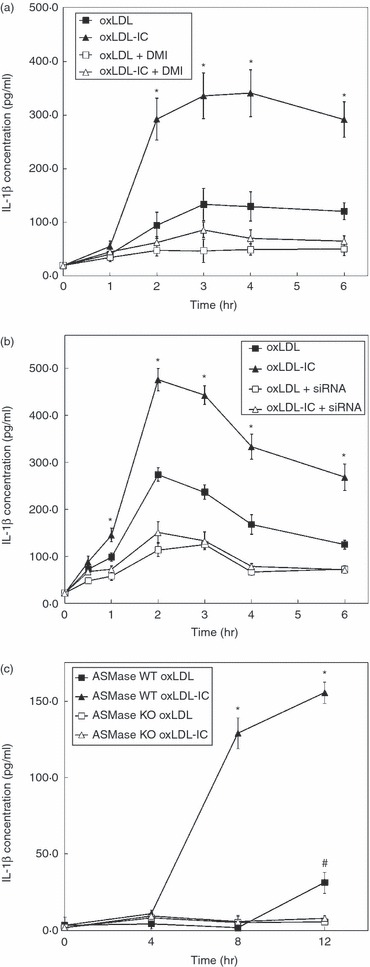
Acid sphingomyelinase (ASMase) activity is required for interleukin-1β (IL-1β) secretion in exsosomes. (a) U937 cells were transfected with ASMase small interfering (si) RNA and treated with oxidized low-density lipoprotein (oxLDL; 75 μg/ml) or oxLDL-immune complexes (oxLDL-IC; 100 μg/ml) and then conditioned media were collected and analysed by ELISA for IL-1β at various time-points. *denotes significant difference between oxLDL-treated cells and oxLDL-IC-treated cells within the control-transfected group at each time-point (P < 0·01). Both oxLDL and oxLDL-IC treatments exhibit significant differences between control and ASMase siRNA-transfected cells (P < 0·01). (b), Monocytes were isolated from ASMase knockout and wild-type mice and treated as mentioned above for U937 cells. *denotes significant difference between oxLDL-treated and oxLDL-IC-treated cells within the wild-type group at each time-point (P < 0·01). #denotes significant difference between ASMase-expressing monocytes and monocytes lacking ASMase expression in oxLDL-treated cells (P < 0·01). Data shown are Mean ± SE from duplicate samples from two independent experiments (n = 4).
Interleukin-1β release was also examined in monocytes from ASMase KO mice and wild-type controls (Fig. 8c). Again, there was no significant release of IL-1β in either oxLDL-IC-treated or oxLDL-treated ASMase KO monocytes compared with controls. The highest levels of released IL-1β in response to oxLDL-IC were at 12 hr after treatment; however, oxLDL began to stimulate the release of IL-1β at 12 hr after treatment (Fig. 8c). These data suggest that ASMase activity is required for IL-1β release in monocytes stimulated with either oxLDL-IC or free oxLDL, and that regulation of IL-1β release may be in part post-translational. Intriguingly, we detected the presence of IL-1β in the isolated exosomes (Fig. 7). Taken together, these data suggest that treatment with oxLDL-IC, and to a lesser extent oxLDL, induces IL-1β-containing exosomes via an ASMase-dependent mechanism.
Discussion
The role of S-ASMase in macrophage activation and development of atherosclerotic plaques has been obscure despite several seminal studies on synthesis and functionality of the enzyme.32,48,49 The source of S-ASMase has often been assumed to have originated from endothelial cells in vivo;50 however, our data suggest that activated monocytes/macrophages may also be an additional source of released S-ASMase. In this study we have provided evidence for a potential mechanism by which uptake of oxLDL-IC, as opposed to oxLDL, can promote and regulate the release of S-ASMase in macrophages. Additionally, we demonstrated that the uptake of oxLDL-IC requires exogenous SMase activity and that uptake and intracellular trafficking are also dependent on L-ASMase activity. Furthermore, we showed that S-ASMase is indeed targeting the insoluble oxLDL-IC cross-linking to the cell membrane receptors. The co-localization of S-ASMase with HSP70B’ extracellularly and in the endosomal compartment in response to oxLDL-IC implies a key role for HSP70B’ in the redistribution of ASMase and in maintaining its enzymatic activity. We also showed that ASMase activity is required for IL-1β secretion in response to oxLDL, in free form or complexed to corresponding antibodies of the IgG isotype. Desipramine inhibited both L-ASMase and the formation of exosomes in oxLDL-IC-treated cells, which suggests that L-ASMase activity is required for exosome release.
It is important to note that although we observed extracellular S-ASMase by confocal microscopy within 30 min of treatment (Fig. 1b), we did not detect S-ASMase activity in the conditioned media before 8 hr (Fig. 2c). Extracellular ASMase was localized to insoluble oxLDL-IC and upon pelleting the treated cells the insoluble oxLDL-IC with associated ASMase would have also been in the pellet, hence removing the majority of S-ASMase present in the conditioned medium. Furthermore, S-ASMase activity requires the presence of additional Zn2+, which is not required for L-ASMase activity;31 therefore, the in vitro assay measuring L-ASMase activity adopted in this study would not detect S-ASMase activity. The increasing S-ASMase activity detected after 8 hr of stimulation with oxLDL-IC suggests that there was no feedback mechanism that could limit the secretion. However, the individual roles of macrophage-derived L-ASMase and S-ASMase on the development of atherosclerosis remain to be determined.
We have previously shown that oxLDL particles begin to be internalized by macrophages at around 30 min post-treatment, whereas oxLDL-IC internalization begins 5 hr after treatment.4 This difference in kinetics of uptake may be attributed to the engagement of Fcγ receptors by immune complexes. In the present study, L-ASMase activity in oxLDL-treated cells decreased below baseline for up to 8 hr after treatment then rose to above baseline activity for the duration of the experiment (Fig. 2d). Whereas the degradation of oxLDL occurs rapidly, there may be a threshold level of intracellular by-products of degradation (such as cholesterol esters) that might increase L-ASMase activity after 8 hr. In contrast, immune complexes induced an initial increase in L-ASMase activity that was maintained for 8 hr, suggesting a role for Fc receptor stimulation (Fig. 2d). Once bound to immune complexes, Fc receptors are internalized and degraded.51 This may eventually lead to a reduction in Fc receptor density on the cell surface, or decrease the extracellular concentration of immune complexes. Either of these mechanisms could result in insufficient Fc receptor-mediated L-ASMase activation after 8 hr of incubation with immune complexes. The increased L-ASMase activity observed as a result of oxLDL-IC treatment may be the result of synergism between both Fc receptor signalling and oxLDL degradation.
Our data suggest that oxLDL-IC requires both exogenous (S-ASMase) and endogenous (L-ASMase) ASMase activity for both internalization and trafficking to the endosomal compartment. In addition, exogenous SMase activity allowed for uptake of oxLDL and oxLDL-IC and their simultaneous trafficking. One effect that SMase activity may have is hydrolysing the SM present in oxLDL, leading to larger particles that can more easily aggregate.33 Larger oxLDL particles may induce phagocytosis, which may involve different uptake mechanisms than the uptake of oxLDL alone, such as via LDL receptor-related protein.52,53 Another non-mutually exclusive possibility is that S-ASMase may be hydrolysing cell surface SM into ceramide to facilitate internalization, which has been previously shown to enhance the internalization of the transferrin receptor.54
Desipramine has been shown to decouple ASMase from the lysosomal compartment causing its deactivation and breakdown.55,56 Inhibition of IL-1β release by desipramine treatment may therefore be the result of the inhibition of L-ASMase activity. Our data show that lack of ASMase (in this instance probably L-ASMase) significantly reduced levels of IL-1β. Inhibition of ASMase using a chemical inhibitor (SMA-7) was also reported to reduce the secretion of inflammatory cytokines including IL-1β in THP-1 macrophages and in an inducible colitis mouse model.57 It is possible that the requirement for ASMase activity in cytokine release is cell-type specific, and also may depend on which cell surface receptors have been stimulated.58 In addition, it is interesting to note that ASMase knockdown inhibited IL-1β release after treatment with both oxLDL-IC and oxLDL, but with different magnitude. We speculate that the early and transient increase in L-ASMase activity after addition of oxLDL could also be sufficient for IL-1β release. It has been shown that the release of IL-1β follows an unconventional pathway involving caspase-1 activation by the inflammasome, an innate immune effector.59 Inflammasome activation and induced autophagy in macrophages were recently found to synergize in secreting IL-1β.60 Further study on any possible inflammasome involvement during oxLDL-IC stimulation would be warranted.
We have previously shown that knockdown of HSP70B’ (gene: HSPA6) completely inhibits oxLDL-IC-induced secretion of IL-1β.12 The findings indicated that HSP70B’ expression is required in the process by which oxLDL-IC augments the secretion of IL-1β. Interestingly, we found that IL-1β mRNA expression is not affected by HSP70B’ siRNA knockdown.12 We have later shown that oxLDL-IC induces up-regulation of both HSP70 and HSP70B’ in RAW 264.7 cells.11 In this study HSP70, but not HSP70B’, was detected in released exosome, which suggests that HSP70B’ and HSP70 play different but complementary roles in macrophages stimulated with oxLDL-IC.
While L-ASMase is by definition located in the lysosomes, our data show that the uptake of oxLDL-IC forced the redistribution of L-ASMase outside the lysosome into the endosomal compartment containing oxLDL-IC. Given that the activity of L-ASMase in response to oxLDL-IC steadily increases, this suggests that ASMase activity is at least partially maintained. Treatment with desipramine did not result in any significant difference in the magnitude of intracellular co-localization of ASMase with HSP70B’, suggesting that ASMase activity is not required for the expression and distribution of HSP70B’ in oxLDL-IC-treated cells. However, large aggregates containing both HSP70B’ and ASMase were detected outside the desipramine-treated cells, indicating that L-ASMase activity is required for the internalization of HSP70B’/ASMase associated with oxLDL-IC. Our results therefore suggest that in oxLDL-IC-treated cells, HSP70B’ up-regulation and co-localization with ASMase may be required for both redistribution of L-ASMase in the endosomal compartment outside lysosomes and maintaining S-ASMase activity. As we found that desipramine-treated oxLDL-IC-stimulated cells also show HSP70B’ associated with ASMase extracellularly, we propose that the role of HSP70B’/S-ASMase complexes in oxLDL-IC-treated macrophages may be in enhancing and maintaining ASMase activity for proper phagocytosis of oxLDL-IC.
The redistribution of ASMase and the differential regulation of ASMase activity suggest different roles in the uptake and processing of oxLDL versus oxLDL-IC. The uptake of oxLDL engages the class A scavenger receptor (SR-A),61 CD36,62 CD14 and Toll-like receptor 4.63 On the other hand, oxLDL-IC engage Fcγ receptor. Hence, the differential trafficking and processing of the particles once engulfed may enable the macrophage to fulfil its innate and acquired immunological roles, and may provide the context for the function of the different processing. Although our data show that ASMase activity is necessary for IL-1β release in oxLDL-IC-treated cells, the effects of free oxLDL are different in untransformed mouse monocytes compared with U937 cells. Our data with wild-type monocytes are broadly in agreement with previously published data.10,64 However, we detected differences in the magnitude of IL-1β release in response to oxLDL in wild-type monocytes and U937 cells. These differences may be attributable to an increased state of activation of the transformed U937 cell line.
Human serum S-ASMase activity can be induced by tumour necrosis factor, IL-1β,50 lipopolysaccharide and oxidative stress.65 Additionally, S-ASMase levels are increased in haemophagic lymphohistiocytosis, a disease where one of the symptoms includes hyper-activated macrophages.66 It has also been reported that MCF-7 cells release S-ASMase upon tumour necrosis factor and IL-1β stimulation, hydrolysing long-chain SM in the plasma membrane to form ceramide.39 Hence, S-ASMase may play a role in producing ceramide in the extracellular compartment, and may therefore enhance cellular stimulation in either an autocrine or a paracrine manner. The released S-ASMase bound to oxLDL-IC in this study might generate ceramide on neighbouring macrophages and stimulate their response to oxLDL-IC, which is supported in part with our data using bSMase. In addition, bacterial infections have been linked to the formation of atherosclerosis.67 Our data showing the increased uptake of oxLDL and oxLDL-IC in the presence of bSMase suggest another mechanism where bacterial infection may promote the formation of atherosclerotic plaques.
In conclusion, our current data show that engagement of Fcγ receptors with modified LDL immune complexes stimulates the release of S-ASMase and may produce a long-term activation of L-ASMase. In addition, up-regulation of HSP70B’ protein appears to be associated with ASMase redistribution. Importantly, we showed that ASMase activity promotes the release of the inflammatory cytokine IL-1β. Activated ASMase may provide the necessary substrates for the downstream synthesis of the bioactive sphingolipid sphingosine-1-phosphate, which is involved with both survival and inflammatory responses. Both enhanced macrophage survival and enhanced release of pro-inflammatory cytokines are likely to play a key role in the perpetuation of the vascular inflammatory disease.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants HL079274, R01 HL079274-04S1 (American Recovery and Investment Act (ARRA)) and the South Carolina COBRE in Lipidomics and Pathobiology (P20 RR17677 from NCRR) to SMH, and P01 HL55782, R01 DK081352, and R01 DK081352-02S1 (ARRA) to MFL-V. This work was also supported by funding from the Department of Veterans Affairs to MFL-V. ASMase KO and ASMase wild-type mice were provided by SC COBRE Animal Pathobiology Core supported by NIH grant P20 RR17677. Special thanks to the Lipidomics Shared Resource facility at MUSC. We thank Dr Richard Kolesnick (Memorial Sloan-Kettering Cancer Center, New York) for anti-ASMase antibodies and Charlyne Chassereau (MUSC) for assistance with lipoprotein isolation.
Disclosure
The authors have no conflicts of interest to declare.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Data S1. Materials and methods.
Video S1. Video depicting involvement of acidsphingomyelinase (ASMase) in phagocytosis of3,3′-dioctadecyloxacarbocyanine perchlorate-labelled oxidizedlow-density lipoprotein-containing immune complexes (oxLDL-IC).
Figure S1. Enzyme activity of secreted acidsphingomy-elinase (ASMase) in conditioned media isZn2+-dependent.
Figure S2. Cellular acid sphingomyelinase (ASMase) protein is not cleaved during incubation with either oxidized low-density lipoprotein (oxLDL) or oxLDL-containing immune complexes (oxLDL-IC).
Figure S3. Addition of bacterial sphingomyelinase (bSMase) has no significant effect on U937 viability.
Figure S4. Extracellular sphingomyelinase (S-ASMase) activity co-aggregates oxidized low-density lipoprotein (oxLDL) and oxLDL-containing immune complexes (oxLDL-IC).
Figure S5. Treatment with acid sphingomyelinase (ASMase) small interfering (si) RNA inhibits ASMase protein expression.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP) Annu Rev Biochem. 1994;63:601–37. doi: 10.1146/annurev.bi.63.070194.003125. [DOI] [PubMed] [Google Scholar]
- 2.Nozaki S, Kashiwagi H, Yamashita S, et al. Reduced uptake of oxidized low density lipoproteins in monocyte-derived macrophages from CD36-deficient subjects. J Clin Investig. 1995;96:1859–65. doi: 10.1172/JCI118231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jerome WG, Cash C, Webber R, Horton R, Yancey PG. Lysosomal lipid accumulation from oxidized low density lipoprotein is correlated with hypertrophy of the Golgi apparatus and trans-Golgi network. J Lipid Res. 1998;39:1362–71. [PubMed] [Google Scholar]
- 4.Al Gadban MM, Smith KJ, Soodavar F, et al. Differential trafficking of oxidized LDL and oxidized LDL immune complexes in macrophages: impact on oxidative stress. PLoS ONE. 2010;5:e12534. doi: 10.1371/journal.pone.0012534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Virella G, Koskinen S, Krings G, Onorato JM, Thorpe SR, Lopes-Virella M. Immunochemical characterization of purified human oxidized low-density lipoprotein antibodies. Clin Immunol. 2000;95:135–44. doi: 10.1006/clim.2000.4857. [DOI] [PubMed] [Google Scholar]
- 6.Virella G, Lopes-Virella MF. Lipoprotein autoantibodies: measurement and significance. Clin Diagn Lab Immunol. 2003;10:499–505. doi: 10.1128/CDLI.10.4.499-505.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Virella G, Munoz JF, Galbraith GM, Gissinger C, Chassereau C, Lopes-Virella MF. Activation of human monocyte-derived macrophages by immune complexes containing low-density lipoprotein. Clin Immunol Immunopathol. 1995;75:179–89. doi: 10.1006/clin.1995.1069. [DOI] [PubMed] [Google Scholar]
- 8.Griffith RL, Virella GT, Stevenson HC, Lopes-Virella MF. Low density lipoprotein metabolism by human macrophages activated with low density lipoprotein immune complexes. A possible mechanism of foam cell formation. J Exp Med. 1988;168:1041–59. doi: 10.1084/jem.168.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang Y, Jaffa A, Koskinen S, Takei A, Lopes-Virella MF. Oxidized LDL-containing immune complexes induce Fcγ receptor I-mediated mitogen-activated protein kinase activation in THP-1 macrophages. Arterioscler Thromb Vasc Biol. 1999;19:1600–7. doi: 10.1161/01.atv.19.7.1600. [DOI] [PubMed] [Google Scholar]
- 10.Saad AF, Virella G, Chassereau C, Boackle RJ, Lopes-Virella MF. OxLDL immune complexes activate complement and induce cytokine production by MonoMac 6 cells and human macrophages. J Lipid Res. 2006;47:1975–83. doi: 10.1194/jlr.M600064-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Smith KJ, Twal WO, Soodavar F, Virella G, Lopes-Virella MF, Hammad SM. Heat shock protein 70B’ (HSP70B’) expression and release in response to human oxidized low density lipoprotein immune complexes in macrophages. J Biol Chem. 2010;285:15985–93. doi: 10.1074/jbc.M110.113605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hammad SM, Twal WO, Barth JL, Smith KJ, Saad AF, Virella G, Argraves WS, Lopes-Virella MF. Oxidized LDL immune complexes and oxidized LDL differentially affect the expression of genes involved with inflammation and survival in human U937 monocytic cells. Atherosclerosis. 2009;202:394–404. doi: 10.1016/j.atherosclerosis.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oksjoki R, Kovanen PT, Lindstedt KA, Jansson B, Pentikainen MO. OxLDL–IgG immune complexes induce survival of human monocytes. Arterioscler Thromb Vasc Biol. 2006;26:576–83. doi: 10.1161/01.ATV.0000201041.14438.8d. [DOI] [PubMed] [Google Scholar]
- 14.Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem. 2000;275:17221–4. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- 15.Jackson RL, Morrisett JD, Gotto AM., Jr Lipoprotein structure and metabolism. Physiol Rev. 1976;56:259–316. doi: 10.1152/physrev.1976.56.2.259. [DOI] [PubMed] [Google Scholar]
- 16.Hurwitz R, Ferlinz K, Vielhaber G, Moczall H, Sandhoff K. Processing of human acid sphingomyelinase in normal and I-cell fibroblasts. J Biol Chem. 1994;269:5440–5. [PubMed] [Google Scholar]
- 17.Liu P, Anderson RG. Compartmentalized production of ceramide at the cell surface. J Biol Chem. 1995;270:27179–85. doi: 10.1074/jbc.270.45.27179. [DOI] [PubMed] [Google Scholar]
- 18.Grassme H, Jendrossek V, Bock J, Riehle A, Gulbins E. Ceramide-rich membrane rafts mediate CD40 clustering. J Immunol. 2002;168:298–307. doi: 10.4049/jimmunol.168.1.298. [DOI] [PubMed] [Google Scholar]
- 19.Scheel-Toellner D, Wang K, Assi LK, Webb PR, Craddock RM, Salmon M, Lord JM. Clustering of death receptors in lipid rafts initiates neutrophil spontaneous apoptosis. Biochem Soc Trans. 2004;32(Pt 5):679–81. doi: 10.1042/BST0320679. [DOI] [PubMed] [Google Scholar]
- 20.Grassme H, Jekle A, Riehle A, Schwarz H, Berger J, Sandhoff K, Kolesnick R, Gulbins E. CD95 signaling via ceramide-rich membrane rafts. J Biol Chem. 2001;276:20589–96. doi: 10.1074/jbc.M101207200. [DOI] [PubMed] [Google Scholar]
- 21.Cremesti A, Paris F, Grassme H, Holler N, Tschopp J, Fuks Z, Gulbins E, Kolesnick R. Ceramide enables Fas to cap and kill. J Biol Chem. 2001;276:23954–61. doi: 10.1074/jbc.M101866200. [DOI] [PubMed] [Google Scholar]
- 22.Schramm M, Herz J, Haas A, Kronke M, Utermohlen O. Acid sphingomyelinase is required for efficient phago-lysosomal fusion. Cell Microbiol. 2008;10:1839–53. doi: 10.1111/j.1462-5822.2008.01169.x. [DOI] [PubMed] [Google Scholar]
- 23.Hauck CR, Grassme H, Bock J, Jendrossek V, Ferlinz K, Meyer TF, Gulbins E. Acid sphingomyelinase is involved in CEACAM receptor-mediated phagocytosis of Neisseria gonorrhoeae. FEBS Lett. 2000;478:260–6. doi: 10.1016/s0014-5793(00)01851-2. [DOI] [PubMed] [Google Scholar]
- 24.Esen M, Schreiner B, Jendrossek V, Lang F, Fassbender K, Grassme H, Gulbins E. Mechanisms of Staphylococcus aureus induced apoptosis of human endothelial cells. Apoptosis. 2001;6:431–9. doi: 10.1023/a:1012445925628. [DOI] [PubMed] [Google Scholar]
- 25.Yu H, Zeidan YH, Wu BX, Jenkins RW, Flotte TR, Hannun YA, Virella-Lowell I. Defective acid sphingomyelinase pathway with Pseudomonas aeruginosa infection in cystic fibrosis. Am J Respir Cell Mol Biol. 2009;41:367–75. doi: 10.1165/rcmb.2008-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Truman JP, Al Gadban MM, Smith KJ, Hammad SM. Acid sphingomyelinase in macrophage biology. Cell Mol Life Sci. 2011;68:3293–305. doi: 10.1007/s00018-011-0686-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheruku SR, Xu Z, Dutia R, Lobel P, Storch J. Mechanism of cholesterol transfer from the Niemann–Pick type C2 protein to model membranes supports a role in lysosomal cholesterol transport. J Biol Chem. 2006;281:31594–604. doi: 10.1074/jbc.M602765200. [DOI] [PubMed] [Google Scholar]
- 28.Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci USA. 2008;105:15287–92. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schissel SL, Schuchman EH, Williams KJ, Tabas I. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J Biol Chem. 1996;271:18431–6. doi: 10.1074/jbc.271.31.18431. [DOI] [PubMed] [Google Scholar]
- 30.Jenkins RW, Canals D, Hannun YA. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal. 2009;21:836–46. doi: 10.1016/j.cellsig.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schissel SL, Keesler GA, Schuchman EH, Williams KJ, Tabas I. The cellular trafficking and zinc dependence of secretory and lysosomal sphingomyelinase, two products of the acid sphingomyelinase gene. J Biol Chem. 1998;273:18250–9. doi: 10.1074/jbc.273.29.18250. [DOI] [PubMed] [Google Scholar]
- 32.Schissel SL, Jiang X, Tweedie-Hardman J, et al. Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral pH. Implications for atherosclerotic lesion development. J Biol Chem. 1998;273:2738–46. doi: 10.1074/jbc.273.5.2738. [DOI] [PubMed] [Google Scholar]
- 33.Marathe S, Kuriakose G, Williams KJ, Tabas I. Sphingomyelinase, an enzyme implicated in atherogenesis, is present in atherosclerotic lesions and binds to specific components of the subendothelial extracellular matrix. Arterioscler Thromb Vasc Biol. 1999;19:2648–58. doi: 10.1161/01.atv.19.11.2648. [DOI] [PubMed] [Google Scholar]
- 34.Devlin CM, Leventhal AR, Kuriakose G, Schuchman EH, Williams KJ, Tabas I. Acid sphingomyelinase promotes lipoprotein retention within early atheromata and accelerates lesion progression. Arterioscler Thromb Vasc Biol. 2008;28:1723–30. doi: 10.1161/ATVBAHA.108.173344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006;103:10340–5. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Virella G, Thorpe SR, Alderson NL, Derrick MB, Chassereau C, Rhett JM, Lopes-Virella MF. Definition of the immunogenic forms of modified human LDL recognized by human autoantibodies and by rabbit hyperimmune antibodies. J Lipid Res. 2004;45:1859–67. doi: 10.1194/jlr.M400095-JLR200. [DOI] [PubMed] [Google Scholar]
- 37.Lopes-Virella MF, Koskinen S, Mironova M, Horne D, Klein R, Chassereau C, Enockson C, Virella G. The preparation of copper-oxidized LDL for the measurement of oxidized LDL antibodies by EIA. Atherosclerosis. 2000;152:107–15. doi: 10.1016/s0021-9150(99)00456-6. [DOI] [PubMed] [Google Scholar]
- 38.Pitas RE, Innerarity TL, Weinstein JN, Mahley RW. Acetoacetylated lipoproteins used to distinguish fibroblasts from macrophages in vitro by fluorescence microscopy. Arteriosclerosis. 1981;1:177–85. doi: 10.1161/01.atv.1.3.177. [DOI] [PubMed] [Google Scholar]
- 39.Jenkins RW, Canals D, Idkowiak-Baldys J, et al. Regulated secretion of acid sphingomyelinase: implications for selectivity of ceramide formation. J Biol Chem. 2010;285:35706–18. doi: 10.1074/jbc.M110.125609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin J, Hou Q, Mullen TD, et al. Ceramide generated by sphingomyelin hydrolysis and the salvage pathway is involved in hypoxia/reoxygenation-induced Bax redistribution to mitochondria in NT-2 cells. J Biol Chem. 2008;283:26509–17. doi: 10.1074/jbc.M801597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammad SM, Taha TA, Nareika A, Johnson KR, Lopes-Virella MF, Obeid LM. Oxidized LDL immune complexes induce release of sphingosine kinase in human U937 monocytic cells. Prostaglandins Other Lipid Mediat. 2006;79:126–40. doi: 10.1016/j.prostaglandins.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 42.Anand PK, Anand E, Bleck CK, Anes E, Griffiths G. Exosomal Hsp70 induces a pro-inflammatory response to foreign particles including mycobacteria. PLoS ONE. 2010;5:e10136. doi: 10.1371/journal.pone.0010136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khatua AK, Taylor HE, Hildreth JE, Popik W. Exosomes packaging APOBEC3G confer human immunodeficiency virus resistance to recipient cells. J Virol. 2009;83:512–21. doi: 10.1128/JVI.01658-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kirkegaard T, Roth AG, Petersen NH, et al. Hsp70 stabilizes lysosomes and reverts Niemann–Pick disease-associated lysosomal pathology. Nature. 2010;463:549–53. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- 45.Lee Y, Sohn WJ, Kim DS, Kwon HJ. NF-κB- and c-Jun-dependent regulation of human cytomegalovirus immediate-early gene enhancer/promoter in response to lipopolysaccharide and bacterial CpG-oligodeoxynucleotides in macrophage cell line RAW 264.7. Eur J Biochem. 2004;271:1094–105. doi: 10.1111/j.1432-1033.2004.04011.x. [DOI] [PubMed] [Google Scholar]
- 46.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 47.Record M, Subra C, Silvente-Poirot S, Poirot M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem Pharmacol. 2011;81:1171–82. doi: 10.1016/j.bcp.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 48.Guarino AJ, Tulenko TN, Wrenn SP. Sphingomyelinase-to-LDL molar ratio determines low density lipoprotein aggregation size: biological significance. Chem Phys Lipids. 2006;142:33–42. doi: 10.1016/j.chemphyslip.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 49.Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–44. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 50.Marathe S, Schissel SL, Yellin MJ, Beatini N, Mintzer R, Williams KJ, Tabas I. Human vascular endothelial cells are a rich and regulatable source of secretory sphingomyelinase. Implications for early atherogenesis and ceramide-mediated cell signaling. J Biol Chem. 1998;273:4081–8. doi: 10.1074/jbc.273.7.4081. [DOI] [PubMed] [Google Scholar]
- 51.Mellman IS. Endocytosis, membrane recycling and Fc receptor function. Ciba Found Symp. 1982;92:35–58. [PubMed] [Google Scholar]
- 52.Llorente-Cortes V, Martinez-Gonzalez J, Badimon L. LDL receptor-related protein mediates uptake of aggregated LDL in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:1572–9. doi: 10.1161/01.atv.20.6.1572. [DOI] [PubMed] [Google Scholar]
- 53.Patel M, Morrow J, Maxfield FR, Strickland DK, Greenberg S, Tabas I. The cytoplasmic domain of the low density lipoprotein (LDL) receptor-related protein, but not that of the LDL receptor, triggers phagocytosis. J Biol Chem. 2003;278:44799–807. doi: 10.1074/jbc.M308982200. [DOI] [PubMed] [Google Scholar]
- 54.Abdel Shakor AB, Atia MM, Kwiatkowska K, Sobota A. Cell surface ceramide controls translocation of transferrin receptor to clathrin-coated pits. Cell Signal. 2012;24:677–84. doi: 10.1016/j.cellsig.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 55.Kornhuber J, Medlin A, Bleich S, Jendrossek V, Henkel AW, Wiltfang J, Gulbins E. High activity of acid sphingomyelinase in major depression. J Neural Transm. 2005;112:1583–90. doi: 10.1007/s00702-005-0374-5. [DOI] [PubMed] [Google Scholar]
- 56.Kornhuber J, Tripal P, Reichel M, Terfloth L, Bleich S, Wiltfang J, Gulbins E. Identification of new functional inhibitors of acid sphingomyelinase using a structure–property–activity relation model. J Med Chem. 2008;51:219–37. doi: 10.1021/jm070524a. [DOI] [PubMed] [Google Scholar]
- 57.Sakata A, Ochiai T, Shimeno H, et al. Acid sphingomyelinase inhibition suppresses lipopolysaccharide-mediated release of inflammatory cytokines from macrophages and protects against disease pathology in dextran sulphate sodium-induced colitis in mice. Immunology. 2007;122:54–64. doi: 10.1111/j.1365-2567.2007.02612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bianco F, Perrotta C, Novellino L, et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009;28:1043–54. doi: 10.1038/emboj.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–31. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 60.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30:4701–11. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leyva FJ, Pershouse MA, Holian A. Modified low density lipoproteins binding requires a lysine cluster region in the murine macrophage scavenger receptor class A type II. Mol Biol Rep. 2010;37:2847–52. doi: 10.1007/s11033-009-9837-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Febbraio M, Silverstein RL. CD36: implications in cardiovascular disease. Int J Biochem Cell Biol. 2007;39:2012–30. doi: 10.1016/j.biocel.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chavez-Sanchez L, Madrid-Miller A, Chavez-Rueda K, Legorreta-Haquet MV, Tesoro-Cruz E, Blanco-Favela F. Activation of TLR2 and TLR4 by minimally modified low-density lipoprotein in human macrophages and monocytes triggers the inflammatory response. Hum Immunol. 2010;71:737–44. doi: 10.1016/j.humimm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 64.Thomas CE, Jackson RL, Ohlweiler DF, Ku G. Multiple lipid oxidation products in low density lipoproteins induce interleukin-1 beta release from human blood mononuclear cells. J Lipid Res. 1994;35:417–27. [PubMed] [Google Scholar]
- 65.Mathias S, Pena LA, Kolesnick RN. Signal transduction of stress via ceramide. Biochem J. 1998;335:465–80. doi: 10.1042/bj3350465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takahashi T, Abe T, Sato T, et al. Elevated sphingomyelinase and hypercytokinemia in hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2002;24:401–4. doi: 10.1097/00043426-200206000-00016. [DOI] [PubMed] [Google Scholar]
- 67.Muhlestein JB. Bacterial infections and atherosclerosis. J Investig Med. 1998;46:396–402. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.