

Abstract
Seven transmembrane receptors (7TMRs) are nature's prototype allosteric proteins made to bind molecules at one location to subsequently change their shape to affect the binding of another molecule at another location. This paper attempts to describe the divergent 7TMR behaviours (i.e. third party allostery, receptor oligomerization, biased agonism) observed in pharmacology in terms of a homogeneous group of allosteric behaviours. By considering the bodies involved as a vector defined by a modulator, conduit and guest, these activities can all be described by a simple model of functional allostery made up of the Ehlert allosteric model and the Black/Leff operational model. It will be shown how this model yields parameters that can be used to characterize the activity of any ligand or protein producing effect through allosteric interaction with a 7TMR.
LINKED ARTICLES
This article is part of a themed section on the Molecular Pharmacology of G Protein-Coupled Receptors (GPCRs). To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.165.issue-6. To view the 2010 themed section on the same topic visit http://onlinelibrary.wiley.com/doi/10.1111/bph.2010.159.issue-5/issuetoc
Keywords: drug nomenclature, receptor agonists and antagonists, drug classification
Seven transmembrane receptors (7TMRs) are proteins residing on the cell surface to transmit chemically coded information to the cell. They do so through ligand-induced changes in conformation that are read by other proteins in the cytosol. This is a general mechanism of protein function referred to as allosterism. By definition, allosteric interactions on proteins occur through the binding of a molecule to affect the free energy of conformation of the protein where this subsequently affects its behaviour towards the cell or other molecules. When a molecule binds to a receptor protein, the energy of the protein changes irrespective of how relatively small the molecule is in relation to the protein. This change in energy can lead to a change in the behaviour of the receptor. This paper will discuss 7TMRs in terms of allosteric protein function in an attempt to unify their complex behaviour through general concepts.
Receptor states and models
Early models of 7TMR function denoted the mechanics of receptor function through the formation of an ‘active’ receptor state. These approaches to describe agonist effect utilized linkage models so called because they identify the protein species present and link them with isoenergetic pathways. The resulting models parsimoniously defined a minimum number of receptor states for lack of data to indicate otherwise; that is, there usually was only a single cellular readout (e.g. tissue response) for the presence of receptor active states. These models were referred to by the number of defined spontaneously formed states (e.g. R and R*) and given identities such as ‘two-state’ models.
As technology progressed to yield assays that measure multiple 7TMR-mediated drug responses, it has become evident that different ligands can ‘traffic’ stimulus to different cellular pathways with varying emphasis. This phenomenon, commonly referred to as functional selectivity or biased agonism (vide infra), has been proposed on theoretical grounds to be due to the agonist-selective formation of different receptor conformations (Kenakin, 1995); at present, the effect has been observed for many receptor types (for reviews, see Gurwitz and Haring, 2003; Hermans, 2003; Kenakin and Miller, 2010). These effects will be discussed later in this paper, but for the present discussion, it is relevant to note that such trafficking phenomena are incompatible with the mediation of 7TMR response through a single receptor active state. This appears to be in conflict with the parsimoniously named ‘two-state’ models used to describe receptor action, but this conflict is only apparent. It should be noted that the two states described in the two-state model refer to spontaneously formed receptors (inactive and active); once a ligand binds to the receptor, a thermodynamically distinct new state of the receptor is formed. Under these circumstances, a condition of multiple receptor active states is actually not incompatible with the existing linkage models of receptor function notwithstanding their limited nomenclature. This is because the thermodynamic parameters defining the changes in receptor reactivity (see Figure 1) actually can define a very large number of receptor states through the modification of receptor affinity for coupling protein such as G-proteins with parameters such as α (for the extended ternary complex model –Samama et al., 1993) and γ and δ (cubic ternary complex model –Weiss et al., 1996a,b,c). Therefore, far from being ‘two-state’ models, these are multi-state models that are capable of describing complex 7TMR behaviours towards ligands and cells. In spite of this fact, it should still be recognized that linkage models are limited in their description of receptor systems due to the requirement that they pre-define the receptor species present in the system. It will be seen that this requirement is inconsistent with protein behaviour as given by molecular dynamics that describe proteins as spontaneously sampling numerous conformations in response to the energy of the system. Thus, proteins can be thought of as existing in systems of multiple conformations such that at any instant, a snapshot of the system would yield a collection of conformations referred to as an ensemble. While linkage models can accommodate multiple ligand states of the receptor, they require pre-identification of multiple spontaneously formed receptor conformations, and this leads to complex models containing many unverifiable parameters; multiple protein conformational ensembles are better seen through probabilities of state formation (Onaran and Costa, 1997; Onaran et al., 2002). However, for the purposes of characterizing allosteric drug parameters, it will be seen that linkage models furnish the tools to ascribe numbers to modulators to guide medicinal chemistry.
Figure 1.

Linkage models for receptor function. Receptors are assumed to exist in two states: active (with respect to coupling to G-proteins to induce response denoted [Ra]) and inactive ([Ri]). The ligand A binds to the receptor thereby co-binding with G-proteins. It can be seen that the presence of ligand A on the receptor changes the interaction constants of the receptor by a factor α for the extended ternary complex model and δγ for the cubic ternary complex model. Since α and δγ values can be unique to the ligand A, this allows each ligand to stabilize a unique conformation. The term ‘two state’ thus refers only to the species Ra and Ri and not to the response capability of the system in the presence of agonists.
7TMRs as allosteric machines
The full spectrum of 7TMR behaviour towards ligands and cells includes complex probe-dependent cooperative interaction between co-binding ligands, oligomerization on the cell surface and biased cell signalling. All of these effects can be described economically in terms of considering 7TMRs as allosteric machines, and it is worth defining the elements of 7TMR signalling systems in this light. A basic tenet of this approach is to view allostery as a transfer of energy, through the protein, between two sites of interaction with external bodies. A useful terminology for this type of system is to view one of the bodies as a ‘modulator’ transmitting energy through a ‘conduit’ (the receptor protein) to a ‘guest’ (the other external body binding to the protein). This transfer of energy is vectorial (i.e. a modulator producing a change in the behaviour of a guest) but must be considered bidirectional; that is, modulators and guests are interchangeable with the nomenclature existing only to define a given 7TM function. Thus, a modulator agonist could increase the affinity of a receptor for a G-protein guest (a vector directed towards the cytosol) or, equally a G-protein modulator could increase the affinity of the receptor for an agonist guest (vector aimed to the extracellular space). The energy flow is of equal magnitude. A representation of such a modulator/conduit/guest system is shown in Figure 2 where a modulator binds to its own site on the receptor (conduit) to alter the effect of a guest, in this case an agonist. It should be noted that all 7TMR agonism is allosteric with all agonists being modulators affecting the response of the protein to cytosolic signalling proteins as guests (vide infra).
Figure 2.

An allosteric system consisting of a modulator binding to the receptor (conduit) to affect the interaction of a guest with the same receptor. The modulator and guest nomenclature is interchangeable since the energy of interaction through the conduit is equal magnitude and bidirectional. The modulator/guest nomenclature identifies a vector for purposes of identifying specific receptor effects.
It is also useful to consider the vectorial nature of allosteric effect to classify 7TMR behaviours. Figure 2 shows guest allostery whereby the reactivity of a protein towards two molecules is affected by the binding of those same molecules; the binding of one of the molecules affects the binding of the other. This is the earliest form of allosterism described as in, for example, the inhibition of metabolic enzymes by structurally diverse products of subsequent enzymes in a metabolic pathway. These effects led to the postulate that proteins can be modulated through binding of molecules at more than one location (Umbarger, 1956). There also are allosteric vectors directed along the plane of the cell membrane, which mediate interactions between two or more receptors to produce receptor oligomers (Bouvier, 2001; Angers et al., 2002; James et al., 2006; Milligan et al., 2006). These effects also can involve a receptor and other membrane bound protein such as a RAMP (receptor activity modifying protein –Hay et al., 2006). There are different kinds of allosteric systems that can result from cell surface receptor oligomerization. For instance, a receptor could itself function as a modulator to modify an existing agonist/receptor/G-protein system (Figure 3A). Alternatively a receptor may dimerize to form a new conduit and thereby construct a system whereby a ligand binding to one protomer affects signalling mediated by the other protomer (Figure 3B). There is a great deal of literature describing the function of 7TMR homodimers and heterodimers, which are beyond the scope of this present paper (for reviews, see Breitwieser 2004; Franco et al. 2008; Giraldo 2008; Gurevich and Gurevich 2008; Milligan 2008). Finally, the allosteric vector may be directed into the cytosol; as discussed previously, every 7TMR agonist is an allosteric modulator. As seen in Figure 4, cytosolic signalling proteins such as G-proteins, GRKs, β-arrestin and other coupling proteins form the array of guests for the modulator/conduit pairs on the cell surface.
Figure 3.
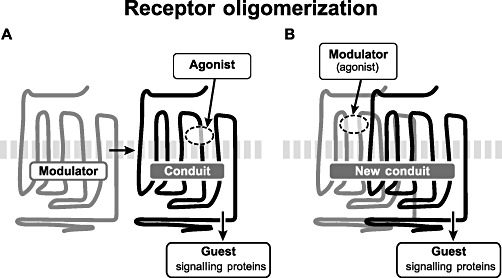
Complexation of receptors along the plane of the cell membrane. (A) Receptor functioning as a modulator to affect the interaction of an agonist with another receptor and signalling guest molecules in the cell. (B) Receptor hetero- or homodimer functioning as a new conduit to allow a modulator (agonist) for one receptor to affect a signalling interaction with the other protomer in the dimer.
Figure 4.

Allosteric system with the allosteric vector directed towards the cytosol. Modulators (agonists) bind to the conduit to transmit energy to cytosolic signalling proteins as guests. The conformation of the conduit dictates differential interaction with each guest to produce possible bias in activation.
The binding of molecules to separate sites leads to a permissive system with unique properties that emanate from the allosterically modulated receptor (with both ligands co-bound). Specifically, the behaviour of the modulator-bound protein can differ for various co-binding ligands, leading to one of the most important features of allostery, namely that of probe dependence. For example, considering the endogenous ligand as a probe of receptor function, a given allosteric modulator can have variable effects on different probes. A second important feature of allostery is saturation of effect. Unlike competitive mechanisms where effects can continue as long as different quantities of the interactants are added to the system, allosteric effects cease when the allosteric site on the protein is saturated. The properties of probe dependence and saturation of effect form the basis of 7TMR complex behaviours discussed in this paper.
There are no pre-set rules for how a given allosteric modulator should affect receptor probes. For example, receptor responsiveness can be reduced to yield allosteric antagonism. An important feature of allosteric antagonists is that they come to a maximal asymptotic effect (when the allosteric site is fully occupied). Under these circumstances, the maximal effect they have on a receptor system is determined by co-operativity factors (vide infra). For example, a surmountable allosteric antagonist produces dextral displacement of agonist dose–response curves that come to a maximal value, causing distinct curvature of the derivative Schild regression (Christopoulos and Kenakin, 2002). Similarly, the principle of probe dependence dictates that different probes may be affected in different ways; that is, an allosteric antagonist may block some agonists but not others. Allosteric modulators also can increase the responsiveness of protein targets; these molecules are referred to as positive allosteric modulators (PAMs). Such potentiation is also subject to probe dependence as seen in the agonist dependent and pathway dependent effects of the PAM NOVO2 (6,7-dichloro2-methylsulfonyhl-3-tert-butylaminoquinoxaline). This modulator produces differential potentiation of the natural agonists GLP-1(7-36)NH2 and oxyntomodulin (vide infra) and can even change the quality of agonist signal produced by the activated receptor. Specifically, while both of these agonists produce signalling through cAMP and calcium, NOVO2 potentiates cAMP but not calcium (Koole et al., 2010). In general, the unique allosteric properties of saturation of effect and probe dependence lead to a highly flexible array of 7TMR-ligand behaviours that allow 7TMRs to be efficient signal transmitters both for natural ligands and also for synthetic molecules aimed at therapeutic effect. It is worth considering how these properties can be exploited in drug discovery.
Unique effects of allosteric modulators
Orthosteric molecules (i.e. antagonists, inverse agonists) preclude access of other molecules such as agonists to the receptor resulting in a pre-emptive system. This leads to a common maximal result for all such antagonists, namely an inactivated (or in the case of partial agonists, a partially activated) receptor to all agonists. In contrast, allosteric molecules are permissive in that they potentially allow interaction of the protein with other molecules. This effect and the property of saturation of effect cause allosteric modulators to have a unique range of activities. These are:
a. Allosteric modulators have the potential to alter the interaction of very large proteins
When two large proteins interact, there often are multiple areas of contact. It would be expected that it would be difficult for an orthosteric ligand, binding to a single location on the protein to disrupt such an interaction through steric hindrance. In this sense, an orthosteric molecule is defined as one binding to the natural binding site for the endogenous ligand. An allosteric ligand binding to a separate site on the protein by definition produces an effect on the orthosteric binding site through a change in protein conformation. There are data to show that allosteric ligands can stabilize new global conformations of the receptor, and this has the potential to alter the position of numerous areas of the protein. Under these circumstances, it would be predicted that multiple binding loci would be affected. An example of this type of effect is the blockade of the interaction of the chemokine CCR5 receptor and the HIV-1 virus coat protein gp120 by the allosteric ligand aplaviroc (a molecule 1/200 the size of the proteins that binds to a site different from those binding chemokines and HIV-1) to prevent HIV-1 infection (Watson et al., 2005).
b. Allosteric modulators have the potential to modulate but not completely activate and/or inhibit receptor function
Allosteric modulators can produce limited effects on target proteins because complete occupancy of the allosteric site produces saturation of effect. Unlike an orthosteric antagonist, an allosteric antagonist modulator can reduce the responsiveness of a target without completely blocking its function. Saturability of effect can lead to another property of modulators, namely the possibility of dissociating the length of time the effect is operative (i.e. target coverage) and the intensity of effect. Under these circumstances, high doses of a limited allosteric antagonist can yield a pool of drug to give long-lasting response without overdose.
c. Allosteric modulators have the potential to preserve physiological patterns
While direct agonism produces blanket activation of systems, PAMs potentiate the existing responses in proportion to the natural physiological tone. This may have relevance to organs such as the brain where failing complex patterns of neurological signalling may need to be augmented (as in diseases such as Alzeimer's).
d. Allosteric modulators may yield therapies with reduced side effects
In therapies where augmentation of physiological effect is required, PAMs would be expected to have a lower side-effect profile. This is because they produce no direct effect but rather have actions only when the system is active through natural presence of the endogenous agonist.
e. Allosteric antagonists can produce texture in antagonism
While orthosteric antagonists all produce a common end product upon saturation of binding (namely an inoperative biological target), allosteric antagonists produce antagonism through alteration of protein target conformation. Since these alterations in conformation need not be identical, different allosteric modulators could produce pharmacological blockade through production of different receptor species. For example, it has been shown that for the CCR5 receptor, the antibody binding profiles of Ab45531 and Ab45523 differ in the presence of the allosteric HIV-1 entry inhibitors TAK779, SCH-C and aplaviroc, suggesting that these modulators produce different tertiary conformations of the CCR5 receptor (Kenakin, 2007).This may be of importance in diseases such as AIDs where it is expected that HIV-1 viral mutation eventually will lead to tolerance to an HIV-1 entry inhibitor as the virus mutates to a form that can utilize the allosterically modified receptor (Trkola et al., 2002; Kuhmann et al., 2004). Therefore, once resistance to one type of modulator is observed, therapy with another modulator that produces a different conformation might be beneficial.
f. Allosteric modulators can have separate effects on agonist affinity and efficacy
Allosteric changes need not be in the same direction (antagonism or potentiation), leading to the possibility that an allosteric modulator could change agonist affinity in one direction and efficacy in another. For example, the CCR5 allosteric modulator aplaviroc minimally affects the binding of the chemokine CCL5 to the receptor but completely blocks CCL5-mediated agonism (Maeda et al., 2004; Watson et al., 2005). Thus, while the high affinity binding of CCL5 is not greatly affected by aplaviroc (suggesting that G-protein binding is not altered by the allosteric effect), the steps leading to G-protein activation and subsequent cellular response are completely blocked. This is consistent with aplaviroc having little effect on CCL5 affinity but a strong negative effect on CCL5 efficacy. This is similar to the effect of the ester CPCCOEt (7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester), which completely blocks the responses to glutamate in CHO cells naturally expressing human GluR1b receptors while not affecting glutamate binding (Litschig et al., 1999). One particular combination of these activities can be useful, namely an allosteric antagonist modulator that increases agonist affinity but decreases agonist efficacy; this combination could lead to a molecule that becomes more potent with higher agonist concentrations. This is because allosteric energy flow is reciprocal in nature; that is, if the modulator increases the affinity of the agonist, then the agonist will also increase the affinity of the modulator in a like manner; this has been shown experimentally (Trankle et al., 1999). Therefore, the presence of higher agonist concentrations will promote allosteric antagonist binding and lead to greater antagonism. This profile can be seen with antagonists such as ifenprodil (for NMDA receptors; Kew et al., 1996) and Org27569 (for cannabinoid receptors – see Price et al., 2005).
g. Allosteric modulators may have an extraordinary selectivity for receptor types
Physiological orthosteric binding sites for endogenous ligands (neurotransmitters, hormones) may be highly conserved between receptor subtypes, making it difficult to attain selectivity through interaction at this site. This conservation is not necessary for allosteric binding sites, and these may be more diverse between subtypes of receptor (Melchiorre et al., 1989; Ellis et al., 1991; Liang et al., 1996; Gnagey et al., 1999; Johnson et al., 2004).
h. Allosteric modulators exercise ‘probe dependence’
Since allosterism involves a change in the shape of the protein, it is possible that a change in shape that is catastrophic to the activity of one probe (i.e. agonist) may have no effect at all on another, especially if those probes bind to different regions of the protein. For instance, the CCR5 allosteric modulator aplaviroc produces very little effect on the binding of the chemokine CCL5 to the receptor but completely blocks the binding of the chemokine CCL3 (Watson et al., 2005). This effect has implications both for the therapeutic application and mode of discovery of allosteric modulators. Regarding how allosteric modulators are discovered and developed, probe dependence dictates that the endogenous ligand targeted therapeutically should be used in the screening and discovery process. For example, a PAM may be of use in augmenting a failing cholinergic neuronal transmission in Alzheimer's disease (Maelicke and Albuquerque, 1996; Krause et al., 1998). However, the natural agonist (acetylcholine) is chemically unsuitable for use in many drug discovery screens and subsequent experiments thus stable analogues, namely carbachol and/or pilocarpine are often used in the screening process. However, PAMs such as LY2033298 have been shown to cause agonist-dependent differential potentiation of different agonists such as acetylcholine and oxotremorine (Suratman et al., 2011). In general, the existing data suggest that if the natural agonist (such as acetylcholine) cannot be used in the screening process, then the modulator should be tested with it as early on in the discovery and development process as possible. This also is relevant to PAMs for targets with multiple natural agonists. For example, while the anti-diabetic PAM NOVO2 produces a fivefold potentiation of one of the natural agonists for the GLP-1 receptor GLP-1(7-36)NH2, it produces a 25-fold potentiation of oxyntomodulin, another natural agonist for this receptor (Koole et al., 2010). These data suggest that all agonists for a given receptor need be tested when studying the effects of allosteric modulators. With regard to the impact of probe dependence on therapeutic application of allosteric modulators, the possibility exists that a given modulator will block or potentiate the endogenous agonist and have no effect on other agonists. For example, HIV-1 utilizes the CCR5 chemokine receptor to cause infection and CCR5 allosteric antagonists block this effect. Interestingly, however, it has been shown that allosteric modulators have varying relative activities of for HIV-1 blockade versus chemokine-induced blockade of CCR5 internalization (Muniz-Medina et al. 2009). Specifically, CCR5 allosteric modulators have differing relative potency as blockers of HIV entry and CCL5-induced CCR5 internalization. This texture of antagonism could be useful in AIDs therapy as it has been shown that the CCR5 chemokine receptor mediates favourable protection in progression to AIDs after HIV-1 infection (Gonzalez et al., 2005). This suggests that a superior allosteric modulator would block the utilization of CCR5 by HIV-1 but otherwise allow normal chemokine function for this receptor; this is possible with allosteric modulators (Muniz-Medina et al. 2009).
Quantifying allosteric effect
Allosteric effects can be quantified by comparing dose-response curves, obtained in the presence of different concentrations of allosteric modulator, to a model. The most simple model for doing this is an amalgam of the Ehlert model of allosteric receptor effect (Ehlert, 1988) and the Black/Leff operational model of receptor function (Black and Leff, 1983). These have been combined to yield a model that can describe the functional effects of allosteric modulators (see Ehlert, 2005; Kenakin, 2005; Price et al., 2005). A schematic diagram relating the relevant elements is given below, showing receptors (R) bind to a receptor probe (agonist, A) and an allosteric modulator (B):
 |
1 |
Allosteric modification of effect is quantified by two co-operativity factors, namely α and β. The term α quantifies the effect of the modulator B on the affinity of the receptor to A (and similarly the reciprocal effect on affinity that A has on the affinity of B). The term β quantifies the effect the modulator has on the efficacy of A. It can be seen from this model that an allosteric modulator cannot be characterized by only affinity (KB−1) but rather will also have a type of ‘efficacy’ for the receptor and its interaction with A in the form of α and β.
A concise and flexible form of this model has been formulated by Leach et al. (2007) to furnish the equation that incorporates salient aspects of direct effects (allosteric agonism) and modification of co-binding agonist effects of allosteric modulators:
 |
2 |
where Em is the maximal response capability of the system, τA is the efficacy of the co-binding probe agonist, KA and KB the equilibrium dissociation constants of the agonist and antagonist–receptor complexes respectively and n a fitting factor for the curves. This model also has the capability of describing direct agonism for the modulator; τB refers to the efficacy for direct agonism of the allosteric modulator.
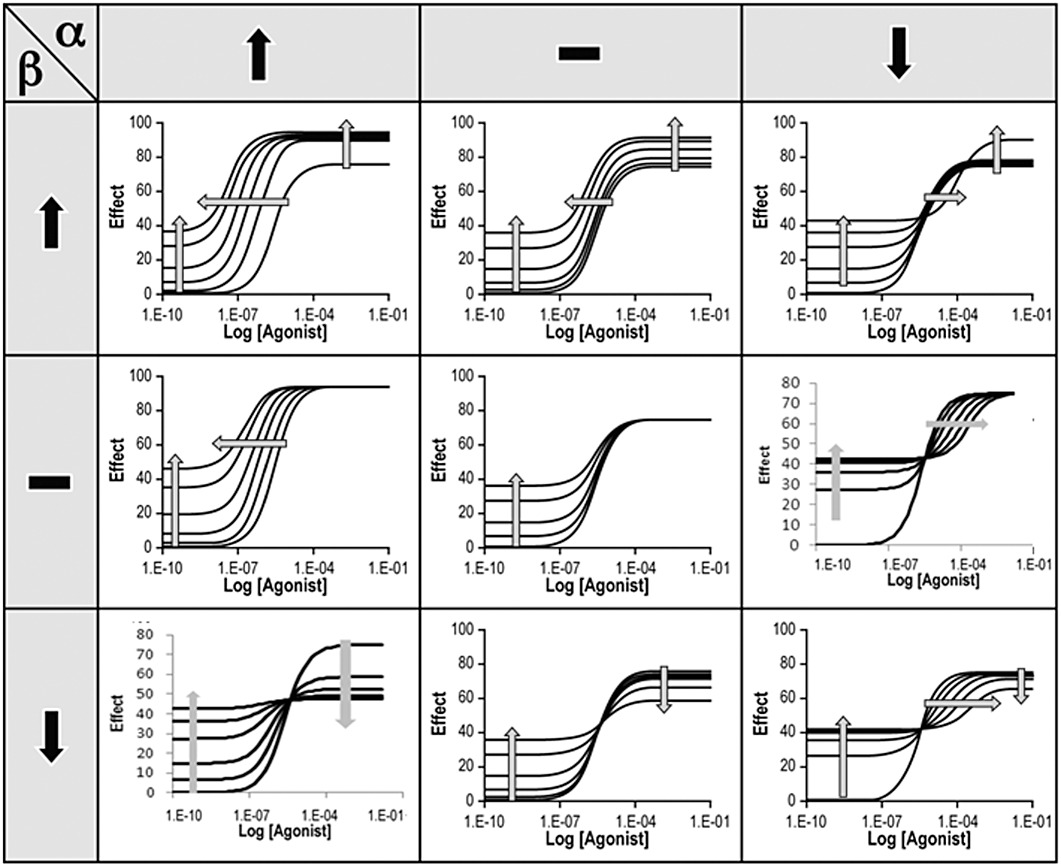
It is worth exploring the capability of this model in terms of the characterization of allosteric modulators. Considering modulators that have no direct agonist effect, there are three possible effects (increase, no effect and decrease) on affinity (α) and efficacy (β) of the agonist. This leads to (3 × 3) − 1 possible combinations of effect; these are shown in Figure 5. It can be seen that every possible effect on dose–response curve location parameter (EC50) and/or maxima can be described by equation 2. It also shows that a full description of the allosteric effects of a modulator requires three estimates: KB (the concentration of modulator that binds to the receptor protein), α (effect on affinity of the co-binding ligand) and β (effect on efficacy of the co-binding ligand). Obviously, some of these will be unique to the particular co-binding ligand.
Figure 5.

Possible outcomes of effect for allosteric modulators with no direct agonist activity; curves calculated with equation 2 (Em= 100; τ= 3; n = 1.6; KA= 10 µM; KB= 10 nM). Panels from top row–left: (α= 30/β= 5) (α= 1/β= 5) (α= 0.01/β= 5), middle row–left (α= 30/β= 1) (α= 30/β= 1) (middle panel, no curves: (α= 1/β= 1) (α= 0.01/β= 1), bottom row–left (α= 30/β= 0.3) (α= 1/β= 0.3) (α= 0.01/β= 0.3).
With regard to modulator binding, the potency of an allosteric modulator activity is described by an affinity constant (pKB) much like other antagonists. However, since allosteric systems describe the energy of interaction between two molecules acting on the protein, the effective affinity of the modulator is modified by the α factor provided by the co-binding ligand. Under these circumstances, it can be seen that different α factors for different ligands can lead to varying affinity; that is, the affinity of the modulator is contingent upon the nature of the co-binding ligand. Similarly, the functional effect of an allosteric modulator also depends upon the value of β since this defines the effect of the modulator on the efficacy of the co-binding agonist. In practice, therapeutically targeted allosteric modulators deal with endogenous agonists; therefore, a single set of pKB, α and β values for the endogenous ligand may be sufficient for characterization. However, in the case of multiple endogenous ligands (such as the situation for some peptide receptors), complications may arise as the activity of the allosteric modulator may vary for different endogenous ligands. These ideas also suggest that it may be difficult to relate binding data to function unless it is assured that the nature of the guests and the relative stoichiometry of the conduit to guest in the experimental system will be identical to the physiological system.
Allosteric modulators stabilize conformational states of proteins, and these may also have direct effects in their own right. Therefore, allosteric modulation also can be associated with direct agonism. A pure allosteric agonist binds to its own site on the protein and stabilizes an active state; this process can be independent of the process of natural agonism produced by the endogenous agonist. Since the allosteric agonist does not interfere with the binding of the endogenous agonist, the resulting allosteric agonism will be additive to endogenous agonism. However, since allosterism may produce a global change in the protein conformation, there is no a priori reason why an allosteric agonist could not also produce a modified endogenous agonist response. If the allosteric modulator has direct agonist effect, then 3 × 3 possibilities exist for unique combinations, the added one being where the modulator has no effect on the co-binding agonist (α=β= 1) but where the allosteric agonism is additive to the system. These various effects are shown in Figure 6.
Figure 6.
Possible outcomes of effect for allosteric modulators with direct agonist activity; curves calculated with equation 2 (Em= 100; τ= 3; n = 1.6; KA= 10 µM; KB= 10 nM; τB= 0.25). Panels from top row–left: (α= 30/β= 5) (α= 1/β= 5) (α= 0.01/β= 5), middle row–left: (α= 30/β= 1) (middle panel; α= 1/β= 1) (α= 0.01/β= 1), bottom row–left (α= 30/β= 0.3) (α= 1/β= 0.3) (α= 0.01/β= 0.3).
Detecting allosteric effect
Allosterism can produce surmountable or insurmountable antagonism that can closely resemble that seen with conventional orthosteric ligands under certain circumstances. It is useful to consider techniques that can differentiate allosteric from orthosteric effects since the identification of ligand mode of action is valuable in predicting therapeutic value. Utilizing quantitative models of antagonism there are three general concepts that are useful in identifying allosteric modes of action:
Prediction of acute effect
Adherence to predicted pattern(s)
Probe/concentration exploration
Clearly, the first prerequisite is to identify a quantitative model that describes the effect of the ligand on the observed agonist dose–response curve (point 1, prediction of effect). However, this is a loose criterion as many allosteric antagonists produce surmountable and/or insurmountable effects identical to those seen with conventional orthosteric blockers. However, fitting data for a range of antagonist concentrations (point 2, adherence to predicted pattern) is a more stringent criterion since the pattern of antagonism produced by allosteric antagonists often differs from that seen with orthosteric antagonists. For instance, a surmountable allosteric antagonist of α= 0.1 produces a maximal 10-fold shift to the right of agonist dose–response curves, while a simple competitive antagonist produces a much larger maximal dextral displacement. This illustrates the basic allosteric property of saturation of effect; that is, the effect ceases when the allosteric site is maximally bound. The third idea is an extension of this one, namely the use of as large a range of concentrations as possible to detect possible saturation of effect. In addition, as many receptor probes as possible (i.e. agonists or radioligands) should be utilized to detect possible probe dependence. Thus, if antagonism is limited or varies with different agonists (or radioligands), then allosterism is indicated. Finally, it should be noted that all of these are ‘one-way’ experiments in that the converse is not necessarily true. If no saturation and/or probe dependence is detected, it still may be that low values of α or β are operative (so that saturation occurs at only very, very high concentrations) or the wrong probes were used in attempts to detect probe dependence. It should be pointed out that definitive evidence of allosteric effect is to show differences in the kinetics of interaction of receptor probes in the presence of the suspected allosteric modulator (i.e. Leach et al. 2011).
Biased signalling in allosteric terms
There are data to show that agonists can vary in their capability to activate different signalling pathways in cells; this has been generally referred to as ‘functional selectivity’ or ‘biased agonism’. These effects can be applied to antagonists as well. Discussion of the applicability of these effects to new drug discovery is beyond the scope of this paper but has been reviewed in numerous papers (Hermans, 2003; Kenakin, 2003, 2011; Perez and Karnick, 2005; Leach et al., 2007; Mailman, 2007; Kenakin and Miller, 2010). It will be assumed that these effects are directed from the receptor and thus will transfer to all tissues possessing that receptor. Under these circumstances, biased signalling, if controlled through medicinal chemistry, has the potential of being a relevant approach to selective pharmacologic therapies. Currently, due to the fact that there are relatively few biased ligands in clinical studies, it is premature to judge the importance of this approach. For the purposes of this discussion, the mechanism of action of biased signalling will be considered.
The first theoretical mechanism proposed for biased signalling suggested that different agonists stabilize different receptor conformations, and that these interact differentially with coupling proteins to induce signalling emphasis in cells (Kenakin, 1995). There are data utilizing a number of experimental approaches to support the existence of numerous ligand-specific receptor conformations (see Kenakin and Miller, 2010 for further discussion) consistent with the conformational hypothesis for biased signalling. Such a mechanism also is consistent with the allosteric nature of 7TMR function. As pointed out previously, the ability of a modulator to affect guests differentially is a known feature of allostery (probe dependence). Therefore, while classical guest allosterism and biased agonism have been described in the literature as an apparently separate phenomenon, it should be noted that functional selectivity is nothing more than a cytosolically directed allosterism.
It is interesting to note how the model describing functional allosterism (equation 2; no direct modulator agonism present, τB= 0) also describes biased agonism. This can be shown by redefining the parameters of equation 2 to the following form:
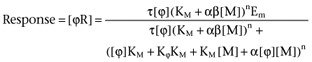 |
3 |
where [ϕ] is the amount of signalling protein and takes the place of agonist [A] in equation 2, response is considered to the amount of receptor-coupling protein complex [ϕR], n is a curve fitting factor and Em the maximal response capability of the system. It is assumed that there is a finite capability of the unliganded receptor to interact with the signalling protein in the form a τ value ([Rt]/KE where [Rt] is the total density of receptors and KE is the Black/Leff equilibrium dissociation constant of the unligand receptor-coupling protein complex). This then allows the ‘agonist’ to be a modulator [M] with an equilibrium dissociation constant for the receptor of KM. The agonist affects the interaction between receptor and signalling protein through the parameters α and β just as any modulator would in a conventional guest allosteric system. Thus, α controls the change in affinity of the receptor and signalling protein produced by the modulator and β the change in signalling quality (efficacy) of the complex in producing response. Simply by redefining the species, it can be seen that signalling bias can be described by the allosteric model as delineated by equation 2.
Figure 7 shows two hypothetical responses (one labelled cAMP and the other β-arrestin complexation) and the responses to two agonists (modulators 1 and 2). For a constant set of system parameters (Em, τ, n), each agonist has a distinct set of KM, α and β values (in accordance with probe dependence). It can be seen that these agonists do not have the same capability to activate each pathway; that is, they are biased towards the pathways with respect to each other. At this point, it is useful to define and differentiate system bias from ligand bias. System bias relates to how well coupled any two signalling pathways are in a cell. All systems will be seen to be biased since the relative activity of ligands for two pathways will depend upon the nature of the cell, the relative stoichiometry of signalling proteins and receptors and how the experiment is conducted. However, what is meant by functional selectivity is that, within a given experimental system bias, some ligands demonstrate a selective ability to activate one of the pathways when compared with another ligand. This can be shown with a bias plot as shown in Figure 7E, which shows the response to one pathway as a function of an observed response to the other pathway. It can be seen that modulator 2 yields a straight line (in this particular case there is little system bias), whereas modulator 1 produces much more β-arrestin response per unit response of cAMP. That is, it is a β-arrestin-biased agonist.
Figure 7.

Biased agonism in terms of the functional allosteric model. (A) The agonist is a modulator labelled M interacting with a receptor conduit, which also interacts with two guests labelled Gα (for a cAMP response) and β-arrestin for an association with β-arrestin. (B) Upon binding of agonist M, the affinity of the conduit changes in accordance with αG and αB for the particular agonist. The quality of action of the couplers also changes in accordance with βG and βB. (C and D) System parameters: Em= 1; [ϕ]= 0.001 for G-protein/0.002 for β-arrestin; n = 1.6; τ= 1 for G-protein / 0.3 for β-arrestin. Kϕ= 0.03 for both G-protein and β-arrestin. For agonist 1 (modulator M1: KM= 10−6 for unliganded receptor, α= 2000; β= 0.3). For agonist 2 (modulator M2: KM= 10–6 for unliganded receptor, α= 1000; β= 0.6). (E) Bias plot of β-arrestin response (ordinates) as a function of cAMP response (abscissa) for agonists 1 and 2 from panels C and D.
Conclusions
This discussion illustrates how the allosteric nature of receptors accounts for all their known behaviours. It also suggests that the characterization of ligand activity on 7TMRs and the nomenclature of 7TMR ligands are complex processes. As distinctive 7TMR ligand profiles are identified, it will be extremely interesting to see how (or even if) they compare with clinical therapeutic phenotypes.
Acknowledgments
I wish to thank numerous colleagues for stimulating discussions on allosterism and biased signalling, including Arthur Christopoulos, Bryan Roth, Marc Caron, Bob Lefkowitz, Christian Watson, Fred Ehlert and Louis Luttrel.
Glossary
- 7TMR
seven transmembrane receptor
- aplaviroc
4-{[4-({(3R)-1-butyl-3-[-R-cyclohexyl(hydroxyl)methyl]-2,5-dioxo-1,4,9-triazaspiro[5,5]undec-9-yl} methyl)phenyl]oxy}benzoic acid hydrochloride
- CCR5
C-C chemokine receptor type 5
- GLP-1(7-36)NH2
Glucagon-like peptide-1 fragment 7-36 amide
- Gp120
HIV-1 envelope glycoprotein GP120
- HIV-1
human immunodeficiency virus type 1
- Ifenprodil
2-(4-benzylpiperidino)-1-(4-hydroxyphenyl)propanol 4-Benzyl-α-(p-hydroxyphenyl)-β-methyl-1-piperidineethanol
- LY2033298
3-amino-5-chloro-6-methoxy-4-methyl-thieno(2,3-b)pyridine-2-carboxylic acid cyclopropylamide
- NOVO2
6,7-dichloro2-methylsulfonyhl-3-tert-butylaminoquinoxaline
- Org
27569, 5-chloro-3-ethyl-1H-indole-2-carboxylic acid [2-(4-piperidin-1-yl-phenyl)-ethyl]-amide
- PAM
positive allosteric modulator
- RAMP
receptor activity modifying protein
- Sch-C
(Z)-(4-bromophenyl) {1′-[(2,4-dimethyl-1-oxido-3-pyridinyl)carbonyl]-4′-bipiperidin-4-yl} methanone O-ethyloxime
- TAK779
N,N-dimethyl-N-[4-[[[2-(4-methylphenyl)-6,7-dihydro-5H-benzocyclohepten-8-yl]carbonyl]amino] benzyl]tetrahydro-2H-pyran-4-aminium chloride
References
- Angers S, Salahpour A, Bouvier M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–435. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonist. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Bouvier M. Oligomerization of G-protein coupled transmitter receptors. Nat Rev Neurosci. 2001;2:274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- Breitwieser G. G protein-coupled receptor oligomerization: implications for G protein activation and cell signaling. Circ Res. 2004;9/23:17–27. doi: 10.1161/01.RES.0000110420.68526.19. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin TP. G-protein coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Ehlert FJ. Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mol Pharmacol. 1988;33:187–194. [PubMed] [Google Scholar]
- Ehlert FJ. Analysis of allosterism in functional assays. J Pharmacol Exp Ther. 2005;315:740–754. doi: 10.1124/jpet.105.090886. [DOI] [PubMed] [Google Scholar]
- Ellis J, Huyler J, Brann MR. Allosteric regulation of cloned M1-M5 muscarinic receptor subtypes. Biochem Pharmacol. 1991;42:1927–1932. doi: 10.1016/0006-2952(91)90591-r. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Cortés A, Mallol J, Ciruela F, Ferré S, et al. G-protein-coupled receptor heteromers: function and ligand pharmacology. Br J Pharmacol. 2008;153:S90–S98. doi: 10.1038/sj.bjp.0707571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldo J. On the fitting of binding data when receptor dimerization is suspected. Br J Pharmacol. 2008;155:17–23. doi: 10.1038/bjp.2008.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnagey AL, Seidenberg M, Ellis J. Site-directed mutagenesis reveals two epitopes involved I nthe subtype selectivity of the allosteric interactions of gallamine at muscarinic acetylcholine receptors. Mol Pharmacol. 1999;56:1245–1253. doi: 10.1124/mol.56.6.1245. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:14334–11440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. How and why do GPCRs dimerize. Trends Pharmacol Sci. 2008;29:234–240. doi: 10.1016/j.tips.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurwitz D, Haring R. Ligand selective signaling and high content screening for GPCR drugs. Drug Discov Today. 2003;8:1108–1109. doi: 10.1016/s1359-6446(03)02897-6. [DOI] [PubMed] [Google Scholar]
- Hay DL, Poyner DR, Sexton PM. GPCR modulation by RAMPS. Pharmacol Ther. 2006;109:173–197. doi: 10.1016/j.pharmthera.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Hermans E. Biochemical and pharmacological control of the multiplicity of coupling at G-protein coupled receptors. Pharmacol Ther. 2003;99:25–44. doi: 10.1016/s0163-7258(03)00051-2. [DOI] [PubMed] [Google Scholar]
- James JR, Oliveira MI, Carmo AM, Iaboni A, Davis SJ. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat Methods. 2006;3:1001–1006. doi: 10.1038/nmeth978. [DOI] [PubMed] [Google Scholar]
- Johnson MP, Nisenbaum ES, Large TH, Emkey R, Baez M, Kingston AE. Allosteric modulators of metabotropic glutamate receptors: leassons learnt from mGlu1, mGlu2, and mGlu5 potentiators and agonists. Biochem Soc Trans. 2004;32:881–887. doi: 10.1042/BST0320881. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Agonist-receptor efficacy II: agonist-trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci. 2003;24:346–354. doi: 10.1016/S0165-6147(03)00167-6. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nat Rev Drug Discov. 2005;4:919–927. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol Sci. 2007;28:407–415. doi: 10.1016/j.tips.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- Kenakin TP, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew JNC, Trube G, Kemp JA. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurons. J Physiol. 1996;497(3):761–772. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koole C, Wooten D, Simms J, Valant C, Sridhar R, Woodman OL, et al. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pthway-selective manner: implications for drug screening. Mol Pharmacol. 2010;78:456–465. doi: 10.1124/mol.110.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause RM, Buisson B, Bertrand S, Corringer P-J, Galzi J-L, Changeux J-P, et al. A positive allosteric effector of the a7 neuronal nicotinic acetylcholine receptor. Mol Pharmacol. 1998;53:283–294. doi: 10.1124/mol.53.2.283. [DOI] [PubMed] [Google Scholar]
- Kuhmann SE, Pugach P, Kunstman KJ, Taylor J, Stanfield RL, Snyder A, et al. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape form small-molecule CCR inhibitor. J Virol. 2004;78:2790–2807. doi: 10.1128/JVI.78.6.2790-2807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive pharmacology. Trends Pharmacol Sci. 2007;28:382–390. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. 2011. Quantification of Allosteric Interactions at G Protein–Coupled Receptors Using Radioligand Binding Assays http://www.currentprotocols.com/protocol/ph0122.
- Liang JS, Carsi-Gabrenas J, Krajesewski JL, McCafferty JM, Parkerson SL, Santiago MP, et al. Anti-muscarinic toxins from dendroaspis angusticeps. Toxicon. 1996;34:1257–1267. doi: 10.1016/s0041-0101(96)00109-2. [DOI] [PubMed] [Google Scholar]
- Litschig S, Gasparini F, Ruegg DF, Stoehr N, Flor PJ, Vranesic I, et al. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol. 1999;55:453–461. [PubMed] [Google Scholar]
- Maeda K, Nakata H, Koh Y, Miyakawa T, Ogata H, Takaoka Y, et al. Spirodiketopiperazine-based CCR5 inhibitor which preserves CC-chemokine/CCR5 interactions and exerts potent activity against R5 human immunodeficiency virus type 1 in vitro. J Virol. 2004;78:8654–8662. doi: 10.1128/JVI.78.16.8654-8662.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelicke A, Albuquerque EX. New approach to drug therapy of Alzheimer's dementia. Drug Discov Today. 1996;1:53–59. [Google Scholar]
- Mailman RB. GPCR functional selectivity has therapeutic impact. Trends Pharmacol Sci. 2007;29:390–396. doi: 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchiorre C, Minarini A, Angeli P, Giardina D, Gulini U, Quaglia W. Polymethylene tetraamines as muscarinic receptor probes. Trends Pharmacol Sci. 1989;10(Suppl.):55–59. [PubMed] [Google Scholar]
- Milligan G. A day in the life of a G protein coupled receptor: the contribution to function of G protein-coupled receptor dimerization. Br J Pharmacol. 2008;153:S216–S229. doi: 10.1038/sj.bjp.0707490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G, Canals M, Pediani JD, Ellis J, Lopez-Gimenez JF. The Role of GPCR dimerisation/oligomerisation in receptor signalling. Ernst Schering Found Symp Proc. 2006;48:145–161. doi: 10.1007/2789_2006_007. [DOI] [PubMed] [Google Scholar]
- Muniz-Medina VM, Jones S, Maglich JM, Galardi C, Hollingsworth RE, Kazmierski WM, et al. The relative activity of ‘function sparing’ HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property? Mol Pharmacol. 2009;75:490–501. doi: 10.1124/mol.108.052555. [DOI] [PubMed] [Google Scholar]
- Onaran HO, Costa T. Agonist efficacy and allosteric models of receptor action. Ann NY Acad Sci. 1997;812:98–115. doi: 10.1111/j.1749-6632.1997.tb48150.x. [DOI] [PubMed] [Google Scholar]
- Onaran HO, Scheer A, Cotecchia S, Costa T. A look at receptor efficacy. From the signaling network of the cell to the intramolecular motion of the receptor. In: Kenakin TP, Angus JA, editors. The Pharmacology of Functional, Biochemical, and Recombinant Systems Handbook of Experimental Pharmacology. Vol. 148. Heidelberg: Springer; 2002. pp. 217–280. [Google Scholar]
- Perez DM, Karnick SS. Multiple signaling states of G-protein coupled receptors. Pharmacol Rev. 2005;57:147–161. doi: 10.1124/pr.57.2.2. [DOI] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the b2-adrenergic receptor: extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- Suratman S, Leach K, Sexton PM, Felder CC, Loiacono RE, Christopoulos A. Impact of species variability and ‘probe-dependence’ on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br J Pharmacol. 2011;162:1659–1670. doi: 10.1111/j.1476-5381.2010.01184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trankle C, Weyand A, Schroter A, Mohr K. Using a radioalloster to test predictions of the cooperativity model for gallamine binding to the allosteric site of muscarinic acetylcholine (m2) receptors. Mol Pharmacol. 1999;56:962–965. doi: 10.1124/mol.56.5.962. [DOI] [PubMed] [Google Scholar]
- Trkola A, Kuhmann SE, Strizki JM, Maxwell E, Ketas T, Morgan T, et al. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc Natl Acad Sci U S A. 2002;99:395–400. doi: 10.1073/pnas.012519099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbarger HE. Evidence for a negative feedback mechanism in the biosynthesis of isoleucine. Science. 1956;123:848. doi: 10.1126/science.123.3202.848. [DOI] [PubMed] [Google Scholar]
- Watson C, Jenkinson S, Kazmierski W, Kenakin TP. The CCR5 Receptor-based mechanism of action of 873140, a potent allosteric non-competitive HIV entry-inhibitor. Mol Pharmacol. 2005;67:1268–1282. doi: 10.1124/mol.104.008565. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor-occupancy model. I. Model description. J Theor Biol. 1996a;178:151–167. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor-occupancy model. II. Understanding apparent affinity. J Theor Biol. 1996b;178:169–182. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor-occupancy model. III. Resurrecting efficacy. J Theor Biol. 1996c;181:381–397. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]