

Abstract
BACKGROUND AND PURPOSE
New antithrombotic agents with the potential to prevent atherothrombotic complications are being developed to target receptors on platelets and other cells involved in plaque growth. The aim of this study was to investigate the antiplatelet effects of F 16618, a new non-peptidic PAR1 (thrombin receptor) antagonist.
EXPERIMENTAL APPROACH
We investigated the inhibitory effect of F 16618 on human platelet aggregation ex vivo, in whole blood and washed platelets, by using a multiple-electrode platelet aggregometer based on impedance and an optical aggregometer, respectively. Its effects on whole-blood haemostasis (clot parameters) were analysed with the ROTEM thromboelastometry device and the platelet function analyser PFA-100. A guinea-pig model of arterial thrombosis was used to investigate its effects on thrombus formation in vivo.
KEY RESULTS
F 16618 inhibited PAR1 agonist peptide (SFLLR-peptide)-induced washed platelet aggregation ex vivo. This effect was concentration-dependent and exhibited a competitive inhibition profile. Washed platelet aggregation, as well as P-selectin expression induced by thrombin, were significantly inhibited by 10 µM F 16618. In whole-blood experiments, 20 µM F 16618 inhibited SFLLR-induced platelet aggregation by 49%. In contrast, it had no effect on whole-blood haemostasis. In the guinea-pig model of carotid thrombosis, 0.32 mg·kg−1 F 16618 doubled the occlusion time.
CONCLUSIONS AND IMPLICATIONS
F 16618 was shown to have strong antithrombotic activity in vivo and moderate antiplatelet effects ex vivo. As these effects were not associated with major effects on physiological haemostasis, this molecule is a good antiplatelet drug candidate for use either alone or in combination with current treatments.
Keywords: PAR1 antagonist, platelets, antiplatelet agent, aggregation, arterial thrombosis
Introduction
Platelet activation plays a key role in haemostasis and in thrombotic diseases such as atherothrombosis (Jennings, 2009). Current antiplatelet drugs such as ADP receptor antagonists, thromboxane A2 pathway inhibitors and glycoprotein (GP)IIb/IIIa antagonists have been shown to prevent cardiovascular events (Gross and Weitz, 2009; Smyth et al., 2009).
Platelets are activated by a variety of agonists, such as thrombin, ADP, thromboxane A2, collagen, 5-HT and adrenaline, leading to their adhesion and aggregation at sites of vascular injury. However, this process not only contributes to bleeding arrest but can also lead to vessel occlusion. Thrombin, the most potent platelet activator, also activates a number of vascular and perivascular cell types, including endothelial cells, leucocytes and smooth muscle cells (Hirano and Kanaide, 2003; Ramachandran and Hollenberg, 2008). Thrombin is a serine protease that cleaves multiple substrates, including clotting factors. The cellular function of thrombin is mediated by the cleavage of so-called ‘proteinase-activated receptors’ (PARs), a seven-transmembrane-domain family of GPCRs. Four PARs have been cloned (PAR1, PAR2, PAR3 and PAR4) and are implicated in multiple physiological and pathological processes, such as angiogenesis, cell proliferation, coagulation and inflammation (Coughlin, 2005; Alexander et al., 2011). Only PAR1, PAR3 and PAR4 are activated by thrombin. A new N-terminal domain starting with the peptidic sequence SFLLR is thus exposed and acts as a ‘tethered ligand’ that binds to the second extracellular loop and thereby activates the receptor. A synthetic peptide PAR1 agonist peptide with a sequence (SFLLR) equivalent to that of the new terminal exodomain mimics the effects of thrombin, independently of receptor cleavage (Vassallo et al., 1992; Ramachandran and Hollenberg, 2008).
Although PARs are ubiquitously distributed, human platelets express only PAR1 and PAR4. PAR1 is the high-affinity receptor while PAR4, which requires higher concentrations of thrombin to be activated, potentiates PAR1 activity (Kahn et al., 1998; Andersen et al., 1999; Chung et al., 2002; Wu et al., 2010). Once activated, each of these receptors is sufficient to trigger platelet aggregation and secretion, although PAR1 and PAR4 show marked differences in their activation kinetics and signalling pathways (Covic et al., 2000; Shapiro et al., 2000; Chung et al., 2002; Wu et al., 2010). PAR1 antagonists, including peptido-mimetic (BMS-200261, RWJ-58259) and non-peptidic molecules (FR-171113, SCH53048, E5555), show promise as new antithrombotic drugs (Bernatowicz et al., 1996; Damiano et al., 2003; Kato et al., 2003; Oestreich, 2009; Serebruany et al., 2009). In a search for new antiplatelet agents, we screened our proprietary library and found an interesting hit that, after a first optimization step, gave rise to a lead compound derived from cinnamoylpiperazine, which was further transformed as described elsewhere (Perez et al., 2009). After extensive optimization of this compound, we identified F 16618, a new non-peptidic, small-molecule PAR1 antagonist and demonstrated its potent antithrombotic activity in vivo (Perez et al., 2009; Letienne et al., 2010) and its inhibitory effect on restenosis (Chieng-Yane et al., 2011). The aim of the present study was to examine the effects of this new PAR1 antagonist on (i) human platelet aggregation ex vivo, using whole blood and washed platelets, and (ii) thrombus formation in vivo, using a guinea-pig model of arterial thrombosis. We found that F 16618 markedly inhibits H-Ser-Phe-Leu-Leu-Arg-OH (SFLLR peptide)-induced human platelet aggregation and shows potent antithrombotic activity in a pre-clinical model of arterial thrombosis, without affecting haemostasis.
Methods
Reagents
The following reagents were used: SFLLR peptide (Bachem, Weil am Rhein, Germany), human thrombin, RGDS peptide, PAR4 agonist peptide (Sigma Chemical, Saint Quentin Fallavier, France), collagen (Horm, Nycomed, Linz, Austria), ADP (Roche, Meylan, France) and arachidonic acid (Helena Bioscience Europe, Saint-Leu La Forêt, France).
F 16618 was synthesized by the Division of Medicinal Chemistry IV (Centre de Recherche Pierre Fabre, Castres, France) and was dissolved in 40% polyethylene glycol 300 in saline for use in the guinea-pig arterial thrombosis model and in 3.6 × 10−3 M HCl for ex vivo platelet experiments. Preliminary experiments showed that this HCl concentration had no effect on platelet aggregation (data not shown).
Human platelet aggregation assays
Venous blood from informed healthy donors was obtained from the French blood bank institute Etablissement Français du Sang (EFS) according to the agreement between Inserm and EFS (CPSL C UNT – 06/EFS/029). It was collected in Vacutainer® tubes containing ACD-A (citric acid-citrate-glucose) (BD Vacutainer®, Becton Dickinson, Le Pont de Claix, France). Washed platelets were prepared within 2 h after sampling. PRP was prepared by centrifugation of whole blood at 215×g for 11 min and then diluted in washing buffer (36 mM citric acid, 5 mM glucose, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 103 mM NaCl, pH 6.5) (2 vol PRP:1 vol buffer) in the presence of prostaglandin E1 (2 × 10−7 M) and apyrase (0.06 U·mL−1). After centrifugation at 1250×g for 12 min, the platelet pellet was resuspended in washing buffer and centrifuged in the same conditions as above. Finally, the platelets were resuspended in HEPES buffer (10 mM HEPES, 140 mM NaCl, 3 mM KCl, 5 mM NaHCO3, 5 mM MgCl2, 10 mM glucose, pH 7.35) and adjusted to 2.5 × 108 platelets·mL−1 in the presence of 2 mM CaCl2.
Washed platelet aggregation was measured with a PAP-8E Biodata® optical aggregometer (Bio/Data Corp, Horsham, PA, USA). Platelets were pre-incubated with F 16618 (0.2–10 µM) or vehicle for 3 min at 37°C with stirring (1200 r.p.m.). Agonists were then added, and aggregation was monitored for 5 min as the change in light transmittance. Results are expressed as the percentage of maximum aggregation ± SEM.
SFLLR-induced platelet aggregation in whole blood was measured with a Multiplate IL® (Instrumentation Laboratory S.A, Paris, France), a multiple-electrode platelet aggregometer based on impedance. Briefly, venous blood was collected in Vacutainer tubes containing 0.109 M citrate and diluted 1:2 with 0.9% NaCl solution. F 16618 (5–20 µM) or vehicle was preincubated for 3 min at 37°C with stirring, and 0.75 or 1.00 µM SFLLR was then added.
Platelet secretion
Platelet secretion was measured with a Becton Dickinson FACScalibur flow cytometer (BD Biosciences, Le Pont de Claix, France). Washed platelets (2.5 × 108 pl·mL−1), in the presence of 200 µM RGDS to avoid aggregation, were pre-incubated with F 16618 or vehicle for 3 min before adding the agonist. After 5 min, platelet activation was stopped by adding 0.2% paraformaldehyde, and 5 µL of sample was diluted in HEPES buffer (1:10) and incubated for 10 min with anti-CD41-FITC and anti-CD62P-PE monoclonal antibodies or with IgG1-PE and -FITC isotypic controls, using saturating concentration of each antibody, as recommended by the manufacturer (Beckman Coulter, Roissy, France). Before analysis, a further dilution step (1:10) was performed, and a total of 10 000 platelet events (CD41-positive) was recorded. The increase in CD62P-positive cells after agonist exposure is expressed as both the percentage of positive cells and mean fluorescence intensity (MFI; arbitrary units) relative to resting platelets.
Clot formation
Clot parameters were analysed with the ROTEM thromboelastometry device (Pentapharm GmbH, Munich, Germany). Citrated blood was incubated with 20 µM F 16618 or its vehicle in a cuvette at 37°C. Clot formation was initiated by adding thromboplastin and calcium (EXTEM plus STARTEM, Pentapharm GmbH) and the reaction was recorded for 150 min. The following clot parameters were measured: the clot formation time (CFT), the clotting time (CT) and the maximum clot firmness (MCF).
Clot retraction
Clot retraction was studied with citrated PRP (adjusted to 2.5 × 108 platelets·mL−1) pre-incubated in a glass tube with 20 µM F 16618 or vehicle. After adding 2 U·mL−1 thrombin in the presence of 2 mM CaCl2, the clots were allowed to retract at 37°C and were photographed at various times. The two-dimensional sizes of retracted clots were quantified with Image J software (NIH Image, Bethesda, MD, USA) and expressed as the percentage reduction in the initial size.
Global exploration of primary haemostasis with the platelet function analyser PFA-100
Citrated blood (800 µL) was incubated with 20 µM F 16618 or its vehicle and then analysed with two PFA-100 cartridges coated with collagen and either adrenaline or ADP. The PFA-100 system (Siemens, Munich, Germany) measures the closure time in seconds.
Arterial thrombosis model
All animal care and experimental procedures were in accordance with the Association for the Assessment and Accreditation of Laboratory Animal Care-accredited facility, in strict compliance with all applicable regulations. The protocol complied with French regulations and local ethics committee guidelines for animal research.
Forty-eight male guinea-pigs weighing 350–400 g at the date of the experiments were purchased from Charles River (Hartley, Crl:HA, L'Arbresle, France). They were housed at the Pierre Fabre Research Center in climate-controlled conditions (20°C ± 2°C) for at least 1 week before use, with free access to food and water.
The animals were anaesthetized with an i.p. injection of sodium pentobarbitone (50–60 mg·kg−1). After tracheotomy, the animals were mechanically ventilated at 60 cycles·min−1 (2.5 mL per respiration, Ventilator Model 683, Harvard Apparatus South Natick, MA, USA). Both jugulars were catheterized to administer Rose Bengal (20 mg·kg−1) and the test compounds. The left carotid artery was gently exposed and a 0.7 mm (VB type) flow probe (Transonic Systems, New York, NY, USA) was placed around the artery to measure blood flow during the entire experiment. Green light transillumination (514 nm) was delivered with a laser (Argon type, Melles Griot, Carlsbad, CA, USA), distally from the blood flow probe and positioned 4 mm above the exposed artery.
After stabilization of carotid blood flow, the treatment (drug/vehicle) was administered i.v. as a bolus over 2 min, followed by Rose Bengal (20 mg·kg−1) over 30 s. The laser was turned on for 3 min, and the treatment was infused simultaneously for 15 min. F 16618 was studied at seven doses (0.04–0.63 mg·kg−1) and a vehicle group [40% polyethylene glycol 300 in sterile saline (0.9%)] was also used. The correspondence between the in vivo dose and the plasma concentration was analysed in a previous study using rat model. The dose administered in vivo was similar to that used in vitro (0.32 mg·kg−1, or 19.4 µM) (Chieng-Yane et al., 2011). The carotid artery was considered to be occluded when blood flow stopped completely. Blood flow sometimes recovered spontaneously after an initial fall, but these cyclic events were not taken into account. In some F 16618-treated groups, the carotid was not completely occluded 33 min after the beginning of the infusion; in this case, the occlusion time was recorded as 33 min. Carotid blood flow is reported as the average of all successive determinations made during a 10 s period. The analogue blood flow signal was digitized (500 Hz), recorded simultaneously and analysed online with interactive software (Notocord-hem 4.1, Notocord Systems, Croissy sur Seine, France).
Calculations and statistical analysis
Intergroup comparisons were based on one-way anova or on one-way anova on ranks (Kruskal–Wallis) followed by Dunnett's or Dunn's test (Sigma Stat, Jandel, Erkrath, Germany), or on Student's t-test for paired comparisons. Differences were considered significant when P < 0.05.
Geometric mean IC50 and EC50 values and their 95% CIs were determined from concentration-response curves fitted with an operational sigmoid model (Origin®, Microcal Software Inc. Northampton, MA, USA), using non-linear curve fitting as follows: y = Emax x (xnH)/[(EC50nH) + (xnH)], where Emax is the maximal effect and nH (Hill coefficient) is the slope.
Results
Effect of F 16618 on SFLLR-induced human washed platelet aggregation
The PAR1 agonist peptide SFLLR induced concentration-dependent platelet aggregation. When washed platelets were pre-incubated with increasing concentrations of F 16618 (2.5–7.5 µM), SFLLR-induced platelet aggregation was inhibited in a concentration-dependent and competitive manner. As shown in Figure 1A, the concentration-response curves were shifted to the right. The competitive inhibition profile yielded a pA2 value of 5.88 (Figure 1B). Figure 1C shows the F 16618 inhibitory concentration-response obtained with 1.5 µM SFLLR. In the absence of the PAR-1 antagonist, this 1.5 µM SFLLR concentration gave a maximal aggregation of 71.1 ± 3.6% (Figure 1C). F 16618 concentrations from 1 µM induced concentration-dependent inhibition of platelet aggregation, with significant inhibition from 2 µM and complete inhibition with 4 µM (Figure 1D). The F 16618 IC50 calculated for SFLLR-induced platelet aggregation was 1.44 µM (95% CI: 1.34–1.53), in agreement with the potency of F 16618 shown in Figure 1B.
Figure 1.
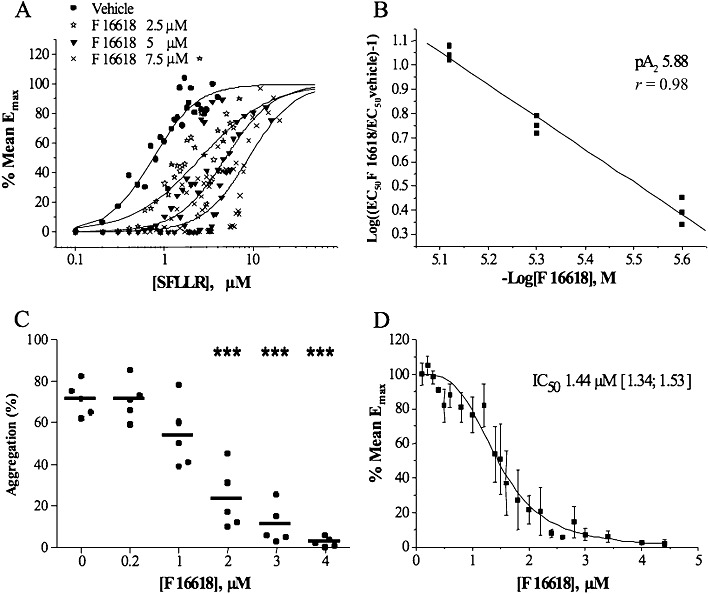
Inhibitory effects of F 16618 on SFLLR-induced human washed platelet aggregation. Platelets were pre-incubated with various concentrations of F 16618 and stimulated with SFLLR 0.1– 50 µM for 5 min. (A) and (B) SFLLR-induced platelet aggregation in the presence of F 16618 (2.5, 5 and 7.5 µM). The percentage of the mean Emax is shown in (A), and the corresponding Schild plot is presented in (B), for the calculation of pA2. (C) SFLLR (1.5 µM)-induced platelet aggregation was significantly inhibited by F 16618 (2, 3 and 4 µM). Each point represents the mean of duplicate tests, and the horizontal bar represents the mean value. (D) SFLLR (1.5 µM)-induced platelet aggregation expressed as the percentage of mean Emax used to calculate the IC50 value. ***P < 0.001 versus untreated group.
Thrombin-induced washed platelet aggregation
The antiplatelet effect of F 16618 was tested on thrombin-induced human washed platelet aggregation. Two concentrations of thrombin were used, 0.07 and 0.1 U·mL−1, both inducing basal aggregation of over 50% at 5 min. In the first set of experiments (0.07 U·mL−1 thrombin), platelet aggregation was inhibited by 50.5 ± 13.1 (P = 0.002; n = 6) and 72.9 ± 7 (P < 0.001; n = 6) at 2 min with 5 µM and 10 µM F 16618, respectively. This inhibition was no longer significant at 5 min with 5 µM F 16618, probably in part owing to a large interdonor variability (38.2 ± 19.2%). However, the inhibition remained statistically significant with 10 µM F 16618 (54.5 ± 14.4%; P = 0.013) (Figure 2A). In the presence of 0.1 U·mL−1 thrombin, platelet aggregation was inhibited by 32.9 ± 12.6 (P = 0.002; n = 14) and 41.1 ± 12.3 (P < 0.0001; n = 14) at 2 min and by 27.0 ± 11.7% (P = 0.011) and 29.1 ± 10.9% (P = 0.001) at 5 min, with 5 and 10 µM F 16618, respectively (Figure 2B).
Figure 2.
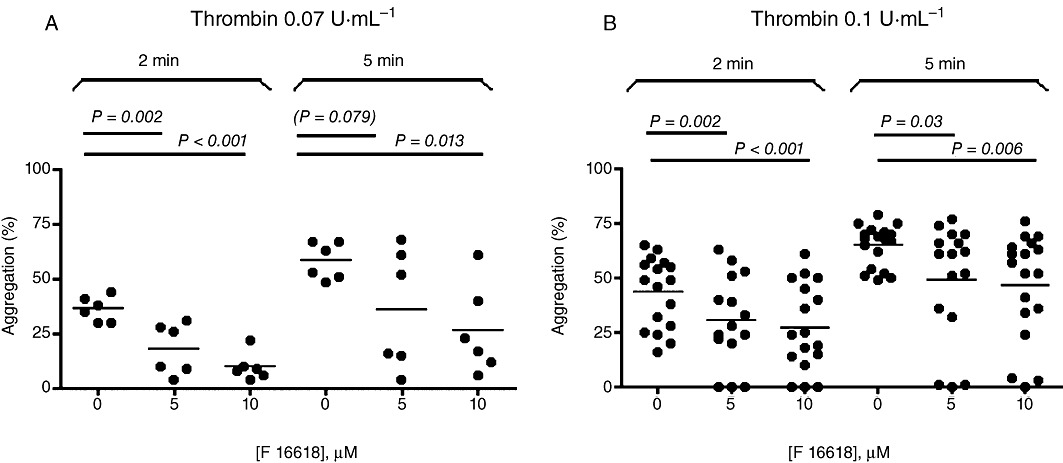
Inhibitory effects of F 16618 (5 and 10 µM) on human washed platelet aggregation induced by thrombin 0.07 U·mL−1 (n = 6) (A) or 0.1 U·mL−1 (n = 14) (B). Aggregation is expressed as a percentage of maximal light transmission measured at 2 min and 5 min. Each point represents the mean of duplicate tests, and the horizontal bar represents the mean value. *P < 0.05, **P < 0.01 versus untreated group.
Collagen- and PAR4 agonist-induced washed platelet aggregation
In order to determine the specificity of F 16618, we investigated the inhibitory effect of 10 µM F 16618 on collagen- and PAR4 agonist peptide-induced washed platelet aggregation. As shown in Figure 3, 5 µg·mL−1 collagen-induced platelet aggregation was slightly but significantly inhibited by 10 µM F 16618 (58.4 ± 2.4% vs. 45.9 ± 3.1%; n = 22; P < 0.001), whereas PAR4 agonist peptide-induced platelet aggregation was similar in the presence and absence of 10 µM F 16618 (74.6 ± 1.2% and 69.9 ± 3.2%, respectively; n = 17; NS).
Figure 3.
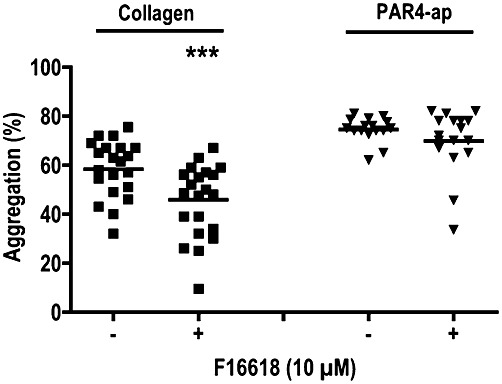
Inhibitory effects of F 16618 (10 µM) on collagen (5 µg·mL−1) (n = 22) and PAR4 agonist peptide (PAR4-ap;100 µM)-induced aggregation of human washed platelets (n = 17). ***P < 0.001 versus untreated group (–).
Effect of F 16618 on washed platelet secretion
To further examine the effects of F 16618 on platelet activation, we investigated α-granule secretion and measured the expression of the α-granule membrane-associated protein P-selectin by means of flow cytometry, in the presence of RGDS peptide to avoid platelet aggregation. In these conditions, less than 8% of resting platelets expressed P-selectin (MFI = 2 ± 0.4). After platelet activation, the proportion of P-selectin-positive cells increased by 44 ± 6% (MFI = 20 ± 4) (n = 4), 50 ± 12% (MFI = 22 ± 5) (n = 5) and 8 ± 1% (MFI = 20 ± 5) (n = 5) in the presence of SFLLR (1.5 µM), thrombin (0.1 U·mL−1) and collagen (5 µg·mL−1) respectively. SFLLR-induced P-selectin expression was strongly inhibited when platelets were pre-incubated with 4 µM F 16618 (5 ± 2%, MFI = 1.8 ± 1.8, P = 0.006, n = 4). However, concentrations of F 16618 up to 10 µM did not inhibit thrombin- or collagen-induced secretion (44 ± 12%, MFI = 19.6 ± 5.8, NS; and 6 ± 2, MFI = 18.8 ± 3, NS, respectively).
Effect of F 16618 on SFLLR-induced platelet aggregation in whole blood
F 16618 (5, 10 and 20 µM) was tested for its effect on 0.75 and 1.00 µM SFLLR-induced whole-blood platelet aggregation in the multiplate device (Figure 4). With SFLLR 0.75 µM, F 16618 at 10 and 20 µM significantly inhibited platelet aggregation in a concentration-dependent manner, by 42.6 ± 11.2% (n = 17, P = 0.007) and 61.3 ± 4.8% (n = 17, P < 0.001), respectively (Figure 4A). As expected, 1 µM SFLLR induced stronger aggregation than 0.75 µM SFLLR (522.6 ± 24.3 AU vs. 359.5 ± 34.9 AU), and 10 and 20 µM F 16618 still significantly inhibited platelet aggregation induced by 1 µM SFLLR (29.0 ± 7.9%, n = 21, P = 0.004; and 48.8 ± 8.4%, n = 20, P < 0.001). In order to evaluate the specificity of F 16618, we induced platelet aggregation with collagen and PAR4-ap, two other conventional platelet agonists. Even at the highest concentration used (20 µM), F 16618 did not inhibit platelet aggregation induced by either agonist (Figure 4C).
Figure 4.
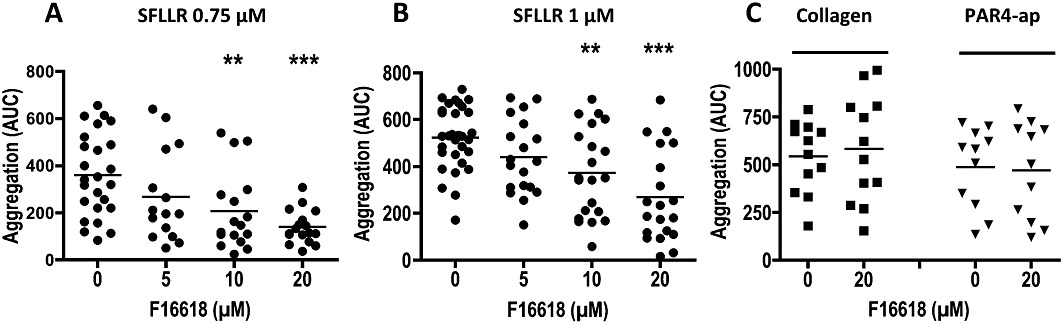
Inhibitory effects of F 16618 (5–20 µM) on human platelet aggregation in whole blood, induced by SFLLR 0.75 µM (n≥ 15) (A), SFLLR 1 µM (n≥ 19) (B), collagen 5 µg·mL−1 (n = 12) or PAR4-ap 100 µM (n = 11) (C). Aggregation is expressed as the area under the curve. (AUC) calculated after 6 min of activation. Each point represents the mean of duplicate tests, and the horizontal bar represents the mean value. **P < 0.01, ***P < 0.001 versus untreated group.
Effect of F 16618 on whole-blood haemostasis
As all current antithrombotic drugs have hemorrhagic effects, we explored the effects of F 16618 on coagulation and haemostasis in whole-blood assays. As shown in Table 1, the CT, CFT and MCF parameters measured with the ROTEM device were not modified by 20 µM F 16618, suggesting that F 16618 does not interfere with the coagulation cascade.
Table 1.
Effect of F 16618 on coagulation and haemostasis in whole blood
| F 16618 (µM) | P value | ||
|---|---|---|---|
| 0 | 20 | ||
| ROTEM-CT (s) | 68 ± 2 (60–76) | 71 ± 3 (61–82) | NS |
| ROTEM-CTF (s) | 117 ± 9 (82–156) | 117 ± 11 (78–167) | NS |
| ROTEM-MCF (mm) | 56 ± 2 (49–67) | 56 ± 2 (49–64) | NS |
| PFA collagen-ADP (s) | 100 ± 8 (75–126) | 99 ± 7 (80–125) | NS |
| PFA collagen-adrenaline (s) | 126 ± 10 (105–166) | 136 ± 14 (103–180) | NS |
Clotting time (CT), clot formation time (CFT) and maximum clot firmness (MCF). All data are means ± SEM (range) of six separate experiments.
We then tested the effects of F 16618 on the ability of platelets to occlude the aperture in a membrane coated either with collagen and adrenaline, or with collagen and ADP, in the PFA-100 device. F 16618 did not affect the closure time in either experimental condition, indicating that F 16618 does not modify platelet function in this assay, which measures both platelet adhesion and aggregation in high-shear conditions (Table 1).
Finally, we assessed the effect of F 16618 on clot retraction, a phenomenon that plays a role in thrombus stability. Clot retraction depends both on fibrin, produced by the coagulation cascade, and on platelet contractile activity. As shown in Table 2, the degree of clot retraction was unaffected by F 16618 at all time points (5, 10 and 30 min).
Table 2.
Effect of F 16618 on clot retraction
| Clot retraction (%) | F 16618 | |
|---|---|---|
| – | 20 µM | |
| 5 min | 26 ± 1 | 29 ± 1 |
| 10 min | 55 ± 1 | 57 ± 1 |
| 30 min | 81 ± 1 | 81 ± 1 |
Results are means ± SEM (n = 3).
Arterial thrombosis model
Initial mean carotid flow was similar in vehicle-treated and F 16618-treated guinea pigs (data not shown). Bolus administration of F 16618 had no effect on mean blood flow, even at the highest tested dose of 2.5 mg·kg−1 (data not shown). At 0.08 mg·kg−1, F 16618 delayed occlusion, but the difference did not reach significance. The first effective dose of F 16618 was 0.16 mg·kg−1, which delayed occlusion from 612 ± 39 s in vehicle controls to 949 ± 174 s (P < 0.05) (Figure 5). Maximal antithrombotic activity was observed at 0.32 mg·kg−1, the occlusion time being significantly increased by 107 ± 35%. A geometric mean ED50 value of 0.13 mg·kg−1 (95% confidence limits: 0.08–0.21 mg·kg−1) was calculated for F 16618 (Figure 5).
Figure 5.
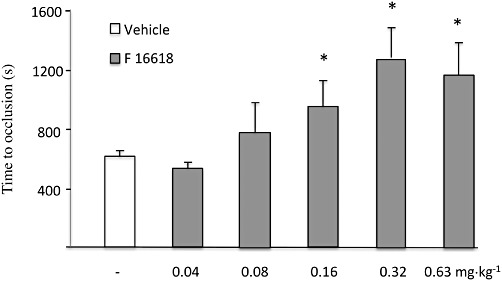
Effects of F 16618 (0.04 mg·kg−1, n = 6; 0.08 mg·kg−1, n = 7; 0.16 mg·kg−1, n = 7; 0.32 mg·kg−1, n = 8; 0.63 mg·kg−1, n = 8) and its vehicle (40% polyethylene glycol 300 in sterile 0.9% saline, n = 12) on the occlusion time in s. Data are mean ± SEM. *P < 0.05 versus vehicle.
Discussion and conclusions
Although the efficacy of clopidogrel, alone or combined with aspirin, has been largely documented, marked interindividual variability in the response to this treatment has led to the concept of ‘biological resistance’, and persistently high on-treatment platelet reactivity in vitro has been linked to an increased risk of cardiovascular events (Snoep et al., 2007; Marcucci et al., 2009). In the recent TRITON trial, the thienopyridine agent prasugrel led to a significant reduction in thrombotic events in acute coronary syndrome (ACS) patients undergoing percutaneous coronary interventions, but at a cost of more frequent major bleeding (Wiviott et al., 2007). New antiplatelet agents with an improved therapeutic index, such as ticagrelor, a reversible P2Y12 inhibitor, have been developed to overcome this risk of bleeding (Wallentin et al., 2009). PAR1 antagonists are good drug candidates in this respect as they inhibit thrombin-mediated platelet activation without affecting the role of thrombin in the coagulation cascade.
Here, we evaluated the ex vivo and in vivo effects of a new PAR1 antagonist, F 16618. We first examined the anti-aggregating effects of this compound ex vivo, on SFLLR-induced human washed platelets, and obtained an IC50 of 1.5 µM. F 16618 thus has more moderate ex vivo activity than other thrombin receptor antagonists. Indeed, with washed platelets, the IC50 values reported for FR171113, RWJ-56110 and SCH-79797 are 0.15, 0.07 and 0.48 µM, respectively (Kato et al., 1999; 2003; Wu and Teng, 2006). However, it is difficult to compare the ex vivo effect of these PAR-1 antagonists, as the published studies used different procedures and agonist peptides (TRAP5, TRAP6, high-affinity TRAP, etc.). The two PAR-1 antagonists SCH530348 and E5555 currently in phase III of development were studied in PRP and showed a better inhibition profile, with respective IC50 values of 0.025 and 0.031 µM (Chackalamannil et al., 2008; Kogushi et al., 2011). However, the thrombin inhibition profiles of these agents are different: FR17113, SCH-530348 and E5555 showed good activity on thrombin-induced aggregation (IC50 0.3, 0.047 and 0.064 µM, respectively), while RWJ-56110 and SCH-79797 inhibit thrombin-induced aggregation by less than 20% (Ahn et al., 2000; Wu and Teng, 2006). F 16618 has an intermediate profile, with moderate inhibition of thrombin-induced aggregation.
Although studies of F 16618 have given promising results, we cannot reliably predict the possible advantages of F 16618 over other drugs already on the market or in development. However, F 16618 has a shorter half-life compared with SCH530348 (2.8 h in rats, Perez et al., 2009; compared with 5.1 h for SCH in the same model, Chackalamannil et al., 2008), which suggests a more rapid offset of action, which is reported to be 5–11 days with vorapaxar. F 16618 is also likely to be cleared more rapidly from the circulation, which may represent an advantage in some specific situations (elderly, renal and hepatic failure, surgical procedures, etc.). Finally, F 16618 has probably an excellent bioavailability illustrated by similar and homogeneous results obtained after both i.v. and oral administration (Perez et al., 2009). The inhibition profile of F 16618 suggests a mechanism involving competitive inhibition, in line with a recent study of the effect of F 16618 on smooth muscle cell contraction (Bocquet et al., 2009).
In a mouse model lacking PAR4, the receptor that mimics human PAR1, PAR4 was required for platelet thrombus growth but not for initial thrombus formation (Vandendries et al., 2007). Moreover, platelet activation by thrombin in this arterial thrombosis model was associated with normal fibrin deposition. The concept that the PAR1 platelet pathway may not be essential for normal haemostasis has been confirmed in pre-clinical studies (Derian et al., 2003). We have recently demonstrated, in a rat model of extracorporeal arteriovenous shunt, that i.v. F 16618 has potent antithrombotic activity, without affecting the bleeding time (Letienne et al., 2010). In a guinea-pig model of arterial thrombosis, the non-peptidic PAR1 antagonist FR171113 was also reported to have no effect on the bleeding time (Kato et al., 2003). More recently, the orally active non-peptidic competitive PAR1 inhibitor SCH-530348 (Chackalamannil et al., 2008), which is currently in phase III clinical trials for the treatment of ACS, in combination with standard therapy, exhibited a good safety profile, both alone and in combination with aspirin and/or clopidogrel (Morrow et al., 2009). Together, these results suggest that the most potent platelet-activating pathway can be inhibited without markedly increasing the bleeding risk.
We also investigated the effect of F 16618 on human washed platelet aggregation and secretion induced by thrombin, the physiological PAR1 agonist. The inhibitory effect of F 16618 was less potent after thrombin-induced aggregation than after SFLLR-induced aggregation, possibly because the two agonists activate PAR1 through different mechanisms. Indeed, SFLLR binding to PAR1 directly activates the receptor and downstream signalling pathways, whereas thrombin first proteolytically cleaves PAR1, thereby exposing a tethered ligand that then interacts with and activates the receptor. A role of PAR4 in platelet activation by thrombin may also be involved (Wu and Teng, 2006). Thus, simultaneous inhibition of PAR1 and PAR4 might improve ex vivo platelet inhibition, but no PAR4 antagonists are currently available. Alternatively, the lower activity on thrombin-activated platelets could be because of an artefact caused by the washing procedure. This is supported by the difference in the inhibitory effect observed in vitro and in vivo. Moreover, in proliferating human aortic smooth muscle cells, a model involving no washing step, F 16618 had the same inhibitory effect on thrombin- and SFLLR-activated cells (chieng-Yane et al., 2011).
F 16618 did not interfere with PAR4 agonist-induced platelet aggregation but reduced collagen-induced aggregation, contrasting with the effects of FR171113 (Kato et al., 2003). However, this effect was observed with washed platelets but not in a system exploring platelet function in high-shear conditions. Moreover, F 16618 did not interfere with collagen-induced granule secretion, or with clot formation or retraction. Interestingly, similar results have been obtained with E5555, which inhibited collagen- and ADP-induced aggregation by 15–20% in PRP, but not in whole blood (Serebruany et al., 2009). Moreover, the moderate inhibitory effect of F 16618 on collagen-induced aggregation is probably not because of a direct effect on GPIIb/IIIa; otherwise, we would have observed an inhibition of aggregation induced by all agonists. It is possible that this non-specific effect is because of a direct effect on collagen or on its receptors. However, the lack of effect on the PFA test, performed with membrane coated with collagen, would support that this effect is restricted to the soluble form of collagen.
In the second part of this study, we tested the effect of F 16618 on platelet aggregation in whole blood, based on impedance. This system, which has recently been shown to be sensitive to aspirin and clopidogrel (Siller-Matula et al., 2009), also proved capable of detecting F 16618 inhibition of platelet aggregation. However, this inhibitory effect was less potent in whole blood than in washed platelets, and further experiments suggested that this difference could be because of an inhibitory effect of albumin (data not shown).
Finally, we used a guinea-pig model to evaluate the preventive effect of F 16618 on arterial thrombus formation in vivo. Thrombotic occlusion of the carotid artery was initiated by a photochemical reaction between transmural green light and i.v. Rose Bengal. Administration of F 16618 i.v. delayed arterial occlusion in a dose-dependent manner. The inhibitory effect of F 16618 was maximal at a dose of 0.32 mg·kg−1, which doubled the occlusion time. This potent antithrombotic effect in vivo, despite relatively moderate platelet inhibition ex vivo, might be related to an effect on other cells involved in thrombus formation. Indeed, PAR1 is widely distributed (Hirano and Kanaide, 2003; Coughlin, 2005) and PAR1 antagonists can inhibit the effects of thrombin not only on platelets but also on vascular cells. As F 16618 can limit smooth muscle cell contraction (Bocquet et al., 2009), it might also regulate endothelial cells and monocyte–macrophage activation, migration and proliferation. PAR1 antagonists are expected to have a beneficial effect on atherosclerotic arteries overexpressing PAR1, potentially providing a new way of modulating vascular repair and restenosis following percutaneous coronary intervention (Andrade-Gordon et al., 2001). An inhibitory effect of F 16618 on restenosis induced by balloon injury in the rat carotid artery has been observed in a study that also demonstrated specific inhibition of PAR1-mediated smooth muscle cell proliferation and migration, two mechanisms involved in restenosis (Chieng-Yane et al., 2011).
In conclusion, we have characterized a new PAR1 antagonist with strong antithrombotic activity in vivo and moderate antiplatelet effects ex vivo. This antithrombotic activity, which is not associated with major effects on physiological haemostasis, makes F 16618 a good drug candidate for use either alone or in combination with current treatments.
Acknowledgments
We are grateful to Véronique Remones and Fouad Dali Ali for their excellent technical assistance.
Glossary
- CFT
clot formation time
- CT
clotting time
- F16618
2-[5-oxo-5-(4-pyridin-2-yl-piperazin-1-yl)-penta-1,3-dienyl]-benzonitrile hydrochloride
- MCF
maximum clot firmness
- PAR
proteinase-activated receptor
- SFLLR peptide
PAR1 agonist peptide
Conflicts of interest
C B-L and P G received a research grant from Pierre Fabre laboratories. F N-W, M P, R L and BLeG are employees of Pierre Fabre laboratories.
References
- Ahn HS, Foster C, Boykow G, Stamford A, Manna M, Graziano M. Inhibition of cellular action of thrombin by N3-cyclopropyl-7-[[4-(1-methylethyl)phenyl]methyl]-7H-pyrrolo[3, 2-f]quinazoline-1,3-diamine (SCH 79797), a nonpeptide thrombin receptor antagonist. Biochem Pharmacol. 2000;60:1425–1434. doi: 10.1016/s0006-2952(00)00460-3. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen H, Greenberg DL, Fujikawa K, Xu W, Chung DW, Davie EW. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc Natl Acad Sci USA. 1999;96:11189–11193. doi: 10.1073/pnas.96.20.11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade-Gordon P, Derian CK, Maryanoff BE, Zhang HC, Addo MF, Cheung W, et al. Administration of a potent antagonist of protease-activated receptor-1 (PAR-1) attenuates vascular restenosis following balloon angioplasty in rats. J Pharmacol Exp Ther. 2001;298:34–42. [PubMed] [Google Scholar]
- Bernatowicz MS, Klimas CE, Hartl KS, Peluso M, Allegretto NJ, Seiler SM. Development of potent thrombin receptor antagonist peptides. J Med Chem. 1996;39:4879–4887. doi: 10.1021/jm960455s. [DOI] [PubMed] [Google Scholar]
- Bocquet A, Letienne R, Sablayrolles S, De Vries L, Perez M, Le Grand B. Effects of a new PAR1 antagonist, F 16618, on smooth muscle cell contraction. Eur J Pharmacol. 2009;611:60–63. doi: 10.1016/j.ejphar.2009.03.056. [DOI] [PubMed] [Google Scholar]
- Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061–3064. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- Chieng-Yane P, Bocquet A, Letienne R, Bourbon T, Sablayrolles S, Perez M, et al. Protease activated Receptor-1 antagonist, F 16618 reduces arterial restenosis by down-regulation of TNF{alpha} and MMP7 expression, and migration and proliferation of vascular smooth muscle cells. J Pharmacol Exp Ther. 2011;336:643–651. doi: 10.1124/jpet.110.175182. [DOI] [PubMed] [Google Scholar]
- Chung AW, Jurasz P, Hollenberg MD, Radomski MW. Mechanisms of action of proteinase-activated receptor agonists on human platelets. Br J Pharmacol. 2002;135:1123–1132. doi: 10.1038/sj.bjp.0704559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry. 2000;39:5458–5467. doi: 10.1021/bi9927078. [DOI] [PubMed] [Google Scholar]
- Damiano BP, Derian CK, Maryanoff BE, Zhang HC, Gordon PA. RWJ-58259: a selective antagonist of protease activated receptor-1. Cardiovasc Drug Rev. 2003;21:313–326. doi: 10.1111/j.1527-3466.2003.tb00124.x. [DOI] [PubMed] [Google Scholar]
- Derian CK, Damiano BP, Addo MF, Darrow AL, D'Andrea MR, Nedelman M, et al. Blockade of the thrombin receptor protease-activated receptor-1 with a small-molecule antagonist prevents thrombus formation and vascular occlusion in nonhuman primates. J Pharmacol Exp Ther. 2003;304:855–861. doi: 10.1124/jpet.102.042663. [DOI] [PubMed] [Google Scholar]
- Gross PL, Weitz JI. New antithrombotic drugs. Clin Pharmacol Ther. 2009;86:139–146. doi: 10.1038/clpt.2009.98. [DOI] [PubMed] [Google Scholar]
- Hirano K, Kanaide H. Role of protease-activated receptors in the vascular system. J Atheroscler Thromb. 2003;10:211–225. doi: 10.5551/jat.10.211. [DOI] [PubMed] [Google Scholar]
- Jennings LK. Role of platelets in atherothrombosis. Am J Cardiol. 2009;103:4A–10A. doi: 10.1016/j.amjcard.2008.11.017. [DOI] [PubMed] [Google Scholar]
- Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- Kato Y, Kita Y, Nishio M, Hirasawa Y, Ito K, Yamanaka T, et al. In vitro antiplatelet profile of FR171113, a novel non-peptide thrombin receptor antagonist. Eur J Pharmacol. 1999;384:197–202. doi: 10.1016/s0014-2999(99)00658-5. [DOI] [PubMed] [Google Scholar]
- Kato Y, Kita Y, Hirasawa-Taniyama Y, Nishio M, Mihara K, Ito K, et al. Inhibition of arterial thrombosis by a protease-activated receptor 1 antagonist, FR171113, in the guinea pig. Eur J Pharmacol. 2003;473:163–169. doi: 10.1016/s0014-2999(03)01973-3. [DOI] [PubMed] [Google Scholar]
- Kogushi M, Matsuoka T, Kawata T, Kuramochi H, Kawaguchi S, Murakami K, et al. The novel and orally active thrombin receptor antagonist E5555 (Atopaxar) inhibits arterial thrombosis without affecting bleeding time in guinea pigs. Eur J Pharmacol. 2011;657:131–137. doi: 10.1016/j.ejphar.2011.01.058. [DOI] [PubMed] [Google Scholar]
- Letienne R, Leparq-Panissie A, Calmettes Y, Nadal-Wollbold F, Perez M, Le Grand B. Antithrombotic activity of F 16618, a new PAR1 antagonist evaluated in extracorporeal arterio-venous shunt in the rat. Biochem Pharmacol. 2010;79:1616–1621. doi: 10.1016/j.bcp.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Marcucci R, Gori AM, Paniccia R, Giusti B, Valente S, Giglioli C, et al. Cardiovascular death and nonfatal myocardial infarction in acute coronary syndrome patients receiving coronary stenting are predicted by residual platelet reactivity to ADP detected by a point-of-care assay: a 12-month follow-up. Circulation. 2009;119:237–242. doi: 10.1161/CIRCULATIONAHA.108.812636. [DOI] [PubMed] [Google Scholar]
- Morrow DA, Scirica BM, Fox KA, Berman G, Strony J, Veltri E, et al. Evaluation of a novel antiplatelet agent for secondary prevention in patients with a history of atherosclerotic disease: design and rationale for the Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2 degrees P)-TIMI 50 trial. Am Heart J. 2009;158:335–341. doi: 10.1016/j.ahj.2009.06.027. [DOI] [PubMed] [Google Scholar]
- Oestreich J. SCH-530348, a thrombin receptor (PAR-1) antagonist for the prevention and treatment of atherothrombosis. Curr Opin Investig Drugs. 2009;10:988–996. [PubMed] [Google Scholar]
- Perez M, Lamothe M, Maraval C, Mirabel E, Loubat C, Planty B, et al. Discovery of novel protease activated receptors 1 antagonists with potent antithrombotic activity in vivo. J Med Chem. 2009;52:5826–5836. doi: 10.1021/jm900553j. [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol. 2008;153(Suppl. 1):S263–S282. doi: 10.1038/sj.bjp.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serebruany VL, Kogushi M, Dastros-Pitei D, Flather M, Bhatt DL. The in-vitro effects of E5555, a protease-activated receptor (PAR)-1 antagonist, on platelet biomarkers in healthy volunteers and patients with coronary artery disease. Thromb Haemost. 2009;102:111–119. doi: 10.1160/TH08-12-0805. [DOI] [PubMed] [Google Scholar]
- Shapiro MJ, Weiss EJ, Faruqi TR, Coughlin SR. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J Biol Chem. 2000;275:25216–25221. doi: 10.1074/jbc.M004589200. [DOI] [PubMed] [Google Scholar]
- Siller-Matula JM, Gouya G, Wolzt M, Jilma B. Cross validation of the Multiple Electrode Aggregometry. A prospective trial in healthy volunteers. Thromb Haemost. 2009;102:397–403. doi: 10.1160/TH08-10-0669. [DOI] [PubMed] [Google Scholar]
- Smyth SS, Woulfe DS, Weitz JI, Gachet C, Conley PB, Goodman SG, et al. G-protein-coupled receptors as signaling targets for antiplatelet therapy. Arterioscler Thromb Vasc Biol. 2009;29:449–457. doi: 10.1161/ATVBAHA.108.176388. [DOI] [PubMed] [Google Scholar]
- Snoep JD, Hovens MM, Eikenboom JC, van der Bom JG, Jukema JW, Huisman MV. Clopidogrel nonresponsiveness in patients undergoing percutaneous coronary intervention with stenting: a systematic review and meta-analysis. Am Heart J. 2007;154:221–231. doi: 10.1016/j.ahj.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Vandendries ER, Hamilton JR, Coughlin SR, Furie B, Furie BC. Par4 is required for platelet thrombus propagation but not fibrin generation in a mouse model of thrombosis. Proc Natl Acad Sci USA. 2007;104:288–292. doi: 10.1073/pnas.0610188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassallo RR, Kieber-Emmons T, Cichowski K, Brass LF. Structure-function relationships in the activation of platelet thrombin receptors by receptor-derived peptides. J Biol Chem. 1992;267:6081–6085. [PubMed] [Google Scholar]
- Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–1057. doi: 10.1056/NEJMoa0904327. [DOI] [PubMed] [Google Scholar]
- Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–2015. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]
- Wu CC, Teng CM. Comparison of the effects of PAR1 antagonists, PAR4 antagonists, and their combinations on thrombin-induced human platelet activation. Eur J Pharmacol. 2006;546:142–147. doi: 10.1016/j.ejphar.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Wu CC, Wu SY, Liao CY, Teng CM, Wu YC, Kuo SC. The roles and mechanisms of PAR4 and P2Y12/phosphatidylinositol 3-kinase pathway in maintaining thrombin-induced platelet aggregation. Br J Pharmacol. 2010;161:643–658. doi: 10.1111/j.1476-5381.2010.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]