

Abstract
BACKGROUND AND PURPOSE
Acute NOS inhibition in humans and animals is associated with hypersensitivity to NO donors. The mechanisms underlying this phenomenon have not been fully elucidated. The purpose of the present study was to assess whether hypersensitivity to NOS-blockade is linked to endothelin-1 (ET-1) signalling.
EXPERIMENTAL APPROACH
Sprague Dawley rats were instrumented with indwelling arterial and venous catheters for continuous assessments of haemodynamic parameters and drug delivery, respectively. Mesenteric arteries were isolated and tested for reactivity by wire myography.
KEY RESULTS
NOS blockade with L-NG-nitroarginine methyl ester (L-NAME) caused a pronounced increase in arterial blood pressure (BP) (∼40 mmHg). In L-NAME-treated animals, the dose of sodium nitroprusside (SNP) required to cause a significant reduction in arterial BP was lower than in vehicle-treated rats (P < 0.001), and the magnitude of the reduction in BP was greater. Similar results were obtained with other NO mimetics, but not isoprenaline; moreover, decreasing the BP back to baseline levels with prazosin after L-NAME treatment did not attenuate the hyper-responsiveness to NO donors. The increased responsiveness to NO donors was abolished by pretreatment with the ETA/B receptor antagonist, PD145065, or the ETA receptor-specific antagonist ABT627. Ex vivo, L-NAME treatment potentiated the constriction induced by big endothelin-1 (bET-1), the precursor to active ET-1, but had no effect on the ET-1-mediated constriction.
CONCLUSIONS AND IMPLICATIONS
These data suggest that the increased sensitivity to NO donors is mediated, at least in part, by ET-1 in vivo, and the mechanism may involve the conversion of bET-1 to ET-1.
Keywords: endothelin-1, hypertension, NO, vasoconstriction
Introduction
NO is an endogenous free radical formed from the terminal guanidino nitrogen of the amino acid L-arginine by the enzyme NOS (Palmer et al., 1988). Among its many physiological functions, NO plays an important role in regulating vascular resistance, and hence arterial BP (Rees et al., 1989; Vallance et al., 1989; Moncada and Higgs, 2006; Bian et al., 2008). Indeed, in vivo inhibition of endogenous NO production with analogues of L-arginine [e.g. L-NG-nitroarginine methyl ester (L-NAME)] results in a marked hypertensive response characterized by pronounced vascular constriction (Rees et al., 1989).
Both humans and animals exhibit increased vascular responsiveness to NO donors following acute NOS inhibition (Alheid et al., 1987; Busse et al., 1989; Flavahan and Vanhoutte, 1989; Luscher et al., 1989; Gardiner et al., 1991; Moncada et al., 1991). This phenomenon remains of interest to investigators because it provides important insights into the mechanisms of acute vascular control. Despite being first described over two decades ago, the mechanisms underlying the increased responsiveness to NO donors following NOS inhibition are still not completely understood. Studies have shown that the increased responsiveness to NO donors may stem, in part, from a vascular sensitization of second messenger cGMP following NOS inhibition (Moncada et al., 1991), although whether this mechanism predominates in vivo is not known.
In addition to directly eliciting vasodilatation (Rees et al., 1989; Vallance et al., 1989), NO has been shown to regulate vascular tone via modulation of vasoconstrictor agents, including endothelin-1 (ET-1) (Banting et al., 1996; Filep, 1997; Cardillo et al., 2000). ET-1 is a potent vasoconstrictor peptide that is produced primarily within the endothelium. It elicits its vascular effects by binding to either ETA or ETB receptor subtypes. ET-1 binding to ETA and ETB receptors, located on smooth muscle cells, causes vasoconstriction, whereas binding to ETB receptors, located on endothelial cells, mediates NO release (Warner et al., 1989). Previous studies in humans and animals have shown that antagonism of ET-1 receptors attenuates the hypertensive effect caused by NOS inhibition (Banting et al., 1996; Filep, 1997; Cardillo et al., 2000), suggesting that ET-1 signalling is an important mechanism by which NO maintains normal vascular tone. On the basis of this work, we hypothesized that the increased responsiveness to NO donors following NOS inhibition could involve ET-1 signalling. More specifically, we reasoned that NOS inhibition would unmask ET-1 actions, and subsequent addition of low doses of NO donors would oppose this effect, resulting in an exaggerated NO responsiveness. As such, the pharmacological antagonism of ET-1 receptors would attenuate this hyper-responsiveness to NO donors. The objectives of the present study were: (i) to assess and characterize the nature of BP responses to vasodilator agents in naïve and L-NAME-treated rats; (ii) to assess the involvement of the ET-1 system in these animals using endothelin receptor antagonists; (iii) to assess isolated vascular responses to vasoconstrictors in the presence of L-NAME treatment and ET-1 antagonists.
Methods
Animals and treatments
All animal care and experimental procedures were performed in accordance with standards established by the Canadian Council of Animal Care, and approved by the University of Alberta and Queen's University Animal Care Committees. The investigation complied with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85–23, revised 1996). Adult male Sprague-Dawley rats (approximately 12 weeks of age) were purchased from Charles River (St. Constant, QC for experiments done at Queen's University; Wilmingham, MA, USA for experiments done at the University of Alberta), and given at least 3 days to acclimatize to their surroundings before experimentation. Rats were housed in an animal care facility, which maintained a 12 h/12 h light/dark cycle and an ambient temperature of 23 ± 1°C. Rats had ad libitum access to food and water.
Conscious BP assessments
BP assessments were performed as described by Banting et al. (1996). Briefly, rats were anaesthetized with ketamine hydrochloride (70 mg·kg−1, i.p.; Rogar/STB, London, Ontario, Canada) and xylazine (5 mg·kg−1, i.p. Bayer, Etobicoke, Ontario, Canada), and a proper depth of anaesthesia was verified using a toe pinch. Rats were then implanted with four catheters (0.3 mm i.d., 0.76 mm O.D. Cole Parmer, Laval, Quebec, Canada); one in the aorta, distal to the kidneys, for BP assessments; two in the inferior vena cava distal to the kidneys and one in the intraperitoneal cavity, for drug administrations. All catheters were made of vinyl, with the exception of the venous catheters used for administration of glyceryl trinitrate (GTN), which were polyethylene (PE50: 0.58 mm i.d., 0.97 mm O.D., Becton Dickson, Sparks, MD, USA) to minimize drug absorption by the tubing. The catheters were filled with heparin-containing saline (10 IU·mL−1), and held in place with a small amount of cyanoacrylate glue at the puncture site. All catheters were exteriorized and sutured into position after passing through the abdominal wall and subcutaneously tunneled towards the nape of the neck. Post-surgical pain was managed by s.c. administration of buprenorphine hydrochloride (3 mg·kg−1, Reckitt and Colman Pharmaceuticals, Richmond, VA, USA). Rats were also administered trimethoprim and sulfadiazine (Tribrissen 24%, 0.3 mg·kg−1, s.c.; Intervet/Schering-Plough, Kirkland, Quebec, Canada) for antibiotic prophylaxis. All BP data presented in this study were obtained from conscious animals. Rats were given at least 3 days to recover from the surgeries; this length of time was found to be sufficient for circulatory responses to recover from anaesthetic effects.
On the day of the experiment, BP was continuously recorded in conscious rats using a Powerlab® data acquisition system (ADInstruments, Colorado Springs, CO, USA), calibrated manually using a sphygmomanometer. Rats were given a washout period of at least 30 min before experimentation and between each series of experiments to establish a steady state baseline. Drugs were infused using a motorized syringe pump (Model 220, KD Scientific, Holliston, MA, USA). For experiments involving pretreatment with either L-NAME (100 mg·kg−1, i.p., Sigma, St. Louis, MO, USA), PD145065 (10 mg·kg−1·min−1 i.v., EMD Chemicals, Gibbstown, NJ, USA), or ABT627 (200 µg·kg−1·min−1 i.v., for 10 min, followed by 100 µg·kg−1·min−1 i.v. thereafter; Abott Laboratories, Abott Park, IL, USA), these maintenance drugs were administered after a steady haemodynamic [heart rate and mean arterial pressure (MAP)] baseline had been achieved, and infusions were continued until all concentration-response curves with vasodilators were obtained. In the experiments where L-NAME and endothelin receptor antagonists were co-administered, endothelin receptor antagonists were administered first, followed by L-NAME. In the experiments where L-NAME and prazosin (2 mg·kg−1 i.p., Sigma, dissolved in 5% ethanol/95% saline) were co-administered, L-NAME was administered first followed by prazosin. Time-controlled rats received saline. Doses of sodium nitroprusside (SNP; 0.8–32·µg·kg−1·min−1 i.v., Sigma), GTN (2–200 µg·kg−1·min−1 i.v., Sabex, Boucherville, Quebec, Canada), methylamine hexamethylene methylamine NONOate (10–60 µg·kg−1·min−1 i.v., Alexis Biochemicals, Plymouth Meeting, PA, USA), 8-bromo-cGMP (8Br-cGMP; 0.1–50 µg·kg−1·min−1 i.v., Sigma), and isoprenaline (0.02–0.8 µg·kg−1·min−1, Sigma) were administered in a non-cumulative manner over a period of 30 s.
For assessments of circulating ET-1, rats were treated with L-NAME (100 mg·kg−1, i.p.) or saline under isofluorance anaesthesia (dosed to effect by inhalation), and blood was collected after 15 min. Blood was kept on ice and allowed to clot, and was then centrifuged at 3650×g for 15 min. ET-1 from serum samples was purified, and quantified by enzyme immunoassay (Caymen Chemical Co., Ann Arbor, MI, USA) according to the manufacturer's instructions. Sample yield was calculated based on recovery of [125I]-ET-1 (>22 uCi·mmol−1; Vitrax Radiochemicals, Placentia, CA, USA) from spiked samples.
Isolated vessel experiments
Rats were anaesthetized with isoflurane (dosed to effect by inhalation), and killed by exsanguination. Mesenteric vessels was rapidly excised and placed in iced HEPES-buffered physiological saline solution (in mmol·L−1: NaCl 142, KCl 4.7, MgSO4 1.17, CaCl2 4.7, K2PO4 1.18, HEPES 10 and glucose 5.5; pH 7.4). Second order mesenteric arteries were carefully isolated from the surrounding adipose tissue using a binocular microscope, and arteries with internal diameters ranging 150–250 µm were mounted in an isometric myograph system (DMT, Copenhagen, Denmark) using 40 µm tungsten wire. Vessels were normalized through a series of stepwise increases in diameter to determine their optimal resting tension, set to 0.8 × IC100 (the internal circumference equivalent to a transmural pressure of 100 mmHg). Vessels were then given 30 min to equilibrate to their optimal resting tension.
Following equilibration, mesenteric arteries were twice treated with phenylephrine (10 µM; Sigma) to assess viability. On the second phenylephrine dose, methacholine (3 µM, Sigma) was administered to all baths to assess integrity of the endothelium; vessels that did not achieve a minimum relaxation of 80% were excluded from all experiments. After several washes, vessels were treated for 30 min with antagonists: L-NAME (100 µM); PD145065 (5 µM). After incubation with antagonists, cumulative dose–response curves were generated for big endothelin-1 (bET-1; Anaspec, Fremont, CA, USA; 10 nM to 300 nM), ET-1 (1 nM to 300 nM), and SNP (0.01 nM to 1 µM). In the case of bET-1 and ET-1, vessels were allowed to incubate with each cumulative dose for a minimum of 5 min. For SNP dose–response curves, vessels were submaximally preconstricted (80% of maximum) using phenylephrine, and cumulative doses were added to the bath when vessels reached a plateau response (<2 min).
Statistical analyses
Data are presented as mean ± SEM. MAP values were calculated as peak responses, averaged over 5 s, within a dosing window. ΔMAP values were obtained by subtracting the MAP after each drug dose from the pretreatment baseline. Data from the dose–response curves were fitted to the Hill equation. The EC50 was determined. For bET-1 dose–response curves, area under the curve was calculated, and compared between groups. MAP measurements were analysed by two-way anova for effects of treatment and dose, followed by Bonferroni post-hoc test. Analyses with two comparisons were conducted using Student's t-test; where three or more comparisons were made, anova was used, followed by Tukey's post hoc analysis. Data were analysed using GraphPad Prism 5 Software (La Jolla, CA, USA). P < 0.05 was considered significant.
Results
NOS inhibition causes enhanced responsiveness to NO donors
Concentration-response curves with the NO-donor SNP were performed in conscious rats in the absence and presence of the NOS inhibitor L-NAME (Figure 1). The doses of SNP were selected because they elicit minimal effects on BP under normal circumstances. As expected, treatment with L-NAME caused a hypertensive response (40 ± 4 mmHg) compared with vehicle (Veh) controls (P < 0.001), and this maximal effect was attained within 15 min of dosing. Subsequent administration of SNP to L-NAME-treated rats produced significant depressor effects at doses of 3.2 µg·kg−1·min−1 (−17.8 ± 3.4 mmHg, P < 0.01; Figure 1) or greater. In contrast, in control rats, the dose of SNP required to produce a significant depressor effect was 8 µg·kg−1·min−1 (−11.4 ± 2.8 mmHg, P < 0.05). The change in MAP from baseline levels is presented in Figure 2. At all doses tested, the magnitude of the decrease in BP induced by SNP was threefold greater in L-NAME-treated rats compared with controls (Figure 2A). Compared with controls, the BP in L-NAME-treated rats also exhibited increased responsiveness to the NO mimetics GTN (Figure 2B) and methylamine-hexamethylene-methylamine-NONOate (Hrabie et al., 1993) (2.6- and 2.1-fold decrease in BP at doses of 40 and 50 µg kg−1·min−1, respectively; P < 0.05 for both parameters). Compared with Veh-treated rats, L-NAME-treated rats also exhibited increased responsiveness to the cGMP analogue 8Br-cGMP (Figure 2C). In contrast, NOS-inhibited rats showed no hyper-responsiveness to isoprenaline (Figure 2D) or PGE1 (data not shown), which cause vasodilatation predominantly via cAMP-dependent signalling mechanisms.
Figure 1.
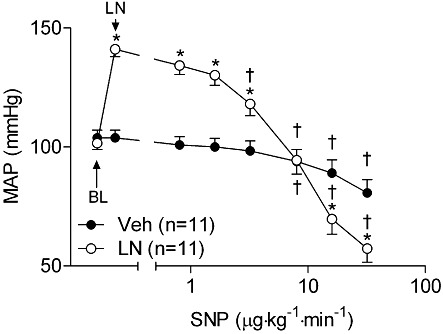
MAP depressor responses to SNP are exaggerated following NOS inhibition in conscious rats. Rats were treated with L-NAME (100 mg·kg−1, i.p.) or saline, and subsequently administered the NO-donor SNP. *P < 0.05 compared with Veh-treated rats at the same dose of vasodilator. †P < 0.05 compared with MAP after administration of either L-NAME or saline in same group. LN, L-NAME.
Figure 2.
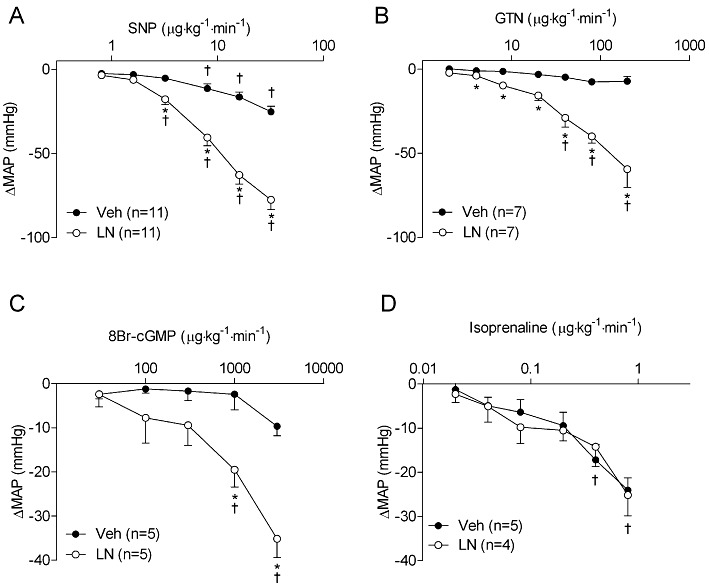
NOS inhibition causes increased BP responsiveness to the vasodilators (A) SNP, (B) GTN, (C) the cGMP analogue 8Br-cGMP, but not (D) isoprenaline. Rats were treated with L-NAME (100 mg·kg−1, i.p.) or saline, and subsequently administered vasodilators. Data are shown as changes from steady state MAP levels after infusion of saline or L-NAME. *P < 0.05 compared with Veh-treated rats at the same dose of vasodilator. †P < 0.05 compared with baseline (BL) in same group. Note that in (D), treatment groups were combined since two-way anova revealed no overall effect of L-NAME; therefore symbols reflect pooled data. GTN, glyceryl trinitrate; LN, L-NAME.
As L-NAME caused a marked hypertensive response, we considered the possibility that the increased responsiveness to NO donors was related to mechanisms associated with this increase in BP (e.g. release of humoral factors, increased parasympathetic outflow, etc.), rather than an absence of NO per se. We therefore sought to determine whether the hyper-responsiveness to NO donors remained when BP was lowered to baseline levels (pre-L-NAME pressures). In a separate cohort, rats were treated with the α-adrenoceptor antagonist prazosin after L-NAME treatment, which lowered BP to a level similar to naïve animals (Figure 3A). Despite the lack of BP difference compared with naïve animals, rats co-treated with L-NAME and prazosin exhibited a similar enhanced responsiveness to infused SNP (Figure 3B) as the rats treated with L-NAME alone (Figure 2A).
Figure 3.
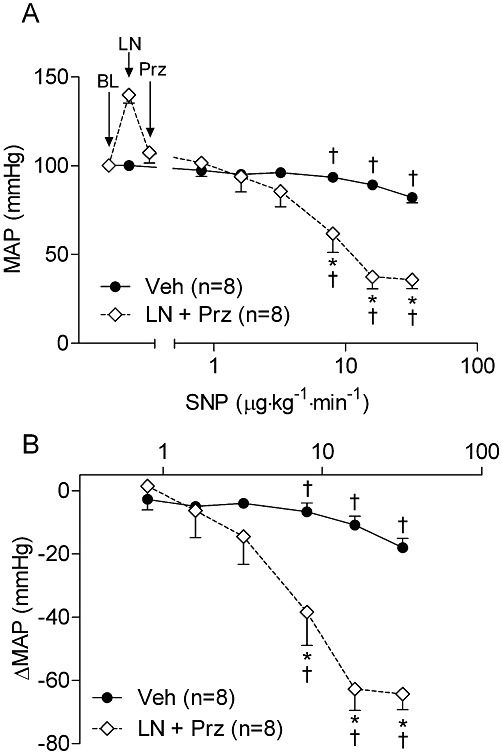
(A) MAP responsiveness to SNP following NOS inhibition is not dependent on initial MAP level. Rats co-treated with α-adrenoceptor antagonist prazosin (Prz; 2 mg·kg−1, i.p.) and L-NAME (100 mg·kg−1, i.p.) had similar MAP levels as Veh-treated rats, and had a similarly exaggerated responsiveness to SNP as L-NAME-treated rats. (B) Data from same experiments in (A) shown as changes from steady state MAP levels after infusion of saline, or combination of L-NAME and prazosin. *P < 0.05 compared with controls at the same dose of vasodilator. †P < 0.05 compared with baseline in same group. BL, baseline MAP; LN, L-NAME.
Increased responsiveness to NO donors following NOS inhibition is dependent on ET-1 signalling
We subsequently investigated the involvement of ET-1 in the hyper-responsiveness to NO donors following NOS inhibition. In rats pretreated with the ETA/B receptor antagonist PD145065, L-NAME had only a modest hypertensive effect, corresponding to a blunting of 68% compared with L-NAME-treated rats (L-NAME: 41 ± 4 mmHg; L-NAME+PD145065: 13 ± 3 mmHg P < 0.05). Treatment with PD145065 also completely abolished the hypersensitivity to NO donors following NOS inhibition with L-NAME, and in fact resulted in diminished depressor responses even compared with controls (Figure 4A). To ascertain whether these effects were mediated by ETA receptors, we conducted similar experiments using the ETA receptor antagonist ABT627. Similar to PD145065, pretreatment with ABT627 attenuated the hypertensive response caused by L-NAME treatment by 53% (L-NAME: 40 ± 6 mmHg; L-NAME+ABT627: 19 ± 2 mmHg; P < 0.05), and abolished the hypersensitivity to NO donors (Figure 4B). Interestingly, we also observed that PD145065 blunted the responsiveness to NO donors in the absence of L-NAME (Figures 4C, P = 0.004 for overall effect by two-way anova). In a separate cohort of rats, we assessed circulating levels of immunoreactive ET-1 following L-NAME or saline treatment. Serum levels of ET-1 were not different between treatment groups at 15 min [saline: 375 ± 12 ng mL−1 (n = 4); L-NAME: 364 ± 11 ng mL−1 (n = 5); P = 0.23].
Figure 4.
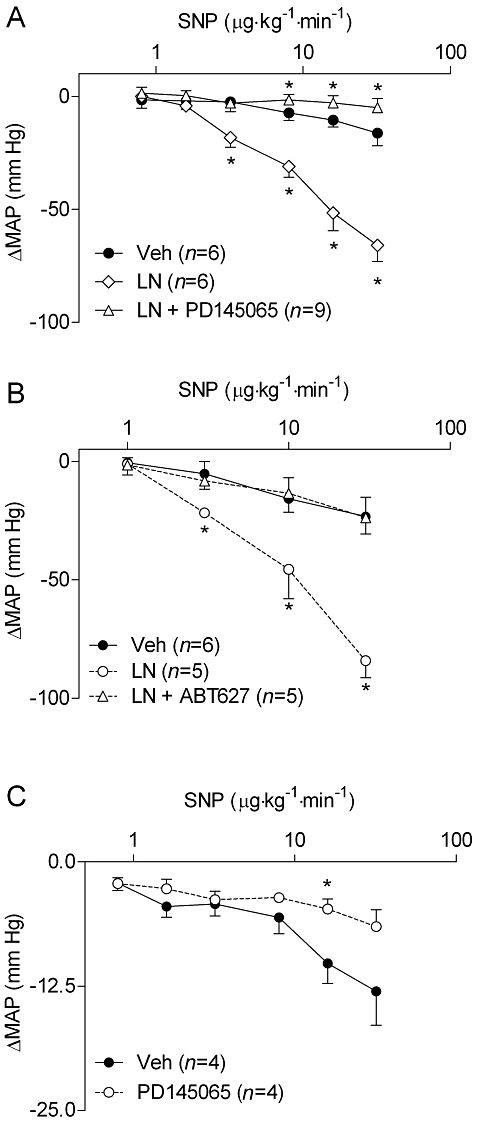
Increased MAP responsiveness to SNP following NOS inhibition with L-NAME (100 mg·kg−1, i.p.) is attenuated after pre-treatment with (A) the dual ETA/B receptor antagonist PD145065 (10 mg kg−1 min−1 i.v.) or (B) the ETA receptor antagonist ABT627 (200 µg·kg−1·min−1 i.v. for 10 min, followed by 100 µg·kg−1·min−1 i.v. thereafter). (C) BP responsiveness to SNP is also attenuated by PD145065 (10 mg·kg−1·min−1 i.v.) in NOS-intact rats. Data are shown as MAP changes from steady state baseline levels after infusion of saline, L-NAME, or combination of L-NAME and ET receptor antagonist. *P < 0.05 compared with controls at the same dose of vasodilator. LN, L-NAME.
To gain insights into the mechanisms by which NO interacts with ET-1, we assessed vascular responses to vasoconstrictors in isolated vessels. Isolated mesenteric arteries were treated with bET-1, the inactive precursor to functional ET-1, in the presence or absence of L-NAME (Figure 5A). Treatment with L-NAME resulted in a potentiation of the bET-1-induced constriction, resulting in a 96% increase in the area under the curve compared with Veh-treated vessels (Figure 5A inset). Treatment with the endothelin converting enzyme phosphoramidon significantly inhibited the constriction induced by bET-1, corresponding to an 80% reduction in the area under the curve (P < 0.001; data not shown), confirming that bET-1 must be converted to active ET-1 to elicit vasoconstriction. These data demonstrate that NO modulates conversion of bET-1 to active ET-1. In contrast, treatment with L-NAME had no effect on the dose–response curve to ET-1 (Figure 5B).
Figure 5.
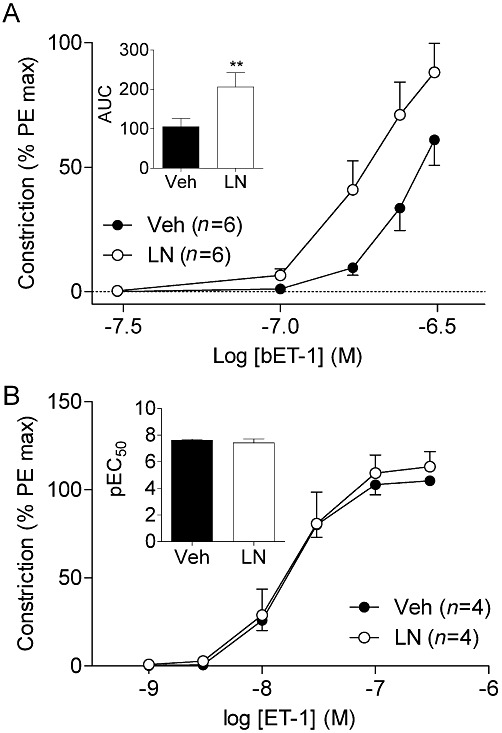
(A) bET-1 conversion to vasoactive ET-1 is increased after L-NAME treatment in isolated vessels. Inset: calculated AUC from bET-1 dose–response curves. *P < 0.01 compared with Veh AUC. (B) ET-1 mediated vasoconstriction is not affected by L-NAME (100 µM) treatment. Inset: pEC50 values were derived from ET-1 dose–response curves. AUC, area under the curve; LN, L-NAME; PE, phenylephrine.
Finally, because the increased responsiveness to NO donors following acute L-NAME treatment was observed in isolated vessels, we tested whether this phenomenon was also dependent on ET-1 signalling. We further assessed the responsiveness of isolated vessels to SNP in the presence and absence of L-NAME (Figure 6). Pretreatment of vessels with L-NAME resulted in an increased responsiveness to SNP, corresponding to a leftward shift in the dose–response curve, and increase in the pEC50 (Figure 6, inset). Responses to SNP in vessels treated with both L-NAME and the endothelin receptor antagonist PD145065 were superimposed on those induced in L-NAME-treated vessels, corresponding to an unaltered pEC50.
Figure 6.
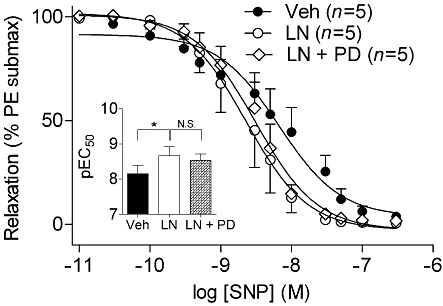
Increased vascular reactivity to SNP following NOS inhibition is not dependent on ET-1 in isolated mesenteric vessels. Isolated vessels were incubated with Veh, L-NAME (100 µM), or a combination of L-NAME (100 µM) and the ETA/B receptor antagonist PD145065 (5 µM). Inset: pEC50 values were derived from dose–response curves. *P < 0.05 compared with Veh-treated vessels. LN, L-NAME, N.S., not significant, PD, PD145065.
Discussion and conclusions
The role of NO as a critical mediator of vascular tone is well established. The importance of NO in the acute regulation of BP is underscored by the fact that NO-mimetics, such as GTN and SNP, represent first-line treatments in a host of circulatory pathologies, including ischaemic heart disease and acute hypertensive crises. As such, hyper-responsiveness to NO-mimetics resulting from diminished NO synthesis could be an important clinical issue. In the present study, we investigated the contribution of ET-1 signalling in this phenomenon, and sought to gain additional insights into the interaction between NO and ET-1 in the regulation of vascular tone, and hence, BP. To summarize, we demonstrated, using a conscious rat model that: (i) acute treatment with L-NAME causes increased sensitivity to NO-mimetics and the cGMP analog 8Br-cGMP, but not to the other vasodilators tested; (ii) hypersensitivity to NO-mimetics is abolished after dual antagonism of ETA/B receptors, or selective antagonism of ETA receptors; furthermore, in isolated vessels, (iii) NOS inhibition potentiated the conversion of bET-1 to active ET-1, but had no effect on ET-1-mediated vasoconstriction; and finally, (iv) whereas treatment of isolated mesenteric resistance arteries vessels results in enhanced responsiveness to NO donors following NOS inhibition, this effect is not dependent on endothelin signalling.
The involvement of ET-1 in the hypertensive response, observed after acute NOS inhibition, has been demonstrated by several groups (Banting et al., 1997; Filep, 1997; Cardillo et al., 2000). The present study extends these findings by demonstrating that low doses of NO, which have little effect on BP in naïve animals, have marked effects on BP in NOS-inhibited animals. These observations may indicate that concentrations of NO below those that directly elicit vasodilatation effectively inhibit ET-1-mediated vasoconstriction. It is possible, therefore, that under normal circumstances, the predominant role for NO is to suppress the actions of vasoconstrictors such as ET-1. This is consistent with the observation that PD145065 inhibited the response to SNP even in the absence of L-NAME (Figure 4C). The observation that circulating ET-1 levels were not altered following L-NAME treatment may reflect the fact that ET-1 release is predominantly abluminal, and therefore changes in circulating levels do not necessarily indicate ET-1 signalling at the level of vascular receptors. It is clear, however, that more extensive studies are needed to fully test this hypothesis. Nevertheless, it is clear that ET-1 is involved in the hyper-responsiveness to NO donors following NOS inhibition. The finding that ABT627 could also attenuate this effect is consistent with the results of Richard et al. (1995) who demonstrated that L-NAME, used at a similar dose as in the present study, induced a hypertensive response that could be attenuated by ETA receptor antagonism.
To gain additional insights into the mechanisms by which NO may modulate the ET-1 system in the vasculature, we performed similar experiments in isolated resistance arteries. We demonstrated that NO and ET-1 directly interact at the level of the vasculature. Specifically, the absence of endogenous NO potentiated the conversion of bET-1 to active ET-1, confirming that NO mediates an inhibitory effect on ET-1 signalling. We considered the possibility that blockade of NOS could simply be potentiating the vasoconstrictor effects of newly formed ET-1 by inhibiting the NO produced following autocrine activation of ETB receptors (Warner et al., 1989). However, NOS blockade failed to potentiate the vasoconstrictor effects of ET-1, which is consistent with other studies (Fernandez et al., 1995; Ferrero et al., 2008), suggesting that NO derived from ETB activation does not appreciably oppose vasoconstriction. In this regard, it is tempting to further speculate that autocrine activation of ETB receptors in vivo produces low quantities of NO that are sufficient to inhibit further ET-1 production, but do not contribute directly to modulation of vascular tone.
The increased responsiveness to NO donors in isolated tissues is consistent with previous reports (Alheid et al., 1987; Busse et al., 1989; Flavahan and Vanhoutte, 1989; Luscher et al., 1989; Moncada et al., 1991). However, the inability of PD145065 to reverse the L-NAME-mediated hyper-responsiveness to SNP in isolated tissues suggests that this mechanism is different when studied in vivo and ex vivo. Whereas the hyper-responsiveness in vivo implicates ET-1, the increased responsiveness to NO donors in isolated tissues does not, and is more likely attributed to enhanced sGC activity and sensitization of downstream pathways following NOS inhibition, which has been reported previously (Moncada et al., 1991; Gupta et al., 2008). The apparent disparity between in vivo and ex vivo involvement of ET-1 in the hyper-responsiveness to NO donors is easily reconciled. As removal of resistance arteries from the rat results in loss of vascular modulatory systems – including sympathetic, parasympathetic and nitrergic innervations as well as actions by local and humoral factors – vascular tone is generated ex vivo‘artificially’ by the addition of PE (i.e. preconstriction). It is therefore perhaps not surprising that hyper-responsiveness observed in the wake of NOS inhibition is not dependent on ET-1 signalling. In contrast, in a fully integrated system (i.e. conscious rat) vascular tone is established by a host of signalling systems acting in concert, resulting in the perpetual release of ET-1 and NO (among several other vasoactive systems). However, we must also acknowledge the possibility that the interaction between NO and ET-1 observed in the present experiments may be occurring in other tissues, or could be due to regional vascular differences in responsiveness to NO donors and NOS inhibition, which has been described previously (Gardiner et al., 1991).
A point that is particularly noteworthy in the context of the present study is the time course in which NOS is inhibited. Although increased responsiveness to NO donors has been observed in both acute and chronic NOS inhibition models (Alheid et al., 1987; Busse et al., 1989; Flavahan and Vanhoutte, 1989; Luscher et al., 1989; Gardiner et al., 1991; 1993; Moncada et al., 1991), the mechanisms underlying these effects may be different (Bourque et al., 2011). The present experiments, which provide evidence for a complex interaction between NO and ET-1, were performed on a relatively short time scale (acute NOS inhibition), and may not be applicable in models of chronic NOS inhibition. Indeed, in studies in which ET-1 antagonists are administered before or immediately after NOS blockade, the hypertension is preventable or reversible (Richard et al., 1995; Banting et al., 1997; Filep, 1997; Cardillo et al., 2000; present study). However, after several weeks of NOS blockade, the hypertension is not reversible by ET-1 antagonism (Bank et al., 1994; Fujita et al., 1995). Similarly, whereas L-arginine, the precursor to NO, readily reverses the hypertension associated with L-NAME when administered simultaneously with the NOS inhibitor (Salazar et al., 1992), it was shown to have progressively less effect when administered several days (Ribeiro et al., 1992) or several weeks after L-NAME treatment began (Erley et al., 1995; Qiu et al., 1995).
In summary, the present study offers insights into the mechanisms by which NOS inhibition causes enhanced sensitivity to NO. Perhaps more importantly, it also offers insights into the general mechanisms of vascular control. That is, the present results, together with other studies, suggest that NO maintains vascular tone largely through an ET-1-dependent signalling mechanism (Bourque et al., 2011). The paradigm we see emerging is one in which vascular control by NO occurs by several mechanisms, including opposing the actions of vasoconstrictor agents, as well as direct vasodilatation, among others.
Acknowledgments
We are indebted to Ms. Corry Smallegange for her technical assistance. This work is supported by grants from the Canadian Institutes of Health Research (CIHR). SLB is supported by salary awards from the CIHR and Alberta Innovates and Health Solutions. STD is an Alberta Heritage Foundation for Medical Research Scientist and Canada Research Chair (Tier 1) in Women's Cardiovascular Health.
Glossary
- ABT627
2R-(4-methoxyphenyl)-4S-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)aminocarbonyl-methyl)-pyrrolidine-3R-carboxylic acid
- bET-1
big endothelin-1
- ET-1
endothelin-1
- GTN
glyceryl trinitrate
- L-NAME
L-NG-nitroarginine methyl ester
- PD145065
N-acetyl-α-[10,11-dihydro-5h-dibenzo[a,d]cycloheptadien-5-yl]-d-gly-leu-asp-ile-ile-trp
- SNP
sodium nitroprusside
- Veh
vehicle
Conflict of interest
None.
References
- Alheid U, Dudel C, Forstermann U. Selective inhibition by gossypol of endothelium-dependent relaxations augments relaxations to glyceryl trinitrate in rabbit coeliac artery. Br J Pharmacol. 1987;92:237–240. doi: 10.1111/j.1476-5381.1987.tb11317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank N, Aynedjian HS, Khan GA. Mechanism of vasoconstriction induced by chronic inhibition of nitric oxide in rats. Hypertension. 1994;24:322–328. doi: 10.1161/01.hyp.24.3.322. [DOI] [PubMed] [Google Scholar]
- Banting JD, Friberg P, Adams MA. Acute hypertension after nitric oxide synthase inhibition is mediated primarily by increased endothelin vasoconstriction. J Hypertens. 1996;14:975–981. [PubMed] [Google Scholar]
- Banting JD, Thompson KE, Friberg P, Adams MA. Blunted cardiovascular growth induction during prolonged nitric oxide synthase blockade. Hypertension. 1997;30:416–421. doi: 10.1161/01.hyp.30.3.416. [DOI] [PubMed] [Google Scholar]
- Bian K, Doursout MF, Murad F. Vascular system: role of nitric oxide in cardiovascular diseases. J Clin Hypertens (Greenwich) 2008;10:304–310. doi: 10.1111/j.1751-7176.2008.06632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque SL, Davidge ST, Adams MA. The interaction between endothelin-1 and nitric oxide in the vasculature: new perspectives. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1288–R1295. doi: 10.1152/ajpregu.00397.2010. [DOI] [PubMed] [Google Scholar]
- Busse R, Pohl U, Mulsch A, Bassenge E. Modulation of the vasodilator action of SIN-1 by the endothelium. J Cardiovasc Pharmacol. 1989;14(Suppl 11):S81–S85. doi: 10.1097/00005344-198906152-00015. [DOI] [PubMed] [Google Scholar]
- Cardillo C, Kilcoyne CM, Cannon RO, 3rd, Panza JA. Interactions between nitric oxide and endothelin in the regulation of vascular tone of human resistance vessels in vivo. Hypertension. 2000;35:1237–1241. doi: 10.1161/01.hyp.35.6.1237. [DOI] [PubMed] [Google Scholar]
- Erley CM, Rebmann S, Strobel U, Schmidt T, Wehrmann M, Osswald H, et al. Effects of antihypertensive therapy on blood pressure and renal function in rats with hypertension due to chronic blockade of nitric oxide synthesis. Exp Nephrol. 1995;3:293–299. [PubMed] [Google Scholar]
- Fernandez N, Monge L, Garcia-Villalon AL, Garcia JL, Gomez B, Dieguez G. Endothelin-1-induced in vitro cerebral venoconstriction is mediated by endothelin ETA receptors. Eur J Pharmacol. 1995;294:483–490. doi: 10.1016/0014-2999(95)00577-3. [DOI] [PubMed] [Google Scholar]
- Ferrero E, Labalde M, Fernandez N, Monge L, Salcedo A, Narvaez-Sanchez R, et al. Response to endothelin-1 in arteries from human colorectal tumours: role of endothelin receptors. Exp Biol Med (Maywood) 2008;233:1602–1607. doi: 10.3181/0802-RM-69. 10.3181/0802-RM-69. [DOI] [PubMed] [Google Scholar]
- Filep JG. Endogenous endothelin modulates blood pressure, plasma volume, and albumin escape after systemic nitric oxide blockade. Hypertension. 1997;30:22–28. doi: 10.1161/01.hyp.30.1.22. [DOI] [PubMed] [Google Scholar]
- Flavahan NA, Vanhoutte PM. Mechanism underlying the inhibitory interaction between the nitrovasodilator SIN-1 and the endothelium. J Cardiovasc Pharmacol. 1989;14(Suppl. 11):S86–S90. [PubMed] [Google Scholar]
- Fujita K, Matsumura Y, Miyazaki Y, Takaoka M, Morimoto S. Role of endothelin-1 in hypertension induced by long-term inhibition of nitric oxide synthase. Eur J Pharmacol. 1995;280:311–316. doi: 10.1016/0014-2999(95)00216-8. [DOI] [PubMed] [Google Scholar]
- Gardiner SM, Kemp PA, Bennett T. Effects of NG-nitro-L-arginine methyl ester on vasodilator responses to acetylcholine, 5′-N-ethylcarboxamidoadenosine or salbutamol in conscious rats. Br J Pharmacol. 1991;103:1725–1732. doi: 10.1111/j.1476-5381.1991.tb09854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner SM, Kemp PA, Bennett T. Effects of chronic treatment with nitric oxide synthase inhibitors on regional haemodynamic responses to vasodilators in conscious Brattleboro rats. Br J Pharmacol. 1993;109:222–228. doi: 10.1111/j.1476-5381.1993.tb13557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PK, Subramani J, Singh TU, Leo MD, Sikarwar AS, Prakash VR, et al. Role of protein kinase G in nitric oxide deficiency-induced supersensitivity to nitrovasodilator in rat pulmonary artery. J Cardiovasc Pharmacol. 2008;51:450–456. doi: 10.1097/FJC.0b013e31816949ca. 10.1097/FJC.0b013e31816949ca. [DOI] [PubMed] [Google Scholar]
- Hrabie JA, Klose JR, Wink DA, Keefer LK. New nitric oxide-releasing zwitterions derived from polyamines. J Org Chem. 1993;58:1472–1476. [Google Scholar]
- Luscher TF, Richard V, Yang ZH. Interaction between endothelium-derived nitric oxide and SIN-1 in human and porcine blood vessels. J Cardiovasc Pharmacol. 1989;14(Suppl 11):S76–S80. [PubMed] [Google Scholar]
- Moncada S, Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp Pharmacol. 2006;176:213–254. doi: 10.1007/3-540-32967-6_7. [DOI] [PubMed] [Google Scholar]
- Moncada S, Rees DD, Schulz R, Palmer RM. Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo. Proc Natl Acad Sci U S A. 1991;88:2166–2170. doi: 10.1073/pnas.88.6.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- Qiu C, Engels K, Baylis C. Endothelin modulates the pressor actions of acute systemic nitric oxide blockade. J Am Soc Nephrol. 1995;6:1476–1481. doi: 10.1681/ASN.V651476. [DOI] [PubMed] [Google Scholar]
- Rees DD, Palmer RM, Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc Natl Acad Sci U S A. 1989;86:3375–3378. doi: 10.1073/pnas.86.9.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro MO, Antunes E, de Nucci G, Lovisolo SM, Zatz R. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension. 1992;20:298–303. doi: 10.1161/01.hyp.20.3.298. [DOI] [PubMed] [Google Scholar]
- Richard V, Hogie M, Clozel M, Loffler BM, Thuillez C. In vivo evidence of an endothelin-induced vasopressor tone after inhibition of nitric oxide synthesis in rats. Circulation. 1995;91:771–775. doi: 10.1161/01.cir.91.3.771. [DOI] [PubMed] [Google Scholar]
- Salazar FJ, Pinilla JM, Lopez F, Romero JC, Quesada T. Renal effects of prolonged synthesis inhibition of endothelium-derived nitric oxide. Hypertension. 1992;20:113–117. doi: 10.1161/01.hyp.20.1.113. [DOI] [PubMed] [Google Scholar]
- Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- Warner TD, Mitchell JA, de Nucci G, Vane JR. Endothelin-1 and endothelin-3 release EDRF from isolated perfused arterial vessels of the rat and rabbit. J Cardiovasc Pharmacol. 1989;13(Suppl. 5):S85–S88. doi: 10.1097/00005344-198900135-00021. discussion S102. [DOI] [PubMed] [Google Scholar]