

Abstract
BACKGROUND AND PURPOSE
Ursolic acid (UA) has been extensively used as an anti-leukaemic agent in traditional Chinese medicine. In the present study, we investigated the ability of UA to induce apoptosis in human leukaemia cells in relation to its effects on caspase activation, Mcl-1 down-regulation and perturbations in stress-induced signalling pathways such as PKB and JNK.
EXPERIMENTAL APPROACH
Leukaemia cells were treated with UA after which apoptosis, caspase activation, PKB and JNK signalling pathways were evaluated. The anti-tumour activity of UA was evaluated using xenograft mouse model.
KEY RESULTS
UA induced apoptosis in human leukaemia cells in a dose- and time-dependent manner; this was associated with caspase activation, down-regulation of Mcl-1 and inactivation of PKB accompanied by activation of JNK. Enforced activation of PKB by a constitutively active PKB construct prevented UA-mediated JNK activation, Mcl-1 down-regulation, caspase activation and apoptosis. Conversely, UA lethality was potentiated by the PI3-kinase inhibitor LY294002. Interruption of the JNK pathway by pharmacological or genetic (e.g. siRNA) attenuated UA-induced apoptosis. Furthermore, UA-mediated inhibition of tumour growth in vivo was associated with induction of apoptosis, inactivation of PKB as well as activation of JNK.
CONCLUSIONS AND IMPLICATIONS
Collectively, these findings suggest a hierarchical model of UA-induced apoptosis in human leukaemia cells in which UA induces PKB inactivation, leading to JNK activation and culminating in Mcl-1 down-regulation, caspase activation and apoptosis. These findings indicate that interruption of PKB/JNK pathways may represent a novel therapeutic strategy in haematological malignancies.
Keywords: ursolic acid, apoptosis, leukaemia, PKB, xenograft
Introduction
In recent years, the use of natural products has become widely accepted as a realistic option for the treatment of malignant tumours, and novel anti-tumour compounds from herbal medicines represent attractive alternatives for drug development. Ursolic acid (UA), an active pentacyclic triterpene acid, has been isolated from many medicinal plants, such as Eriobotrya japonica, Rosmarinus offıcinalis and Glechoma hederaceae (Ohigashi et al., 1986; Abe et al., 2002; Taniguchi et al., 2002). This compound is also the major biologically active constituent in Hedyotis diffusa, which is comprehensively used for anti-leukaemic activity in traditional Chinese medicine. UA has been used in traditional Asian medicine for centuries as an antibacterial, antifungal, anti-inflammatory as well as an anti-cancer agent (Ikeda et al., 2008; Shai et al., 2008). The naturally occurring triterpene has recently attracted a great attention as it has been shown to display a wide range of anti-cancer activities, including inhibition of cell growth, induction of cell differentiation and apoptosis. It has been reported to inhibit cell growth and induce apoptosis in various cancer cells by multiple mechanisms including intracellular Ca2+ release (Baek et al., 1997), down-regulations of c-IAPs and Bcl-2 (Choi et al., 2000; Kassi et al., 2009), activation of p53 (Manu and Kuttan, 2008), activation of stress-related signalling (Liu and Jiang, 2007) and inactivation of cytoprotective pathways (Li et al., 2010). Preclinical data have indicated that UA could have potential as an anti-cancer agent, and it would be meaningful and challenging to develop this compound to be a novel anti-tumour drug.
Signalling transduction pathways are believed to be potential therapeutic targets in several cancers including leukaemia (Wenner, 2010). The PI-3-kinase/PKB (PI3K/PKB) pathway plays an important role in the development of various human cancers (Vivanco and Sawyers, 2002). PKB activation leads to phosphorylation of several downstream molecules that are involved in cell cycle, glycogen synthesis, cell death and cell survival. Phosphatase and tensin homologue (PTEN), which acts as a lipid phosphatase for PI at the 3′ end and depletes PIP3 (Maehama and Dixon, 1998), is a negative regulator of the PI3K/PKB cell survival pathway (Stanbolic et al., 1998). Loss of PTEN activity by mutations and deletions at high frequency in many primary and metastatic human cancers leads to constitutive levels of PKB signalling (Blanco-Aparicio et al., 2007).
JNKs, also known as stress-activated protein kinases, were originally identified by their ability to phosphorylate the N-terminal of the transcription factor c-Jun and by their activation in response to a variety of stresses. JNK are multifunctional kinases involved in many physiological processes. The JNK pathway has been shown to play a major role in apoptosis in many cell death paradigms. For example, the JNK pathway is required for cancer cell death induced by many apoptotic stimuli, including DNA damage (Xia et al., 2009), oxidative stress (Shen and Liu, 2006), u.v. irradiation (Sun et al., 2004) and TNFα (De Smaele et al., 2001). It has been noted that the JNK–AP-1 pathway is involved in the increased expression of pro-apoptotic genes such as TNFα, Fas-L and Bak (Fan and Chambers, 2001). JNK can both phosphorylate and regulate the expression of several members of the Bcl-2 protein family, such as BAX (Lei and Davis, 2003), Bcl-2 antagonist of cell death (BAD and Mcl-1) (Donovan et al., 2002; Inoshita et al., 2002), as well as 14-3-3 proteins (Yoshida et al., 2005). Phosphorylation of 14-3-3 proteins by JNK releases pro-apoptotic proteins, such as BAX and FOXO transcription factors, from inactive complexes, thereby facilitating JNK-mediated apoptosis.
Studies investigating the role of signalling cascades in UA-related lethality have primarily focused on those related to oxidative stress and cell signalling pathways. For example in NTUB1 human bladder cancer cells, UA induces G2/M cell cycle arrest and apoptosis through induction of reactive oxygen species (ROS) (Tu et al., 2009). In LNCaP prostate cancer cells, it has been reported that UA induces apoptosis in association with activation of JNK-induced Bcl-2 phosphorylation and degradation (Zhang et al., 2010a). UA has also been shown to inhibit NF-κB activation and enhance the apoptosis induced by the chemotherapeutic agent Taxol(Li et al., 2010). Furthermore, in prostate cancer cells, inactivation of PKB has been implicated in UA-induced apoptosis (Zhang et al., 2010b). However, the detailed functional role of cell signalling pathways, including PKB and JNK in UA-induced apoptosis in human leukaemia cells, has not yet been explored and little is known about the roles of these pathways in UA-mediated anti-leukaemic activity in vivo. The purpose of the present study was to characterize the functional role of PKB and related pathways in the lethal effects of UA on human leukaemia cells. Furthermore, the effect of UA on tumour growth in vivo was evaluated in xenograft mouse model. Our results indicate a hierarchical model of UA-induced lethality in human leukaemia cells characterized by inactivation of the cytoprotective PKB pathway, resulting in JNK activation and culminating in Mcl-1 down-regulation. UA-inhibited tumour growth in vivo was associated with inactivation of PKB and activation of JNK. Taken together, the results of the present study demonstrate that UA could be effective in the therapy of leukaemia and possibly other haematological malignancies.
Methods
Cells and reagents
U937, HL-60 and Jurkat cells were provided by the American Type Culture Collection (ATCC, Manassas, VA) and maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS). The constitutive active form of PKB (PKB-CA) and the dominant-negative PKB mutant (PKB-DN) were kindly provided by Dr Richard Roth (Stanford University, School of Medicine, Stanford, CA) and were subcloned into the pcDNA3.1. U937 cells were stably transfected with PKB-CA and PKB-DN using the Amaxa nucleofectorTM (Cologne, Germany) as recommended by the manufacturer. Stable single cell clones were selected in the presence of 400 µg mL–1 geneticin. Thereafter, the expression of PKB from each cell clone was assessed by Western blot as described below.
Peripheral blood samples for the in vitro studies were obtained from 12 patients with newly diagnosed or recurrent acute myeloid leukaemia (AML) and six patients with acute lymphoma leukaemia (ALL) after informed consent. Approval was obtained from the Southwest Hospital (Chongqing, China) institutional review board for these studies. AML and ALL blasts were isolated by density gradient centrifugation over Histopaque-1077 (Sigma Diagnostics, St. Louis, MO) at 400×g for 38 min. Isolated mononuclear cells were washed and assayed for total number and viability using trypan blue exclusion. Blasts were suspended at 8 × 105 mL–1 and incubated in RPMI 1640 medium containing 10% FBS in 24-well plates. Fresh normal bone marrow mononuclear cells were purchased from Allcells (Emeryvill, CA). After being washed and counted, cells were suspended at 8 × 105 mL–1 before being treated.
UA was purchased from Sigma (St. Louis, MO). LY294002, SP600125 and Z-VAD-FMK were purchased from EMD Biosciences (La Jolla, CA). Antibodies against PKB, phospho-JNK, JNK and β-actin were from Santa Cruz Biotechnology (Santa Cruz, CA); cleaved caspase-3, cleaved caspase-7, cleaved caspase-9, phospho-PKB (Ser473), Bcl-xL, PP2A-B and PP2A-C were from Cell Signaling Technology (Beverly, MA); XIAP, Mcl-1, Bax and Bad were from PharMingen (San Diego, CA); PARP was from Biomol (Plymouth Meeting, PA); caspase-8 was from Alexis (Carlsbad, CA); Bcl-2 was from Dako (Carpinteria, CA); Bim was from EMD Biosciences.
RNA interference and transfection
U937 cells (1.5 × 106) were transfected with 1 µg JNK1-annealed dsRNAi oligonucleotide 5′-CGUGGGAUUUAUGGUCUGUGTT-3′/3′-TTGCACCUAAAUACCAGACAC-5′ (Orbigen, San Diego, CA) using the Amaxa nucleofectorTM as recommended by the manufacturer. After incubation at 37°C for 24 h, transfected cells were treated with UA and subjected to determinations of apoptosis and JNK expression using Annexin V/PI staining and Western blot respectively.
Detection of apoptosis
The extent of apoptosis in leukaemia cells was evaluated by flow cytometric analysis using FITC-conjugated Annexin V/ propidium iodide (PI) (BD PharMingen) staining as per the manufacturer's instruction as previously described (Gao et al., 2009). Both early apoptotic (Annexin V-positive, PI-negative) and late apoptotic (Annexin V-positive and PI-positive) cells were included in cell death determinations.
Western blot analysis
The total cellular samples were washed twice with ice-cold PBS and lysed in 1× NuPAGE LDS sample buffer supplemented with 50 mM dithiothreitol. The protein concentration was determined using Coomassie Protein Assay (Pierce, Rockford, IL); 30 µg protein was separated by SDS-PAGE and transferred to nitrocellulose membrane. Membranes were blocked with 5% fat-free dry milk in 1× Tris-buffered saline (TBS) and incubated with antibodies. Protein bands were detected by incubating with horseradish peroxidase-conjugated antibodies (Kirkegaard and Perry Laboratories, Gaithersburg, MD) and visualized with enhanced chemiluminescence reagent (Perkin Elmer, Boston, MA).
In vivo mouse xenograft assay
NOD/SCID mice (5 weeks old) were purchased from Vital River Laboratories (VRL, Beijing, China). All animal care and experimental procedures were conducted according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University. U937 cells (2 × 106 0.2 mL−1·per mouse) were suspended in sterile PBS and injected s.c. into the right flank of the mice. Mice were randomized into two groups with 10 mice per group. Three days after tumour inoculation, the treatment group received UA (50 mg·kg−1, i.p. for 20 days). The control group received an equal volume of solvent control. Tumour size and body weight were measured after treatment at various time intervals throughout the study. At the termination of the experiment, mice were killed 24 h after the last administration of UA. The tumours were excised and weighed. Tumours were collected at selected times and fixed in 10% paraformaldehyde. Paraffin-embedded tissues were sectioned and processed for haematoxylin and eosin (H&E), terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) and immunohistochemical staining.
TUNEL assay
The apoptotic cells in tissue samples were detected using an In Situ Cell Death Detection kit (Roche, Mannheim, Germany) according to the manufacturer's instructions. After deparaffinization and permeabilization, the tissue sections were incubated with proteinase K for 15 min at room temperature. The sections were then incubated with the TUNEL reaction mixture that contains terminal deoxynucleotidyl transferase (TdT) and fluorescein-dUTP at 37°C for 1 h. After being washed three times with PBS, the sections were incubated with the Converter-POD that contains anti-fluorescein antibody conjugated with horseradish peroxidase (POD) at room temperature for 30 min. The sections were again washed three times with PBS before being incubated with 0.05% 3-3′-diaminobenzidine tetrahydrochloride (DAB) and analysed under a light microscope.
Histological and immunohistochemical evaluation
At the termination of the experiments, tumour tissues from representative mice were sectioned, embedded in paraffin and stained with H&E for histopathological evaluation. For immunohistochemical analysis, tissue sections of 4 µm in thickness were dewaxed and rehydrated in xylene and graded alcohols. Antigen retrieval was performed with 0.01 M citrate buffer at pH 6.0 for 20 min in a 95°C water bath. Slides were allowed to cool for another 20 min followed by sequential rinsing in PBS and TBS-T buffer. Endogenous peroxidase activity was quenched by incubation in TBS-T containing 3% hydrogen peroxide. Each incubation step was carried out at room temperature and was followed by three sequential washes (5 min each) in TBS-T. After being blocked with 10% goat serum for 1 h, sections were incubated with primary antibodies, washed three times in PBS, incubated with biotinylated secondary antibody for 1 h followed by incubation with a streptavidin–peroxidase complex for another 1 h. After three additional washes in PBS, diaminobenzidine working solution was applied. Finally, the slides were counterstained in haematoxylin.
Statistical analysis
Tumour volumes, body weights and percentage of apoptotic cells are presented as mean ± SD. The statistical significance of the difference between control and UA-treated groups was evaluated using Student's t-test. P < 0.05 or P < 0.01 was considered significant.
Results
UA induces apoptosis in U937 leukaemia cells in a dose- and time-dependent manner
U937 leukaemia cells were treated with various concentrations of UA for 6 and 12 h, after which apoptosis was assessed by Annexin V/PI analysis. As shown in Figure 1A, modest degrees of apoptosis were noted with 10 µM UA; this increased substantially with 15 µM UA and became very extensive with 20 µM UA. A time course study of cells treated with 20 µM UA revealed a moderate increase in apoptosis as early as 3 h after drug treatment. These events became apparent after 6 and 12 h of drug treatment and reached near-maximal levels after 24 h of drug treatment (Figure 1B).
Figure 1.
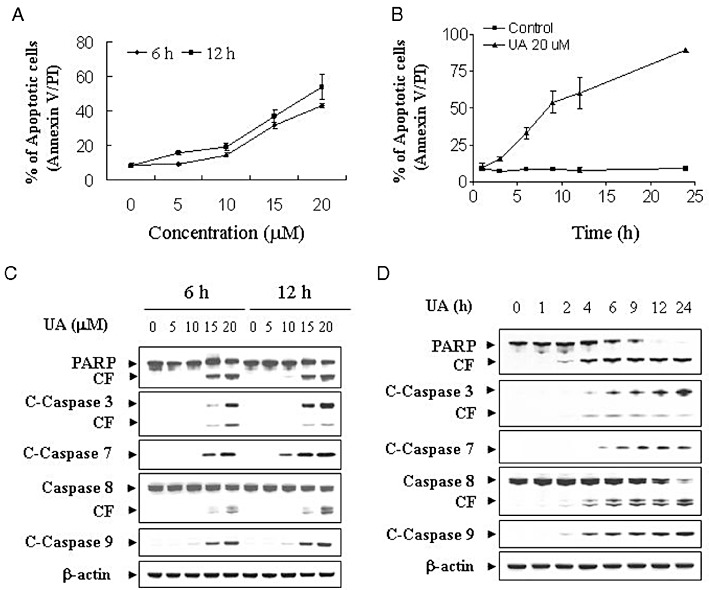
UA induces apoptosis in U937 human leukaemia cells in a dose- and time-dependent manner. (A) U937cells were treated without or with various concentrations of UA as indicated for 6 and 12 h. (B) U937 cells were treated with 20 µM UA for 1, 3, 6, 9, 12 and 24 h. The cells were stained with Annexin V/ PI, and apoptosis was determined using flow cytometry as described in Methods. (C) U937cells were treated without or with various concentrations of UA as indicated for 6 and 12 h. (D) U937 cells were treated without or with 20 µM UA for 1, 2, 4, 6, 9, 12 and 24 h. After treatment of U937 cells with the indicated UA concentration or the indicated time interval, total cellular extracts were prepared and subjected to Western blot analysis using antibodies against PARP, cleaved caspase-3 (C-Caspase-3), caspase-8, cleaved caspase-7 (C-Caspase-7) and cleaved caspase-9 (C-Caspase-9). Each lane was loaded with 30 µg protein. Blots were subsequently stripped and re-probed with antibody against β-actin to ensure equivalent loading and transfer. Two additional studies yielded equivalent results.
Western blot analysis revealed that treatment of U937 cells with 10 µM UA resulted in a slight increase in cleavage/activation of caspase-3, -7, -8 and -9 as well as PARP degradation and a marked increase at concentrations ≥15 µM (Figure 1C). A time course study of cells treated with 20 µM UA revealed moderate increases in cleavage/activation of caspase-3, -7, -8 and -9, as well as PARP degradation after 4 and 6 h treatment. These effects were more apparent after 12 and 24 h treatment (Figure 1D).
Treatment of human leukaemia cells with UA results in down-regulation of Mcl-1
Dose- and time-dependent effects of UA were then examined in relation to expression of various Bcl-2 family members. A dose-dependent study demonstrated that treatment of U937 cells with UA for 6 and 12 h at a concentration of 5 or 10 µM resulted in a slight decrease of Mcl-1 expression. This effect was more evident at concentrations ≥15 µM (Figure 2A). A time course study revealed that treatment of U937 cells with 20 µM UA resulted in a reduction of Mcl-1 expression level 2 h after drug treatment. This effect was more apparent after 6 h of drug treatment. The complete blockage of Mcl-1 was noted at 24 h after drug treatment (Figure 2B). In contrast, exposure to these doses of UA for the various times resulted in little or no change in the expressions of XIAP, Bcl-2, Bcl-xL, Bax and Bad.
Figure 2.
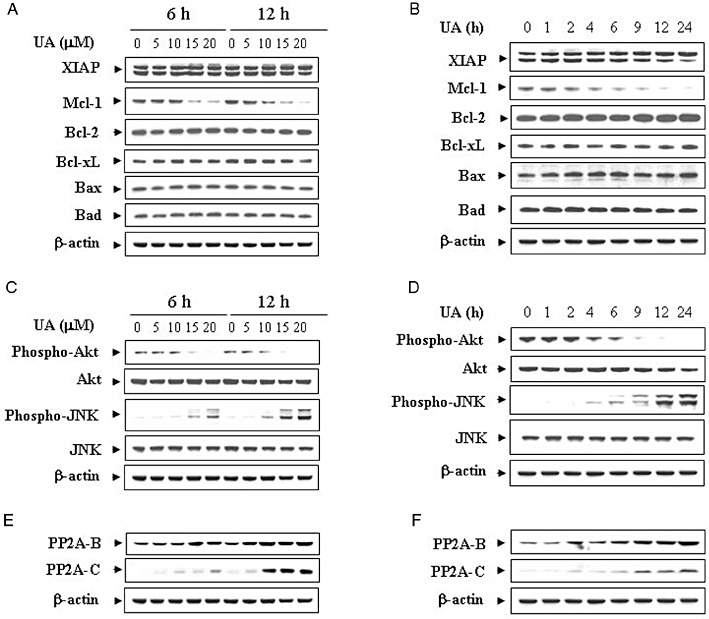
Effects of UA on apoptosis-related gene expression and various signal transduction pathways. (A) U937 cells were treated with the indicated concentrations of UA for 6 and 12 h. (B) U937 cells were treated with 20 µM UA for 1, 2, 4, 6, 9, 12 and 24 h. Total cellular extract were prepared and subjected to Western blot analysis using antibodies against apoptosis-related proteins including XIAP, Mcl-1, Bcl-2, Bcl-xL, Bax and Bad. U937 cells were treated with UA at the indicated concentrations (C) or for the indicated time intervals (D) as described above. Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against phospho-PKB (Ser473), PKB, phospho-JNK and JNK. U937 cells were treated with UA at the indicated concentrations (E) or for the indicated time intervals (F) as described above. Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against PP2A-B and PP2A-C. Each lane was loaded with 30 µg protein. Blots were subsequently stripped and re-probed with antibody against β-actin to ensure equivalent loading and transfer. Two additional studies yielded equivalent results.
Treatment of human leukaemia cells with UA results in a pronounced reduction in levels of phospho-PKB and a marked activation of JNK
Effects of UA were also determined in relation to changes in various signal transduction pathways implicated in apoptosis regulation. Western blot analysis indicated that treatment of U937 cells with UA resulted in a dose-dependent reduction in levels of phospho-PKB (Ser473) but had no significant effects on total PKB (Figure 2C). In addition, treatment of cells with UA caused a dramatic increase of JNK phosphorylation but had no effect on the total JNK. A time course study showed that treatment of U937 cells with 20 µM UA caused a marked decrease in levels of phospho-PKB and a modest increase in the phosphorylation of JNK as early as 4 h after drug treatment; near-maximal effects were obtained at 12 or 24 h (Figure 2D). In contrast, UA had little or no effect on the expression of total or phospho-mTOR, phospho-ERK and phospho-p38 (data not shown). To delineate the mechanism by which UA dephosphorylates PKB, we determined the expressions of phosphatases such as PHLPP and PP2A. The dose and time course study revealed that UA up-regulated the expression of PP2A (subunits B and C) (Figure 2E and F), while the expression of PHLPP did not change (data not shown). These results suggest that a pronounced inactivation of PKB and activation of JNK may play important roles in UA-induced apoptosis in U937 human leukaemia cells.
UA had similar effects on apoptosis in other human leukaemia cell lines as well as primary human leukaemia cells
To determine whether the induction of apoptosis induced by UA was confined to U937 cells, parallel studies were performed with other human leukaemia cell lines, including Jurkat T-lymphoblastic and HL-60 promyelocytic leukaemia cells. UA exhibited similar apoptotic effects on these cells, except that on HL-60 cells (Figure 3A). Also, UA exhibited comparable degrees of caspase-3, -7, -8 and -9 activation and PARP degradation in Jurkat and HL-60 cells (Figure 3B). Similarly, UA down-regulated the expression of Mcl-1 but had no effect on the expressions of XIAP, Bcl-2, Bcl-xL, Bax and Bad in Jurkat and HL-60 cells(data not shown) (Figure 3C). Lastly, the ability of UA to trigger inactivation of PKB and activation of JNK was essentially identical in Jurkat and HL-60 cells to observed in U937 cells (Figure 3D). These results indicate that the effects of UA are not cell type-specific.
Figure 3.
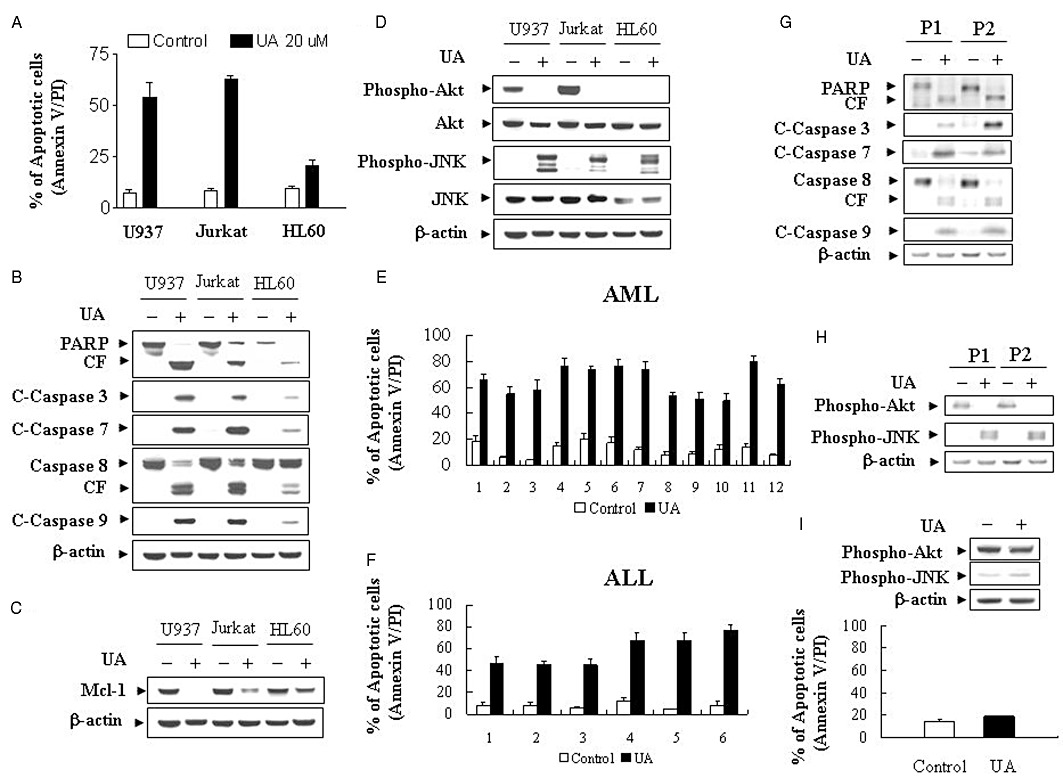
UA induces apoptosis in U937, Jurkat and HL-60 cells and in primary AML and ALL blasts. (A) U937, Jurkat and HL-60 cells were treated with 20 µM UA for 12 h, after which the percentage of apoptotic cells was determined by Annexin V/PI staining and flow cytometry as described in Methods. The values obtained from Annexin V/PI assays represent the mean ± SD for three separate experiments. (B) Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against PARP, C-Caspase-3, C-Caspase-7, caspase-8 and C-Caspase-9. (C) Total cellular extracts were also prepared and subjected to Western blot analysis using an antibody against Mcl-1. (D) Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against phospho-PKB (Ser473), PKB, phospho-JNK and JNK. Blasts from 12 patients with AML (E) and six patients with ALL (F) were isolated as described in Methods. After washing and counting, isolated mononuclear cells were treated without or with 20 µM UA for 24 h, after which the percentage of apoptotic cells was determined by Annexin V/PI staining and flow cytometry as described in Methods. (G) Total cellular extracts of blasts from two AML patients were prepared and subjected to Western blot analysis using antibodies against PARP, C-Caspase-3, C-Caspase-7, caspase-8, C-Caspase-9 as well as cell signalling protein including phospho-PKB (Ser473) and phospho-JNK (H). (I) Normal bone marrow mononuclear cells were treated without or with 20 µM UA for 24 h, after which the percentage of apoptotic cells was determined by Annexin V/PI staining and flow cytometry as described in Methods. For apoptosis assay, values represent the means ± SD for 3 replicate determinations. Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against phospho-PKB (Ser473) and phospho-JNK. For Western blot analysis, each lane was loaded with 30 µg of protein; blots were subsequently stripped and re-probed with antibody against β-actin to ensure equivalent loading. Two additional studies yielded equivalent results.
To determine whether effects of UA on apoptosis were restricted to human leukaemia cell lines, we utilized primary human leukaemia cells isolated from 12 AML and 6 ALL patients. Primary human leukaemia cells were treated without or with 20 µM UA for 24 h, after which apoptosis was determined by Annexin V/PI analysis. As shown in Figure 3E and F, treatment of AML and ALL cells with UA resulted in pronounced increase in apoptosis. Consistent with these results, treatment of primary leukaemia cells from two AML patients with 20 µM UA for 24 h caused cleavage/activation of caspases-9, -8, -7 and -3, as well as PARP degradation (Figure 3G). These effects were also accompanied by inactivation of PKB and activation of JNK (Figure 3H). A parallel study was also performed with normal bone marrow mononuclear cells. As shown in Figure 3I, the UA regimen induced relatively little apoptosis and had no effect on the expression of phospho-PKB and phospho-JNK in normal bone marrow mononuclear cells. Together, these findings indicate that UA selectively kills human leukaemia cell lines and primary leukaemia cells but not normal haematopoietic cells.
UA lethality was associated with the caspase-independent inactivation of PKB and activation of JNK
To determine whether UA-induced inactivation of PKB and activation of JNK were secondary to caspase-mediated degradation, U937 cells were incubated with 20 µM of UA in the presence or absence of the broad-spectrum caspase inhibitor Z-VAD-FMK (10 µM). Addition of Z-VAD-FMK significantly blocked UA-induced apoptosis (Figure 4A), caspase-3, -7, -8 and -9 activation, as well as PARP degradation (Figure 4B), partially blocked UA-induced down-regulation of Mcl-1 (Figure 4C). In contrast, Z-VAD-FMK failed to prevent PKB inactivation and JNK activation induced by UA (Figure 4D). Together, these findings indicate that UA-mediated PKB inactivation and JNK activation represent primary rather than caspase-dependent effects, suggesting that these effects may be involved in UA-mediated caspase activation and lethality.
Figure 4.
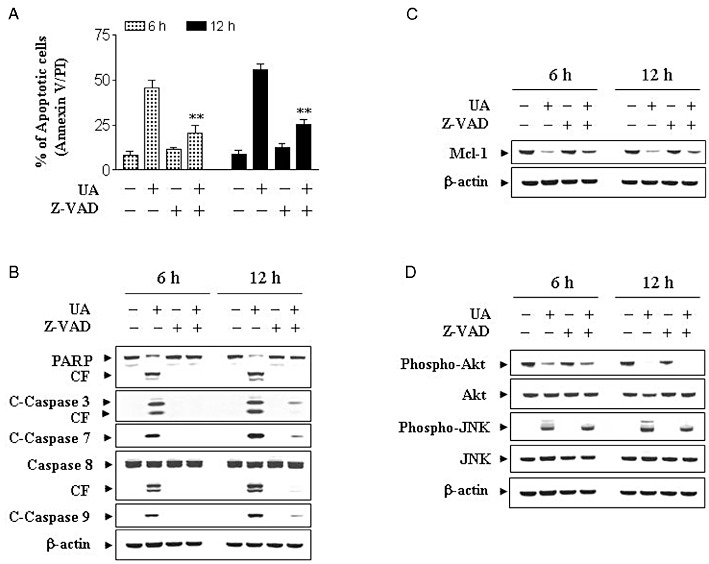
UA-induced lethality was associated with down-regulation of the cytoprotective PKB pathway and reciprocal activation of the stress-related JNK pathway. (A) U937 cells were pretreated with the caspase inhibitor Z-VAD-FMK (10 µM) for 1 h followed by treatment with 20 µM of UA for 6 and 12 h. Cells were stained with Annexin V/PI. Apoptosis was determined using flow cytometry as described in Methods. The values obtained from Annexin V assays represent the means ± SD for three separate experiments. *Values for cells treated with UA and Z-VAD-FMK were significantly reduced compared with values obtained for UA alone by Student's t-test; P < 0.01. (B) U937cells were pretreated with the caspase inhibitor Z-VAD-FMK (10 µM) for 1 h followed by treatment with 20 µM UA for 6 and 12 h. Total protein extracts were prepared and subjected to Western blot assay using antibodies against PARP, C-Caspase-3, C-Caspase-7, caspase-8 and C-Caspase-9. Total protein extracts were also prepared and subjected to Western blot assay using antibodies against Mcl-1 (C) and cell signalling proteins including phospho-PKB (Ser473), PKB, phospho-JNK and JNK (D). For Western blot analysis, each lane was loaded with 30 µg of protein; blots were subsequently stripped and re-probed with antibody against β-actin to ensure equivalent loading.
PKB inactivation plays an important functional role in UA-mediated JNK activation, Mcl-1 down-regulation, caspase activation and apoptosis
The preceding findings implied that inactivation of PKB might play an important role in UA-mediated lethality. To test this possibility, cells were co-exposed to UA and the PI3K inhibitor LY294002, and apoptosis was monitored. As shown in Figure 5A, co-administration of a non-toxic concentration of LY294002 (i.e. 20 µM) with a modestly toxic concentration of UA (10 µM; ∼15% for 12 h) resulted in a pronounced increase in apoptosis (i.e. to ∼50%). Western blot analysis revealed that co-administration of UA and LY294002 resulted in a pronounced increase in the activation of caspases-3, -7, -8 and -9, and PARP degradation (Figure 5B), and potentiation of Mcl-1 down-regulation, but not UA or LY294002 alone (Figure 5C). In addition, co-administration of UA and LY294002 resulted in the abolition of PKB expression/activation and a pronounced increase in JNK activation (Figure 5D). Together, these findings suggest that inactivation of PKB plays a critical role in regulating the lethality of UA in human leukaemia cells.
Figure 5.
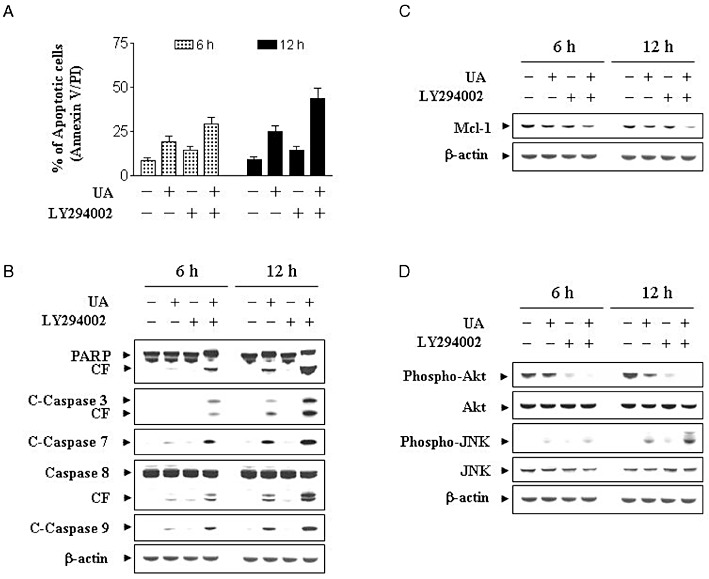
Effects of the pharmacological inhibitor of PI3K/PKB on apoptosis induced by UA in U937 cells. U937 cells were pretreated with 20 µM of LY for 1 h followed by the addition of 10 µM of UA for 12 h. (A) Cells were stained with Annexin V/PI, and apoptosis was determined using flow cytometry as described in Methods. The values obtained from Annexin V/PI assays represent the means ± SD for three separate experiments. **Values for cells treated with UA and LY in combination were significantly greater than those for cells treated with UA alone by Student's t-test; P < 0.01. (B) Total cellular extracts were prepared as described in Methods and subjected to Western blot analysis using antibodies against PARP, C-Caspase-3, C-Caspase-7, caspase-8 and C-Caspase-9. Total cellular extracts were also prepared and subjected to Western blot assays using antibodies against Mcl-1 (C), and cell signalling proteins including phospho-PKB (Ser473), PKB, phospho-JNK and JNK (D). For the Western blot assay, each lane was loaded with 30 µg of protein; blots were subsequently stripped and re-probed with antibody against β-actin to ensure equivalent loading. Two additional studies yielded equivalent results.
To further assess the functional significance of PKB inactivation in UA-mediated lethality, U937 cells ectopically expressing the constitutive active form of PKB and the dominant-negative PKB were employed. As shown in Figure 6A, a clone displaying constitutive PKB activation was markedly less sensitive to UA-induced apoptosis than pcDNA3.1 vector control cells (P < 0.01). However, UA-induced apoptosis in PKB dominant-negative (PKB-DN) cells was similar to that in pcDNA3.1 vector control cells. Consistent with these findings, UA was considerably less effective in triggering activation of caspase-3, -7, -8, -9 and PARP degradation in PKB constitutively active cells compared with pcDNA3.1 vector control and PKB-DN cells (Figure 6B). In addition, enforced activation of PKB effectively blocked UA-mediated Mcl-1 down-regulation (Figure 6C). Western blot analysis displayed marked increase in levels of total and phospho-PKB in cells expressing constitutive active PKB. UA failed to induce inactivation of PKB in these PKB constitutively active cells (Figure 6D). Interestingly, the ability of UA to induce JNK activation was essentially abolished in the cells expressing constitutive active PKB (Figure 6D). These findings indicate that inactivation of the PKB pathway plays a critical role in UA-induced apoptosis, and that this event lies upstream of Mcl-1 down-regulation and JNK activation.
Figure 6.
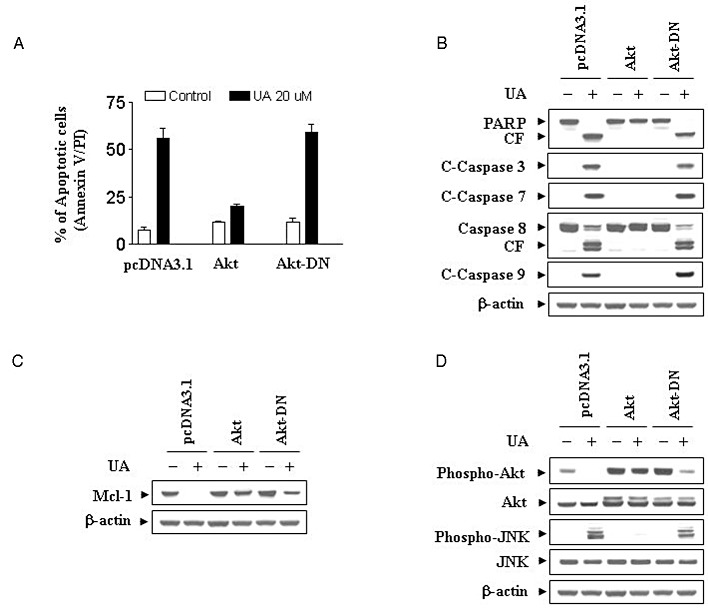
Induction of activated PKB markedly protected cells from UA-induced apoptosis. U937 cells were stably transfected with constitutively active forms of PKB (PKB-CA), dominant-negative PKB mutant (PKB-DN) and an empty vector (pcDNA3.1) as described in Methods. All cells were then treated with 20 µM of UA for 12 h. (A) After treatment, apoptosis was determined using Annexin V–FITC assay as described in Methods. **Values for PKB-CA cells treated with UA were significantly decreased compared with those for pcDNA3.1 cells by Student's t-test; P < 0.01. (B) Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against PARP, C-Caspase-3, C-Caspase-7, caspase-8 and C-Caspase-9. Total cellular extract were also prepared and subjected to Western blot analysis using antibodies against Mcl-1 (C) and cell signalling proteins including phospho-PKB (Ser473), PKB, phospho-JNK and JNK (D). For Western blot assay, each lane was loaded with 30 µg of protein; blots were subsequently stripped and re-probed with antibody against β-actin to ensure equivalent loading. Two additional studies yielded equivalent results.
JNK activation plays an important functional role in UA-induced Mcl-1 down-regulation, caspase activation and apoptosis
The functional significance of JNK activation in UA lethality was then investigated using both pharmacological and genetic approaches. U937 cells were pretreated with JNK inhibitor SP600125 (10 µM) followed by treatment with UA (20 µM) for 6 and 12 h, apoptosis was detected using Annexin V/PI. As shown in Figure 7A, pretreatment with SP600125 essentially abolished UA-induced lethality. Co-administration of SP600125 also blocked UA-induced caspase-3, -7, -8 and -9 activation, and PARP degradation (Figure 7B). Antagonism of UA-mediated Mcl-1 down-regulation by SP600125 was also noted (Figure 7C). SP600125 also markedly reduced UA-induced phosphorylation of JNK (Figure 7D). To further understand the functional significance of JNK activation in UA-induced apoptosis, a genetic approach utilizing JNK1 siRNA was employed. As shown in Figure 7E, transient transfection of U937 cells with JNK1 siRNA reduced the expression of total JNK1 and phospho-JNK, and resulted in a significant reduction in UA-mediated apoptosis. Collectively, these findings indicate that UA-induced JNK activation plays an important functional role in UA-related lethality.
Figure 7.
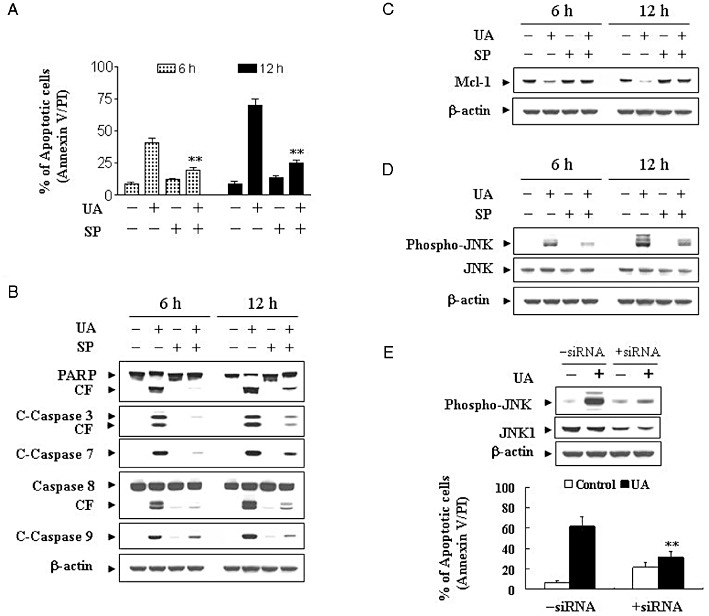
Pharmacological inhibition of JNK and transfection of JNK1 siRNA significantly protect cells from UA-induced apoptosis. U937cells were pretreated with 10 µM of the JNK inhibitor, SP600125 (SP), for 1 h, followed by the addition of 20 µM of UA for 12 h. (A) Cells were stained with Annexin V/PI, and apoptosis was determined using flow cytometry as described in Methods. The values obtained from Annexin V/PI assays represent the means ± SD for three separate experiments. **Values for cells treated with UA and SP were significantly less than those obtained for cells treated with UA alone by Student's t-test; P < 0.01. After treatment, total cellular extracts were prepared and subjected to Western blot analysis using antibodies against PARP, C-Caspase-3, C-Caspase-7, caspase-8 and C-Caspase-9 (B), Mcl-1 (C), as well as cell signalling proteins including phospho-JNK and JNK (D). (E) U937 cells were transiently transfected with JNK1 siRNA oligonucleotides or controls and incubated for 24 h at 37°C, after which cells were treated with 20 µM of UA for 12 h. Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against phospho-JNK and JNK1. Apoptosis was determined using the Annexin V–FITC assay as described in Methods. **Values for cells treated with UA after transfection with JNK1 siRNA oligonucleotides were significantly decreased compared to those for control cells treated with UA by Student's t-test; P < 0.01.
UA exhibits significant anti-leukaemic activity in immunodeficient mice
The ability of UA to kill both leukaemia cell lines and primary human leukaemia cells in vitro led us to evaluate its anti-leukaemic activity in vivo. NOD/SCID mice were inoculated i.p. with U937 cells, after which mice received injections of vehicle or UA (50 mg·kg−1, i.p.) for 20 days starting 3 days after the injection of U937 human leukaemia cells. Tumour growth was measured once every 5 days, and tumour volume (V) was calculated as V = (L×W2) × 0.5, where L is the length, and W is the width of a tumour. As shown in Figure 8A, treatment with UA significantly inhibited tumour growth. The mean volume of tumours in mice treated with UA was much smaller than that of tumours in the vehicle-control mice (P < 0.01). There was no significant difference in the body weights between the UA treatment and vehicle-control group (Figure 8B), indicating that no severe toxicity was observed.
Figure 8.
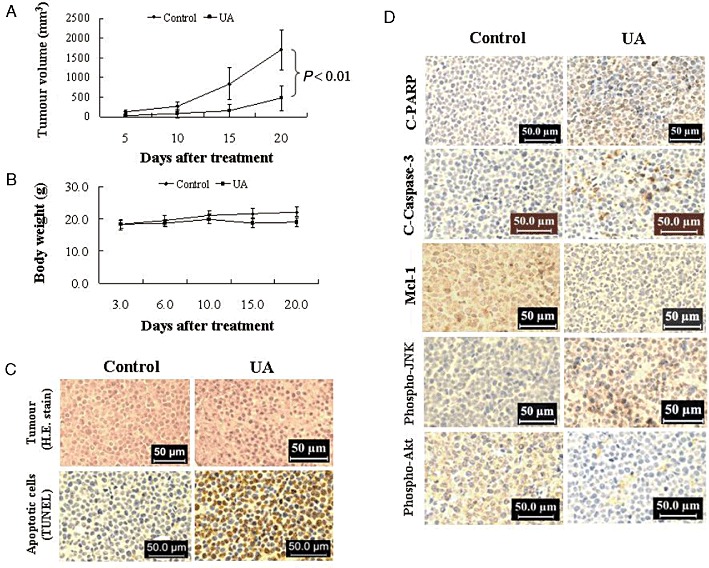
In vivo anti-leukaemic activity of UA in mouse tumour xenografts. Twenty NOD/SCID mice were inoculated with U937 cells (2 × 106 cells·per mouse, s.c.) and randomly divided into two groups (10 per group) for treatment with UA (50 mg·kg−1, i.p., daily, five times per week) or with vehicle control solvent as described in Methods. (A) Average tumour volume in vehicle control mice and mice treated with 50 mg·kg−1 UA. Data are mean ± SE (n = 20; 10 mice·per group with tumours implanted on right flank of each mouse). P < 0.01, significantly different compared with vehicle control by Student's t-test. (B) Body weight changes of mice during the 20 days of study. Statistical analysis of body weight changes showed no significant differences between UA treatment and vehicle control groups. (C) Representative photographs of biopsy samples from mice treated for 20 days with UA (50 mg·kg−1) or vehicle control. Original magnification, ×400. (D) Expression of C-PARP, C-Caspase-3, Mcl-1, phospho-JNK and phospho-PKB (Ser473) in tumour tissues of xenograft mouse treated with UA and vehicle control. After treatment with UA, tumour tissues were sectioned and subjected to immunohistochemistry using antibodies against C-PARP, C-Caspase-3, Mcl-1, phospho-PKB (Ser473) and phospho-JNK. The sections were lightly counterstained with haematoxylin and photographed with a Scan Scope.
To evaluate whether administration of UA results in morphological changes and induction of apoptosis in U937 cells in vivo, isolated tumour samples were sectioned and stained with H&E or TUNEL as described in Methods. As shown in Figure 8C, the sections of U937 xenografts from mice treated with UA showed that cancer cells were markedly decreased, with signs of necrosis with infiltration of inflammatory cells (i.e. phagocytic cells), fibrosis, as well as apoptotic regions, identified by their amorphous shape and condensed nuclei. Characteristic features of apoptosis were also observed in the tumour sections stained with TUNEL. The sections of tumour from UA-treated mice showed numerous dark brown-coloured apoptotic cells.
To gain insights into the mechanisms involved in the increased apoptosis in tumours of UA-treated mice, the tumours were analysed for the expression of cleaved caspase-3 and cleaved PARP (C-PARP) using an immunohistochemistry assay. As shown in Figure 8D, the number of C-PARP-positive cells in tissue sections of xenografts from mice treated with UA was increased substantially compared with vehicle-control, with morphological evidence of nuclear fragmentation showed positive staining for C-PARP. Consistent with these findings, treatment with UA resulted in a marked increase in the expression of cleaved caspase-3 in tissue sections of tumours.
The preceding findings imply that down-regulation of Mcl-1, inactivation of PKB and activation of JNK might play important roles in UA-mediated lethality in U937 cells in vitro. To test this possibility, immunohistochemistry analysis was performed to evaluate the expression of Mcl-1, phospho-PKB and phospho-JNK in tissue sections of xenografts. As expected, tumours from vehicle-treated control mice stained strongly for Mcl-1 and phospho-PKB (Figure 8D), which were immunolocalized to the cytoplasm of cancer cells. Treatment with UA resulted in a marked decrease in the expression of Mcl-1 and phospho-PKB in tissue sections of tumours. Immunostaining of tumours from mice treated with UA showed that phospho-JNK was markedly increased.
Overall, these findings demonstrate that UA administration significantly inhibits tumour growth in xenograft mouse model. UA-mediated anti-leukaemic activity in vivo is associated with inactivation of PKB and activation of JNK.
Discussion
In the present study, we demonstrated that UA dramatically induces apoptosis in diverse human leukaemia cell lines as well as in primary human AML or ALL blast cells in a dose- and time-dependent manner. Additionally, our results indicate molecular mechanism involved in these pro-apoptotic effects of UA in human leukaemia cells (i.e. by inhibiting phosphorylation of PKB and expression of Mcl-1 and by inducing phosphorylation of JNK). In recent studies, UA was shown to induce apoptosis in many cancer cells such as prostate (Choi et al., 2000), breast (Kassi et al., 2009), melanoma (Manu and Kuttan, 2008), leukaemia cells (Liu and Jiang, 2007) as well as multiple myeloma (Pathak et al., 2007) through diverse cell signalling pathways including ROS, PKB, NF-κB and JNK (Tu et al., 2009, Li et al., 2010; Zhang et al., 2010a,b). However, these studies did not explore the functional role of the PKB and JNK pathway in UA-induced lethality in human leukaemia cells. The present study demonstrates that UA-mediated caspase activation and subsequent lethality in human leukaemia cells are associated with PKB inactivation and JNK activation.
PKB is a serine–threonine kinase intimately involved in the regulation of cell survival (Kim et al., 2001). It is activated by recruitment to the cell membrane through the actions of PI3K, which in turn is regulated negatively by the PTEN phosphatase (Stanbolic et al., 1998), mutations of which are among the most commonly encountered in human cancers (Mao et al., 2004). However, the fact that Jurkat and U937 cells do not express wild-type PTEN (Liu et al., 2000; Shan et al., 2000), argue against this notion. A more likely possibility is that UA, through a mechanism yet to be elucidated, blocks the actions of PI3K. PKB represents a major downstream target of PI3K (Franke et al., 1997) and has been linked, through both indirect and direct mechanisms, to a wide variety of anti-apoptotic functions (Nicholson and Anderson, 2002), and we found that LY294002, an inhibitor of PI3K, enhanced the lethality of UA by blocking the activation of PKB and one or more of its downstream targets (i.e. Mcl-1). Caspase-dependent down-regulation of PKB is a well-described phenomenon (Yang and Widmann, 2002). Our results indicate that UA induces apoptosis by activating both caspase-8 and -9, along with caspase-3 and -7, raising the possibility that PKB inactivation might represent a consequence of engagement of the caspase cascade. In the present study, co-treatment of U937 cells with the pan-caspase inhibitor Z-VAD-FMK, abolished UA-induced activation of caspases (i.e. caspase-3, -7, -8 and -9) and apoptosis, but failed to prevent PKB inactivation, JNK activation, arguing strongly that factors other than caspase-mediated events are involved in this phenomenon. In addition, PKB can be dephosphorylated and thus inactivated by phosphatases such as PP2A and PHLPP (Sato et al., 2000 and Gao et al., 2005). The dose- and time-dependent increase in PP2A-B and PP2A-C levels suggest that these phosphatases might be involved in UA-induced dephosphorylation of PKB. Additional mechanistic studies are required to demonstrate the role of these phosphatases in UA-induced lethality in leukaemia cells. Others and our unpublished data have shown that UA induces oxidative stress in leukaemia cells. Therefore, it is possible that oxidative stress may be responsible for UA-induced dephosphorylation of PKB. In fact, in a recent study, Cao et al. (2009) showed that 4-HPR (N-(-4-hydroxyphenyl) retinamide)-induced oxidative stress in human leukaemia cells caused a conformational change in PKB. 4-HPR induced an ROS-mediated conformational change in PKB via the formation of an intracellular disulphide bond in PKB thus inhibiting the formation of the PKB–Hsp90 complex and increasing the dephosphorylation of PKB by PP2A. The same phenomenon might be true for UA-induced dephosphorylation of PKB.
A body of evidence suggests that in human leukaemia cells, UA-induced PKB inactivation plays a critical functional role in mediating UA lethality. Significantly, enforced activation of PKB largely reversed the lethal consequences of UA exposure, including caspase activation, PARP cleavage and apoptosis. It is of interest that UA exposure resulted in the down-regulation of Mcl-1, an anti-apoptotic protein that may play a particularly important role in regulating apoptosis in malignant haematopoietic cells (Rinkenberger et al., 2000). It has been reported that the anti-apoptotic gene Mcl-1 is up-regulated by the PI3K/PKB signalling pathway (Wang et al., 1999; Kuo et al., 2001), and down-regulation of Mcl-1 by inhibition of the PI3K/PKB pathway is required for cell death (Araki et al., 2002). The finding that enforced activation of PKB largely blocked UA-mediated down-regulation of Mcl-1 suggests it may significantly contribute to UA-mediated lethality.
Induction of caspase activation and apoptosis by UA was also associated with activation of the stress-related JNK pathway. JNK belongs to the superfamily of MAPKs that are involved in the regulation of cell proliferation, differentiation and apoptosis (Dhanasekaran and Reddy, 2008). The critical role of JNK has been demonstrated in the lethal effects of diverse cytotoxic stimuli, including ceramide (Verheij et al., 1996), Fas ligand (Wilson et al., 1996) and u.v. light (Zanke et al., 1996). The finding that pharmacological and genetic interruption of the JNK pathway attenuated UA-mediated lethality indicates that stress pathways play a critical functional role in the induction of apoptosis by this agent. Interestingly, co-administration of UA with the PI3K inhibitor LY294002, which potentiates inactivation of PKB, enhanced the JNK activation and apoptosis induced by UA. Furthermore, enforced activation of PKB not only blocked UA-mediated caspase activation and apoptosis but also prevented the striking increase in JNK activation, raising the possibility that one of the mechanisms by which PKB protects cells from UA lethality is by opposing JNK activation. This phenomenon might be explained by the following lines of evidence: firstly, ASK-1, the protein that activates JNK, is a target of PKB inhibitory phosphorylation. Phosphorylation by PKB inhibits JNK activity, which is mediated by ASK1, providing the direct link between PKB and JNK. Secondly, the interaction between PKB and JIP1 inhibits JIP1-mediated potentiation of JNK activity by decreasing JIP1 binding to specific JNK pathway kinases, suggesting that PKB interaction with JIP1 acts as a negative switch for JNK activation (Kim et al., 2002).
Recent studies have shown that UA is strong inducer of apoptosis in human leukaemia cells in vitro (Liu and Jiang, 2007). It has been shown that UA induces apoptosis through the JNK activation pathway in human leukaemia K562 cells. In a recent study, we found that UA induces apoptosis in various human leukaemia cell lines (i.e. U937, Jurkat and HL-60) and primary human AML or ALL blast cells, and showed that cell signalling pathways including PKB inactivation and JNK activation are involved in these events. However, it is still unclear whether the UA-induced apoptosis of human leukaemia cells in vivo occurs in a similar to the response to treatment with this compound in vitro. In the present study, we showed that UA did stimulated the induction of apoptosis in vivo, which could be responsible for its inhibitory effects on tumour growth. In particular, immunohistochemistry analysis showed that UA-induced apoptosis in vivo involved caspase-3 activation and PARP cleavage, suggesting that apoptotic pathways involving caspase-3 and PARP are the likely major molecular target(s) in the apoptotic responses to UA treatment. To elucidate the possible mechanisms that trigger the apoptotic signalling, we also determined the expression levels of phospho-PKB, phospho-JNK and Mcl-1 in tissue sections using immunohistochemistry analysis. Consistent with the in vitro data, our results indicate that suppression of phospho-PKB and Mcl-1, and induction of phospho-JNK are closely correlated with the reduction of tumour volume. Because UA selectively kills leukaemia cells without a significant toxic effect on normal peripheral blood mononuclear cells, UA could be effective in the therapy of leukaemia and possibly other haematological malignancies.
In summary, the present study indicates that UA induces apoptosis, caspase activation and PARP cleavage in both human leukaemia cells and mouse tumour xenografts via inactivation of PKB, activation of JNK and down-regulation of Mcl-1. Collectively, these observations suggest a hierarchy of events in UA-induced lethality in which PKB inactivation represents the primary insult, leading in turn to JNK activation, resulting in Mcl-1 down-regulation and culminating in caspase activation and apoptosis. Further efforts to understand the mechanism(s) by which UA induces apoptosis in human leukaemia cells and tumour xenografts could provide a more rational basis for attempts to incorporate such agents into anti-leukaemic regimens. Accordingly, such investigations are currently underway in our laboratory.
Acknowledgments
This work was supported by awards NIH Grants RO1 ES015375 (X S), 1R21 ES019249 (ZZ) and National Natural Science Foundation of China (No. 30971288).
Glossary
- AML
acute myeloid leukaemia
- ALL
acute lymphoma leukaemia
- Bad
Bcl-2 associated agonist of cell death
- Bax
Bcl-2 associated X protein
- Bcl-2
B-cell lymphoma
- FITC
fluorescein isothiocyanate
- H&E
haematoxylin and eosin
- Mcl-1
myeloid cell leukaemia sequence 1
- mTOR
mammalian target of rapamycin
- NOD/SCID
non-obese diabetic/severe combined immunodeficiency
- PI
propidium iodide
- PI3K
phosphoinositide-3-kinase
- PTEN
phosphatase and tensin homologue
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labelling
- UA
ursolic acid
- XIAP
X-linked inhibitor of apoptosis protein
Conflict of interest
None of the listed authors has any financial or other interests that could be of conflict.
References
- Abe F, Yamauchi T, Nagao T, Kinjo J, Okabe H, Higo H, et al. Ursolic acid as a trypanocidal constituent in rosemary. Biol Pharm Bull. 2002;25:1485–1487. doi: 10.1248/bpb.25.1485. [DOI] [PubMed] [Google Scholar]
- Araki T, Hayashi M, Watanabe N, Kanuka H, Yoshino J, Miura M, et al. Down regulation of Mcl-1 by inhibition of the PI3-K/Akt pathway is required for cell shrinkage-dependent cell death. Biochem Biophys Res Commun. 2002;290:1275–1281. doi: 10.1006/bbrc.2002.6329. [DOI] [PubMed] [Google Scholar]
- Baek JH, Lee YS, Kang CM, Kim JA, Kwon KS, Son HC, et al. Intracellular Ca2+ release mediates ursolic acid–induced apoptosis in human leukemic HL-60 cells. Int J Cancer. 1997;73:725–728. doi: 10.1002/(sici)1097-0215(19971127)73:5<725::aid-ijc19>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28:1379–1386. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- Cao J, Danqing X, Wang D, Wu R, Zhang L, Zhu H, et al. ROS-driven Akt dephosphorylation at Ser-473 is involved in 4-HPR-mediated apoptosis in NB4 cells. Free Radic Biol Med. 2009;47:536–547. doi: 10.1016/j.freeradbiomed.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Choi YH, Baek JH, Yoo MA, Chung HY, Kim ND, Kim KW. Induction of apoptosis by ursolic acid through activation of caspases and down-regulation of c-IAPs in human prostate epithelial cells. Int J Oncol. 2000;17:565–571. [PubMed] [Google Scholar]
- De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, et al. Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signaling. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran DN, Reddy EB. JNK signaling in apoptosis. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan N, Becker EB, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem. 2002;277:40944–40949. doi: 10.1074/jbc.M206113200. [DOI] [PubMed] [Google Scholar]
- Fan M, Chambers TC. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist Updat. 2001;4:253–267. doi: 10.1054/drup.2001.0214. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Gao N, Budhraja A, Cheng S, Yao H, Zhang Z, Shi X. Induction of apoptosis in human leukemia cells by grape seed extract occurs via activation of c-Jun NH2-Terminal kinase. Clin Cancer Res. 2009;15:140–149. doi: 10.1158/1078-0432.CCR-08-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt promotes apoptosis and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Murakami A, Ohigashi H. Ursolic acid: an anti- and pro-inflammatory triterpenoid. Mol Nutr Food Res. 2008;52:26–42. doi: 10.1002/mnfr.200700389. [DOI] [PubMed] [Google Scholar]
- Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J, et al. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem. 2002;277:43730–43734. doi: 10.1074/jbc.M207951200. [DOI] [PubMed] [Google Scholar]
- Kassi E, Sourlingas TG, Spiliotaki M, Papoutsi Z, Pratsinis H, Aligiannis N, et al. Ursolic acid triggers apoptosis and Bcl-2 downregulation in MCF-7 breast cancer cells. Cancer Invest. 2009;27:723–733. doi: 10.1080/07357900802672712. [DOI] [PubMed] [Google Scholar]
- Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AH, Yano H, Cho H, Meyer D, Monks B, Margolis B, et al. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron. 2002;35:697–709. doi: 10.1016/s0896-6273(02)00821-8. [DOI] [PubMed] [Google Scholar]
- Kuo ML, Chuang SE, Lin MT, Yang SY. The involvement of PI 3-K/Akt-dependent up-regulation of Mcl-1 in the prevention of apoptosis of Hep3B cells by interleukin-6. Oncogene. 2001;20:677–685. doi: 10.1038/sj.onc.1204140. [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xing D, Chen Q, Chen WR. Enhancement of chemotherapeutic agent-induced apoptosis by inhibition of NF-κB using ursolic acid. Int J Cancer. 2010;127:462–473. doi: 10.1002/ijc.25044. [DOI] [PubMed] [Google Scholar]
- Liu TC, Lin PM, Chang JG, Lee JP, Chen TP, Lin SF. Mutation analysis of PTEN/MMAC1 in acute myeloid leukemia. Am J Hematol. 2000;63:170–175. doi: 10.1002/(sici)1096-8652(200004)63:4<170::aid-ajh2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Liu XS, Jiang J. Induction of apoptosis and regulation of the MAPK pathway by ursolic acid in human leukemia K562 cells. Planta Med. 2007;73:1192–1194. doi: 10.1055/s-2007-981597. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3, 4, 5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Manu KA, Kuttan G. Ursolic acid induces apoptosis by activating p53 and caspase-3 gene expressions and suppressing NF-κB mediated activation of bcl-2 in B16F-10 melanoma cells. Int Immunopharmacol. 2008;8:974–981. doi: 10.1016/j.intimp.2008.02.013. [DOI] [PubMed] [Google Scholar]
- Mao JH, To MD, Perez-Losada J, Wu D, Del Rosario R, Balmain A. Mutually exclusive mutations of the Pten and ras pathways in skin tumor progression. Genes Dev. 2004;18:1800–1805. doi: 10.1101/gad.1213804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- Ohigashi H, Takamura H, Koshimizu K, Tokuda H, Ito Y. Inhibitory effects of ursolic and oleanolic ancid on skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Lett. 1986;30:143–151. doi: 10.1016/0304-3835(86)90067-4. [DOI] [PubMed] [Google Scholar]
- Pathak AK, Bhutani M, Nair AS. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Mol Cancer Res. 2007;5:943–955. doi: 10.1158/1541-7786.MCR-06-0348. [DOI] [PubMed] [Google Scholar]
- Rinkenberger JL, Horning S, Klocke B, Roth K, Roth K, Korsmeyer SJ. Unique biology of Mcl-1: therapeutic opportunities in cancer. Genes Dev. 2000;14:23–27. [PMC free article] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai LJ, McGaw LJ, Aderogba MA, Mdee LK, Eloff JN. Four pentacyclic triterpenoids with antifungal and antibacterial activity from Curtisia dentata (Burm.f) C.A. Sm. Leaves. J Ethnopharmacol. 2008;119:238–244. doi: 10.1016/j.jep.2008.06.036. [DOI] [PubMed] [Google Scholar]
- Shan X, Czar MJ, Bunnell SC, Liu P, Liu Y, Schwartzberg PL, et al. Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasma membrane and hyperresponsiveness to CD3 stimulation. Mol Cell Biol. 2000;20:6945–6957. doi: 10.1128/mcb.20.18.6945-6957.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40:928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- Stanbolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Sun Y, Yuan J, Liu H, Shi Z, Baker K, Vuori K, et al. Role of gab1 in UV-induced c-Jun NH2-terminal kinase activation and cell apoptosis. Mol Cell Biol. 2004;24:1531–1539. doi: 10.1128/MCB.24.4.1531-1539.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi S, Imayoshi Y, Kobayashi E, Takamatsu Y, Ito H, Hatano T, et al. Production of bioactive triterpenes by Eriobotrya japonica calli. Phytochemistry. 2002;59:315–323. doi: 10.1016/s0031-9422(01)00455-1. [DOI] [PubMed] [Google Scholar]
- Tu HY, Huang AM, Wei BL, Gan KH, Hour TC, Yang SC, et al. Ursolic acid derivatives induce cell cycle arrest and apoptosis in NTUB1 cells associated with reactive oxygen species. Bioorg Med Chem. 2009;17:7265–7274. doi: 10.1016/j.bmc.2009.08.046. [DOI] [PubMed] [Google Scholar]
- Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Wang JM, Chao JR, Chen W. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-Kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol. 1999;19:6195–6206. doi: 10.1128/mcb.19.9.6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner CE. Cell signaling and cancer-possible targets for therapy. J Cell Physiol. 2010;223:299–308. doi: 10.1002/jcp.22021. [DOI] [PubMed] [Google Scholar]
- Wilson DJ, Fortner KA, Lynch DH, Mattingly RR, Macara IG, Posada JA, et al. JNK, but not MAPK, activation is associated with Fas-mediated apoptosis in human T cells. Eur J Immunol. 1996;26:989–994. doi: 10.1002/eji.1830260505. [DOI] [PubMed] [Google Scholar]
- Xia Y, Ongusaha P, Lee SW, Liou YC. Loss of Wip1 sensitizes cells to stress- and DNA damage-induced apoptosis. J Biol Chem. 2009;284:17428–17437. doi: 10.1074/jbc.M109.007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Widmann C. The RasGAP N-terminal fragment generated by caspase cleavage protects cells in a Ras/PI3K/Akt-dependent manner that does not rely on NFkappa B activation. J Biol Chem. 2002;277:14641–14646. doi: 10.1074/jbc.M111540200. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat Cell Biol. 2005;7:278–285. doi: 10.1038/ncb1228. [DOI] [PubMed] [Google Scholar]
- Zanke BW, Boudreau K, Rubie E, Winnett E, Tibbles LA, Zon L, et al. The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Curr Biol. 1996;6:606–613. doi: 10.1016/s0960-9822(02)00547-x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kong C, Zeng Y, Wang L, Li Z, Wang H, et al. Ursolic acid induces PC3 cell apoptosis via activation of JNK and inhibition of Akt pathways in vitro. Mol Carcinog. 2010b;49:374–385. doi: 10.1002/mc.20610. [DOI] [PubMed] [Google Scholar]
- Zhang YX, Kong CZ, Wang LH, Li JY, Liu XK, Xu B, et al. Ursolic acid overcomes Bcl-2-mediated resistance to apoptosis in prostate cancer cells involving activation of JNK-induced Bcl-2 phosphorylation and degradation. J Cell Biochem. 2010a;109:764–773. doi: 10.1002/jcb.22455. [DOI] [PubMed] [Google Scholar]