

Abstract
BACKGROUND AND PURPOSE
Lung macrophages are critically involved in respiratory diseases. This study assessed the effects of the PDE4 inhibitor roflumilast and its active metabolite, roflumilast N-oxide on the release of a range of chemokines (CCL2, 3, 4, CXCL1, 8, 10) and of TNF-α, from human lung macrophages, stimulated with bacterial lipopolysaccharide LPS.
EXPERIMENTAL APPROACH
Lung macrophages isolated from resected human lungs were incubated with roflumilast, roflumilast N-oxide, PGE2, the COX inhibitor indomethacin, the COX-2 inhibitor NS-398 or vehicle and stimulated with LPS (24 h). Chemokines, TNF-α, PGE2 and 6-keto PGF1α were measured in culture supernatants by immunoassay. COX-2 mRNA expression was assessed with RT-qPCR. PDE activities were determined in macrophage homogenates.
KEY RESULTS
Expression of PDE4 in lung macrophages was increased after incubation with LPS. Roflumilast and roflumilast N-oxide concentration-dependently reduced the LPS-stimulated release of CCL2, CCL3, CCL4, CXCL10 and TNF-α from human lung macrophages, whereas that of CXCL1 or CXCL8 was not altered. This reduction by the PDE4 inhibitors was further accentuated by exogenous PGE2 (10 nM) but abolished in the presence of indomethacin or NS-398. Conversely, addition of PGE2 (10 nM), in the presence of indomethacin restored inhibition by roflumilast. LPS also increased PGE2 and 6-keto PGF1α release from lung macrophages which was associated with an up-regulation of COX-2 mRNA.
CONCLUSIONS AND IMPLICATIONS
Roflumilast and roflumilast N-oxide reduced LPS-induced release of CCL2, 3, 4, CXCL10 and TNF-α in human lung macrophages.
Keywords: roflumilast, lung macrophages, chemokines, TNF-α, LPS
Introduction
PDEs are a group of metallophosphohydrolases regulating the breakdown of intracellular cAMP and cGMP. Eleven major families (PDE1–11) and at least 21 subtypes with numerous splice variants have currently been identified and are distinguished by substrate specificity, sensitivity to inhibitors and allosteric activators and their N-terminal regulatory domains that largely govern recruitment to scaffolding proteins, hence subcellular compartmentation as well as post-translational regulation of enzymatic activity (Bender and Beavo, 2006).
Increasing intracellular cAMP may be attributed to enhanced synthesis related to receptor-triggered adenylate cyclase activation or attenuated breakdown by cAMP-hydrolysing PDEs. PDE4 is an isoenzyme specifically degrading cAMP and prominently expressed in inflammatory cells. As a corollary, selective PDE4 inhibitors were found to curb inflammatory responses in cells and experimental animals. Consequently, PDE4 inhibitors have been considered to represent a new therapeutic concept of non-steroid, anti-inflammatory remedies, potentially tailored to mitigate the specific airway inflammatory pattern in respiratory disorders such as chronic obstructive pulmonary disease (COPD) (Houslay et al., 2005; Fan Chung, 2006; Spina, 2008). Many PDE4 inhibitors have been synthesized and extensively analysed in cellular and animal studies. A few of them such as roflumilast, cilomilast, apremilast or CDP 840 have been in clinical development for mostly COPD or asthma but also psoriasis (apremilast). One of these, roflumilast, recently received approval as an oral, once daily tablet for the maintenance treatment of severe COPD (Giembycz and Field, 2010). Indeed, roflumilast was shown to improve lung function and, importantly, reduce the rate of exacerbations (Calverley et al., 2009; Fabbri et al., 2009) in this condition.
In the liver, roflumilast is converted by CYP3A4 and 1A2 to its active metabolite, roflumilast N-oxide, which has potency as a PDE4 inhibitor similar to that of the parent compound and maintains a high degree of selectivity towards other PDEs. In man, roflumilast N-oxide accounts for about 90% of the overall PDE4 inhibition, with the remainder being attributed to the parent compound roflumilast (Lahu et al., 2010). Following repeated, once daily, oral administration of roflumilast at the clinical dose of 500 µg·day−1, the plasma levels of roflumilast N-oxide are maintained in a range of about 1–2 nM (free, not bound to plasma proteins) over the dosing interval (Bethke et al., 2007). Over this concentration range, roflumilast N-oxide affected numerous functions of human cells involved in COPD. In addition, in vivo treatment with roflumilast modified inflammatory and structural remodelling responses in key disease models related to COPD. For example, roflumilast reduced the lung infiltration by CD4+ and CD8+ T cells, B cells and macrophages in mice exposed to tobacco smoke over 6 months (Martorana et al., 2008). Taken together, these results provided a mechanistic rationale for the efficacy of roflumilast in COPD (Hatzelmann et al., 2010).
Macrophages play a critical role in the pathophysiology of COPD and asthma (Barnes, 2004; Peters-Golden, 2004). Human lung macrophages orchestrate lung inflammation by releasing an array of leukocyte-recruiting chemokines (Barnes, 2009), as well as TNF-α. The precursors of macrophages are circulating monocytes and the differentiation of human peripheral blood monocytes to macrophages in vitro is accompanied by a change in the pattern of PDE isoenzymes. While PDE4 is the predominant isoform in human monocytes, in monocyte-derived macrophages PDE3 and PDE1 are increased along with a decline in PDE4 activity (Gantner et al., 1997). This PDE profile is similar to that described for human alveolar macrophages (Tenor et al., 1995).
Roflumilast and its active metabolite reduce LPS-induced TNF-α release from human blood monocytes. In agreement with the down-regulation of PDE4 during in vitro differentiation, LPS-stimulated TNF-α production in monocyte-derived macrophages is scarcely affected by PDE4 inhibitors (Gantner et al., 1997; Hatzelmann and Schudt, 2001).
The current study was designed to analyse the inhibitory effects of roflumilast and roflumilast N-oxide on the release of some chemokines and TNF-α from human lung macrophages and how such a response to the PDE4 inhibitors might be modulated by PGE2, which is increased in exhaled air of patients with COPD (Montuschi et al., 2003). We assayed for the CC and CXC chemokines involved in the recruitment of T lymphocytes [CCL3 (MIP-1α), CCL4 (MIP-1β), CXCL10 (IP-10)], monocytes [CCL2 (MCP-1)] and neutrophils [CXCL1 (GRO-α), CXCL8 (IL-8); chemokine nomenclature follows Alexander et al., 2011] in the airways and which are enhanced in COPD (Keatings et al., 1996; Capelli et al., 1999; Traves et al., 2002; Freeman et al., 2007; Costa et al., 2008) and in the airways of mice chronically exposed to tobacco smoke that develop lung infiltration by T and B cells, macrophages and neutrophils (Bracke et al., 2007; Nie et al., 2008; Braber et al., 2010).
The major outcome of the study was that the PDE4 inhibitors roflumilast and roflumilast N-oxide partially reduced the release of CCL2, 3, 4, CXCL10 and TNF-α from LPS-stimulated human lung macrophages, which was supported by the presence of endogenous prostanoids, such as PGE2 and PGI2.
Methods
Isolation and culture of human lung macrophages
The use of resected lung tissues for research purposes was approved by the Regional Ethics Committee for Biomedical Research (Boulogne-Billancourt, France). Lung tissues were obtained from 22 patients (age: 62 ± 2 years, 13 males, 9 females, FEV1/FVC ratio: 0.85 ± 0.03, smokers/ex-smokers 7/11, pack years: 39 ± 9 and 4 non-smokers), undergoing surgical resection for lung carcinoma and who had not received preoperative anti-cancer chemotherapy or radiotherapy. Lung macrophages were isolated from finely minced peripheral tissues (far from the tumour) by adherence as described previously (Buenestado et al., 2010). Briefly, the collected fluid from several washings of the minced peripheral lung tissues was centrifuged (300× g, 10 min), and the cell pellet was re-suspended in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mM l-glutamine and antibiotics. Re-suspended viable cells were then plated at 106 cells·mL−1. Following incubation for at least 1 h at 37°C (5% CO2 humidified atmosphere) non-adherent cells were removed by gentle washings. The adherent population of cells (198 ± 18 × 103 cells per well of a 24-well plate) were >95% pure macrophages, as determined by May–Grünwald–Giemsa staining and CD68 immunocytochemistry. Cell viability exceeded 95% as assessed by Trypan blue dye exclusion.
Incubation of lung macrophages with PDE4 inhibitors, PGE2 and COX inhibitors
The 24-well plates with adherent lung macrophages were washed with warm medium, without FCS, and 1 mL of fresh medium supplemented with 1% FCS was added per well. Lung macrophages were pre-incubated with roflumilast (0.1–1000 nM) or roflumilast N-oxide (0.1–1000 nM) or vehicle dimethyl sulphoxide (DMSO) for 30 min before stimulation with LPS at a concentration of 10 ng·mL−1. Following a 24 h incubation period, cell culture supernatants were collected and stored at −80°C for later analyses of chemokines and TNF-α. The optimal LPS concentration (10 ng·mL−1) and incubation time (24 h) were selected from preliminary time and concentration studies (data not shown). For example, concentration-dependent effect curves revealed a maximum stimulation of CCL2 and TNF-α release from human alveolar macrophages at 100–1000 ng·mL−1 LPS. At 1 ng·mL−1 LPS, the mean release of CCL2 and TNF-α amounted to 20% and 15% of the maximum, while at 10 ng·mL−1, optimal inhibition levels of 85% and 75% of the maximum were achieved. Consequently, the latter LPS concentration (10 ng·mL−1) was selected as the stimulus in the experiments. In some experiments, either PGE2 (10 nM), a non-selective COX inhibitor (indomethacin, 1 µM) or a COX-2 selective inhibitor (NS-398, 30 µM) were added in the presence or absence of the PDE4 inhibitors. Roflumilast and roflumilast N-oxide selectively inhibit PDE4 up to a concentration of 1 µM (Hatzelmann and Schudt, 2001). Stock solutions (10 mM) of PDE4 inhibitors, indomethacin and NS-398 were prepared in DMSO. PGE2 stock solution (10 mM) was prepared in ethanol. All subsequent dilutions were prepared daily in complete medium. The maximal DMSO concentration applied to cells in culture did not exceed 0.1%. Neither the vehicle nor any of the compounds used in this study altered the viability of the macrophages. All wells were run in duplicate for each series of experiments performed with lung macrophages isolated from a single lung sample.
Measurements of PDE1–5 isoenzyme activities
Lung macrophages (106 cells), with or without incubation with LPS for 24 h, were washed twice in PBS and re-suspended in 1 mL homogenization buffer (137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.5 mM KH2PO4, 10 mM HEPES, 1 mM EGTA, 1 mM MgCl2, 1 mM β-mercaptoethanol, 5 µM pepstatin A, 10 µM leupeptin, 50 µM PMSF, 10 µM soybean trypsin inhibitor, 2 mM benzamidine, pH 8.2). Cells were disrupted by sonication (Branson sonifier, 3 × 15 s), and lysates were immediately used for PDE activity measurements as previously described (Tenor et al., 1995), following a standard protocol (Thompson et al., 1979) with some modifications (Bauer and Schwabe, 1980). The assay mixture (final volume 200 µL) comprised 30 mM Tris–HCl, pH 7.4, 4 mM MgCl2, 0.5 µM of either cAMP or cGMP as substrate including [3H]cAMP or [3H]cGMP (about 30 000 c.p.m. per well), 100 µM EGTA, PDE isoenzyme-specific activators and inhibitors as described below and cell lysate. Incubations were performed for 20 min at 37°C, and reactions were terminated by adding 50 µL of 0.2 M HCl per well. Assays were left on ice for 10 min before 25 µg of 5′- nucleotidase (Crotalus atrox) was added (50 µL in 400 mM Tris–HCl, pH 8.5). Following an incubation period for 10 min at 37°C, assay mixtures were loaded onto QAE-Sephadex A25 columns (1 mL bed volume). Columns were eluted with 2 mL of 30 mM ammonium formate (pH 6.0), and radioactivity in the eluate was counted. Results were corrected for blank values (measured in the presence of denatured protein) that were below 2% of total radioactivity. cAMP or cGMP degradation did not exceed 25% of the amount of substrate added. The final DMSO concentration was 0.3% in all assays. Selective inhibitors and activators of PDE isoenzymes were used to determine activities of PDE1-5 families as described previously with modifications. Briefly, PDE4 was calculated as the difference of PDE activities at 0.5 µM cAMP in the presence and absence of 1 µM piclamilast. The difference between piclamilast-inhibited cAMP hydrolysis in the presence and absence of 10 µM motapizone was defined as PDE3. The fraction of cGMP (0.5 µM) hydrolysis in the presence of 10 µM motapizone that was inhibited by 100 nM sildenafil reflects PDE5. At the concentrations used in the assay piclamilast (1 µM), motapizone (10 µM) and sildenafil (100 nM) completely blocked PDE4, PDE3 and PDE5 activities without interfering with activities from other PDE families. PDE1 was defined as the increment of cAMP hydrolysis (in the presence of 1 µM piclamilast and 10 µM motapizone) or cGMP hydrolysis induced by 1 mM Ca2+ and 100 nM calmodulin. The increase of cAMP (0.5 µM) degrading activity in the presence of 1 µM piclamilast and 10 µM motapizone induced by 5 µM cGMP represented PDE2. The PDE2 inhibitor BAY 60–7550 (100 nM) completely inhibited this cGMP -induced activity increment, further confirming this activity as PDE2.
Chemokine and cytokine assays
Release of selected chemokines, TNF-α and IFN-γ was assessed by measuring their concentrations in the lung macrophage culture supernatants with elisa, according to the manufacturer's instructions (Duoset Development System, R&D Systems Europe, Lille, France). The supernatants were diluted with RPMI as appropriate, and the optical density was determined at 450 nm with an MRX II microplate reader from Dynex Technologies (Saint-Cloud, France). Cytokine concentrations are expressed as ng per 106 lung macrophages. The detection limits of these assays were 4 pg·mL−1 for CCL3, 8 pg·mL−1 for TNF-α, IFN-γ, CCL2 and CCL4 and 16 pg·mL−1 for CXCL1, CXCL8 and CXCL10.
PGE2 and 6-keto PGF1αassays
PGE2 and 6-keto PGF1α (the stable hydrolysis product of PGI2) in lung macrophage culture supernatants were determined using commercially available EIA kits according to the manufacturer's instructions (Cayman Chemical Europe, Tallinn, Estonia). The detection limits were 36 and 6 pg·mL−1 respectively. PGE2 and 6-keto PGF1α concentrations are expressed as nM.
Quantitative RT-PCR analysis
Lung macrophages, stimulated or not with LPS (10 ng·mL−1) for 24 h, were harvested in RNAlater®, and total RNA was extracted with RNeasy Mini Kit (Qiagen, Courtabeuf, France). After DNAse I treatment (DNAse set, Qiagen), 1 µg of total RNA was used to generate single-stranded cDNA (cDNA Archive Kit, Applied Biosystems, Courtabeuf, France). Reactions without reverse transcriptase were systematically run in parallel. Absence of contaminating genomic DNA was assessed by amplifying 24 housekeeping genes (18S, ACTB, AGPAT1, B2M, EEF1A2, GAPDH, GUSB, HDAC1, HMBS, HPRT1, ILF2, PMM1, POLR2H, PPIA, PSMA1, RPL13, RPL37A, SDHA, TAX1BP1, TBP, TPT1, UBC, VIL2, YWHAZ).
A specific TaqMan® low-density array, based on pre-designed reagents (Assay-on Demand, Applied Biosystems), was set up to evaluate expression of COX-2 (PTGS2, Hs00153133_m1). Real-time quantitative PCR was carried out with 100 ng of cDNA in 2 µL of final reaction volume in 384-well microfluidic plates preloaded with probes and primers on the ABI PRISM 7900 Sequence Detection System using TaqMan® Universal PCR Master Mix (Applied Biosystems). The fold changes in mRNA expression were calculated according to the ΔΔCt method (Livak and Schmittgen, 2001). Sample profiles were obtained for the 24 housekeeping genes to determine the best set of genes leading to the most accurate normalization. The mean values of TPT1, PPIA and UBC were selected for further normalization of data. Genes were considered to be significantly expressed, and their transcript was measurable if their corresponding threshold cycle (Ct) value was ≤35.
Statistical analysis
Data are expressed as means ± SEM; n represents the number of patients from whom lung macrophages preparations were obtained. The concentration–effect curves were analysed using non-linear regression GraphPad Prism® Version 5.01 (GraphPad Software Inc., San Diego, CA), and sigmoidal curves were plotted to analyse the effects of PDE4 inhibitors on cytokine production and calculate EC50 and maximum efficacy. Statistical analyses used one-way repeated-measures anova followed by Dunnett's post-tests. Significance was defined as P < 0.05.
Materials
Penicillin–streptomycin, DMSO, l-glutamine, FCS, RNAlater®, Trypan blue dye, HEPES, EGTA, β-mercaptoethanol, pepstatin, leupeptin, PMSF, soybean trypsin inhibitor, benzamidine, cAMP, cGMP and LPS (from Escherichia coli serotype 0111:B4, batch 029 K4022), indomethacin, PGE2 were purchased from Sigma (St. Louis, MO). RPMI 1640 medium, PBS and BSA were from Eurobio Biotechnology (Les Ulis, France). The COX-2 inhibitor, NS-398, was purchased from Tocris Biosciences (Bristol, UK). Roflumilast and roflumilast N-oxide were synthesized at Nycomed GmbH (Konstanz, Germany). Piclamilast and sildenafil were synthesized at the chemical facilities of Nycomed essentially as described in the corresponding patents. Motapizone was a generous gift from Rhone-Poulenc Rorer (Cologne, Germany). [5,8-3H]cAMP, [8-3H]cGMP and [methyl-3H]thymidine were purchased from GE Lifesciences (Freiburg, Germany). All other chemicals were of analytical grade and were obtained from Merck (Darmstadt, Germany). All cell culture plastics were from CML (Nemours, France).
Results
Activities of PDE1-5 isoenzymes in human lung macrophages
In the absence of LPS, basal cAMP and cGMP hydrolysing PDE activities in human lung macrophages cultured for 24 h were 288 ± 69 pmol·min−1 per 108 cells and 78 ± 20 pmol·min−1 per 108 cells (n = 4), respectively, at 0.5 µM substrate concentrations. Based on the use of specific activators and inhibitors of PDE isoenzymes (Ca++-calmodulin-stimulated) PDE1, PDE3 and PDE4 were the predominant activities detected (Figure 1A). Exposure of lung macrophages to LPS (10 ng·mL−1) for 24 h resulted in a 1.6-fold increase in total cAMP hydrolysis. The increased cAMP hydrolysis was entirely attributed to a 3.5-fold up-regulation of PDE4 activity (P < 0.05; Figure 1B), while other PDE isoenzymes were not affected (data not shown).
Figure 1.

PDE1–5 isoenzyme activities in human lung macrophages and LPS-induced up-regulation of PDE4. Lung macrophages (purity > 95%) were homogenized, and PDE activities assessed in whole lysates at 0.5 µM cAMP or cGMP substrate concentrations. (A) Enzymatic activities reflecting basal cAMP and cGMP hydrolyses and PDE1–5 are shown. PDE1 activities are depicted as cGMP or cAMP hydrolysis stimulated by Ca++–calmodulin. (B) Lung macrophages were incubated with 10 ng·mL−1 LPS over 24 h. Enzymic activities reflecting basal cAMP and cGMP hydrolysis as well as PDE4 are depicted. Results from four different donors are shown as the means ± SEM. *P < 0.05, significantly different from control.
Roflumilast and its active metabolite, roflumilast N-oxide, reduced the release of chemokines and TNF-α from LPS-stimulated human lung macrophages
Among the major CC and CXC chemokines and cytokines generated by human lung macrophages in response to stimulation with LPS for 24 h are CCL2, CCL3, CCL4, CXCL1, CXCL8, CXCL10 and TNF-α (Buenestado et al., 2010). On the other hand, IFN-γ was not detected, either at baseline or after stimulation with LPS (n = 6).
The effects of the PDE4 inhibitors roflumilast and roflumilast N-oxide on the LPS-induced release of these chemokines and TNF-α from human lung macrophages were investigated. LPS strongly increased the release of all of the investigated chemokines and TNF-α (Tables 1 and 2). Roflumilast and roflumilast N-oxide (0.1 nM–1 µM) concentration-dependently and partly reduced the LPS-triggered release of the explored CC chemokines, CXCL10 and TNF-α (Table 3, Figures 2 and 3) but not that of CXCL1 and CXCL8 (Table 2). Maximum inhibition (efficacy) was comparable between the two PDE4 inhibitors for each of the responsive analytes, as expected, and strongest for CCL2, followed by CXCL10, TNF-α and then CCL3 and CCL4 (Table 3). Half-maximum inhibition (potency, EC50) was around or below 1 nM for the CC chemokines, CXCL10 and TNF-α with a trend towards somewhat higher potency for roflumilast compared with the active metabolite (with the exception of CCL4) and was in accord with the potencies of these PDE4 inhibitors against PDE4 enzymic activity (Hatzelmann et al., 2010). Roflumilast N-oxide at 1 nM, a concentration in the range of clinically relevant plasma levels, reduced the release of LPS-stimulated CCL2 and TNF-α by about 30% (P < 0.05) and that of CCL3, CCL4 and CXCL10 to a more moderate extent (Table 3), similar to findings in other cellular systems (Hatzelmann et al., 2010). Basal release of these CC, CXC chemokines and TNF-α from unstimulated lung macrophages, was not altered by the two PDE4 inhibitors (n = 3).
Table 1.
The effect of LPS on CCL2, 3, 4, CXCL10 and TNF-α release in culture supernatants of human lung macrophages
| Chemokine/cytokine | |||||
|---|---|---|---|---|---|
| CCL2 (n = 13) | CCL3 (n = 12) | CCL4 (n = 14) | CXCL10 (n = 10) | TNF-α (n = 14) | |
| Basal | 0.4 ± 0.1 | 0.3 ± 0.1 | 2.2 ± 0.5 | 0.01 ± 0.003 | 0.02 ± 0.01 |
| +LPS | 23 ± 7 | 385 ± 66 | 467 ± 90 | 1.0 ± 0.4 | 60 ± 15 |
Table 2.
Roflumilast and roflumilast N-oxide do not influence LPS-stimulated release of CXCL1 and CXCL8 from human lung macrophages
| LPS + | ||||||
|---|---|---|---|---|---|---|
| Roflumilast | Roflumilast N-oxide | |||||
| Cytokine | Basal | LPS only | 1 nM | 100 nM | 1 nM | 100 nM |
| CXCL1 | 0.3 ± 0.1 | 220 ± 56 | 219 ± 42 | 184 ± 28 | 243 ± 50 | 186 ± 33 |
| CXCL8 | 7.2 ± 1.1 | 597 ± 115 | 592 ± 110 | 529 ± 99 | 613 ± 77 | 500 ± 82 |
Macrophages were incubated with medium (basal) or LPS (10 ng·mL−1) in the absence (LPS only) or presence of roflumilast (1 nM or 100 nM) or roflumilast N-oxide (1 nM or 100 nM) or vehicle before being stimulated with LPS for 24 h. Cell culture supernatants were collected and analysed for CXCL1 and CXCL8 by elisa. Data are expressed in ng per 106 cells. Results are shown as the means ± SEM of five to seven independent experiments.
Table 3.
Potency and efficacy of the PDE4 inhibitors roflumilast and roflumilast N-oxide to reduce TNF-α, CCL2, CCL3, CCL4 and CXCL10 release from LPS-stimulated human lung macrophages in presence and absence of 10 nM PGE2
| PDE4 inhibitor | Roflumilast | Roflumilast N-oxide | ||||
|---|---|---|---|---|---|---|
| Calculated parameter | EC50 | Efficacy | EC50 | Efficacy | Inhibition at 1 nM | |
| Analyte | Additive | nM [CI 95] | % | nM [CI 95] | % | % |
| TNF-α | Vehicle | 0.1 [0.03–0.5] | 38 ± 3 | 0.4 [0.1–1.4] | 45 ± 3 | 31 ± 8 |
| 10 nM PGE2 | 0.2 [0.06–0.4] | 57 ± 3 | 0.3 [0.1–0.9] | 60 ± 4 | 43 ± 8 | |
| CCL2 | Vehicle | 0.2 [0.09–0.6] | 58 ± 3 | 1.0 [0.4–2.2] | 63 ± 3 | 30 ± 6 |
| 10 nM PGE2 | 0.2 [0.06–0.6] | 63 ± 4 | 0.7 [0.3–1.5] | 70 ± 3 | 42 ± 6 | |
| CCL3 | Vehicle | 0.5 [0.07–3.1] | 29 ± 3 | 1.5 [0.5–4.4] | 32 ± 2 | 13 ± 2 |
| 10 nM PGE2 | 0.3 [0.09–1.0] | 49 ± 3 | 0.7 [0.2–2.3] | 48 ± 3 | 29 ± 4 | |
| CCL4 | Vehicle | 0.5 [0.1–2.5] | 32 ± 3 | 0.4 [0.04–4.3] | 26 ± 4 | 17 ± 6 |
| 10 nM PGE2 | 0.7 [0.08–2.8] | 45 ± 3 | 0.6 [0.2–2.0] | 46 ± 3 | 29 ± 3 | |
| CXCL10 | Vehicle | 0.1 [0.04–0.4] | 49 ± 3 | 1.6 [0.2–10] | 45 ± 6 | 18 ± 9 |
| 10 nM PGE2 | 0.1 [0.02–0.6] | 60 ± 5 | 1.0 [0.2–3.5] | 58 ± 5 | 30 ± 11 | |
Values of EC50 are shown with 95% confidence intervals (CI 95). The maximum efficacy (mean ± SEM) and the percentage of inhibition achieved by roflumilast N-oxide at 1 nM (mean ± SEM) were calculated from concentration-dependent inhibition curves given in Figure 2 and 3. TNF-α, CCL2, 3 or 4 and CXCL10 release in presence or absence of 10 nM PGE2 were both set as 100%. Effects of 10 nM PGE2 on the release of the chemokines and cytokines are given in the text.
Figure 2.
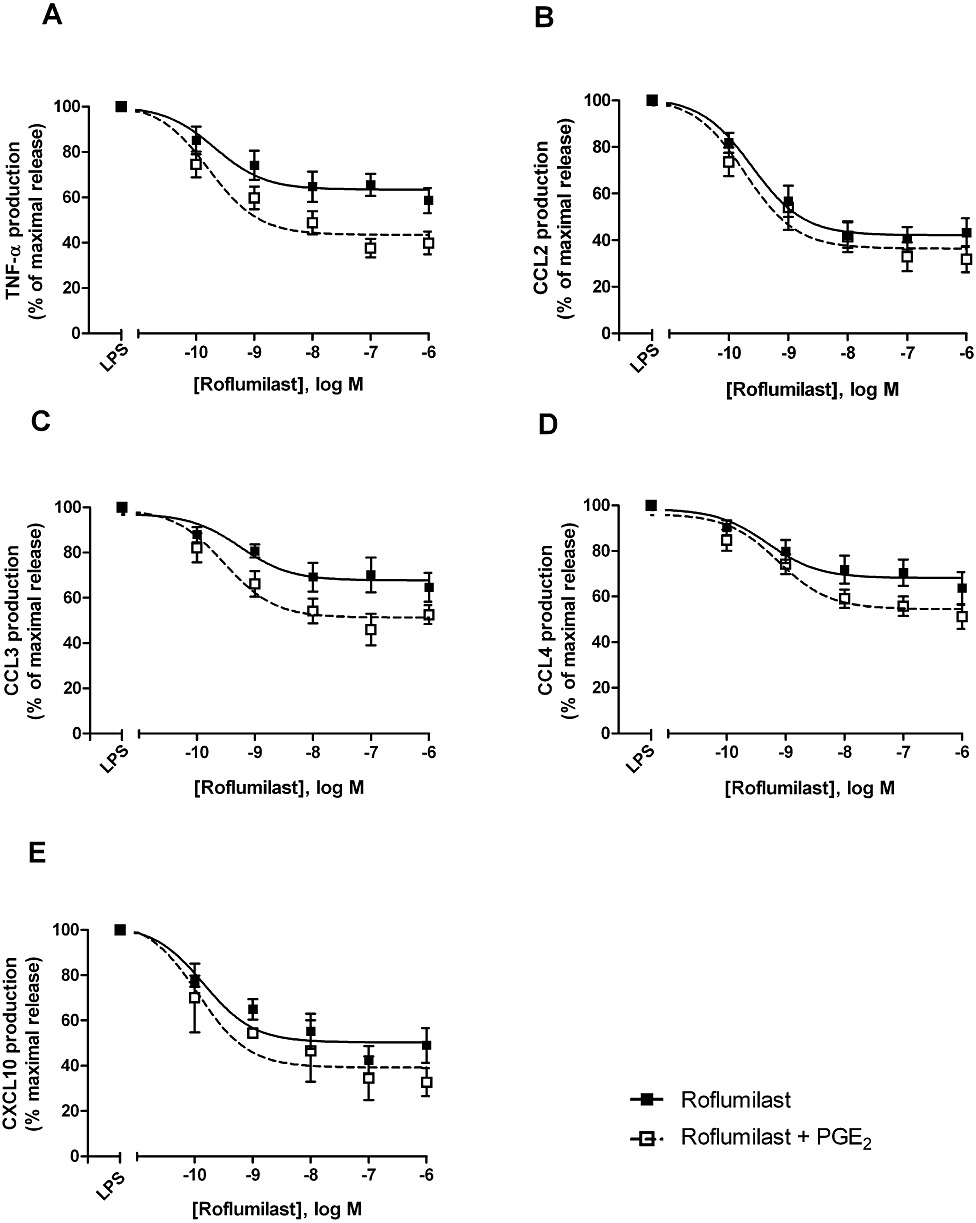
Effects of roflumilast on the release of TNF-α (A), CCL2 (B), CCL3 (C), CCL4 (D) and CXCL10 (E) from LPS-stimulated human lung macrophages, with or without PGE2. Lung macrophages were pre-incubated with roflumilast (0.1–1000 nM), PGE2 (10 nM) or vehicle, before LPS (10 ng·mL−1) was added. Chemokines and TNF-α were determined in culture supernatants collected after a 24 h incubation time. Results are shown as the means ± SEM of five to nine independent experiments and shown as % of maximum release of the respective chemokine or TNF-α related to LPS alone or LPS and PGE2, each defined as maximum (100%).
Figure 3.
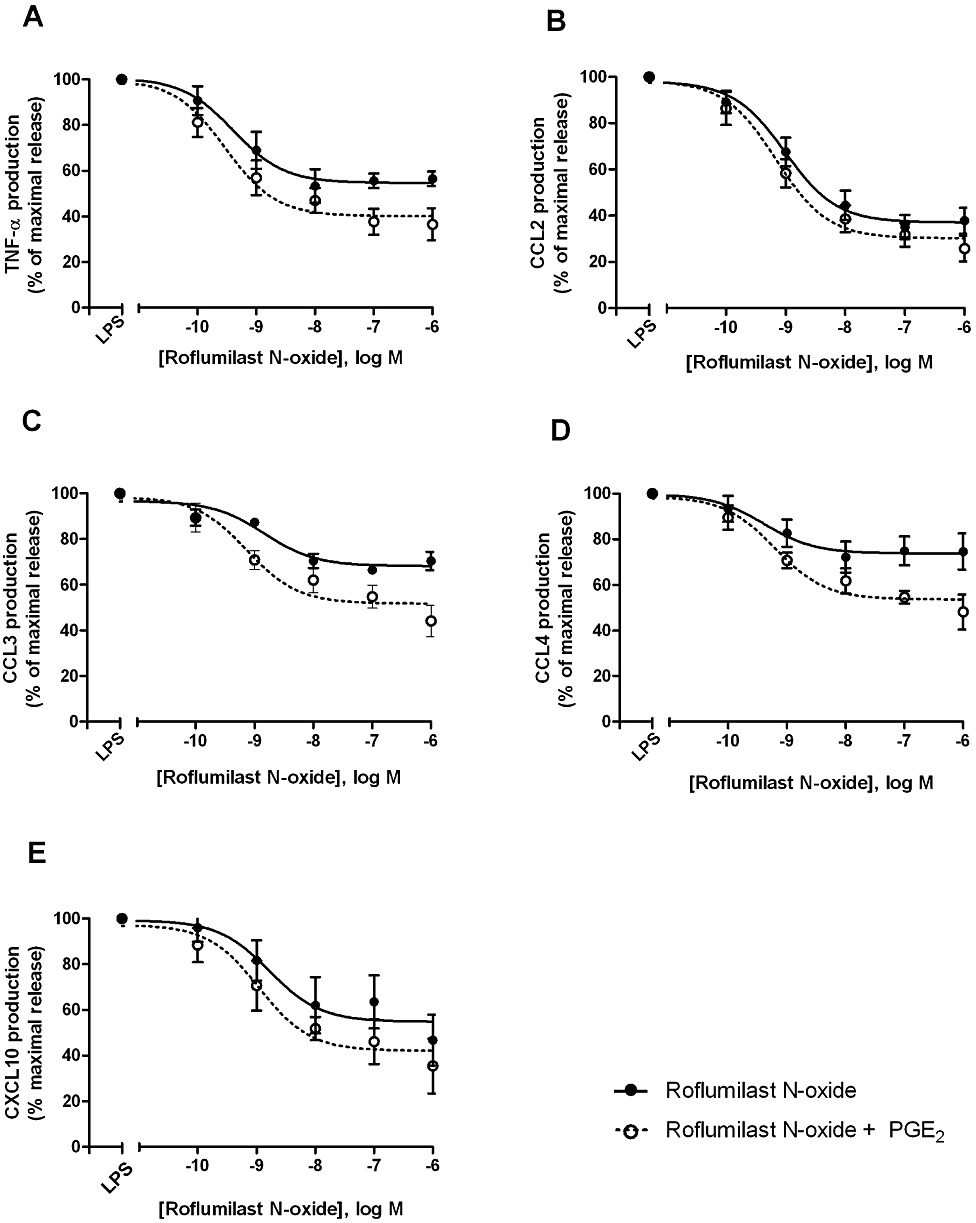
Effects of roflumilast N-oxide on the release of TNF-α (A), CCL2 (B), CCL3 (C), CCL4 (D) and CXCL10 (E) by LPS-stimulated human lung macrophages, without or with PGE2. Lung macrophages were pre-incubated with roflumilast N-oxide (0.1–1000 nM), PGE2 (10 nM) or vehicle before LPS (10 ng·mL−1) was added. Chemokines and TNF-α were determined in culture supernatants collected after a 24 h incubation time. Results are shown as the means ± SEM of five to nine independent experiments and depicted as % of maximum release of the respective chemokine or TNF-α related to LPS alone or LPS and PGE2, each defined as maximum (100%).
Role of PGE2 in the inhibition of LPS-induced chemokine and TNF-α release by roflumilast and roflumilast N-oxide
Previous studies in human monocyte-derived macrophages have shown that the presence of PGE2 (10 nM) (to increase cAMP formation) was required to allow a moderate reduction of LPS-stimulated TNF-α release by PDE4 inhibitors from these cells (Gantner et al., 1997; Hatzelmann and Schudt, 2001). In the current study with LPS-stimulated human lung macrophages, PDE4 inhibitors were able on their own to curb the release of CCL2, 3, 4 and CXCL10 chemokines and TNF-α, as detailed in the previous section (Figures 2 and 3). In this context, the role of PGE2 was analysed next. First of all, the addition of PGE2 (10 nM) did not change the release of CCL2, 3 and 4 but diminished that of TNF-α by 43% and CXCL10 by 48%, following stimulation of lung macrophages with LPS (data not shown). Next, the effects of 10 nM PGE2 were assessed on the concentration-dependent decrease of LPS-stimulated CCL2, 3, 4, CXCL10 and TNF-α release by the PDE4 inhibitors. Overall, the prostanoid did not substantially affect the potencies (EC50) of the PDE4 inhibitors to limit the release of CCL2, 3, 4, CXCL10 and TNF-α from LPS-stimulated lung macrophages (Figures 2 and 3, Table 3). There was a moderate gain in efficacy in the presence of PGE2 for LPS-stimulated release of CC chemokines. However, taking into account the reduction by PGE2 of TNF-α and CXCL10 release (see above), the efficacy versus LPS (in absence of PGE2) reached 71 ± 6% and 76 ± 12% with roflumilast and PGE2 respectively. Release of CXCL1 and CXCL8 remained unaffected by PDE4 inhibitors, even when 10 nM PGE2 was present.
LPS-stimulated human alveolar macrophages are a source of PGE2, following an up-regulation of COX-2 (Hempel et al., 1994). Furthermore, peritoneal macrophages released 6-keto PGF1α following their exposure to LPS, indicating PGI2 production (Rouzer et al., 2004). To unravel a putative role of endogenously produced prostanoids in transducing PDE4 inhibition into reduced mediator release from the LPS-stimulated lung macrophages, the effect of the COX inhibitor indomethacin was explored. Strikingly, indomethacin abolished the inhibitory effects of roflumilast on the release of CCL2, 3, 4, CXCL10 and TNF-α (Figure 4). Substantial amounts of PGE2 and 6-keto PGF1α accumulated in culture supernatants of lung macrophages incubated with LPS (10 ng·mL−1) over 24 h. The increased release of PGE2 and 6-keto PGF1αwas accompanied by an about 70-fold increase of COX-2 transcripts in human lung macrophages, after LPS stimulation (Table 4). Concurrent incubation with indomethacin or the COX-2 selective inhibitor NS-398 abolished the accumulation of PGE2 and 6-keto PGF1α (Figure 5). On the other hand, roflumilast at 1 nM or 100 nM did not affect LPS-induced release of PGE2 and 6-keto PGF1α (Figure 5).
Figure 4.
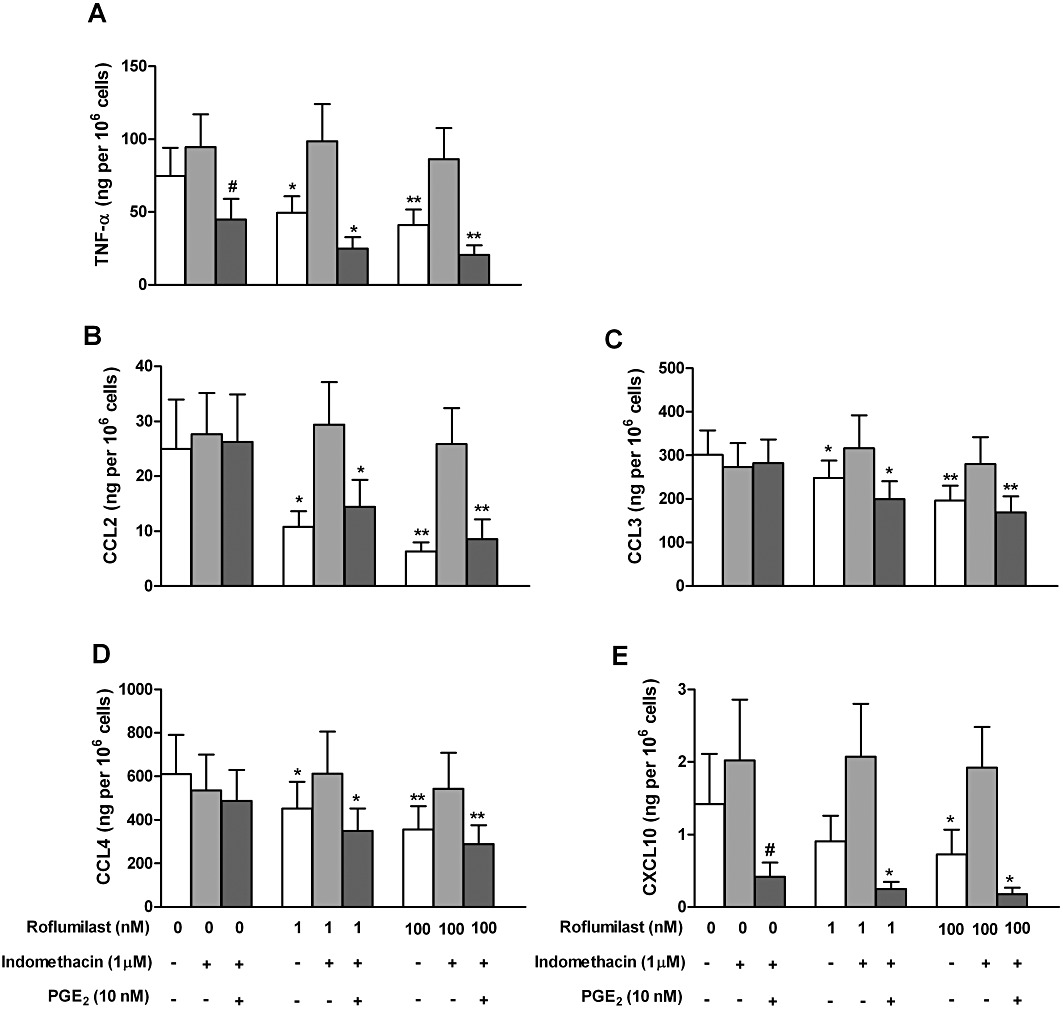
Role of prostanoids in the suppression of LPS-induced TNF-α (A), CCL2 (B), CCL3 (C), CCL4 (D) and CXCL10 (E) release from human lung macrophages by roflumilast. Lung macrophages were pre-incubated with indomethacin (1 µM) or vehicle over 30 min and then with roflumilast (0, 1 or 100 nM), PGE2 (10 nM) or vehicle for another 30 min before being stimulated with LPS (10 ng·mL−1) for 24 h. Results are shown as the means ± SEM of five to six independent experiments, *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from respective controls. #P < 0.05, significantly different from LPS alone. Unstimulated release of chemokines and TNF-α was low and did not exceed 2% of that observed following stimulation with LPS.
Table 4.
Effects of LPS on expression of COX-2 (PTGS2) transcripts in human lung macrophages
| Relative expression in control (mean ± SEM) × 1000 | Fold change | Range of fold change | |
|---|---|---|---|
| PTGS2 | 0.53 ± 0.13 | 68.6 ± 28.6 | 4.0–137.9 |
The relative expression of PTGS2 was determined by real-time quantitative RT-PCR and calculated from the expression of the target gene versus the mean expression of housekeeping genes (TPT1, PPIA, UBC). Results following LPS (10 ng·mL−1) incubation were compared with those under control conditions over 24 h, and the fold change in the relative expression attributed to LPS was calculated. Results are shown as mean ± SEM from five independent experiments.
Figure 5.
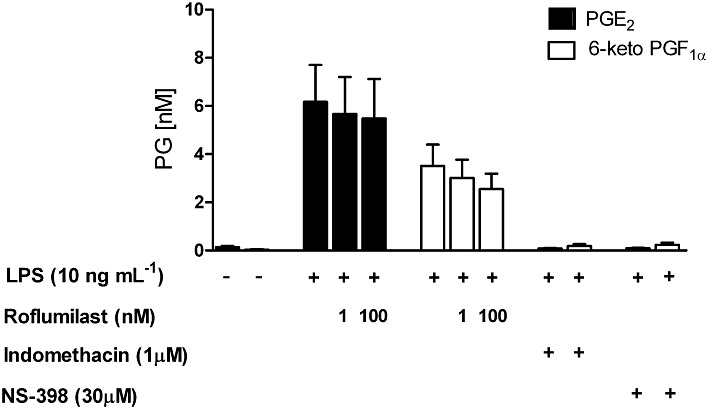
LPS-induced release of PGE2 and 6-keto PGF1α, the stable hydrolysis product of PGI2, from human lung macrophages. Effects of the PDE4 inhibitor roflumilast, the COX inhibitor indomethacin and the COX-2 selective inhibitor NS-398. Human lung macrophages were pre-incubated with roflumilast (1 nM or 100 nM), indomethacin (1 µM), NS-398 (30 µM) or vehicle for 30 min and then stimulated with LPS (10 ng·mL−1). Accumulation of PGE2 or 6-keto PGF1α in cell culture supernatants was measured after 24 h using commercially available EIA. Results are given as the mean ± SEM from at least five independent experiments.
Adding exogenous PGE2 (10 nM) restored the inhibition of mediator release by roflumilast, when indomethacin was present (Figure 4). The presence of the COX inhibitor in the incubation medium (which by itself did not affect cytokine release) induced a synergism between roflumilast (1 nM or 100 nM) and PGE2 (10 nM, a concentration close to that in culture medium following LPS stimulation) to diminish CCL2, 3 or 4 release, while each on their own was ineffective (Figure 4B–D).
In the presence of indomethacin, release of TNF-α and CXCL10 were more sensitive to inhibition by PGE2 at 10 nM, resulting in an about 50% and 85% reduction of their respective release after LPS (Figure 4A, E).
Taken together, these results suggest that the COX-2-dependent formation of prostanoids (such as PGE2 or PGI2) acts together with the PDE4 inhibitors to reduce LPS-stimulated release of cytokines such as CCL2, 3, 4, CXCL10 and TNF-α from lung macrophages.
Discussion and conclusions
In human lung macrophages, (i) PDE1, PDE3 and PDE4 activities were present and PDE4 was up-regulated by LPS; (ii) the PDE4 inhibitors, roflumilast and its active metabolite roflumilast N-oxide, reduced the LPS-stimulated release of the CC chemokines CCL2, CCL3, CCL4, the CXC chemokine CXCL10 (but not that of CXCL1 and CXCL8) and of TNF-α; and (iii) endogenous prostanoids (PGE2 and PGI2) produced following LPS-induced COX-2 up-regulation may have supported the reduced mediator release by PDE4 inhibitors.
The PDE (1–5) isoenzyme pattern in human lung macrophages is similar to that in human monocyte-derived macrophages (Gantner et al., 1997), alveolar macrophages (Tenor et al., 1995) or macrophage-like U937 cells (Shepherd et al., 2004). Previously, PDE7A1 protein has been described in human lung macrophages but a PDE7 inhibitor only marginally reduced LPS-induced TNF-α release from these cells, although it was at least additive with a PDE4 inhibitor (Smith et al., 2004). In the current study, the presence of some PDE7 activity contributing to overall cAMP hydrolysis in human lung macrophages cannot be excluded. However, under the current experimental conditions, PDE3 and PDE4 account for about 85% of total cAMP hydrolysis.
Incubation of human lung macrophages with LPS over 24 h resulted in an increased cAMP-hydrolysing activity, entirely attributable to PDE4, as previously shown in monocytic MonoMac6 cells (Verghese et al., 1995). LPS-induced up-regulation of PDE4 (primarily PDE4B) was also shown in human monocytes (Ma et al., 1999; Wang et al., 1999; Jin and Conti, 2002) and mouse peritoneal macrophages (Jin et al., 2005). An increase of PDE4 activity was reported in alveolar macrophages from smokers with COPD compared with those without COPD (Barber et al., 2004), suggesting the clinical relevance of PDE4 regulation.
The increase in PDE4 found in the present study may increase the functional role this PDE isoenzyme plays in LPS-stimulated human lung macrophages. The PDE4 inhibitors roflumilast and its active metabolite, roflumilast N-oxide reduced the release of the chemokines CCL2, CCL3, CCL4 and CXCL10 as well as of TNF-α from LPS-stimulated lung macrophages. The fact that only partial inhibition was achieved, at concentrations completely and selectively blocking PDE4, is likely to indicate a functional role for the remaining PDE1, PDE3, and PDE7 isoenzymes (Gantner et al., 1997; Hatzelmann and Schudt, 2001).
In contrast to findings in lung macrophages (Smith et al., 2004 and the current study), PDE4 inhibitors did not suppress LPS-induced TNF-α release from human monocyte-derived macrophages but required the addition of PGE2 (Gantner et al., 1997; Hatzelmann and Schudt, 2001). Differences in LPS-induced PGE2 release between these macrophages may account for this inconsistency because earlier reports indicate that endogenous release of PGE2 is restricted to human alveolar macrophages, compared with monocyte-derived macrophages (Takayama et al., 2002; Ratcliffe et al., 2007).
In the current study, roflumilast and roflumilast N-oxide displayed rather comparable potency (EC50) to suppress LPS-induced release of the assayed chemokines and TNF-α with a trend towards a somewhat higher potency for roflumilast. This is in line with the similar potency of roflumilast and roflumilast N-oxide to inhibit PDE4 as reported for neutrophils (IC50 of 0.8 and 2 nM, respectively; Hatzelmann and Schudt, 2001) and PDE4 isoenzymes (Hatzelmann et al., 2010). This potency to inhibit PDE4 is in the range of the functional effects found with the PDE4 inhibitors in lung macrophages.
Roflumilast and roflumilast N-oxide differentially affected chemokine release as the PDE4 inhibitors did not influence the release of the neutrophil attractants CXCL1 and CXCL8 from LPS-stimulated human lung macrophages. This observation may appear inconsistent with evidence that roflumilast mitigates airway neutrophil accumulation in vivo (Hatzelmann et al., 2010) and in patients with COPD (Grootendorst et al., 2007). However, composite effects of the PDE4 inhibitors on neutrophils and on other inflammatory cells, such as T cells, potentially controlling neutrophil migration may reconcile such discrepancies (Sanz et al., 2007; Hatzelmann et al., 2010).
In the current study, several lines of evidence indicated that autocrine prostanoids such as PGE2 or prostacyclin may have supported the PDE4 inhibitor to reduce cytokine release from LPS-stimulated human lung macrophages most likely by an amplified cAMP response emanating from simultaneous activation of cAMP synthesis (prostanoids) and inhibition of its degradation (roflumilast). First of all, inhibitors of COX (indomethacin) and COX-2 (NS-398) abolished the reduced release of the cytokines by roflumilast. Second, LPS up-regulated COX-2 expression and enhanced the release of PGE2 and 6-keto PGF1α, that was blocked by indomethacin or NS-398. The LPS-induced up-regulation of COX-2 in alveolar macrophages is well documented (Hempel et al., 1994) and also occurs following in vitro exposure of these cells to tobacco smoke extract (Profita et al., 2010). In induced sputum of smokers with COPD, the macrophage represents a major COX-2-expressing cell (Profita et al., 2010). Third, replenishing PGE2 (in the presence of indomethacin) at a concentration corresponding to the one accumulating following LPS restored suppression of chemokines and TNF-α by the PDE4 inhibitor. Fourth, roflumilast did not affect the LPS-induced release of PGE2 and 6-keto PGF1α from human lung macrophages. In addition, in presence of indomethacin, both PGE2 (10 nM) and roflumilast (100 nM) were inactive on LPS-induced CCL2, 3 and 4 release from lung macrophages, while adding the prostanoid and the PDE4 inhibitor together elicited a strongly reduced release of these chemokines. Such a synergistic effect indicates that the PDE4 inhibitor acts by enhancing cAMP in this experimental system. In this context, we recently reported that activation of A2A adenosine receptors, which results in enhanced cAMP synthesis, curbed the release of CCL2, CCL3, CCL4, CXCL10 and TNF-α but not CXCL1 and CXCL8 from LPS-stimulated human lung macrophages, corresponding to the pattern observed for the PDE4 inhibitors (Buenestado et al., 2010).
In pulmonary fibroblasts, the in vitro effects of PDE4 inhibitors may also be supported by endogenous PGE2 (Kohyama et al. (2002) as, in human lung fibroblasts, indomethacin reversed the suppressive effects of the PDE4 inhibitors roflumilast and rolipram on fibroblast-driven collagen gel contraction and fibronectin-induced chemotactic response (Togo et al., 2009). Furthermore, proliferation of human peripheral blood mononuclear cells (PBMC) induced by phytohaemagglutinin, was decreased by the PDE4 inhibitors rolipram and CDP840 and this was partially reversed by indomethacin. However, in contrast to the present findings, where LPS-induced release of PGE2 or 6-keto PGF1α from human lung macrophages was not affected by roflumilast, the PDE4 inhibitors further augmented the release of TGFβ1-induced PGE2 in human lung fibroblasts (Togo et al., 2009; Sabatini et al., 2010) and LPS-induced PGE2 in PBMC (Banner et al., 1999). While these different observations reflect cell context-specific effects of PDE4 inhibitors on PGE2 release (Klein et al., 2007; Togo et al., 2009), another possible explanation is that in the protocols with human lung fibroblasts and PBMC (Banner et al., 1999; Togo et al., 2009), PDE4 inhibition partly acts by increasing PGE2 release, in contrast to lung macrophages in the current study.
Although not addressed in this study, endogenous PGI2 produced by human alveolar macrophages following LPS may represent another candidate to synergise with PDE4 inhibitors. Indeed, stable analogues of PGI2 reduce the release of IL-6, TNF-α, GM-CSF and IL-1β from LPS-stimulated human alveolar macrophages (Raychaudhuri et al., 2002; Aronoff et al., 2007).
Increased cAMP translates into reduced synthesis of many pro-inflammatory cytokines and chemokines by human monocytes or macrophages, although the precise mechanisms for each have not yet been elucidated (Kimata et al., 1998; Delgado and Ganea, 2001; Takayama et al., 2002; Bryn et al., 2006). The early PDE4 inhibitor rolipram inhibited, concentration-dependently, the LPS-induced TNF-α release from human lung macrophages (Smith et al., 2004). Maximum inhibition (efficacy) of about 35% was in accord with findings in the current study with roflumilast and roflumilast N-oxide, and the reported potency (EC50 of 150 nM) was close to inhibition of PDE4 catalytic activity by rolipram (Smith et al., 2004). Apremilast, a PDE4 inhibitor in development for psoriasis, reduced LPS-induced release of CCL2, CCL3 and CXCL10 from human mononuclear cells (Schafer et al., 2010).
CXCL10, also known as IFN-γ-inducible protein 10 (IP-10), is produced by macrophages in response to IFN-γ but also to LPS as recently shown in human monocyte-derived macrophages (Kent et al., 2009). However, under our experimental conditions using human lung macrophages, IFN-γ remains undetectable even in LPS-stimulated lung macrophages and is therefore unlikely involved in the regulation of CXCL10 in our system.
The regulation of CXCL8 by cAMP depends on the experimental system. Interventions enhancing cAMP are reported to reduce (Au et al., 1998; Muhl et al., 2000; Slater et al., 2006; Kaur et al., 2008), increase (Linden, 1996; Kavelaars et al., 1997) or not affect (Zhong et al., 1995; Yoshimura et al., 1997; Donnelly et al., 2010; Schafer et al., 2010) the release of CXCL8. In the current study with human lung macrophages, PDE4 inhibition, even in presence of PGE2, had no effect on LPS-induced CXCL8 release in line with findings from our laboratory with A2A receptor agonist (Buenestado et al., 2010) and in LPS-stimulated monocytes using apremilast (Schafer et al., 2010). As found for roflumilast, activation of adenosine A2A receptors did not affect CXCL1 release (Buenestado et al., 2010).
Taking our results together, the PDE4 inhibitors roflumilast and its active metabolite, roflumilast N-oxide partially reduced the LPS-induced release of CCL2, CCL3, CCL4, CXCL10 and TNF-α from human lung macrophages. This reduction was supported by endogenously released prostanoids such as PGE2 or PGI2. However, the release of two other chemokines CXCL1 and CXCL8 remained unchanged.
Acknowledgments
Dr Buenestado was supported by a DIM SEnT-Research Grant of the Conseil Régional d'Ile-de-France. We thank Patricia Tchepikoff for qRT-PCR technical assistance.
Glossary
- BAL
bronchoalveolar lavage
- COPD
chronic obstructive pulmonary disease
- DMSO
dimethyl sulphoxide
- EIA
enzyme immunoassay
- FCS
fetal calf serum
- PDE
phosphodiesterase
Conflicts of interest
Herman Tenor is an employee of Nycomed GmbH, Konstanz, Germany.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronoff DM, Peres CM, Serezani CH, Ballinger MN, Carstens JK, Coleman N, et al. Synthetic prostacyclin analogs differentially regulate macrophage function via distinct analog-receptor binding specificities. J Immunol. 2007;178:1628–1634. doi: 10.4049/jimmunol.178.3.1628. [DOI] [PubMed] [Google Scholar]
- Au BT, Teixeira MM, Collins PD, Williams TJ. Effect of PDE4 inhibitors on zymosan-induced IL-8 release from human neutrophils: synergism with prostanoids and salbutamol. Br J Pharmacol. 1998;123:1260–1266. doi: 10.1038/sj.bjp.0701723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner KH, Hoult JR, Taylor MN, Landells LJ, Page CP. Possible Contribution of Prostaglandin E2 to the antiproliferative effect of phosphodiesterase 4 inhibitors in human mononuclear cells. Biochem Pharmacol. 1999;58:1487–1495. doi: 10.1016/s0006-2952(99)00223-3. [DOI] [PubMed] [Google Scholar]
- Barber R, Baillie GS, Bergmann R, Shepherd MC, Sepper R, Houslay MD, et al. Differential expression of PDE4 cAMP phosphodiesterase isoforms in inflammatory cells of smokers with COPD, smokers without COPD, and nonsmokers. Am J Physiol Lung Cell Mol Physiol. 2004;287:L332–L343. doi: 10.1152/ajplung.00384.2003. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD. 2004;1:59–70. doi: 10.1081/COPD-120028701. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2009;41:631–638. doi: 10.1165/rcmb.2009-0220TR. [DOI] [PubMed] [Google Scholar]
- Bauer AC, Schwabe U. An improved assay of cyclic 3′,5′-nucleotide phosphodiesterases with QAE-Sephadex columns. Naunyn Schmiedebergs Arch Pharmacol. 1980;311:193–198. doi: 10.1007/BF00510259. [DOI] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- Bethke TD, Bohmer GM, Hermann R, Hauns B, Fux R, Morike K, et al. Dose-proportional intraindividual single- and repeated-dose pharmacokinetics of roflumilast, an oral, once-daily phosphodiesterase 4 inhibitor. J Clin Pharmacol. 2007;47:26–36. doi: 10.1177/0091270006294529. [DOI] [PubMed] [Google Scholar]
- Braber S, Henricks PA, Nijkamp FP, Kraneveld AD, Folkerts G. Inflammatory changes in the airways of mice caused by cigarette smoke exposure are only partially reversed after smoking cessation. Respir Res. 2010;11:99. doi: 10.1186/1465-9921-11-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracke KR, D'hulst AI, Maes T, Demedts IK, Moerloose KB, Kuziel WA, et al. Cigarette smoke-induced pulmonary inflammation, but not airway remodelling, is attenuated in chemokine receptor 5-deficient mice. Clin Exp Allergy. 2007;37:1467–1479. doi: 10.1111/j.1365-2222.2007.02808.x. [DOI] [PubMed] [Google Scholar]
- Bryn T, Mahic M, Enserink JM, Schwede F, Aandahl EM, Tasken K. The cyclic AMP-Epac1-Rap1 pathway is dissociated from regulation of effector functions in monocytes but acquires immunoregulatory function in mature macrophages. J Immunol. 2006;176:7361–7370. doi: 10.4049/jimmunol.176.12.7361. [DOI] [PubMed] [Google Scholar]
- Buenestado A, Grassin Delyle S, Arnould I, Besnard F, Naline E, Blouquit-Laye S, et al. The role of adenosine receptors in regulating production of tumour necrosis factor-alpha and chemokines by human lung macrophages. Br J Pharmacol. 2010;159:1304–1311. doi: 10.1111/j.1476-5381.2009.00614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009;374:685–694. doi: 10.1016/S0140-6736(09)61255-1. [DOI] [PubMed] [Google Scholar]
- Capelli A, Di Stefano A, Gnemmi I, Balbo P, Cerutti CG, Balbi B, et al. Increased MCP-1 and MIP-1beta in bronchoalveolar lavage fluid of chronic bronchitics. Eur Respir J. 1999;14:160–165. doi: 10.1034/j.1399-3003.1999.14a27.x. [DOI] [PubMed] [Google Scholar]
- Costa C, Rufino R, Traves SL, Lapa ESJR, Barnes PJ, Donnelly LE. CXCR3 and CCR5 chemokines in induced sputum from patients with COPD. Chest. 2008;133:26–33. doi: 10.1378/chest.07-0393. [DOI] [PubMed] [Google Scholar]
- Delgado M, Ganea D. Inhibition of endotoxin-induced macrophage chemokine production by vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide in vitro and in vivo. J Immunol. 2001;167:966–975. doi: 10.4049/jimmunol.167.2.966. [DOI] [PubMed] [Google Scholar]
- Donnelly LE, Tudhope SJ, Fenwick PS, Barnes PJ. Effects of formoterol and salmeterol on cytokine release from monocyte-derived macrophages. Eur Respir J. 2010;36:178–186. doi: 10.1183/09031936.00158008. [DOI] [PubMed] [Google Scholar]
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, Bundschuh DS, Brose M, Martinez FJ, et al. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet. 2009;374:695–703. doi: 10.1016/S0140-6736(09)61252-6. [DOI] [PubMed] [Google Scholar]
- Fan Chung K. Phosphodiesterase inhibitors in airways disease. Eur J Pharmacol. 2006;533:110–117. doi: 10.1016/j.ejphar.2005.12.059. [DOI] [PubMed] [Google Scholar]
- Freeman CM, Curtis JL, Chensue SW. CC chemokine receptor 5 and CXC chemokine receptor 6 expression by lung CD8+ cells correlates with chronic obstructive pulmonary disease severity. Am J Pathol. 2007;171:767–776. doi: 10.2353/ajpath.2007.061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner F, Kupferschmidt R, Schudt C, Wendel A, Hatzelmann A. In vitro differentiation of human monocytes to macrophages: change of PDE profile and its relationship to suppression of tumour necrosis factor-alpha release by PDE inhibitors. Br J Pharmacol. 1997;121:221–231. doi: 10.1038/sj.bjp.0701124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giembycz MA, Field SK. Roflumilast: first phosphodiesterase 4 inhibitor approved for treatment of COPD. Drug Des Devel Ther. 2010;4:147–158. doi: 10.2147/dddt.s7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootendorst DC, Gauw SA, Verhoosel RM, Sterk PJ, Hospers JJ, Bredenbroker D, et al. Reduction in sputum neutrophil and eosinophil numbers by the PDE4 inhibitor roflumilast in patients with COPD. Thorax. 2007;62:1081–1087. doi: 10.1136/thx.2006.075937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzelmann A, Schudt C. Anti-inflammatory and immunomodulatory potential of the novel PDE4 inhibitor roflumilast in vitro. J Pharmacol Exp Ther. 2001;297:267–279. [PubMed] [Google Scholar]
- Hatzelmann A, Morcillo EJ, Lungarella G, Adnot S, Sanjar S, Beume R, et al. The preclinical pharmacology of roflumilast–a selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2010;23:235–256. doi: 10.1016/j.pupt.2010.03.011. [DOI] [PubMed] [Google Scholar]
- Hempel SL, Monick MM, Hunninghake GW. Lipopolysaccharide induces prostaglandin H synthase-2 protein and mRNA in human alveolar macrophages and blood monocytes. J Clin Invest. 1994;93:391–396. doi: 10.1172/JCI116971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10:1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- Jin SL, Conti M. Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-alpha responses. Proc Natl Acad Sci USA. 2002;99:7628–7633. doi: 10.1073/pnas.122041599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SL, Lan L, Zoudilova M, Conti M. Specific role of phosphodiesterase 4B in lipopolysaccharide-induced signaling in mouse macrophages. J Immunol. 2005;175:1523–1531. doi: 10.4049/jimmunol.175.3.1523. [DOI] [PubMed] [Google Scholar]
- Kaur M, Holden NS, Wilson SM, Sukkar MB, Chung KF, Barnes PJ, et al. Effect of beta2-adrenoceptor agonists and other cAMP-elevating agents on inflammatory gene expression in human ASM cells: a role for protein kinase A. Am J Physiol Lung Cell Mol Physiol. 2008;295:L505–L514. doi: 10.1152/ajplung.00046.2008. [DOI] [PubMed] [Google Scholar]
- Kavelaars A, Van De Pol M, Zijlstra J, Heijnen CJ. Beta 2-adrenergic activation enhances interleukin-8 production by human monocytes. J Neuroimmunol. 1997;77:211–216. doi: 10.1016/s0165-5728(97)00076-3. [DOI] [PubMed] [Google Scholar]
- Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153:530–534. doi: 10.1164/ajrccm.153.2.8564092. [DOI] [PubMed] [Google Scholar]
- Kent LM, Smyth LJ, Plumb J, Clayton CL, Fox SM, Ray DW, et al. Inhibition of lipopolysaccharide-stimulated chronic obstructive pulmonary disease macrophage inflammatory gene expression by dexamethasone and the p38 mitogen-activated protein kinase inhibitor N-cyano-N'-(2-{[8-(2,6-difluorophenyl)-4-(4-fluoro-2-methylphenyl)-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidin-2-yl]amino}ethyl)guanidine (SB706504) J Pharmacol Exp Ther. 2009;328:458–468. doi: 10.1124/jpet.108.142950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata M, Shichijo M, Daikoku M, Inagaki N, Mori H, Nagai H. Pharmacological modulation of LPS-induced MIP-1 alpha production by peripheral blood mononuclear cells. Pharmacology. 1998;56:230–236. doi: 10.1159/000028202. [DOI] [PubMed] [Google Scholar]
- Klein T, Shephard P, Kleinert H, Komhoff M. Regulation of cyclooxygenase-2 expression by cyclic AMP. Biochim Biophys Acta. 2007;1773:1605–1618. doi: 10.1016/j.bbamcr.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Kohyama T, Liu X, Wen FQ, Zhu YK, Wang H, Kim HJ, et al. PDE4 inhibitors attenuate fibroblast chemotaxis and contraction of native collagen gels. Am J Respir Cell Mol Biol. 2002;26:694–701. doi: 10.1165/ajrcmb.26.6.4743. [DOI] [PubMed] [Google Scholar]
- Lahu G, Hunnemeyer A, Diletti E, Elmlinger M, Ruth P, Zech K, et al. Population pharmacokinetic modelling of roflumilast and roflumilast N-oxide by total phosphodiesterase-4 inhibitory activity and development of a population pharmacodynamic-adverse event model. Clin Pharmacokinet. 2010;49:589–606. doi: 10.2165/11536600-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Linden A. Increased interleukin-8 release by beta-adrenoceptor activation in human transformed bronchial epithelial cells. Br J Pharmacol. 1996;119:402–406. doi: 10.1111/j.1476-5381.1996.tb16000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Ma D, Wu P, Egan RW, Billah MM, Wang P. Phosphodiesterase 4B gene transcription is activated by lipopolysaccharide and inhibited by interleukin-10 in human monocytes. Mol Pharmacol. 1999;55:50–57. doi: 10.1124/mol.55.1.50. [DOI] [PubMed] [Google Scholar]
- Martorana PA, Lunghi B, Lucattelli M, De Cunto G, Beume R, Lungarella G. Effect of roflumilast on inflammatory cells in the lungs of cigarette smoke-exposed mice. BMC Pulm Med. 2008;8:17. doi: 10.1186/1471-2466-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montuschi P, Kharitonov SA, Ciabattoni G, Barnes PJ. Exhaled leukotrienes and prostaglandins in COPD. Thorax. 2003;58:585–588. doi: 10.1136/thorax.58.7.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhl H, Chang JH, Huwiler A, Bosmann M, Paulukat J, Ninic R, et al. Nitric oxide augments release of chemokines from monocytic U937 cells: modulation by anti-inflammatory pathways. Free Radic Biol Med. 2000;29:969–980. doi: 10.1016/s0891-5849(00)00389-0. [DOI] [PubMed] [Google Scholar]
- Nie L, Xiang R, Zhou W, Lu B, Cheng D, Gao J. Attenuation of acute lung inflammation induced by cigarette smoke in CXCR3 knockout mice. Respir Res. 2008;9:82. doi: 10.1186/1465-9921-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters-Golden M. The alveolar macrophage: the forgotten cell in asthma. Am J Respir Cell Mol Biol. 2004;31:3–7. doi: 10.1165/rcmb.f279. [DOI] [PubMed] [Google Scholar]
- Profita M, Sala A, Bonanno A, Riccobono L, Ferraro M, La Grutta S, et al. Chronic obstructive pulmonary disease and neutrophil infiltration: role of cigarette smoke and cyclooxygenase products. Am J Physiol Lung Cell Mol Physiol. 2010;298:L261–L269. doi: 10.1152/ajplung.90593.2008. [DOI] [PubMed] [Google Scholar]
- Ratcliffe MJ, Walding A, Shelton PA, Flaherty A, Dougall IG. Activation of E-prostanoid4 and E-prostanoid2 receptors inhibits TNF-alpha release from human alveolar macrophages. Eur Respir J. 2007;29:986–994. doi: 10.1183/09031936.00131606. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri B, Malur A, Bonfield TL, Abraham S, Schilz RJ, Farver CF, et al. The prostacyclin analogue treprostinil blocks NFkappaB nuclear translocation in human alveolar macrophages. J Biol Chem. 2002;277:33344–33348. doi: 10.1074/jbc.M203567200. [DOI] [PubMed] [Google Scholar]
- Rouzer CA, Kingsley PJ, Wang H, Zhang H, Morrow JD, Dey SK, et al. Cyclooxygenase-1-dependent prostaglandin synthesis modulates tumor necrosis factor-alpha secretion in lipopolysaccharide-challenged murine resident peritoneal macrophages. J Biol Chem. 2004;279:34256–34268. doi: 10.1074/jbc.M402594200. [DOI] [PubMed] [Google Scholar]
- Sabatini F, Petecchia L, Boero S, Silvestri M, Klar J, Tenor H, et al. A phosphodiesterase 4 inhibitor, roflumilast N-oxide, inhibits human lung fibroblast functions in vitro. Pulm Pharmacol Ther. 2010;23:283–291. doi: 10.1016/j.pupt.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Sanz MJ, Cortijo J, Taha MA, Cerda-Nicolas M, Schatton E, Burgbacher B, et al. Roflumilast inhibits leukocyte-endothelial cell interactions, expression of adhesion molecules and microvascular permeability. Br J Pharmacol. 2007;152:481–492. doi: 10.1038/sj.bjp.0707428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer PH, Parton A, Gandhi AK, Capone L, Adams M, Wu L, et al. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. 2010;159:842–855. doi: 10.1111/j.1476-5381.2009.00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd MC, Baillie GS, Stirling DI, Houslay MD. Remodelling of the PDE4 cAMP phosphodiesterase isoform profile upon monocyte-macrophage differentiation of human U937 cells. Br J Pharmacol. 2004;142:339–351. doi: 10.1038/sj.bjp.0705770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater DM, Astle S, Woodcock N, Chivers JE, De Wit NC, Thornton S, et al. Anti-inflammatory and relaxatory effects of prostaglandin E2 in myometrial smooth muscle. Mol Hum Reprod. 2006;12:89–97. doi: 10.1093/molehr/gal005. [DOI] [PubMed] [Google Scholar]
- Smith SJ, Cieslinski LB, Newton R, Donnelly LE, Fenwick PS, Nicholson AG, et al. Discovery of BRL 50481 [3-(N,N-dimethylsulfonamido)-4-methyl-nitrobenzene], a selective inhibitor of phosphodiesterase 7: in vitro studies in human monocytes, lung macrophages, and CD8+ T-lymphocytes. Mol Pharmacol. 2004;66:1679–1689. doi: 10.1124/mol.104.002246. [DOI] [PubMed] [Google Scholar]
- Spina D. PDE4 inhibitors: current status. Br J Pharmacol. 2008;155:308–315. doi: 10.1038/bjp.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K, Garcia-Cardena G, Sukhova GK, Comander J, Gimbrone MA, Jr, Libby P. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J Biol Chem. 2002;277:44147–44154. doi: 10.1074/jbc.M204810200. [DOI] [PubMed] [Google Scholar]
- Tenor H, Hatzelmann A, Kupferschmidt R, Stanciu L, Djukanovic R, Schudt C, et al. Cyclic nucleotide phosphodiesterase isoenzyme activities in human alveolar macrophages. Clin Exp Allergy. 1995;25:625–633. doi: 10.1111/j.1365-2222.1995.tb01110.x. [DOI] [PubMed] [Google Scholar]
- Thompson WJ, Terasaki WL, Epstein PM, Strada SJ. Assay of cyclic nucleotide phosphodiesterase and resolution of multiple molecular forms of the enzyme. Adv Cyclic Nucleotide Res. 1979;10:69–92. [PubMed] [Google Scholar]
- Togo S, Liu X, Wang X, Sugiura H, Kamio K, Kawasaki S, et al. PDE4 inhibitors roflumilast and rolipram augment PGE2 inhibition of TGF-{beta}1-stimulated fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2009;296:L959–L969. doi: 10.1152/ajplung.00508.2007. [DOI] [PubMed] [Google Scholar]
- Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax. 2002;57:590–595. doi: 10.1136/thorax.57.7.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese MW, Mcconnell RT, Lenhard JM, Hamacher L, Jin SL. Regulation of distinct cyclic AMP-specific phosphodiesterase (phosphodiesterase type 4) isozymes in human monocytic cells. Mol Pharmacol. 1995;47:1164–1171. [PubMed] [Google Scholar]
- Wang P, Wu P, Ohleth KM, Egan RW, Billah MM. Phosphodiesterase 4B2 is the predominant phosphodiesterase species and undergoes differential regulation of gene expression in human monocytes and neutrophils. Mol Pharmacol. 1999;56:170–174. doi: 10.1124/mol.56.1.170. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kurita C, Nagao T, Usami E, Nakao T, Watanabe S, et al. Effects of cAMP-phosphodiesterase isozyme inhibitor on cytokine production by lipopolysaccharide-stimulated human peripheral blood mononuclear cells. Gen Pharmacol. 1997;29:633–638. doi: 10.1016/s0306-3623(96)00580-0. [DOI] [PubMed] [Google Scholar]
- Zhong WW, Burke PA, Drotar ME, Chavali SR, Forse RA. Effects of prostaglandin E2, cholera toxin and 8-bromo-cyclic AMP on lipopolysaccharide-induced gene expression of cytokines in human macrophages. Immunology. 1995;84:446–452. [PMC free article] [PubMed] [Google Scholar]