

Abstract
The cellular innate immune response is essential for recognizing and defending against viral infection. Retinoic acid-inducible gene-I (RIG-I) and virus-induced signaling adaptor (VISA) mediated immune signalling is critically involved in RNA-virus-induced innate immune responses. Here we demonstrate that the complement C1qA interacts with different RIG-I pathway components and enhances RIG-I-VISA-mediated signalling pathway as well as TBK1-mediated activation of interferon-β (IFN-β) promoter. Our data show that over-expression of C1qA up-regulates RIG-I-mediated activation of IFN-stimulated responsive element (ISRE) and nuclear factor-κB reporters and IFN-β transcription, but not IFN regulatory factor-3-mediated and inhibitor of κB kinase-mediated activation of ISRE and nuclear factor-κB promoter. In addition, C1qA can counteract the function of the C1q receptor gC1qR in RIG-I-mediated signalling. Our results reveal the important role of complement C1qA in the innate immune response.
Keywords: complement C1qA, innate immune response, retinoic acid inducible gene I, VISA
Introduction
During the viral infection, the induction of type I interferons (IFN-α/β) is critical for the host cell establishing an antiviral state. Many IFN-induced or IFN-activated genes such as protein kinase R, 2′,5′-oligoadenylate synthetase (OAS) and RNaseL, immune serum globulins, nitric oxide synthase and MHC proteins mediate the inhibition of virus replication and clearance of virus-infected cells, leading to the development of a virus-resistant state and modulation of adaptive immunity.1 Viral genome RNAs are important pathogen-associated molecular patterns, which are recognized by pattern recognition receptors of the host innate immune system. The pattern recognition receptors sense viral RNAs and initiate signalling for type I IFN induction.2 The main pattern recognition receptors include membrane-bound Toll-like receptors (TLR3, TLR7, TLR8) and cytosolic retinoic acid-inducible gene-I (RIG-I) -like receptors [RLRs; including RIG-I and melanoma-differentiation-associated gene 5 (MDA5)].3
The induction of type I IFNs is primarily regulated at transcriptional level. Multiple transcriptional activators are cooperatively involved in the induction of type I IFNs. There are four regulatory cis-elements (the positive regulatory domains, PRD I, II, III and IV) in the promoter region of the IFN-β gene. The PRD I and III (also known as the IFN-stimulated responsive element, or ISRE) are activated by phosphorylated IFN regulatory factors (IRFs), such as IRF-3 and IRF-7. The PRD II and IV are activated by nuclear factor-κB (NF-κB) and ATF-2/c-Jun respectively.4
Much progress has been made to understand the mechanisms of RLR-mediated induction of type I IFNs. RIG-I and MDA5 selectively recognize different viral RNAs by their C-terminal DexD/H RNA helicase domain and induce an ATP-dependent conformational change5 that allows their N-terminal caspase recruitment domain (CARD) to interact with the N-terminal domain of VISA, a critical adaptor protein localized on the outer membrane of mitochondria.6–9 The interactions between RIG-I or MDA5 and VISA lead to the activation of VISA. The activated VISA then activates IκB kinases (IKKs) and TANK-binding kinase 1 (TBK1), which are responsible for the activation of NF-κB and IRF-3 respectively. NF-κB, IRF-3 and other transcriptional factors then cooperatively induce the expression of type I IFNs.10
Many molecules have been found to interact with RLRs’ critical components and to regulate the RLR-mediated immune pathway. NLRX1, a member of the nucleotide-binding oligomerization domain-like receptor (NLR) family, negatively regulates VISA activity through its interaction with the CARD domain of VISA.11 Suppressor of IKKε (SIKE) interacts with IKKε/TBK1 to block these kinases from interacting with RIG-I or IRF-3.12 Many E3 ubiquitin ligases catalyse lys63-linked or lys48-linked ubiquitination of RIG-I, MDA5, MITA or VISA to up-regulate or down-regulate the activity of the RLR pathway. For example, ring finger protein 5 (RNF5) interacts with MITA (also known as STING) and mediates its ubiquitination and degradation to inhibit RIG-I pathway-mediated IRF-3 activation13 and TRIM25 catalyses the ubiquitination of RIG-I and enhances its downstream signalling.14 The NS1 protein of influenza A virus interacts with RIG-I and VISA and acts as an inhibitory factor in the RIG-I-mediated anti-virus response.15
Complements are considered a vital part of the immune system and are responsible for the destruction of invading microorganisms and elimination of immune complex. The complements also link innate and adaptive immunity. Complements are required for tissue regeneration and repair; clinical trials also indicate the critical role of the complement system in immunodeficiency disorders and various inflammatory diseases.16,17 However, research on complements has mostly focused on their function in the serum; the role of complement in the cytoplasm is rarely considered. In this study, we found that the first component of the complement classical activation pathway, C1qA, interacted with RIG-I and was involved in the RLR-mediated signalling pathway. C1qA enhanced RIG-I-mediated activation of ISRE, NF-κB and IFN-β gene transcription. C1qA also enhanced the TBK1-mediated activation of ISRE and IFN-β promoters but not IRF-3-mediated activation of ISRE promoter and IKKβ-mediated activation of NF-κB promoter. These results suggested that C1qA specifically enhanced IFN-β activation at the TBK1 level.
Materials and methods
Plasmids, antibodies and reagents
The mammalian expression plasmids of VISA, RIG-I and IRF-3 were generous gifts from H. Shu (Wuhan University, China). TBK1 was cloned into mammalian expression vector pCMV-myc. ISRE, NF-κB and the IFN-β promoter luciferase reporter plasmids were from J. Yan and H. Tang (Chinese Academy of Sciences), RIG-I, IKKβ and TBK1 were subcloned into pET30a for bacterial expression. The pCMV-FLAG-TRAF3 and pCMV-FLAG-TRAF6 were provided by Z. Xu (Chinese Academy of Sciences); pCMV FLAG-IKKβ was provided by G. Brosnan (Viral Immune Evasion Group School of Biochemistry and Immunology, Trinity College, Dublin, Ireland); pcDNA3.1-FLAG-MDA5 was provided by J. Rehwinkel (Immunobiology Laboratory, Cancer Research UK, London Research Institute); and pCMV myc-C1qA was constructed as previously described.18
Mouse anti-FLAG monoclonal antibodies, anti-FLAG M2 agarose antibodies, and anti-mouse or anti-rabbit IgG conjugated with horseradish peroxidase and poly I:C were obtained from Sigma (St. Louis, MO). Goat anti-C1qA polyclonal antibody and mouse anti-myc monoclonal antibody (9E10) were from Santa Cruz Biotechnology (Santa Cruz, CA). Dulbecco's modified Eagle's medium (DMEM) was from Invitrogen (Grand Island, NY). Polyethylenimine was from Polysciences (El Dorado, Panama); fetal bovine serum was from PAA (Queensland, Australia); the luciferase assay system was from Promega (Madison, WI) and TPCK-treated trypsin was from Sigma.
Cell culture and virus infection
The 293T and MDCK cells were maintained in Madin-Darby canine kidney cells (MDCK) supplemented with 10% fetal bovine serum. Influenza virus strain A/WSN/33 (H1N1) was generated by reverse genetics.19 For virus infection, the cells were washed with PBS and infected with virus in serum-free medium for 2 hr, then washed with PBS and cultured in DMEM supplemented with 10% fetal bovine serum. For Sendai virus (SeV) infection, 293T cells were washed with PBS and infected with virus in serum-free medium for 6 hr.
Plaque assay
The MDCK cells were cultured in six-well plates to form monolayered cells and were infected with H1N1 virus for 2 hr. The unabsorbed viruses were removed by washing with serum-free DMEM, and the plate was then overlaid with DMEM supplemented with 3% low-melting agarose and TPCK-treated trypsin (2 μg/ml). After 3 days of incubation, visible plaques were counted and the virus titres were calculated. All data were expressed as the mean of triplicate samples.
GST pull-down and co-immunoprecipitation
Glutathione S transferase (GST)-C1qA and His-tagged RIG-I, IKKβ and TBK1 were purified and the GST-pull-down assay was performed as described previously.18 In brief, the GST or GST-C1qA proteins were incubated with indicated His-tagged proteins for 2 hr and the glutathione beads were added and incubated for 1 hr. The associated proteins were analysed by immunoblotting with anti-His antibody. For immunoprecipitation assay, 293T cells were transfected with FLAG-tagged and myc-tagged expression plasmids for 24 hr, then lysed in cell lysis buffer (1% TritonX-100, 150 mm NaCl, 20 mm HEPES (pH7·5), 10% glycerol, 1 mm EDTA) with protease inhibitor (Roche, Germany). The lysates were immunoprecipitated with anti-FLAG beads at 4° for 3 hr and subject to immunoblotting with anti-myc antibody.
Reverse transcription-PCR
The total RNA of HEK 293T cell was extracted using TRIzol (Invitrogen) according to the manufacturer's instructions. Complementary DNA was synthesized by AMV reverse transcriptase (Promega) with oligo-dT primer (Takara, Japan). The PCR was performed with gene-specific primers (β-actin, forward: 5′-GCGGGAAATCGTGCGTGACATT, reverse: 5′-GATGGAGTTGAAGGTAGTTTCGTG; ISG56, forward: ACGGCTGCCTAATTTACAGC, reverse: AGTGGCTGATATCTGGGTGC; ISG54, forward: AATGCCATTTCACCTGGAACTTG, reverse: GTGATAGTAGACCCAGGCATAGT; A20, forward: TTTTGTACCCTTGGTGACCCTG, reverse: TTAGCTTCATCCAACTTTGCGG; IFN-β, forward: CACGACAGCTCTTTCCATGA, reverse: AGCCAGTGCTCGATGAATCT).
Luciferase reporter assay
The HEK 293T cells (1 × 105) were seeded in 24-well plates and transfected with the indicated plasmids with pCMV β-gal as internal control for 24 hr. The cell lysates were harvested and subjected to luciferase assay. All luciferase assays were performed with triplicate samples.
Results
C1qA enhances ISRE, NF-κB and IFN-β luciferase reporter activity in 293T cells
When analysing the role of C1qA in influenza A virus infection,18 we observed that the virus replication in C1qA over-expressed 293T cells was much lower than in control cells (Fig. 1a). It has been reported that the C1q receptor (gC1qR) that recognizes the globular region of C1q functions as a negative modulator of the innate RLR-mediated anti-viral response.20 These observations prompted us to investigate whether C1qA was involved in innate immune signalling. Considering the central role of type I IFNs in innate immunity, first we examined whether C1qA affected the expression of relevant genes. 293T cells were transfected with C1qA expression plasmid and ISRE, NF-κB and IFN-β luciferase reporter plasmids. The data from the luciferase assay indicated that C1qA enhanced the activity of ISRE, NF-κB and IFN-β promoter reporters (Fig. 1b–d). We then transfected 293T cells with C1qA and ISRE luciferase reporter together with poly I:C, which was used as the mimic of viral dsRNA to stimulate RLR pathway-mediated IFN-β production.20 Consistently, we found that C1qA can enhance the activation of ISRE reporter mediated by poly I:C (Fig. 1e). In SeV-infected 293T cells, C1qA can also promote the activation of IFN-β promoter (Fig. 1f).
Figure 1.
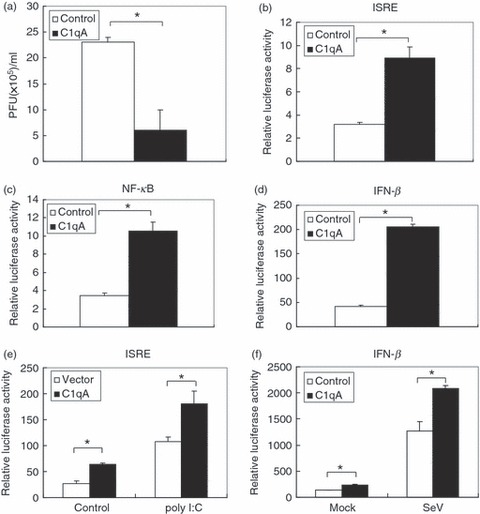
C1qA inhibits influenza virus replication and enhances interferon-stimulated responsive element (ISRE), nuclear factor-κB (NF-κB) and interferon-β (IFN-β) derived luciferase reporter. (a) Plaque assay. 293T cells were transfected with pCMV myc-C1qA or pcDNA3.1 as control for 24 hr then infected with influenza virus (A/WSN/33) at an multiplicity of infection of 0·1 for 16 hr. The supernatants were harvested and subjected to plaque assay. (b–d) Luciferase assay. The 293T cells were transfected with pCMV myc-C1qA together with the indicated luciferase reporter and pCMV β-gal as internal control. The luciferase assays were performed 24 hr after transfection. (e) The 293T cells were transfected with the indicated plasmids together with poly I:C for 24 hr. The cell lysates were harvested and subjected to luciferase assay. (f) 293T cells were transfected with the indicated plasmids for 24 hr then infected with SeV for 6 hr. The cell lysates were harvested for luciferase assay. Each assay was performed with triplicate samples. Graphs show mean ± SD, n = 3, *P < 0·01.
C1qA enhances RIG-I- and VISA-mediated innate immunity
Besides its important role in the classical complement activation pathway that provides protection against pathogen infection, C1q also functions as a regulatory factor in the immune system by interacting with various molecules. It was reported that C1q binds to C-reactive protein bound to apoptotic cells to trigger complement activation and enhance phagocytosis. C1q can bind with CD91 and mediate phagocytosis of apoptotic cells.21 It also interacted with gC1qR, which was recently found to be a negative regulator in RLR-mediated innate immunity through interacting with VISA.20 We wondered whether C1qA was also involved in the RLR-mediated immune pathway. To address this question, we transfected 293T cells with RIG-I or VISA in the presence or absence of C1qA together with ISRE, NF-κB or IFN-β luciferase reporters. The data showed that C1qA significantly enhanced both RIG-I-mediated and VISA-mediated activation of ISRE, NF-κB and IFN-β promoters (Fig. 2a–c).
Figure 2.
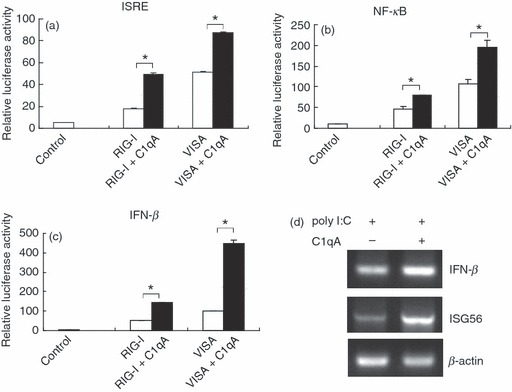
C1qA enhances RIG-I and VISA triggered activation of interferon-stimulated responsive element (ISRE), nuclear factor-κB (NF-κB) and interferon-β (IFN-β) reporter and poly I:C induced transcription of IFN-β, ISG56. (a–c) 293T cells were transfected with retinoic acid-inducible gene-I (RIG-I) and VISA expression plasmids and indicated luciferase reporter in the presence or absence of C1qA for 24 hr. The cell lysates were subjected to luciferase assay. (d) 293T cells were transfected with poly I:C in the presence or absence of C1qA for 24 hr. The RNA was prepared and subjected to reverse transcription-PCR. Graphs show mean ± SD, n = 3, *P < 0·01.
Then we transfected 293T cells with poly I:C with or without C1qA and analysed the transcription of IFN-β and ISG56 by RT-PCR. As shown in Fig. 2(d), the over-expression of C1qA obviously enhanced the transcription of IFN-β and ISG56 as expected.
C1qA is involved in RLR-mediated signalling by interacting with RLR pathway components
Many molecules participated in the regulation of RLR-mediated IFN-β induction by interacting with the critical components in the RLR pathway.5 To understand the mechanism by which C1qA regulates the RLR-mediated signalling pathway, we first examined whether C1qA interacted with RIG-I, VISA or other components in the RLR pathway. 293T cells were co-transfected with C1qA and RIG-I, VISA, TBK1, TRAF3, TRAF6, IKKβ or IRF-3 expression plasmids. The cell lysates were harvested for co-immunoprecipitation analysis. As shown in Fig. 3(a–c), C1qA strongly interacted with RIG-I, VISA and TBK1, and had weak interaction with IKKβ and TRAF6. There were no interactions between C1qA and MDA5, TRAF3 or IRF-3. (Fig. 3d,e). To further analyse which protein was involved in the direct interaction with C1qA, we performed a GST pull-down assay. The data indicated that C1qA directly interacted with RIG-I but not TBK1 (Fig. 3f). Surprisingly, although IKKβ weakly interacted with C1qA in a co-immunoprecipitation assay, it bound with C1qA well in the pull-down assay (Fig. 3f, lower panel). These results suggested that C1qA was involved in the RLR pathway by interacting with the RLR components. To investigate whether C1qA could interact with RLR components endogenously, first immunoprecipitation was performed with the over-expressed C1qA in 293T cells, then immunoblotting with VISA antibody was used to confirm whether the endogenous VISA could interact with over-expressed C1qA. Consistent with previous results, we found that the over-expressed C1qA interacted with endogenous VISA (Fig. 3g). To further investigate whether this interaction occurred naturally in endogenous conditions, and because C1qA and VISA were highly conserved both in mice and humans, we immunoprecipitated C1qA from C1q-expressing Raw264.7 cells (a mouse macrophage line) and THP1 cells (a human acute monocytic leukaemia cell line), then immunoblotted with VISA and found that C1qA interacted with VISA endogenously (Fig. 3h,i).
Figure 3.
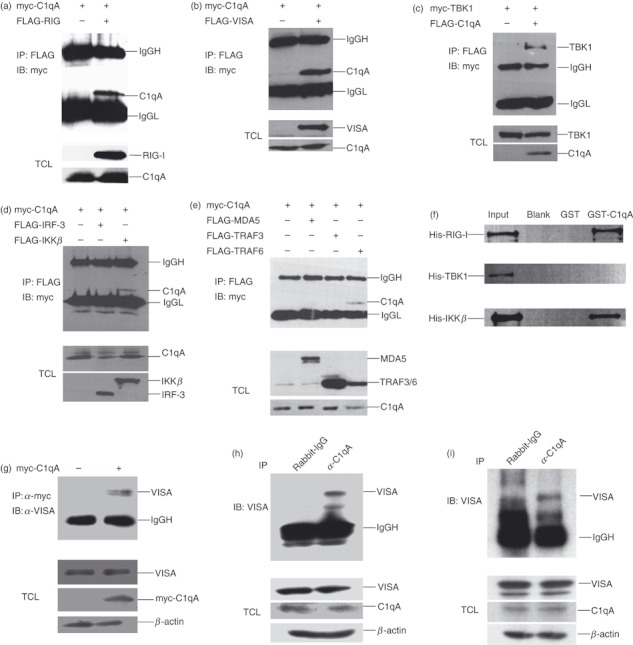
C1qA interacts with different components in the RLR pathway. (a–e) 293T cells were transfected with the indicated plasmids for 24 hr. The total cell lysates (TCL) were harvested and subjected to immunoprecipitation with FLAG antibody followed by immunoblotting with myc antibody. (f) His-tagged retinoic acid-inducible gene-I (RIG-I), TANK-binding kinase 1 (TBK1) and inhibitor of κB kinase (IKKβ) were incubated with GST or GST-tagged C1qA. Proteins associated with C1qA were pulled down with glutathione beads and detected with anti-his antibody. (g) 293T cells were transfected with control or myc-tagged C1qA expression plasmids for 24 hr. The TCL were harvested and subjected to immunoprecipitation with myc antibody followed by immunoblotting with VISA antibody. (h,i) The total cell lysates of Raw 264.7 (h) or THP1 (i) were harvested and subjected to immunoprecipitation with C1qA antibody followed by immunoblotting with VISA antibody.
C1qA enhances RIG-I mediated immune signalling at the TBK1 level
RLR-mediated signalling can be divided into three steps: recognition of exogenous RNA by RIG-I or MDA5, recruitment of RLRs to mitochondria through CARD–CARD interaction with VISA and formation of a VISA-centred platform, and activation of downstream effective molecules such as TBK1/IKKi and IKKα/β and induction of IFN-β.5 To determine which step C1qA plays in promoting RLR-mediated IFN-β activation. We expressed C1qA with different components in the RLR pathway to investigate whether C1qA enhanced the molecule-induced activation of ISRE, NF-κB or IFN-β promoter reporters. It was shown in Fig. 2 that expression of C1qA enhanced RIG-I-mediated and VISA-mediated activation of IFN-β. In addition, we found that C1qA also enhanced TBK1-mediated activation of ISRE and IFN-β promoter reporter (Fig. 4a,b). We did not observe any enhancement of C1qA on IRF-3- or IKKβ-mediated ISRE and NF-κB promoter activation (Fig. 4c,d). These results suggested that C1qA may enhance RIG-I-mediated immune signalling at TBK1 level.
Figure 4.
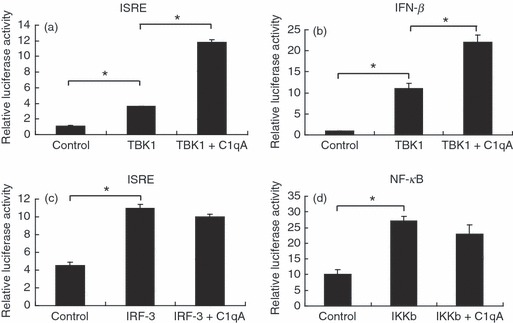
C1qA enhances the retinoic acid-inducible gene-I-like receptor (RLR) pathway at TANK-binding kinase 1 (TBK1) level. (a–d) 293T cells were transfected with the indicated plasmids and luciferase reporters for 24 hr. The cell lysates were harvested for luciferase assay. Graphs show mean ± SD, n = 3, *P < 0·01.
C1qA counteracts with the function of C1q receptor gC1qR in RIG-I-mediated signalling
C1qA was one of the subunits of C1q and contained an N-terminal collagen region and a C-terminal globular region. The C1q globular region receptor gC1qR was reported to function as a physical inhibitor of the RIG-I-mediated and MDA5-mediated immune pathway,20,22 so we wondered whether the inhibitory function gC1qR of on RLR signalling pathway can be counteracted by the expression of C1qA. First we examined how gC1qR interacted with VISA in 293T cells and was located on mitochondria, which was consistent with previous reports (data not shown). Importantly, we found that C1qA interacted with gC1qR in 293T cells (Fig. 5a). Then, we analysed whether C1qA had any influence on the inhibitory effect of gC1qR on RLR signalling using an ISRE luciferase reporter assay. As shown in Fig. 5(b), C1qA counteracted the inhibitory effect of gC1qR on ISRE promoter reporter to control levels. To exclude the possibility that co-transfection of C1qA may affect the expression of gC1qR and lead to the same result as observed in our experiment, we compared the expression of gC1qR in the presence or absence of C1qA. Results showed that co-transfection of C1qA did not affect the expression of gC1qR (Fig. 5c).
Figure 5.

C1qA counteracts the function of gC1qR (a) C1qA interacts with gC1qR. 293T cells were transfected with indicated plasmids for 24 hr. The cell lysates were harvested and subjected to immunoprecipitation with FLAG antibody followed by immunoblotting with myc antibody. (b) 293T cells were transfected with interferon-stimulated responsive element (ISRE) luciferase reporter and the indicated expression plasmids. The luciferase assay was performed 24 hr after transfection. Graphs show mean ± SD, n = 3, *P < 0·01. (c) 293T cells were transfected with gC1qR with or without C1qA expression plasmid for 24 hr. The immunoblot was performed with the indicated antibodies.
Discussion
Innate immunity is the first line of defence against viral infections. The pattern recognition receptors such as TLRs, NLRs and RLRs are activated by their ligands, then initiate a series of signal transductions that lead to the production of type I IFNs.3 Through the activation of the Janus kinase–signal transducer and activator of transcription pathway, type I IFNs induce the transcription of a wide range of antiviral genes.23 These signalling events are critical for establishment by the cell of an antiviral state to limit pathogen intrusion and regulate adaptive immunity. In the long-term, the host has evolved two major mechanisms for the recognition of viral RNA, which are the TLR-mediated and RLR-mediated viral RNA recognition. Although TLRs such as TLR3, TLR7 and TLR8 play an important role in the restriction of RNA viral infection and the development of adaptive immunity, they only exist in limited cell types, such as macrophages and dendritic cells. In contrast, RLRs exist in almost all cell types, so RLR-mediated antiviral signalling plays a wider role in controlling RNA viral infection.5 During the viral infection and replication, the exposed viral RNA is recognized by RLRs in the cytosol. A recent study using a RIG-I pathway reconstitution system showed that RIG-I can detect as few as 20 molecules of 5′-ppp RNA per cell. Theoretically, this means that a cell can respond to the presence of several viruses in the cytosol through RLR-mediated signalling.24 Hence, to a certain extent, the RLRs give non-immune cells an antiviral ability.
The complement is considered one of the main effectors of antibody-mediated immunity. Extensive studies have been aimed at understanding the role of complement in defending against pathogen infection, bridging innate and adaptive immunity, clearing immune complexes and apoptotic cells, and in autoimmune diseases, whereas the intracellular role of complement is barely known.16,17 Our results show that the first component of complement C1qA expressed in 293T cells acts as a positive regulator in RIG-I-mediated innate immune signalling. The positive role of C1qA in RIG-I signalling may be correlated with the function of the C1 receptor gC1qR, which functioned as a negative regulator of RLR-mediated innate immunity. It was reported that gC1qR impaired interaction between RIG-I, MDA5 and VISA, and this provided an explanation for its inhibitory role in the RLR pathway.20 Like gC1qR, we also found that C1qA interacted with the RIG-I and VISA. It is possible that C1qA competitively inhibited the interaction of gC1qR with RIG-I or VISA and so released the inhibitory effect of gC1qR on the RLR signalling pathway. The detailed mechanism needs further study.
Macrophages, monocyte-derived dendritic cells and some cell lines such as THP1 are the main producers of C1qA.25,26 We also reported that C1qA interacted with VISA in THP1 cells. Whether the endogenous C1qA in these cells also possesses similar function needs further study. Our studies revealed that expression of C1qA in 293T cells enhanced RIG-I-mediated antiviral responses. Considering that the RLR pathway exists in various cell types, it is possible that C1qA plays a similar role in physical condition. As a subunit of C1q, C1qA has similar structure to C1qB and C1qC, whether C1qB and C1qC also exhibit similar function as C1qA needs further investigation.
Disclosures
This work was supported by The Ministry of Science and Technology of China (2012CB519003, 2011CB504705, 2009ZX10004-101), and Chinese Academy of Sciences Innovation projects (KSCX2-EW-J-6). Xin Ye is a principal investigator of the Innovative Research Group of the National Natural Science Foundation of China (81021003).
References
- 1.Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 2.Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J. 2009;420:1–16. doi: 10.1042/BJ20090272. [DOI] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 4.Honda K, Yanai H, Takaoka A, Taniguchi T. Regulation of the type I IFN induction: a current view. Int Immunol. 2005;17:1367–78. doi: 10.1093/intimm/dxh318. [DOI] [PubMed] [Google Scholar]
- 5.Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- 6.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–72. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 7.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–82. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–8. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 9.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–40. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 10.Ding SW. RNA-based antiviral immunity. Nat Rev Immunol. 2010;10:632–44. doi: 10.1038/nri2824. [DOI] [PubMed] [Google Scholar]
- 11.Allen I, Moore C, Schneider M, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity. 2011;34:854–65. doi: 10.1016/j.immuni.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, Liu T, Xu LG, Chen D, Zhai Z, Shu HB. SIKE is an IKK epsilon/TBK1-associated suppressor of TLR3- and virus-triggered IRF-3 activation pathways. EMBO J. 2005;24:4018–28. doi: 10.1038/sj.emboj.7600863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bo Z, Lu Z, Cao QL, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. 2009;30:397–407. doi: 10.1016/j.immuni.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 14.Gack MU, Shin YC, Joo CH, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–20. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 15.Mibayashi M, Martínez-Sobrido L, Loo YM, Cárdenas WB, Gale M, Jr, García-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–24. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 17.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–4. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 18.Zhang JJ, Li G, Liu XL, Wang ZF, Liu WJ, Ye X. Influenza A virus M1 blocks the classical complement pathway through interacting with C1qA. J Gen Virol. 2009;90:2751–8. doi: 10.1099/vir.0.014316-0. [DOI] [PubMed] [Google Scholar]
- 19.Abed Y, Goyette N, Boivin G. Generation and characterization of recombinant influenza A (H1N1) viruses harboring amantadine resistance mutations. Antimicrob Agents Chemother. 2005;49:556–9. doi: 10.1128/AAC.49.2.556-559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu L, Xiao N, Liu F, Ren H, Gu J. Inhibition of RIG-I and MDA5-dependent antiviral response by gC1qR at mitochondria. Proc Natl Acad Sci USA. 2009;106:1530–5. doi: 10.1073/pnas.0811029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu JH, Teh BK, Wang L, Wang YN, Tan YS, Lai MC, Reid KB. The classical and regulatory functions of C1q in immunity and autoimmunity. Cell Mol Immunol. 2008;5:9–21. doi: 10.1038/cmi.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim BL, Reid KB, Ghebrehiwet B, Peerschke EI, Leigh LA, Preissner KT. The binding protein for globular heads of complement C1q, gC1qR. Functional expression and characterization as a novel vitronectin binding factor. J Biol Chem. 1996;271:26739–44. doi: 10.1074/jbc.271.43.26739. [DOI] [PubMed] [Google Scholar]
- 23.Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–63. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 24.Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–30. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castellano G, Woltman AM, Nauta AJ, et al. Maturation of dendritic cells abrogates C1q production in vivo and in vitro. Blood. 2004;103:3813–20. doi: 10.1182/blood-2003-09-3046. [DOI] [PubMed] [Google Scholar]
- 26.Walker DG. Expression and regulation of complement C1q by human THP-1-derived macrophages. Mol Chem Neuropathol. 1998;34:197–218. doi: 10.1007/BF02815080. [DOI] [PubMed] [Google Scholar]