

Abstract
BACKGROUND AND PURPOSE
Hydrogen sulphide (H2S) and prostaglandins are both involved in inflammation, cancer and bone turnover, and non-steroidal anti-inflammatory drugs (NSAIDs) and H2S donors exhibit anti-inflammatory and anti-tumour properties. H2S-releasing diclofenac (S-DCF) derivatives are a novel class of NSAIDs combining the properties of a H2S donor with those of a conventional NSAID.
EXPERIMENTAL APPROACH
We studied the effects of the S-DCF derivatives ACS15 and ACS32 on osteoclast and osteoblast differentiation and activity in vitro, human and mouse breast cancer cells support for osteoclast formation and signalling in vitro, and osteolysis ex vivo.
KEY RESULTS
The S-diclofenac derivatives ACS15 and ACS32 inhibited the increase in osteoclast formation induced by human MDA-MB-231 and MCF-7 and mouse 4T1 breast cancer cells without affecting breast cancer cell viability. Conditioned media from human MDA-MB-231 cells enhanced IκB phosphorylation and osteoclast formation and these effects were significantly inhibited following treatment by ACS15 and ACS32, whereas the parent compound diclofenac had no effects. ACS15 and ACS32 inhibited receptor activator of NFκB ligand-induced osteoclast formation and resorption, and caused caspase-3 activation and apoptosis in mature osteoclasts via a mechanism dependent on IKK/NFκB inhibition. In calvaria organ culture, human MDA-MB-231 cells caused osteolysis, and this effect was completely prevented following treatment with ACS15 and ACS32.
CONCLUSIONS AND IMPLICATIONS
S-diclofenac derivatives inhibit osteoclast formation and activity, suppress breast cancer cell support for osteoclastogenesis and prevent osteolysis. This suggests that H2S-releasing diclofenac derivatives exhibit anti-resorptive properties, which might be of clinical value in the treatment of osteolytic bone disease.
Keywords: diclofenac, NSAID, hydrogen sulphide, osteoclast, osteoblast, breast cancer, MDA-MB-231, MCF7, 4T1
Introduction
Hydrogen sulphide (H2S) is involved in the regulation of vasodilatation, neuromodulation, cardiovascular homeostasis and inflammation (Lefer, 2007; Li et al., 2009). A number of preclinical studies reported that H2S-releasing compounds such as garlic-related products exert anti-tumour effects. For example, natural components of garlic such as diallyl sulphide, diallyl disulphide and diallyl trisulphide inhibit proliferation and induce apoptosis of colon, breast, lung and pancreatic tumour cells in vitro and attenuate tumour growth in animal models of cancer (Dipaolo and Carruthers, 1960; Hui et al., 2008; Lei et al., 2008; Oommen et al., 2004; Seki et al., 2008; Sriram et al., 2008; Gayathri et al., 2009; Kim and Kwon, 2009). Studies also showed that S-allylmercapto-L-cysteine, a by-product of naturally occurring garlic derivatives, inhibited growth and induced apoptosis of human colon and prostate cells in vitro and in vivo and suppressed tumorigenesis in a xenograft model of gastric cancer (Pinto et al., 2000; Shirin et al., 2001; Lee et al., 2011). The beneficial effect of these compounds is mostly attributed to H2S production due to catabolism of the polysulphide group in allicin, the major biologically active component of garlic (Seki et al., 2008). There is also evidence to suggest that H2S donors affect bone remodelling. Garlic extract inhibited serum markers of bone turnover and significantly reduced bone loss in rodent models of sex hormone deficiency (Mukherjee et al., 2004; 2006). Whilst these studies suggest that H2S donors may exhibit anti-resorptive and anti-tumour properties, very little research has been conducted on the direct effects of these agents on bone and breast cancer cell proliferation, survival, activity and crosstalk.
H2S-releasing non-steroidal anti-inflammatory drugs (S-NSAIDs) are a novel class of NSAID in which the core structure of the parent compound was modified by the addition of a H2S-releasing moiety (reviewed in Li et al., 2009; Fiorucci and Santucci, 2011). The rationale behind the development of S-NSAIDs is based on the notion that the vasodilator effects of the H2S moiety are likely to counteract the gastrointestinal (GI) toxicity associated with inhibition of cyclo-oxygenase (COX-1) activity and prostaglandin production (Lefer, 2007; Li et al., 2007; 2009; Wallace, 2007; Fiorucci and Santucci, 2011). A number of studies demonstrated that S-NSAIDs retain the desired anti-inflammatory and analgesic properties of conventional NSAIDs, but it has yet to be established whether these agents are less likely to cause GI toxicity (Lefer, 2007; Wallace, 2007; Li et al., 2009; Fiorucci and Santucci, 2011).
H2S-releasing diclofenac (S-diclofenac) derivatives, such as 2-[(2,6-dichlorophenyl)amino] benzeneacetic acid 4-(3-thioxo-3H-1,2-dithiol-5-yl) phenyl ester (ACS15) and [3-(2,6-dichloro-phenylamino)-phenyl]-acetic acid 2-methanesulphonylsulphanyl-ethyl ester (ACS32) (Figure 1), combine the properties of a H2S donor with those of the parent compound. S-diclofenac derivatives slowly release H2S in vitro and in vivo, protect from oxidative damage, inhibit NFκB activation in vitro and in vivo and reduce production of pro-inflammatory cytokines in animal models of arthritis through a mechanism that involves H2S production (Li et al., 2007; Sidhapuriwala et al., 2007; Moody et al., 2010). Recent studies also showed that the S-diclofenac derivative ACS15 (Figure 1) inhibited prostaglandin production and cell proliferation in cultured lung cancer cells in vitro and reduced tumour growth in a xenograft model of lung cancer (Moody et al., 2010). As H2S and prostaglandins are known to exert modulatory effects on both tumorigenesis and bone remodelling, we hypothesized that S-diclofenac derivatives may exert an inhibitory effects on breast cancer-induced osteoclastogenesis and osteolysis.
Figure 1.
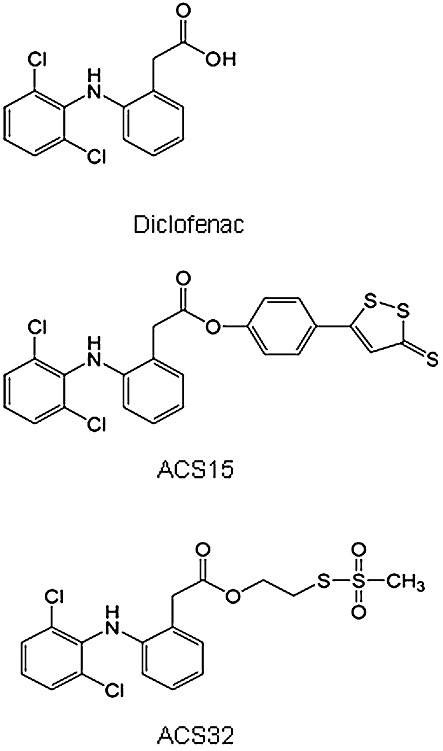
Chemical structure of the NSAID diclofenac and the S-diclofenac derivatives, ACS15 and ACS32.
In this study, we showed that the S-diclofenac derivatives ACS15 and ACS32 inhibited the increase in osteoclast formation induced by human and mouse breast cancer cells in vitro and prevented MDA-MB-231-induced osteolysis ex vivo. ACS-15 and ACS-32 inhibited NFκB activity in mature osteoclasts and their precursors, significantly suppressed osteoclast formation and bone resorption and caused mature osteoclast apoptosis. This suggests that H2S-releasing diclofenac derivatives may be of clinical value for the prevention and treatment of skeletal complications associated with osteolytic bone disease.
Methods
Synthesis of ACS32
A 1N solution of dicyclohexylcarbodiimide (2.8 mL) in dichloromethane was added to a solution of diclofenac (758 mg; 2.56 mmol), methanethiosulphonic acid S-(2-hydroxyethyl) ester (400 mg, 2.56 mmol) and dimethylaminopyridine (15 mg) in dichloromethane (40 mL) and the mixture was stirred at room temperature, under nitrogen for 1.5 h (Foong et al., 1997). After filtration of the dicyclohexylurea, the solution was extracted with a saturated solution of NaHCO3 and then with cold water. The organic phase was dried on anhydrous sodium sulphate and evaporated to dryness. The residue was purified by flash chromatography on silica gel (cyclohexane/ethylacetate; 80:20) and crystallized from ether. Yield was 51.5%. Melting point (Bűchi apparatus) was 41–44°C. 1H NMR (Varian Mercuri 300VX spectrometer (Milan, Italy); CDCl3): δ 7.30 (d, 2H); 7.20 (t, 2h); 7.10 (t, 1H); 6.90–7.05 (q, 2H), 6.65 (s, 1H, collapses with D2O); 4.40 (t, 2H), 3.80 (s, 2H); 3.40 (t, 2H); 3.30 (s, 3H).
RANKL and M-CSF mouse osteoclast culture
All animal care and experimental procedures complied with UK Home Office regulations and were approved by the ethical review board of the University of Edinburgh. Osteoclast formation, apoptosis and resorption were studied using RANKL and M-CSF-generated mouse osteoclasts, as described in detail by Idris et al. (2010). Briefly, female C57BL/6 mice (Harlan Laboratories, UK), 3 to 5 months old, were killed by cervical dislocation and bone marrow cells were flushed from the long bones. These bone marrow cells were plated into Petri dishes and incubated for 48 h in standard αMEM (αMEM supplemented with 10% fetal calf serum, penicillin and streptomycin) and mouse M-CSF (100 ng·mL−1). For osteoclast generation, the resulting M-CSF-dependent osteoclast precursor cells were plated into tissue culture plates (96-well plates, 15 × 103 cells per well; 12-well plates, 150 × 103 cells per well) in standard αMEM supplemented with M-CSF (25 ng·mL−1) and RANKL (100 ng·mL−1). The cultures were terminated by fixation in 4% paraformaldehyde and stained for tartrate-resistant acid phosphatase (TRAcP).
Bone marrow–breast cancer cell co-culture
Mouse M-CSF-dependent osteoclast precursors generated as described above were plated into 96-well plates (10 × 103 cells per well) in standard αMEM supplemented with M-CSF (25 ng·mL−1) and RANKL (100 ng·mL−1) for 6 h before addition of human MDA-MB-231, human MCF-7 or mouse 4T1 breast cancer cells (300 cells per well). For studies involving conditioned media, human MDA-MB-231 breast cancer cells were cultured in standard αMEM and allowed to grow to 80% confluence. Media were removed and replaced with serum free αMEM and then the cells were allowed to grow for a further 24 h. The conditioned media from these cultures were removed, filtered (0.2 µm filter diameter) and then added to osteoclast cultures at a concentration of 10% (v/v) in standard αMEM supplemented with M-CSF (25 ng·mL−1) and RANKL (100 ng·mL−1). The cultures were terminated by fixation in 4% paraformaldehyde and stained for TRAcP.
TRAcP staining
TRAcP staining was used to identify multi-nucleated osteoclasts. At the end of the culture, osteoclast cultures were fixed in 4% paraformaldehyde, washed with PBS and incubated with naphthol-ASBI-phosphate, pararosanilin and tartrate in acetate buffer (30 µM) at 37°C for 45 min as previously described (van't Hof, 2003). TRAcP positive cells with three or more nuclei were considered to be osteoclasts and manually counted on a Zeiss Axiovert light microscope using a 10× objective lens.
Quantification of resorption area
Resorption pits were visualized by reflected light microscopy and the area resorbed was quantified by image analysis using custom software developed using Aphelion ActiveX objects (ADCIS, 14280 Saint-Contest, France) as previously described (van't Hof, 2003).
Osteoblast culture
Primary osteoblasts were isolated from the calvarial bones of 2 day-old mice by sequential collagenase digestion as previously described (Bakker and Klein-Nulend, 1988). For bone nodule assay, osteoblasts were seeded into 12-well plates (10 × 105 cells per well) in standard αMEM supplemented with β-glycerol phosphate (10 mM) and L-ascorbic acid (50 µg·mL−1) for up to 21 days. Osteoblast number, differentiation and bone nodule formation were determined by Alamar Blue assay, alkaline phosphatase (ALP) assay and alizarin red (ALZ) staining respectively.
Alamar Blue assay
Alamar Blue assay was used to measure the number of osteoblast and breast cancer cells (Nakayama et al., 1997). At the end of the culture period, Alamar Blue reagent (10%, v/v) was added to each well, cells were incubated for a further 3 h and then fluorescence was measured (excitation, 530 nm, emission 590 nm) using a Biotek Synergy HT plate reader (Bedfordshire, UK).
ALP assay
ALP activity was used to assess the differentiation of mouse calvarial osteoblasts (Bakker and Klein-Nulend, 1988). Osteoblasts were homogenized in ALP lysis buffer [1 M diethanolamine, 1 mM MgCl2, 0.05% Triton X-100 (Sigma Aldrich, Dorset, UK)] and cell lysate was mixed with an equal volume of p-nitrophenol phosphate (20 mM). Absorbance was measured at 414 nm, and ALP activity was normalized to cell number as determined by the Alamar Blue assay.
ALZ staining
ALZ staining was used to assess bone nodule formation in calvarial osteoblast cultures. Briefly, osteoblast cultures were fixed in 70% cold ethanol for 1 h and immersed in 40 mM ALZ solution (pH 4.2) for 20 min at room temperature on an orbital rotator. Cultures were incubated in destaining solution (10% (w/v) cetylpyridinium chloride in 10 mM sodium phosphate) for 15 min. Absorbance of the extracted stain was then measured at 562 nm using a Biotek Synergy HT plate reader and compared with an ALZ standard curve. The values were normalized to cell number as determined by the Alamar Blue assay.
Human breast cancer cell–mouse calvarial co-culture
Neonatal mouse calvarias were isolated from 7 day-old mice and incubated in standard αMEM for 24 h as previously described (Garrett, 2003). Each mouse calvaria was divided into two halves along the median sagittal suture. Each half was placed in organ culture as free-floating bones on stainless steel rafts in 48-well plates containing standard media (Figure 6A, top) and treated for 7 days as follows: group 1, one half treated with DMSO (veh1) and the other half treated with veh1 in the presence of MDA-MB-231 cells (10 × 103 cells per well) (veh2); group 2, one half treated with 0.1% DMSO in the presence of MDA-MB-231 cells (10 × 103 cells per well) and the other half treated with diclofenac (10 µM) in the presence of MDA-MB-231 cells (10 × 103 cells per well); group 3, one half treated with 0.1% DMSO in the presence of MDA-MB-231 cells (10 × 103 cells per well) and the other half treated with ACS15 (10 µM) in the presence of MDA-MB-231 cells (10 × 103 cells per well); group 4, one half treated with 0.1% DMSO in the presence of MDA-MB-231 cells (10 × 103 cells per well) and the other half treated with ACS32 (10 µM) in the presence of MDA-MB-231 cells (10 × 103 cells per well). Osteolysis was assessed by measuring bone volume using micro-computed tomography (Skyscan 1172 scanner, Skyscan, Antwerp, Belgium) at a resolution of 5 µm.
Figure 6.
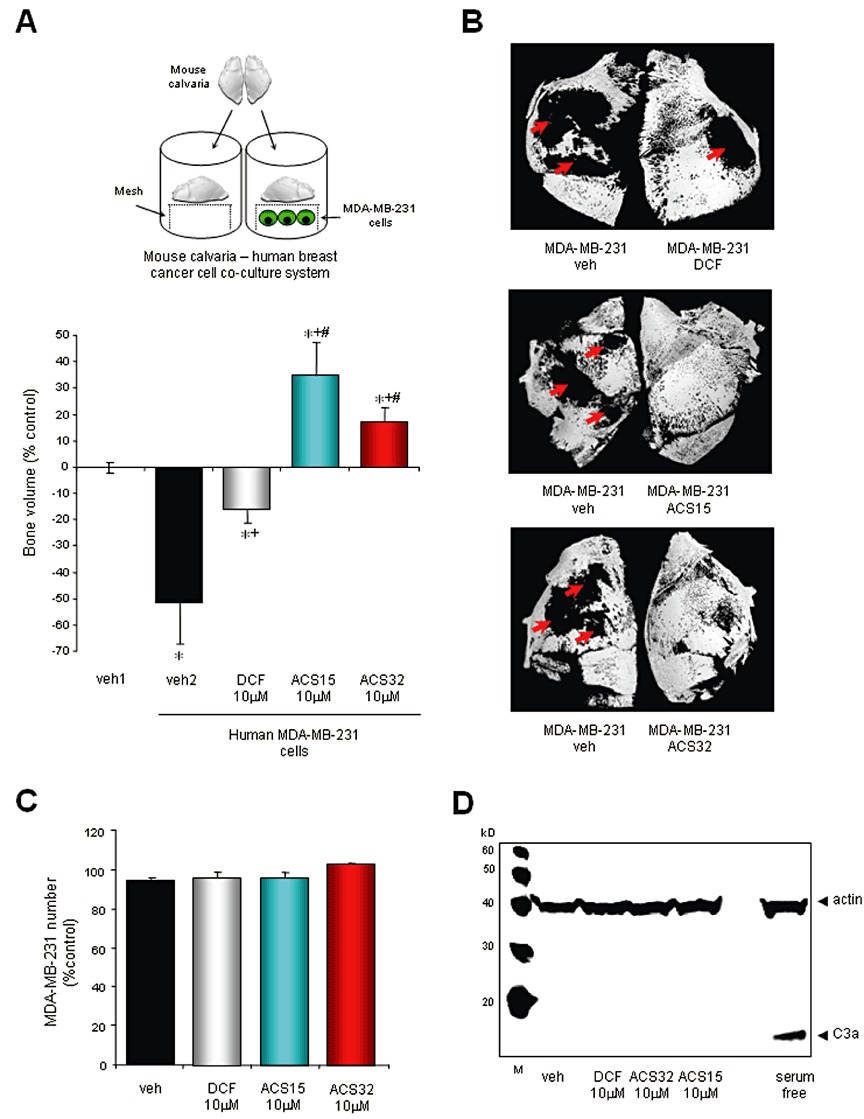
ACS15 and ACS32 prevent MDA-MB-231-induced osteolysis in mouse calvaria organ cultures. (A, upper part) Diagram of the human MDA-MB-231–mouse calvaria co-culture system. (A, lower part) Bone volume in neonatal mouse calvarias isolated from 7 day-old mice and then cultured in standard tissue culture medium for 24 h prior exposure to vehicle (veh; 0.1% DMSO) or test compounds at the indicated concentrations for 7 days. Calvarias were analysed using microCT scanning and changes in bone volume were normalized to vehicle-treated calvarial half that were unexposed to MDA-MB-23, and values were expressed as percent change. Values in each panel are means ± SD from seven calvarias. *P < 0.05, significantly different from vehicle-treated, +P < 0.05 , significantly different from vehicle-treated MDA-MB-231–calvarial organ cultures and #P < 0.05 , significantly different from diclofenac-treated MDA-MB-231–calvarial organ cultures. (B) Representative photomicrographs of mouse calvarias from the experiment described earlier following microCT analysis. Red arrows denote focal osteolysis. (C) MDA-MB-231 cell number in co-cultures described earlier as assessed by Alamar Blue assay. (D) Caspase-3 activity in human MDA-MB-231 breast cancer cells from the MDA-MB-231–mouse calvaria co-cultures described earlier. The results shown are representative of three independent experiments.
Western blotting
Western blot analysis was used to detect protein expression and phosphorylation in cultured bone and breast cancer cells. Briefly, cells were seeded in 12-well plates and maintained in standard αMEM until confluent. Before stimulation with test agents or vehicle, cells were incubated in serum-free αMEM medium for 60 min. Test agents or vehicle were prepared in serum free αMEM medium and were then added for the desired period of time. The cells were then gently scraped in standard lysis buffer [0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycholate, 1% Triton X-100, 1 mM EDTA, 2% (v/v) protease inhibitor cocktail, 10 µM of sodium fluoride and 2% (v/v) phosphatase inhibitor cocktail]. The lysate was incubated on ice for 10 min and centrifuged at 14 000× g at 4°C for 5 min. The supernatant was collected and protein concentration was determined using BCA assay (Thermo Fisher Scientific, Northumberland, UK). Total protein (30–100 µg) was resolved by SDS-PAGE on 12% polyacrylamide SDS gels, transferred onto PVDF membranes (BioRAD, Herts, UK) and immunoblotted with antibodies according to manufacturer's instructions. The immuno-complexes were visualized by an enhanced chemiluminescence detection kit (Pierce, USA) using horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and then visualized using chemiluminescence (Amersham, Buckinghamshire, UK) on a Syngene GeneGnome imaging system.
Morphological assessment of apoptosis
Apoptosis in mature osteoclast cultures was detected by the characteristic changes in nuclear morphology following 4,6-diamidino-2-phenylindole (DAPI) staining. Briefly, osteoclasts were generated in 48-well plates as described earlier and then treated with test agents for the desired period of time. At the end of the culture period, both adherent and non-adherent cells were collected, fixed with 4% paraformaldehyde, cytospun into glass slides and immersed in DAPI solution for 10 min at room temperature. An average of six microscopic fields per treatment group were analysed at 10 × magnification. Number of apoptotic osteoclasts was quantified and expressed as percentage of total osteoclast number as identified by TRAcP staining.
Measurement of NFκB activity
Osteoclasts were generated in six-well plates as described earlier and treated with test compounds for the desired period of time. Following the incubation period, nuclear extracts were prepared using a nuclear extract kit (Active Motif, Rixensart, Belgium) and DNA binding was measured using TransAM® Transcription Factor ELISA for p65 NFκB (Active Motif, Rixensart, Belgium), according to the manufacturer's instructions.
Statistical analysis
Differences between groups were assessed by one-way anova followed by Bonferroni post-test using SPSS for Windows version 11 (Chicago, IL, USA). A P-value value of 0.05 or below was considered statistically significant. The concentration that produced 50% of response IC50 was calculated using GraphPad Prism 4 for Windows (GraphPad Software, La Jolla, CA, USA).
Materials
Diclofenac was purchased from Sigma Aldrich (Dorset, UK). The diclofenac derivative ACS15 (Figure 1) was prepared as previously described (Isenberg et al., 2007), and ACS32 (Figure 1) was prepared as described above. Human MDA-MB-231 and MCF-7 and mouse 4T1 breast cancer cell lines were purchased from ATCC (Manassas, VA, USA). Mouse macrophage colony stimulating factor (M-CSF) was obtained from R&D Systems (Abingdon, UK) and receptor activator of NFκB ligand (RANKL) was a gift from Patrick Mollat (Galapagos SASU, Romainville, France) and was prepared as previously described (Idris et al., 2010). All primary antibodies were purchased from Cell Signalling Biotechnology (Danvers, MA, USA), unless otherwise stated. Tissue culture medium (Minimum Essential Medium Alpha; αMEM) was obtained from Invitrogen (Paisley, UK). All other reagents were purchased from Sigma-Aldrich unless otherwise stated.
Results
The S-diclofenac derivatives ACS15 and ACS32 inhibit human and mouse breast cancer cell support for osteoclast formation
We first investigated the effects of the S-diclofenac derivatives ACS15 and ACS32 (Figure 1) on osteoclast formation in bone marrow–breast cancer cell co-cultures. As shown in Figure 2, the human MDA-MB-231 (Figure 2A) and MCF-7 (Figure 2C) and mouse 4T1 (Figure 2D) breast cancer cells increased osteoclast number, and these effects were significantly inhibited by ACS15 and ACS32 with the maximum response observed at concentrations equal to and greater than 10 µM (Figure 2A–D). The parent compound diclofenac had no effect on osteoclast formation at concentration up to 30 µM (Figure 2A, C and D). None of the compounds tested had any significant effects on human breast cancer cell viability or growth at concentrations that inhibited osteoclast formation (Figure S1).
Figure 2.
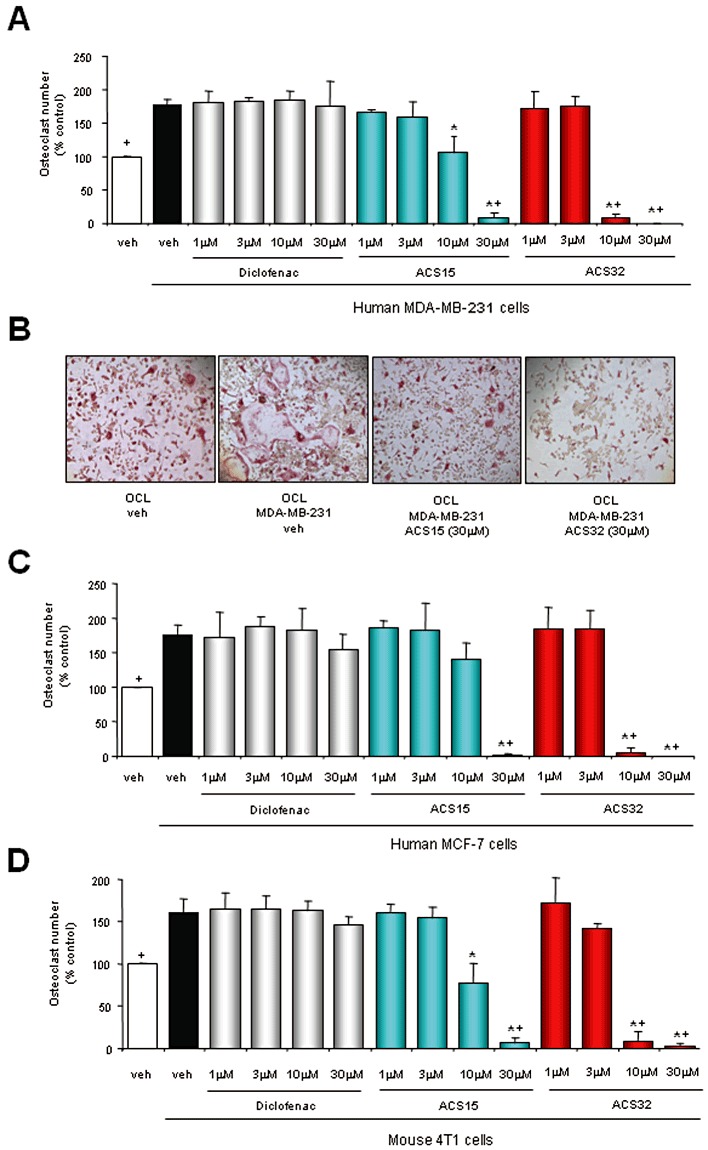
ACS15 and ACS32 inhibit osteoclast formation induced by mouse and human breast cancer cells. (A, C, D) Number of mouse osteoclasts following treatment with vehicle (veh; 0.1% DMSO) or test compounds at the indicated concentrations for 48 h in the presence and absence of (A) human MDA-MB-231, (C) human MCF-7 or (D) mouse 4T1 cells. Osteoclast numbers were assessed by counting TRAcP-positive multi-nucleated cells. Representative photomicrographs from osteoclast–MDA-MB-231 cells co-cultures are shown in panel B. +P < 0.05 from vehicle treated osteoclast – cancer cell co-cultures and *P < 0.05 from vehicle treated osteoclast cultures.
ACS15 and ACS32 inhibit IκB phosphorylation and osteoclast formation induced by human breast cancer cell derived factors
To gain an insight into the mechanism by which these S-diclofenac derivatives suppressed breast cancer cells support for osteoclastogenesis, we next examined the effects of ACS15 and ACS32 on osteoclast formation and signalling induced by factors derived from breast cancer cells. As shown in Figure 3A, ACS15 and ACS32 inhibited the increase in osteoclast number induced by conditioned media from human MDA-MB-231 breast cancer cells. To investigate the mechanism of action by which S-diclofenac derivatives inhibit osteoclast formation, we examined the effects of ACS15 and ACS32 (30 µM) on NFκB activity, a common pathway affected by H2S, diclofenac, S-diclofenac and RANKL (Feng, 2005; Li et al., 2007; Zhi et al., 2007; Karakawa et al., 2009). As shown in Figure 3B and C, ACS15 and ACS32 (30 µM) suppressed IκB phosphorylation induced by conditioned media from human MDA-MB-231 cells. The parent compound diclofenac had no significant effects on MDA-MB-231-induced osteoclast formation or IκB phosphorylation at concentrations up to 30 µM (Figure 3A–C).
Figure 3.
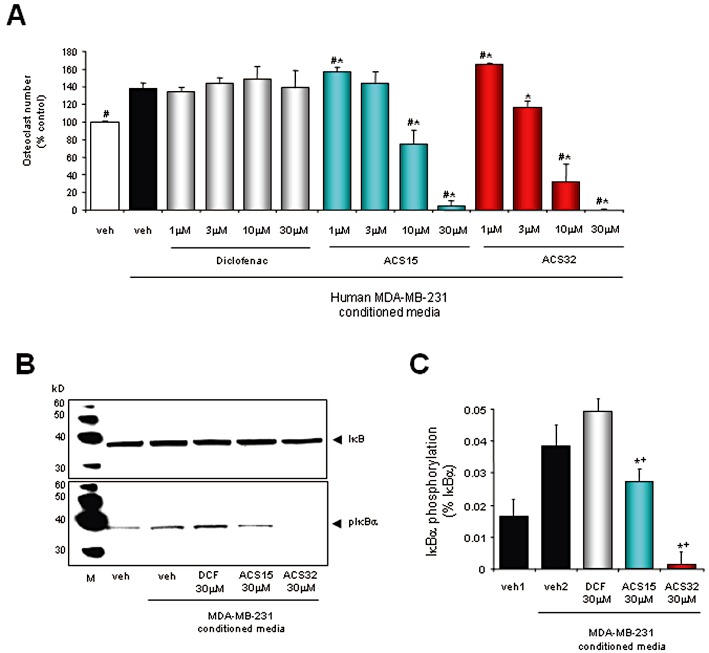
ACS15 and ACS32 inhibit osteoclast formation and IκB phosphorylation induced by human MDA-MB-231 breast cancer cell-derived factors. (A) Number of mouse osteoclasts following treatment with vehicle (veh; 0.1% DMSO) or test compounds at the indicated concentrations for 48 h in the presence and absence of MDA-MB-231 conditioned media (10% v/v in αMEM). Osteoclast numbers were assessed by counting TRAcP-positive multinucleated cells. (B) Western blot of IκB phosphorylation in mouse M-CSF-dependent osteoclast precursors treated with vehicle (veh; 0.1% DMSO) or test compounds (ACS15; ACS32; diclofenac, DCF) at the indicated concentrations for 3 h prior stimulation with MDA-MB-231 conditioned media (20% v/v in αMEM) for 15 min. The results shown are representative of three independent experiments. (C) Quantification of phospho-IκBα from blots in (B) as a ratio to total IκBα. #P < 0.05 from vehicle treated osteoclast–MDA-MB-231 conditioned media cultures; *P < 0.05 from vehicle treated osteoclast cultures; +P < 0.05 from DCF treated osteoclast cultures.
ACS15 and ACS32 inhibit RANKL-induced osteoclast formation and resorption
We next examined the effects of ACS15 and ACS32 in RANKL and M-CSF-induced osteoclast formation and activity. As shown in Figure 4A, ACS15 (3–30 µM) and ACS32 (1–30 µM) strongly inhibited RANKL-stimulated osteoclast formation in a concentration-dependent manner. The mean (±SD) concentration of these compounds, which half maximally inhibited osteoclast formation (IC50), was 4.2 ± 0.7 µM for ACS15; 2.9 ± 1.2 µM for ACS32. ACS15 and ACS32 also inhibited RANKL-induced bone resorption at concentrations that suppressed osteoclast formation (10 µM; P < 0.05) (Figure 4C). In contrast, the parent compound diclofenac exerted only weak inhibition of osteoclast number (30 µM; 17% ± 1.4, P < 0.05) and resorption (30 µM; 18.7% ± 11.2, P < 0.05) (Figure 4A and C).
Figure 4.
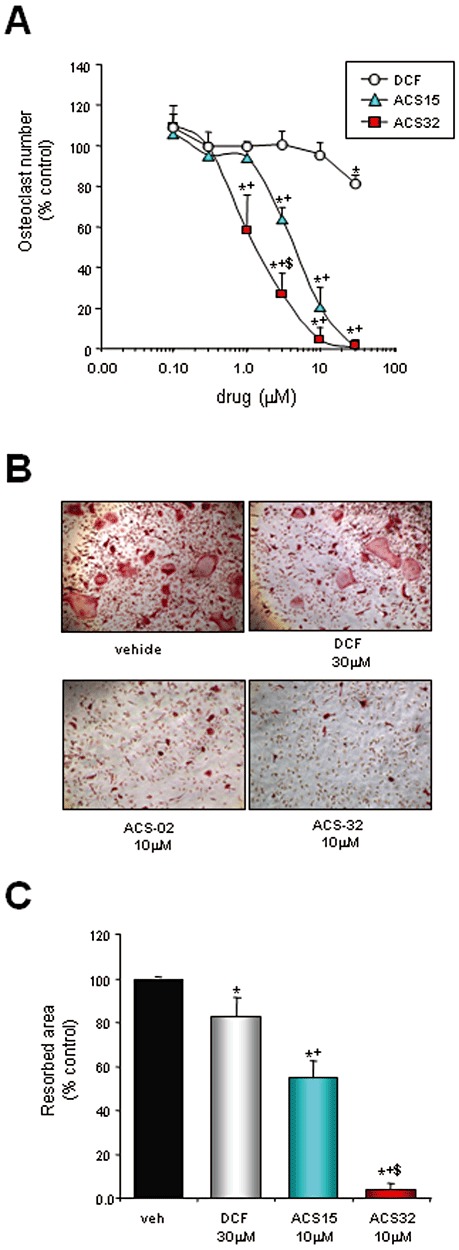
ACS15 and ACS32 inhibit RANKL-induced osteoclast formation and bone resorption. (A) Number of osteoclasts cultured in M-CSF (25 ng·mL−1) and RANKL (100 ng·mL−1) in presence and absence of vehicle (veh; 0.1% DMSO) or test compounds (ACS15; ACS32; diclofenac, DCF) for 48 h at the indicated concentrations. Osteoclast numbers were assessed by counting TRAcP-positive multinucleated cells. Representative photomicrographs from mouse osteoclast cultures are shown in (B). (C) Resorbed area in mouse osteoclasts cultured for 6–8 days on dentine slices and treated with vehicle (veh; 0.1% DMSO) or test compounds at the indicated concentrations for 4 days. Resorbed area was measured using reflective microscopy. Data are mean ± SD of three independent experiments. *P < 0.05, significantly different from vehicle control; +P < 0.05, significantly different from DCF-treated cultures; $P < 0.05, significantly different from ACS15-treated cultures.
ACS15 and ACS32 inhibit NFκB nuclear translocation and induce apoptosis in mature osteoclasts
To further assess the mechanisms responsible for osteoclast inhibition, we investigated the effects of diclofenac, ACS15 and ACS32 on apoptosis in mature osteoclasts cultured in RANKL and M-CSF for 6–7 days prior to drug treatment. ACS15 and ACS32 induced activation of caspase-3 within 18 h (Figure 5B) and caused apoptosis in a dose-dependent manner after 32 h of continuous treatment (Figure 5A; IC50= 22.1 ± 2.1 µM for ACS15 and 5.3 ± 0.5 µM for ACS32). A small increase in the number of apoptotic osteoclasts was observed following treatment with the parent compound diclofenac at concentration of 30 µM (Figure 5A). In order to investigate the mechanisms by which these compounds induce osteoclast apoptosis, we studied the effects of diclofenac, ACS15 and ACS32 on RANKL-induced IκB phosphorylation and NFκB DNA binding, key signalling events for osteoclast survival and activity (Feng, 2005). ACS15 and ACS32 inhibited RANKL-induced IκB phosphorylation (Figure 5C) and NFκB DNA binding (Figure 5D), indicating a strong inhibitory effect on NFκB activity in osteoclasts. ACS15 and ACS32 (30 µM) also showed a non-significant trend towards enhancing M-CSF-induced ERK1/2 phosphorylation in osteoclast cultures (4% ± 3 for ACS15 and 6% ± 4 for ACS32, P < 0.05) (Figure S2). The parent compound diclofenac had no significant effects on NFκB or ERK1/2 activity in mature osteoclasts at concentrations up to 30 µM (Figure 5C and D, Figure S2).
Figure 5.
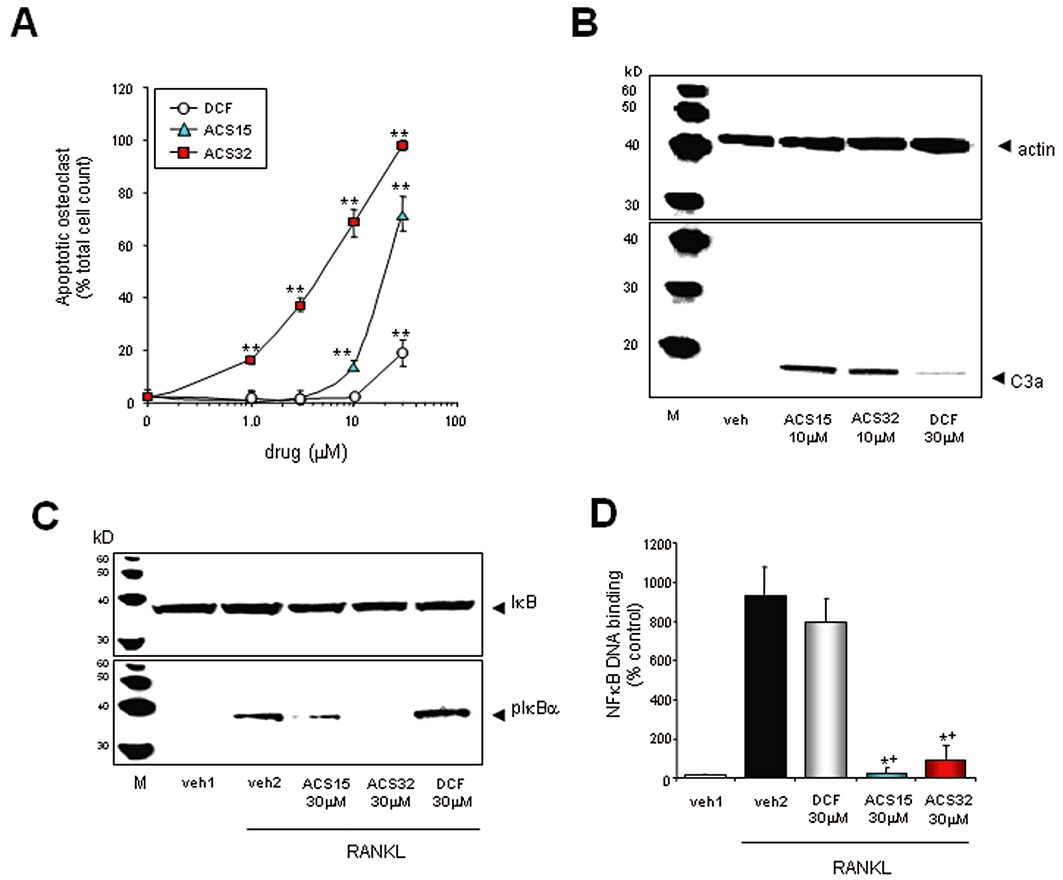
ACS15 and ACS32 inhibit NFκB activation and cause apoptosis in mature osteoclasts. (A) Number of apoptotic mouse osteoclasts following treatment with vehicle (0.1% DMSO) or test compounds (ACS15; ACS32; diclofenac, DCF) at the indicated concentrations for 32 h (A). Apoptotic osteoclasts were visualized by DAPI staining. Values in the graphs are means ± SD and are obtained from three independent experiments. **P < 0.01, significantly different from vehicle (0.1% DMSO)-treated cultures. (B) Western blot analysis of caspase-3 activation in mature mouse osteoclasts exposed to test compounds at the concentrations indicated for 18 h. The results shown are representative of three independent experiments. (C) Western blot analysis of IκBα phosphorylation in mouse osteoclasts exposed to test compounds at the concentrations indicated for 180 min prior stimulation with vehicle (veh2; 0.3% BSA) or RANKL (150 ng·mL−1) for 5 min. (D) NFκB DNA binding in mouse osteoclasts exposed to test compounds at the concentrations indicated for 60 min prior stimulation with vehicle (veh1; 0.3% BSA) or RANKL for 30 min. Nuclear extracts were prepared and NFκB DNA-binding activity analysed using a TRANSAM active motif elisa kit. Data are mean ± SD of three independent experiments. *P < 0.05, significantly different from vehicle-treated (veh2; 0.1% DMSO) and +P < 0.05, significantly different from diclofenac-treated (DCF, 30 µM).
ACS15 and ACS32 protect against osteolysis ex vivo
We next studied the effects of diclofenac, ACS15 and ACS32 on osteolysis using the human MDA-MB-231–mouse calvarial co-culture system (Figure 6A, top). As shown in Figure 6A and B, human MDA-MB-231 breast cancer cells caused osteolysis characterized by a significant loss in bone volume (P < 0.05) when co-cultured with mouse calvaria for 7 days, and these effects were completely prevented by ACS15 and ACS32 (10 µM). In fact, treatment with ACS15 and ACS32 (10 µM) caused a significant gain in bone volume in comparison with vehicle-treated cultures (P < 0.05; Figure 6A). A small reduction in osteolytic bone loss was observed in co-cultures treated with the parent compound diclofenac (10 µM) (P < 0.05). None of the compounds tested affected cell number or caspase-3 activity in human MDA-MB-231 breast cancer cells (Figure 6C and D).
Diclofenac, ACS15 and ACS32 inhibit differentiation and bone nodule formation of calvarial osteoblasts
We next investigated the effects of diclofenac, ACS15 and ACS32 on osteoblast differentiation and activity in primary mouse calvarial osteoblast cultures using ALP and bone nodule assays respectively. At concentrations that inhibited MDA-MB-231-induced osteoclast formation in vitro (Figure 2A) and osteolysis ex vivo (Figure 6A), none of the compound tested affected calvarial osteoblast number, differentiation or bone nodule formation (Figure 7A–C). At higher concentrations (30 µM), diclofenac, ACS15 and ACS32 reduced ALP activity (Figure 7A) and bone nodule formation (Figure 7B) despite causing a significant increase in osteoblast number (Figure 7C).
Figure 7.
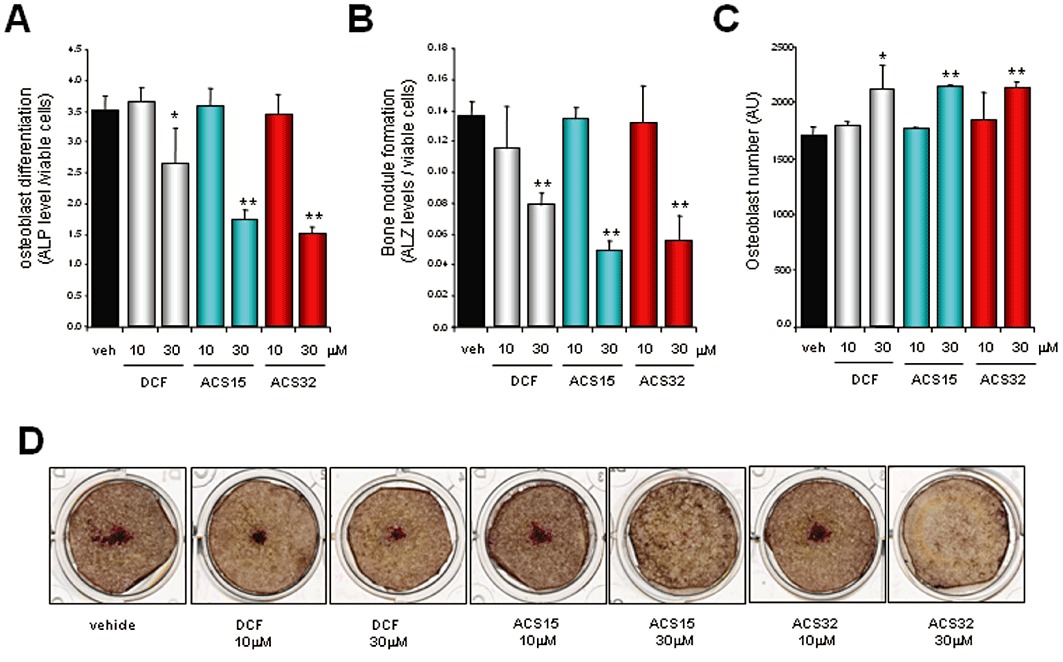
Diclofenac (DCF), ACS15 and ACS32 inhibit osteoblast differentiation and bone nodule formation in calvarial osteoblast cultures. (A) Alkaline phosphatase (ALP) levels in mouse calvarial osteoblast cells cultured in osteogenic medium for 21 days in the presence of vehicle (veh; 0.1% DMSO) or test compounds at the concentrations indicated. Osteoblast differentiation was assessed by alkaline phosphatase assay. (B) Quantification of bone nodule formation in mouse osteoblasts cultured and treated as described earlier. Bone nodule formation was assessed by alizarin red assay. (C) Osteoblast number in mouse osteoblasts cultured and treated as described earlier. Osteoblast number was measured using Alamar Blue assay. Values in the graphs are mean ± SD *P < 0.05 and **P < 0.01 , significantly different from vehicle (veh; 0.1% DMSO)-treated cultures. Representative photomicrographs from mouse calvarial osteoblast cultures are shown in (D).
Discussion and conclusions
Non-steroidal anti-inflammatory drugs and H2S donors exhibit anti-inflammatory, anti-resorptive and anti-tumour properties. These observations provided the rationale for the present study in which we investigated the effects of the S-diclofenac derivatives ACS15 and ACS32 on bone and breast cancer cell proliferation, activity and crosstalk. Studies in vitro using BM–breast cancer cell co-cultures showed that ACS15 and ACS32 inhibited the increase in osteoclast formation induced by human and mouse breast cancer cells. We excluded non-specific effects of these compounds on breast cancer cell growth and death by studying the effects of diclofenac, ACS15 and ACS32 on breast cancer cell number and apoptosis. In these studies, none of the compounds tested exerted any significant effects on MDA-MB-231 growth or caspase-3 activation at concentrations that inhibited osteoclast formation. ACS15 and ACS32 inhibited IκB phosphorylation and osteoclast formation induced by conditioned media from human breast cancer cells. This indicates that ACS15 and ACS32 inhibit osteoclast formation via a mechanism dependent on IKK/NFκB inhibition in osteoclasts. The possibility that suppression of COX-1/2 activity may contribute to these effects was excluded since the parent compound diclofenac was found to be completely ineffective in inhibiting breast cancer-induced IκB phosphorylation and osteoclast formation despite the fact that it is a more potent inhibitor of prostaglandin production than its H2S-releasing derivatives (Rossoni et al., 2008). Previous studies showed that S-NSAIDs, including ACS15, release H2S in vitro and in vivo (Li et al., 2007; Sidhapuriwala et al., 2007; Moody et al., 2010). Taken together, these findings imply that ACS15 and ACS32 exert their anti-resorptive effects by a mechanism mediated by H2S production. However, further studies are needed to determine if the amount of H2S released by these compounds in vitro and in vivo is sufficient to influence osteoclastogenesis.
Studies in osteoclast cultures showed that ACS15 and ACS32 inhibited RANKL- and M-CSF-induced osteoclast formation and caused osteoclast apoptosis in mature osteoclasts. This confirms that these agents inhibit osteoclast formation and survival acting directly on osteoclasts and their precursors. ACS15 and ACS32 also inhibited bone resorption, but these effects were likely to be due to induction of apoptosis and cell death rather than a specific effect on osteoclast function. Further mechanistic studies in mature osteoclasts showed that the mechanism of action by which ACS15 and ACS32 exerted their anti-resorptive effects involved inhibition of RANKL-induced NFκB nuclear translocation and DNA binding, an essential pathway for osteoclast formation and survival (Feng, 2005). Non-specific effects in M-CSF signalling were excluded by the fact that ACS15 and ACS32 did not inhibit M-CSF-induced ERK1/2 MAPK activation (Figure S2) or viability of M-CSF-dependent osteoclast precursors (data not shown). Interestingly, the parent compound diclofenac caused a weak inhibitory effect on osteoclast formation in RANKL- and M-CSF-stimulated cultures, even though it failed to inhibit RANKL- or M-CSF-induced signalling. We speculate that this effect was probably due to inhibition of COX activity and prostaglandin production, which are both known to regulate osteoclast formation (Li et al., 2000; Okada et al., 2000).
In order to examine the effects of S-diclofenac derivatives on osteolysis, we studied the effects of ACS15 and ACS32 on osteolytic bone destruction using the human MDA-MB-231–mouse calvarial co-culture system. Treatment with ACS15 and ACS32 completely prevented focal osteolysis induced by the human breast cancer cells MDA-MB-231, whereas the parent compound was partially active. These findings are entirely consistent with the effects of these agents in vitro and demonstrate the efficiency of S-diclofenac derivatives in inhibiting breast cancer support for osteoclastic bone resorption. At concentrations that prevented osteolysis, none of the compound tested affected the survival of MDA-MB-231 breast cancer cells or calvarial osteoblast survival, differentiation and activity. These results clearly demonstrate the specificity of these agents towards osteoclasts in this model. It is important to note, however, that diclofenac, ACS15 and ACS32 inhibited ALP activity and bone nodule formation at higher concentrations (30 µM), although they increased osteoblast numbers. Therefore, further long term in vivo studies are needed to ascertain whether, and to what extent, inhibition of osteoblast differentiation may limit the long-term usefulness of these agents as anti-resorptive drugs.
In summary, we have shown that S-diclofenac derivatives suppressed osteoclast formation via a mechanism dependent on IKK/NFκB inhibition. Our studies also demonstrated that these S-diclofenac derivatives had additional advantages over conventional NSAID in that they exerted potent inhibitory effects on breast cancer-induced osteoclastogenesis. We speculate that H2S released by S-diclofenac derivatives in the bone micro-environment may inhibit osteolysis, but further in vivo studies will need to be performed to investigate this possibility. Nonetheless, the data presented in this study suggest that H2S-releasing NSAIDs may be of value in the prevention and treatment of skeletal complications associated with osteolytic bone disease.
Acknowledgments
These studies were supported in part by a ECTS/AMGEN fellowship grant to Dr. A.I.Idris.
Glossary
- ALP
alkaline phosphatase
- ALZ
alizarin red
- BM
bone marrow
- BV
bone volume
- COX
cyclo-oxygenase
- DAPI
4,6-diamidino-2-phenylindole
- GI
gastrointestinal
- H2S
hydrogen sulphide
- M-CSF
mouse macrophage colony stimulating factor
- NSAID
non-steroidal anti-inflammatory drugs
- RANKL
receptor activator of NFκB ligand
- S-diclofenac
H2S-releasing diclofenac
- S-NSAID
H2S-releasing NSAID
- TRAcP
tartrate-resistant acid phosphatase
Conflicts of interest
Piero Del Soldato is a shareholder of CTG Pharma S.r.l., Viale Gran Sasso 17, 20131 Milano, Italy. Patrick Mollat is an employee of Galapagos SASU (102 Avenue Gaston Roussel, 93230 Romainville, France).
Supporting information
Figure S1 Diclofenac and S-diclofenacderivatives ACS15 and ACS32 have no effects on human MDA-MB-231breast cancer cell viability. Panel A shows the number of humanMDA-MB-231 breast cancer cells treated with vehicle (veh; 0.1%DMSO) or test compounds at the concentrations indicated for 48 h.Cell number was measured using Alamar Blue assay. Values in thegraphs are mean ± SD and are obtained from three independent experiments. Representative photomicrographs from MDA-MB-231 cultures are shown in panel B.
Figure S2 ACS15 and ACS32 enhance M-CSF-inducedERK1/2 activation in osteoclast cultures. Mouse osteoclasts werecultured in the presence of M-CSF (25ng·mL−1) and RANKL (100ng·mL−1) for 5–6 days and thenexposed to vehicle (veh2; 0.1% DMSO) or test compounds at theindicated concentrations for 180 min prior stimulation with vehicle(veh1; 0.3% bovine serum albumin) or M-CSF (150ng·mL−1) for 10 min. Total cellular proteinwas subjected to Western blot analysis (50μg·lane−1) using rabbitanti-phospho-ERK1/2 (Cell Signalling Biotechnology, USA) or rabbitanti-actin (Sigma-Aldrich, UK). Identical experiments have beenrepeated three times.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Bakker A, Klein-Nulend J. Osteoblast isolation from murine calvariae and long bones. In: Helfrich MH, Ralston SH, editors. Bone Research Protocols. Totowa, NJ: Humana Press; 1988. pp. 19–28. [Google Scholar]
- Dipaolo JA, Carruthers C. The effect of allicin from garlic on tumor growth. Cancer Res. 1960;20:431–434. [PubMed] [Google Scholar]
- Feng X. RANKing intracellular signaling in osteoclasts. IUBMB Life. 2005;57:389–395. doi: 10.1080/15216540500137669. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Santucci L. Hydrogen sulfide-based therapies: focus on H2S releasing NSAIDs. Inflamm Allergy Drug Targets. 2011;10:133–140. doi: 10.2174/187152811794776213. [DOI] [PubMed] [Google Scholar]
- Foong LY, You S, Jaikaran DC, Zhang Z, Zunic V, Woolley GA. Development of a novel thiol reagent for probing ion channel structure: studies in a model system. Biochemistry. 1997;36:1343–1348. doi: 10.1021/bi9624907. [DOI] [PubMed] [Google Scholar]
- Garrett R. Assessing bone formation using mouse calvarial organ cultures. In: Helfrich M, Ralston S, editors. Bone Research Protocols. Totowa, NJ: Humana Press; 2003. pp. 183–198. [DOI] [PubMed] [Google Scholar]
- Gayathri R, Gunadharini DN, Arunkumar A, Senthilkumar K, Krishnamoorthy G, Banudevi S, et al. Effects of diallyl disulfide (DADS) on expression of apoptosis associated proteins in androgen independent human prostate cancer cells (PC-3) Mol Cell Biochem. 2009;320:197–203. doi: 10.1007/s11010-008-9903-5. [DOI] [PubMed] [Google Scholar]
- van't Hof RJ. Osteoclast formation in the mouse coculture assay. In: Helfrich MH, Ralston SH, editors. Bone Research Protocols. Totowa, NJ: Humana Press; 2003. pp. 145–152. [DOI] [PubMed] [Google Scholar]
- Hui C, Jun W, Ya LN, Ming X. Effect of Allium sativum (garlic) diallyl disulfide (DADS) on human non-small cell lung carcinoma H1299 cells. Trop Biomed. 2008;25:37–45. [PubMed] [Google Scholar]
- Idris AI, Krishnan M, Simic P, Landao-Bassonga E, Mollat P, Vukicevic S, et al. Small molecule inhibitors of I{kappa}B kinase signaling inhibit osteoclast formation in vitro and prevent ovariectomy-induced bone loss in vivo. FASEB J. 2010;24:4545–4555. doi: 10.1096/fj.10-164095. [DOI] [PubMed] [Google Scholar]
- Isenberg JS, Jia Y, Field L, Ridnour LA, Sparatore A, Del Soldato P, et al. Modulation of angiogenesis by dithiolethione-modified NSAIDs and valproic acid. Br J Pharmacol. 2007;151:63–72. doi: 10.1038/sj.bjp.0707194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakawa A, Fukawa Y, Okazaki M, Takahashi K, Sano T, Amano H, et al. Diclofenac sodium inhibits NFkappaB transcription in osteoclasts. J Dent Res. 2009;88:1042–1047. doi: 10.1177/0022034509346147. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kwon O. Garlic intake and cancer risk: an analysis using the Food and Drug Administration's evidence-based review system for the scientific evaluation of health claims. Am J Clin Nutr. 2009;89:257–264. doi: 10.3945/ajcn.2008.26142. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kim H, Lee J, Kim K. Anticancer activity of S-allylmercapto-L-cysteine on implanted tumor of human gastric cancer cell. Biol Pharm Bull. 2011;34:677–681. doi: 10.1248/bpb.34.677. [DOI] [PubMed] [Google Scholar]
- Lefer DJ. A new gaseous signaling molecule emerges: cardioprotective role of hydrogen sulfide. Proc Natl Acad Sci U S A. 2007;104:17907–17908. doi: 10.1073/pnas.0709010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei XY, Yao SQ, Zu XY, Huang ZX, Liu LJ, Zhong M, et al. Apoptosis induced by diallyl disulfide in human breast cancer cell line MCF-7. Acta Pharmacol Sin. 2008;29:1233–1239. doi: 10.1111/j.1745-7254.2008.00851.x. [DOI] [PubMed] [Google Scholar]
- Li L, Rossoni G, Sparatore A, Lee LC, Del Soldato P, Moore PK. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic Biol Med. 2007;42:706–719. doi: 10.1016/j.freeradbiomed.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Li L, Hsu A, Moore PK. Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation – a tale of three gases! Pharmacol Ther. 2009;123:386–400. doi: 10.1016/j.pharmthera.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Li X, Okada Y, Pilbeam CC, Lorenzo JA, Kennedy CR, Breyer RM, et al. Knockout of the murine prostaglandin EP2 receptor impairs osteoclastogenesis in vitro. Endocrinology. 2000;141:2054–2061. doi: 10.1210/endo.141.6.7518. [DOI] [PubMed] [Google Scholar]
- Moody TW, Switzer C, Santana-Flores W, Ridnour LA, Berna M, Thill M, et al. Dithiolethione modified valproate and diclofenac increase E-cadherin expression and decrease proliferation of non-small cell lung cancer cells. Lung Cancer. 2010;68:154–160. doi: 10.1016/j.lungcan.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee M, Das AS, Mitra S, Mitra C. Prevention of bone loss by oil extract of garlic (Allium sativum Linn.) in an ovariectomized rat model of osteoporosis. Phytother Res. 2004;18:389–394. doi: 10.1002/ptr.1448. [DOI] [PubMed] [Google Scholar]
- Mukherjee M, Das AS, Das D, Mukherjee S, Mitra S, Mitra C. Effects of garlic oil on postmenopausal osteoporosis using ovariectomized rats: comparison with the effects of lovastatin and 17beta-estradiol. Phytother Res. 2006;20:21–27. doi: 10.1002/ptr.1795. [DOI] [PubMed] [Google Scholar]
- Nakayama GR, Caton MC, Nova MP, Parandoosh Z. Assessment of the Alamar Blue assay for cellular growth and viability in vitro. J Immunol Methods. 1997;204:205–208. doi: 10.1016/s0022-1759(97)00043-4. [DOI] [PubMed] [Google Scholar]
- Okada Y, Lorenzo JA, Freeman AM, Tomita M, Morham SG, Raisz LG, et al. Prostaglandin G/H synthase-2 is required for maximal formation of osteoclast-like cells in culture. J Clin Invest. 2000;105:823–832. doi: 10.1172/JCI8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oommen S, Anto RJ, Srinivas G, Karunagaran D. Allicin (from garlic) induces caspase-mediated apoptosis in cancer cells. Eur J Pharmacol. 2004;485:97–103. doi: 10.1016/j.ejphar.2003.11.059. [DOI] [PubMed] [Google Scholar]
- Pinto JT, Qiao C, Xing J, Suffoletto BP, Schubert KB, Rivlin RS, et al. Alterations of prostate biomarker expression and testosterone utilization in human LNCaP prostatic carcinoma cells by garlic-derived S-allylmercaptocysteine. Prostate. 2000;45:304–314. doi: 10.1002/1097-0045(20001201)45:4<304::aid-pros4>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Rossoni G, Sparatore A, Tazzari V, Manfredi B, Del Soldato P, Berti F. The hydrogen sulphide-releasing derivative of diclofenac protects against ischaemia-reperfusion injury in the isolated rabbit heart. Br J Pharmacol. 2008;153:100–109. doi: 10.1038/sj.bjp.0707540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, Hosono T, Hosono-Fukao T, Inada K, Tanaka R, Ogihara J, et al. Anticancer effects of diallyl trisulfide derived from garlic. Asia Pac J Clin Nutr. 2008;17(Suppl. 1):249–252. [PubMed] [Google Scholar]
- Shirin H, Pinto JT, Kawabata Y, Soh JW, Delohery T, Moss SF, et al. Antiproliferative effects of S-allylmercaptocysteine on colon cancer cells when tested alone or in combination with sulindac sulfide. Cancer Res. 2001;61:725–731. [PubMed] [Google Scholar]
- Sidhapuriwala J, Li L, Sparatore A, Bhatia M, Moore PK. Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative, on carrageenan-induced hindpaw oedema formation in the rat. Eur J Pharmacol. 2007;569:149–154. doi: 10.1016/j.ejphar.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Sriram N, Kalayarasan S, Ashokkumar P, Sureshkumar A, Sudhandiran G. Diallyl sulfide induces apoptosis in Colo 320 DM human colon cancer cells: involvement of caspase-3, NF-kappaB, and ERK-2. Mol Cell Biochem. 2008;311:157–165. doi: 10.1007/s11010-008-9706-8. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol Sci. 2007;28:501–505. doi: 10.1016/j.tips.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Zhi L, Ang AD, Zhang H, Moore PK, Bhatia M. Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK-NF-kappaB pathway. J Leukoc Biol. 2007;81:1322–1332. doi: 10.1189/jlb.1006599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.