

Abstract
BACKGROUND AND PURPOSE
Inhibition of the renin angiotensin system (RAS) has been consistently demonstrated to reduce atherosclerosis. However, there has been no direct comparison among the three available pharmacological modes of inhibiting the RAS, which are inhibitors of renin, ACE and angiotensin II type 1 receptor. The purpose of this study was to determine the relative effects of these three modes of pharmacological RAS inhibition in reducing atherosclerosis by determining the dose–response relationships.
EXPERIMENTAL APPROACH
Male LDL receptor -/- mice were administered either vehicle or any of three doses of aliskiren, enalapril or losartan through s.c. infusion for 12 weeks. All mice were fed a saturated fat-enriched diet during drug infusions. Systolic and diastolic BPs were measured during the study using a non-invasive tail-cuff system. Plasma cholesterol and renin concentrations, atherosclerotic lesions, and renal angiotensin II concentrations were determined at the termination of the study.
KEY RESULTS
Plasma renin concentrations were increased by all three drugs. None of the drugs changed plasma cholesterol concentrations. All drugs produced a dose-related decrease in BP. All three drugs also profoundly reduced atherosclerosis in a dose-dependent manner. The highest dose of each drug markedly attenuated lesion size, with no significant differences between the different drugs. The highest dose of each drug also similarly reduced renal angiotensin II concentrations.
CONCLUSION AND IMPLICATIONS
Drugs that inhibit the RAS, irrespective of their mode of inhibition, profoundly affect atherosclerotic lesion development in a dose-dependent manner.
Keywords: atherosclerosis, renin angiotensin system, aliskiren, enalapril, losartan
Introduction
The renin angiotensin system (RAS) contains a single precursor, angiotensinogen, that is cleaved by renin to form angiotensin (Ang)I. Ang I is subsequently cleaved by angiotensin-converting enzyme (ACE) to generate Ang II, the major bioactive peptide in the RAS. Renin and ACE are the critical enzymes for the synthesis of Ang II, while the Ang II type 1 (AT1) receptor is the major receptor for the physiological and pathophysiological effects of Ang II. Over the last decade, the classic RAS has been expanded by the identification of alternative enzymes to synthesize or catabolize Ang II, a spectrum of other bioactive angiotensin peptides, and an increasing number of receptors that have been recognized to interact with these angiotensin peptides (Ferrario, 2002; Carey and Siragy, 2003; Santos et al., 2003). Therefore, the RAS is a complex system exerting an array of responses, some being antagonistic to others.
Despite the complexity of the RAS, it has been consistently demonstrated that Ang II contributes to the development of atherosclerosis (Daugherty and Cassis, 1999; Daugherty et al., 2000; Weiss et al., 2001; Bruemmer et al., 2003). Conversely, pharmacological inhibition of the classic RAS components decreases experimental atherosclerosis (Rader and Daugherty, 2008). Renin is the rate-limiting enzyme in the generation of angiotensin peptides. Previous studies have demonstrated that aliskiren, a renin inhibitor, blocks the generation of all angiotensin peptides (Lu et al., 2008) and reduces atherosclerosis in animal models (Imanishi et al., 2008; Lu et al., 2008; Nussberger et al., 2008; Weiss and Taylor, 2008). Studies have also consistently demonstrated that ACE or AT1 receptor inhibition profoundly reduces atherosclerosis in a variety of animal models (Daugherty et al., 2001; Candido et al., 2002; 2004; Wassmann et al., 2004; da Cunha et al., 2005; Grothusen et al., 2005). However, the mechanisms by which ACE inhibition or AT1 receptor antagonism reduces atherosclerosis may be complex. For example, inhibition of ACE results in the accumulation of Ang I, which can thus be converted into Ang (1-7) through an ACE2-dependent pathway. In addition, ACE inhibitors also block the degradation of bradykinin, which has potent vasodilator properties. As to AT1 receptor antagonists, although the direct inhibition of Ang II AT1 receptor signalling could account for its anti-atherosclerotic effect, there may also be a contribution from the continuous presence of angiotensin peptides that interact with other receptors such as AT2 or mas receptors. Therefore, inhibition of different sites within the RAS may reduce atherosclerosis through distinct mechanisms, indicating differences in anti-atherosclerotic effects depending on the mode of RAS inhibition.
Despite the consistent demonstration that inhibition of the RAS profoundly reduces atherosclerosis (Daugherty et al., 2001; Candido et al., 2002; 2004; Wassmann et al., 2004; da Cunha et al., 2005; Imanishi et al., 2008; Lu et al., 2008; Nussberger et al., 2008; Rader and Daugherty, 2008; Weiss and Taylor, 2008), no studies have determined the maximal effect or directly compared the relative anti-atherosclerotic effects of the three different modes for pharmacological inhibition of the RAS. Hypercholesterolaemia is critical for the development of atherosclerosis (Rader and Daugherty, 2008). RAS activation is a critical contributor to hypercholesterolaemia-induced atherosclerosis in mice as demonstrated in our previous studies (Daugherty et al., 2004; Lu et al., 2008). In this study, we used LDL receptor -/- male mice fed a saturated fat-enriched diet to determine the dose-related response of the three modes of pharmacological inhibition of the RAS on the reduction of atherosclerosis.
Methods
Mice and diet
LDL receptor -/- (B6.129S7-Ldlrtm1Her; Stock #002207) male mice that have been backcrossed ten times into the C57BL/6 background were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All mice were maintained in a barrier facility and fed with a normal mouse laboratory diet. To induce hypercholesterolaemia, mice were fed with a diet supplemented with saturated fat (milk fat 21% wt/wt) and cholesterol (0.2% wt/wt; Diet# TD.88137; Harlan Teklad, WI, USA). At the end of the experiment, the mice were anaesthetized by an i.p. injection of ketamine (100 mg·kg−1) and xylazine (10 mg·kg−1). Blood was collected with EDTA (1.8 mg·mL−1) via right ventricular puncture, kept on ice, centrifuged (376 g× 20 min) at 4°C and stored at −80°C. After collection of blood, the right atrium was incised, and saline was infused through a left ventricular puncture to remove blood. Aortas were removed and kept in 10% formalin, while other organs such as livers and kidneys were snap- frozen in liquid nitrogen and stored at −80°C. All animal care and experimental procedures were performed with the approval of the University of Kentucky Institutional Animal Care and Use Committee.
Drug administration
Model 2004 Alzet mini-osmotic pumps (Durect Corporation, Cupertino, CA, USA) were implanted into LDL receptor -/- male mice at the age of 8 weeks, and replaced every 4 weeks to continuously deliver drugs for a total of 12 weeks (Daugherty and Cassis, 1999; Daugherty et al., 2000). Ten groups of mice (n = 15 per group) were studied as follows: vehicle (PBS); aliskiren 2.5, 12.5 or 25 mg·kg−1·day−1; enalapril 0.25, 1.25 or 2.5 mg·kg−1·day−1; and losartan 2.5, 12.5 or 25 mg·kg−1·day−1. Doses of each drug were chosen based on estimates that would encompass a range of partial to complete inhibition of their respective targets (Daugherty et al., 2001; Lu et al., 2008). Aliskiren was provided by Novartis. Enalapril (Cat# E6888) and losartan (Cat# 61188) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All study mice were fed the saturated fat-enriched diet, which was started 1 day after the pump implantation throughout the drug infusions.
BP measurements
Systolic and diastolic BPs were measured using a non-invasive tail-cuff system (Coda 8; Kent Scientific Corporation, Torrington, CT, USA) (Daugherty et al., 2009). The measurements were performed for four sequential days prior to and at every 4 weeks during drug infusions.
Measurement of plasma components
Plasma cholesterol concentrations and lipoprotein–cholesterol distributions were determined as described previously (Daugherty et al., 2000). Plasma renin concentrations were measured by incubation of plasma samples (8 µL) with an excess of rat angiotensinogen in the presence of EDTA (0.02 M) for 30 min at 37°C. Ang I generated in the samples was quantified by radioimmunoassay using a commercially available kit (Cat# 1553; DiaSorin, MN, USA) (Lu et al., 2008).
Quantification of atherosclerosis
Atherosclerosis was quantified on aortic intima of arches, thoracic aortas and abdominal aortas by an en face method as described previously (Daugherty and Whitman, 2003; Daugherty and Rateri, 2005). Lesion size was measured in the aortic arch region that included ascending aorta, arch and part of descending aorta (from the aortic orifice of left subclavian artery to 3 mm below), the thoracic aortic region that was defined as from the end of the aortic arch region to the last intercostal arteries, and the abdominal aortic region from the last intercostal arteries to the aortic bifurcation.
Characterization of atherosclerotic tissues
Serial cross sections in aortic roots were cut on a cryostat as described previously (Daugherty and Whitman, 2003). Oil Red O staining was performed to visualize lipid-laden macrophages and collagen was stained using Gomori Trichrome. Immunostaining of smooth muscle alpha actin was performed using a rabbit polyclonal antibody (Cat# ab5694; Abcam, Cambridge, MA, USA) as described previously (Lu et al., 2007b).
Kidney Ang II measurements
Kidney samples (n = 5 per group) from the study mice were weighed and homogenized in 10 volumes of ice-cold buffer containing HCl (0.1N), ethanol (80%), o-phenanthroline (0.5 mM), pepstatin (0.1 mM) and captopril (10 µM). Homogenates were centrifuged at 20 000×g for 20 min at 4°C. The supernatant was stored at −20°C for 12 h, centrifuged and diluted (1:1) with orthophosphoric acid (0.1%). Samples were stored at 4°C for 6 h, centrifuged, and the supernatant diluted (1:1) with orthophosphoric acid (0.02%). Angiotensin peptides were partially purified using C18 mini-columns equilibrated with methanol (4 mL) and water (8 mL). Samples were applied to columns using gentle pressure, columns were washed twice with water (4 mL), and peptides were eluted with methanol (3 mL). Eluate was vacuum evaporated and reconstituted in the buffer for radioimmunoassay using a rabbit anti-AngII antibody (Cat# T-4005; Bachem/Peninsula Laboratories, San Carlos, CA, USA).
Statistical analyses
Version 9.2 of SAS (SAS Institute Inc., Cary, NC, USA) and version 11 of SigmaPlot (Systat Software Inc., San Jose, CA, USA) were used for statistical analyses. A P < 0.05 was considered significant except as noted below. To compare study groups on continuous responses assessed once on each specimen, we employed one-way anova followed by pairwise comparisons with P- values adjusted by the Tukey–Kramer method; observations were weighted, and when necessary, square-root transformed to justify application of one-way anova. To determine the association between two continuous responses assessed once, we computed a Pearson correlation. We also estimated a standardized coefficient for a linear regression model relating the two variables while controlling for group membership. To compare groups on continuous responses assessed repeatedly, we fit a linear mixed model expressing the mean response as a function of group membership and time; because the software did not adjust the P-values in pairwise comparisons, we required a P-value < 0.01 to declare statistical significance. Data are presented as mean ± SEM.
Results
Characteristics of study mice
All doses of the three drugs were well-tolerated as determined by daily visual inspection and steadily body weight gain (Table 1). Plasma cholesterol concentrations (Table 1) and lipoprotein–cholesterol distributions (data not shown) were not influenced by any dose or mode of the RAS inhibition compared with the vehicle. While all doses of enalapril and losartan increased plasma renin concentrations, only the highest dose of aliskiren increased plasma renin concentrations (Figure 1). However, there was no significant difference in plasma renin concentrations among the three doses of aliskiren (Figure 1A). Both enalapril (Figure 1B) and losartan (Figure 1C) dose-dependently increased plasma renin concentrations. The magnitude of change in plasma renin concentrations was equivalent in mice infused with the highest doses of enalapril and losartan.
Table 1.
Characteristics of mice
| Body weight (g) | ||||
|---|---|---|---|---|
| Infusion | Dose (mg·kg−1·day−1) | Baseline | Final | Plasma cholesterol concentrations (mg·mL−1) |
| Vehicle | 24.2 ± 0.5 | 34.1 ± 1.1 | 15.8 ± 1.1 | |
| Aliskiren | 2.5 | 24.6 ± 0.5 | 37.0 ± 1.0 | 15.9 ± 0.7 |
| 12.5 | 24.3 ± 0.4 | 33.9 ± 0.5 | 16.9 ± 0.8 | |
| 25 | 24.1 ± 0.5 | 33.4 ± 0.9 | 15.4 ± 0.9 | |
| Enalapril | 0.25 | 23.7 ± 0.3 | 36.1 ± 0.9 | 17.2 ± 0.8 |
| 1.25 | 23.6 ± 0.4 | 36.3 ± 0.9 | 16.1 ± 0.5 | |
| 2.5 | 24.2 ± 0.5 | 30.8 ± 1.5 | 15.8 ± 0.6 | |
| Losartan | 2.5 | 24.3 ± 0.5 | 38.4 ± 1.0 | 16.4 ± 0.7 |
| 12.5 | 24.7 ± 0.5 | 36.2 ± 1.1 | 15.8 ± 0.8 | |
| 25 | 24.7 ± 0.4 | 34.3 ± 1.0 | 15.9 ± 1.3 | |
Values are presented as mean ± SEM. Comparisons of body weight and plasma cholesterol concentrations among the 10 study groups (n = 8–15 per group) were performed by one-way anova.
Figure 1.
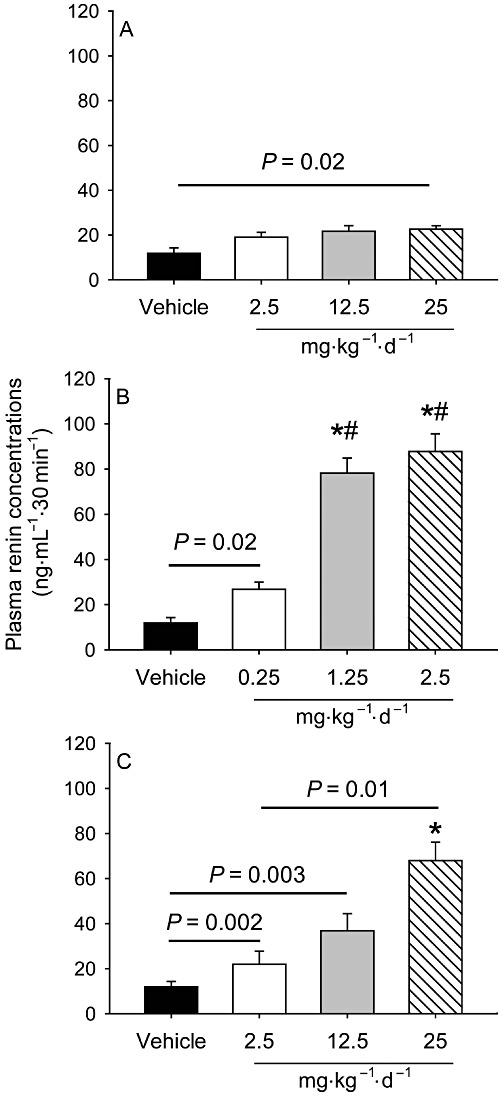
Comparison of different modes of RAS inhibition on changes in plasma renin concentrations. Plasma renin concentrations were measured using a radioimmunoassay kit (n = 7 per group). Histograms represent means and bars represent SEM. *P < 0.0001 versus the vehicle, and #P < 0.0001 versus enalapril 0.25 mg·kg−1·day−1. (A) Effects of aliskiren; (B) enalapril and (C) losartan.
Comparison of three modes of pharmacological RAS inhibition on systolic and diastolic BP reduction
Changes in systolic and diastolic BP (at week 12 during drug infusion vs. baseline) were compared. Each dose of all three drugs produced dose-related decreases in both systolic and diastolic BPs (Figures 2 and 3). The highest doses of each drug reduced both systolic and diastolic BPs to a similar level.
Figure 2.
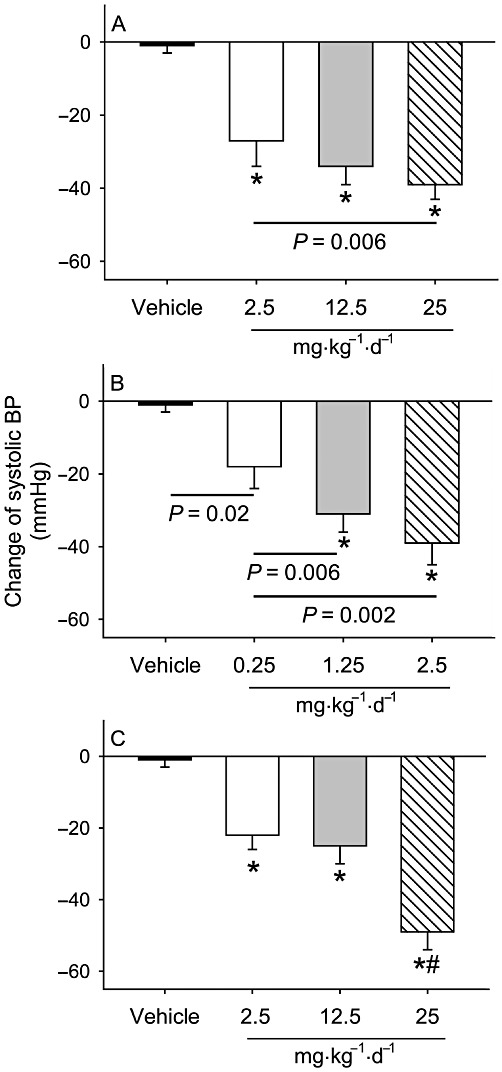
Comparison of different modes of RAS inhibition on changes in systolic BP. Changes in systolic BP were compared in LDL receptor -/- mice between 1 week before pump implantation (baseline) and at 12 weeks after drug administration at the indicated doses (n = 8–14 per group). Histograms represent means and bars represent SEM. *P < 0.0001 versus the vehicle, and #P < 0.0001 versus the two lower doses of losartan (2.5 and 12.5 mg·kg−1·day−1). (A) Effects of aliskiren; (B) enalapril and (C) losartan.
Figure 3.
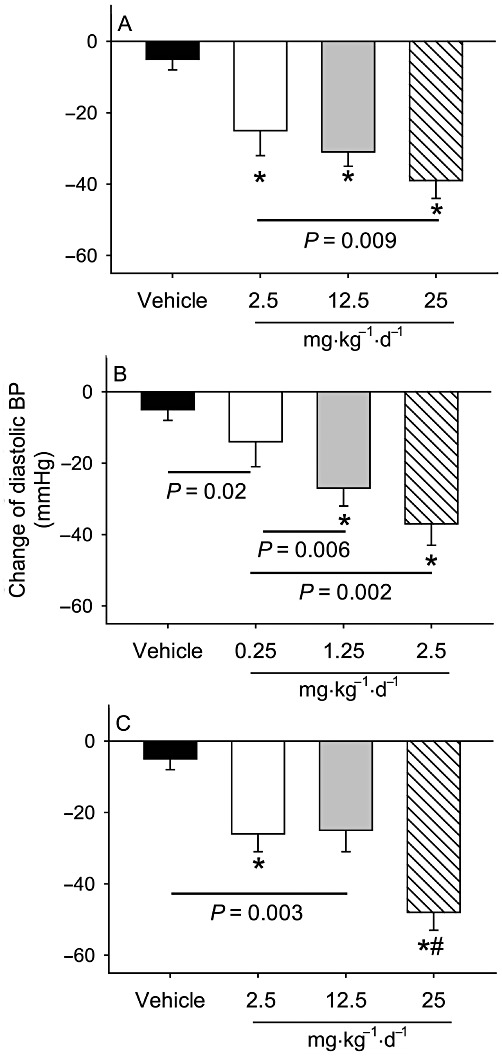
Comparison of different modes of RAS inhibition on changes of diastolic BP. Changes in diastolic BP were compared in LDL receptor -/- mice between 1 week before pump implantation (baseline) and at 12 weeks after drug administration at the indicated doses (n = 8–14 per group). Histograms represent means and bars represent SEM. *P < 0.0001 versus the vehicle, and #P < 0.0001 versus the two lower doses of losartan (2.5 and 12.5 mg·kg−1·day−1). (A) Effects of aliskiren; (B) enalapril and (C) losartan.
Comparison of three modes of pharmacological RAS inhibition on atherosclerosis
Atherosclerotic lesions were quantified as % lesion area in aortic arches, thoracic aortas and abdominal aortas by en face measurement. LDL receptor -/- mice fed the saturated fat-enriched diet for 12 weeks developed readily discernable lesions in aortic arches, modestly sized lesions in thoracic aortas, and minimal lesions in abdominal aortas (data not shown). All three modes of pharmacological RAS inhibition profoundly reduced hypercholesterolaemia-induced atherosclerosis in a dose-dependent manner in both aortic arches (Figure 4 and Supporting Information Figure S1) and thoracic aortas (Supporting Information Figure S2). There were no differences in the maximal reductions in lesion size between the groups.
Figure 4.
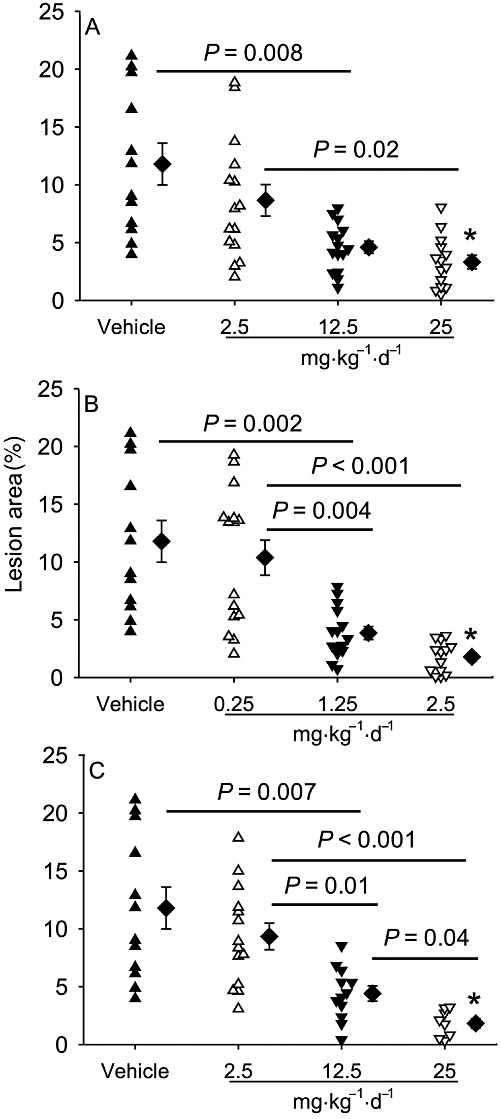
Comparison of different modes of RAS inhibition on atherosclerotic lesion size in aortic arches. The lesion area (as a % of whole area) was measured on the intimal surface of aortic arches (n = 8–14 per group). Triangles represent the values for individual mice, diamonds represent means, and bars are SEM. *P < 0.001 versus the vehicle. (A) Effects of aliskiren; (B) enalapril and (C) losartan.
Oil Red O staining demonstrated the predominance of lipid-laden macrophages in atherosclerotic lesions. The presence of collagen was not readily distinguishable, whereas only sparse positive staining of smooth muscle actin was detected in atherosclerotic lesions (data not shown).
Correlation analysis between BP changes and lesion size
We performed Pearson correlation analyses to determine whether % lesion area in aortic arches was correlated with systolic or diastolic BP changes. The Pearson correlation of % lesion area with systolic BP was −0.421 (P < 0.0001), and with diastolic BP was −0.360 (P = 0.0002). However, if group membership was controlled by fitting a linear regression model with BP changes as the dependent variable, and both % lesion area and group membership as the independent variables, the estimated standardized regression coefficient for % lesion area with either systolic or diastolic BP was −0.093 (P = 0.43 and 0.45, respectively).
Comparisons of renal Ang II concentrations in study mice
We were unable to directly measure Ang II concentrations in aortas. Instead, we measured Ang II concentrations in kidney tissues from mice infused with either PBS or the highest dose of each drug. There was a significant but equivalent reduction in kidney Ang II concentrations in mice infused with the highest dose of each drug, compared with the PBS-infused control mice (Supporting Information Figure S3).
Discussion and conclusions
The present study demonstrated that inhibition of the RAS by drugs that operate through three distinct pharmacological modes reduces atherosclerotic lesion size in a dose-dependent manner in hypercholesterolaemic mice. The dose–response curves showed no marked differences in the relative anti-atherosclerotic effects for these three different modes of RAS inhibition. The RAS inhibition was achieved by s.c. infusion of the drugs through osmotic minipumps to ensure continuous and constant delivery during a 12 week period. To our knowledge, this is the first study to directly compare the three currently available pharmacological modes of inhibition of the RAS on atherosclerosis using multiple doses of each drug within a single study.
The effects of renin inhibition on atherosclerosis have been determined recently in a number of studies (Imanishi et al., 2008; Lu et al., 2008; Nussberger et al., 2008; Weiss and Taylor, 2008; Poss et al., 2010). Conversely, many studies have used experimental models to demonstrate the consistent anti-atherosclerotic effects of ACE inhibitors and AT1 receptor antagonists (Aberg and Ferrer, 1990; Charpiot et al., 1993; Schuh et al., 1993; Strawn et al., 2000; Johnstone et al., 2004; Wassmann et al., 2004; Weiss and Taylor, 2008). Although all three modes of inhibiting the RAS reduced lesion size, it is unclear whether there are differences in efficacy. Renin is the rate-limiting enzyme that acts on its unique substrate of angiotensinogen in the formation of angiotensin peptides. Unlike renin, ACE has both RAS-related and non-RAS-related substrates (Nishimoto et al., 2001). In addition, ACE inhibition influences other substrates of ACE such as bradykinin, which also affects experimental atherosclerosis (Merino et al., 2009). AT1 receptor antagonism prevents the actions of Ang II by inhibiting its binding to the AT1 receptor. However, it simultaneously increases plasma concentrations of Ang II (Muller et al., 1997; Strawn et al., 2000) and possibly other angiotensin peptides. Increased plasma Ang II concentrations during AT1 receptor antagonism may stimulate AT2 receptors; however, the role of AT2 receptors in atherosclerosis is unclear (Daugherty et al., 2001; 2004; Iwai et al., 2005; Johansson et al., 2005; Sales et al., 2005; Hu et al., 2008; Koitka et al., 2010).
Given the different modes of inhibiting the RAS, it is possible that ACE and AT1 receptor inhibition reduces atherosclerosis through different pathways from renin inhibition. There have been reports that ACE inhibition and AT1 receptor antagonism have differential effects on atherosclerosis (Schuh et al., 1993), while several other studies have demonstrated that single doses of drugs targeting different sites within the RAS produce comparable reductions in atherosclerotic lesion size (Ortlepp et al., 2002; Imanishi et al., 2008; Nussberger et al., 2008; Weiss and Taylor, 2008). To directly compare the anti-atherosclerotic effects and thus assist in clarifying the conflicting findings in the literature, the present study, via applying multiple doses of each drug concurrently, provided strong evidence that the three modes of pharmacological RAS inhibition similarly reduce atherosclerosis in mice.
While the reduction in intrarenal Ang II concentration, as measured in the present study, may represent the effectiveness of the three modes of RAS inhibition, it does not imitate the changes in Ang II production in aortas that may directly contribute to the development of atherosclerosis. Unfortunately, measurement of Ang II levels in mouse aortas is not technically feasible (Weiss and Taylor, 2008). We have demonstrated previously that angiotensinogen, renin and ACE are present in atherosclerotic lesions of LDL receptor -/- mice (Daugherty et al., 2004). Angiotensinogen and renin are predominantly co-localized with lipid-laden macrophages, whereas ACE is present in all the major cell types in lesions. Although all these components of the RAS are present in atherosclerotic lesions, the contribution of the local expression of angiotensinogen or ACE to lesion formation has not been defined. Conversely, deletion of renin from bone marrow-derived cells decreases atherosclerosis (Lu et al., 2008). Ang II exerts its effects on atherosclerosis by binding to AT1A receptors (Daugherty et al., 2004; Wassmann et al., 2004). However, we did not find that AT1A receptors on bone marrow-derived cells played a critical role in the development of hypercholesterolaemia-induced atherosclerosis (Lu et al., 2008), although contradictory findings have been reported in different mouse models (Cassis et al., 2007; Fukuda and Sata, 2008; Kato et al., 2008; Koga et al., 2008; Tsubakimoto et al., 2009; Endtmann et al., 2011).
It has been demonstrated consistently, in both human and animal studies, that plasma renin concentrations are increased by the three pharmacological approaches we used to inhibit the RAS (Wiggins and Kelly, 2009). While some human studies have shown that plasma renin concentrations were increased more profoundly in patients treated with aliskiren, compared with ACE inhibitors or AT1 receptor antagonists (Nussberger et al., 2002; 2007; Uresin et al., 2007; Wiggins and Kelly, 2009), others did not find differences in plasma renin concentrations between patients administered aliskiren and ACE inhibitors or AT1 receptor antagonists (Azizi et al., 2004; Menard et al., 2006). In this study, different magnitudes of plasma renin increases may reflect factors inherent within the mode of the assay and the method of blood collection in anaesthetized mice. For example, it has been reported that monoclonal antibodies used in some commercial renin assays may directly interact with renin inhibitors, and factors such as incubation duration may also affect the values from such renin assays (Menard et al., 2006). In the present study, plasma renin concentrations were determined by measuring generation of Ang I from the addition of an excess of rodent angiotensinogen (Cassis et al., 2004). The lower concentrations of plasma renin in mice infused with aliskiren might have resulted from residual aliskiren in the assay that inhibited the production of Ang I. In addition, it is well recognized that anaesthesia can increase plasma renin concentrations (Oates and Stokes, 1974). Therefore, the stated measurements of plasma renin may be overestimated.
While the end point of this study was atherosclerotic lesions, we also delineated the relative role of the different modes of pharmacological RAS inhibition on systolic and diastolic BPs. Consistent with their effects on atherosclerosis, all three modes of inhibiting the RAS also dose-dependently reduced BP. Pearson correlation analyses of lesion size with BP changes indicates that these two parameters represent two distinct manifestations, and both are similarly affected by the three modes of RAS inhibition. Furthermore, while these analyses infer an associative link between BP changes and lesion size, this does not provide a conclusive demonstration that changes in BP per se directly contribute to the mechanisms of atherosclerosis (Lu et al., 2007a). The role of BP has been approached in several studies that have compared the anti-atherosclerotic effects of RAS inhibition to other classes of drugs that lower BP. These include comparisons between irbesartan and hydralazine and between aliskiren and hydralazine, which all produced comparable BP reductions in apoE -/- mice, but only aliskiren and irbesartan reduced atherosclerotic lesions (Wassmann et al., 2004; Poss et al., 2010). Similarly, AT1 receptor antagonism by irbesartan or candesartan and calcium antagonism by amlodipine equivalently reduced BP, but only AT1 receptor blockade markedly reduced atherosclerosis (Candido et al., 2004; Doran et al., 2007). The findings from the literature and this study infer that the link between BP lowering effects of pharmacological RAS inhibition and their anti-atherosclerotic effects may involve a complex mechanism.
This study confirms the important effect of the RAS in the development of experimental atherosclerosis. Using a mouse model in which drug administration can be constantly maintained for 3 months, this study demonstrated that inhibitors of renin, ACE and AT1 receptors markedly reduce atherosclerotic lesion formation in a similar dose-dependent manner.
Acknowledgments
This study was supported by Novartis Institute for Biomedical Research and the National Institutes of Health (HL062846). We acknowledge the skilled technical assistance of Jessica J. Moorleghen, Debra L. Rateri, Victoria English and William R. Caudill.
Glossary
- ACE
angiotensin-converting enzyme
- Ang
angiotensin
- apoE
apolipoprotein E
- AT1
angiotensin II type 1 receptor
- BP
blood pressure
- LDL
low-density lipoprotein
- PCR
polymerase chain reaction
- RAS
renin angiotensin system
Conflict of interest
Aliskiren was provided by Novartis. Gene Liau was an employee of Novartis.
Supporting information
Figure S1 Representative en face images ofaortic arches that were used for quantification of atheroscleroticlesions. Images are provided for examples of atherosclerosis inmice infused with PBS, aliskiren 25mg·kg−1·d−1,enalapril 2.5mg·kg−1·d−1 orlosartan 25mg·kg−1·d−1.
Figure S2 Comparison of RAS inhibition onatherosclerotic lesions in thoracic aorta. Percent lesion area wasmeasured on the intimal surface of thoracic aortas (n =8–14 per group). Triangles represent the values forindividual mice, diamonds represent means, and bars are SEM. *denotes P < 0.001 versus the vehicle.
Figure S3 Comparison of RAS inhibition on renalAngII concentrations. Renal AngII concentrations (n = 5 pergroup) were measured using radioimmunoassay method. Histobarsrepresent means and bars represent SEM. * denotes P < 0.05 versus the vehicle.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aberg G, Ferrer P. Effects of captopril on atherosclerosis in cynomolgus monkeys. J Cardiovasc Pharmacol. 1990;15:S65–S72. [PubMed] [Google Scholar]
- Azizi M, Menard J, Bissery A, Guyenne TT, Bura-Riviere A, Vaidyanathan S, et al. Pharmacologic demonstration of the synergistic effects of a combination of the renin inhibitor aliskiren and the AT1 receptor antagonist valsartan on the angiotensin II-renin feedback interruption. J Am Soc Nephrol. 2004;15:3126–3133. doi: 10.1097/01.ASN.0000146686.35541.29. [DOI] [PubMed] [Google Scholar]
- Bruemmer D, Collins AR, Noh G, Wang W, Territo M, Arias-Magallona S, et al. angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest. 2003;112:1318–1331. doi: 10.1172/JCI18141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candido R, Jandeleit-Dahm KA, Cao ZM, Nesteroff SP, Burns WC, Twigg SM, et al. Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation. 2002;106:246–253. doi: 10.1161/01.cir.0000021122.63813.32. [DOI] [PubMed] [Google Scholar]
- Candido R, Allen TJ, Lassila M, Cao Z, Thallas V, Cooper ME, et al. Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation. 2004;109:1536–1542. doi: 10.1161/01.CIR.0000124061.78478.94. [DOI] [PubMed] [Google Scholar]
- Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev. 2003;24:261–271. doi: 10.1210/er.2003-0001. [DOI] [PubMed] [Google Scholar]
- Cassis LA, Huang J, Gong MC, Daugherty A. Role of metabolism and receptor responsiveness in the attenuated responses to angiotensin II in mice compared to rats. Regul Pept. 2004;117:107–116. doi: 10.1016/j.regpep.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Cassis LA, Rateri DL, Lu H, Daugherty A. Bone marrow transplantation reveals that recipient AT1a receptors are required to initiate angiotensin II-induced atherosclerosis and aneurysms. Arterioscler Thromb Vasc Biol. 2007;27:380–386. doi: 10.1161/01.ATV.0000254680.71485.92. [DOI] [PubMed] [Google Scholar]
- Charpiot P, Rolland PH, Friggi A, Piquet P, Scalbert E, Bodard H, et al. ACE inhibition with prindopril and atherogenesis-induced structural and functional changes in minipig arteries. Arterioscler Thromb. 1993;13:1125–1138. doi: 10.1161/01.atv.13.8.1125. [DOI] [PubMed] [Google Scholar]
- da Cunha V, Tham DM, Martin-McNulty B, Deng G, Ho JJ, Wilson DW, et al. Enalapril attenuates angiotensin II-induced atherosclerosis and vascular inflammation. Atherosclerosis. 2005;178:9–17. doi: 10.1016/j.atherosclerosis.2004.08.023. [DOI] [PubMed] [Google Scholar]
- Daugherty A, Cassis LA. Chronic angiotensin II infusion promotes atherogenesis in low density lipoprotein receptor -/- mice. Ann NY Acad Sci. 1999;892:108–118. doi: 10.1111/j.1749-6632.1999.tb07789.x. [DOI] [PubMed] [Google Scholar]
- Daugherty A, Rateri DL. Development of experimental designs for atherosclerosis studies in mice. Methods. 2005;36:129–138. doi: 10.1016/j.ymeth.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Daugherty A, Whitman SC. Quantification of atherosclerosis in mice. Methods Mol Biol. 2003;209:293–309. doi: 10.1385/1-59259-340-2:293. [DOI] [PubMed] [Google Scholar]
- Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty A, Manning MW, Cassis LA. Antagonism of AT2 receptors augments angiotensin II-induced abdominal aortic aneurysms and atherosclerosis. Br J Pharmacol. 2001;134:865–870. doi: 10.1038/sj.bjp.0704331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty A, Rateri DL, Lu H, Inagami T, Cassis LA. Hypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the AT1A receptor. Circulation. 2004;110:3849–3857. doi: 10.1161/01.CIR.0000150540.54220.C4. [DOI] [PubMed] [Google Scholar]
- Daugherty A, Rateri DL, Lu H, Balakrishnan A. Measuring blood pressure in mice using volume pressure recording, a tail-cuff method. J Vis Exp. 2009;27:1291. doi: 10.3791/1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran DE, Weiss D, Zhang Y, Griendling KK, Taylor WR. Differential effects of AT(1) receptor and Ca(2+) channel blockade on atherosclerosis, inflammatory gene expression, and production of reactive oxygen species. Atherosclerosis. 2007;195:39–47. doi: 10.1016/j.atherosclerosis.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endtmann C, Ebrahimian T, Czech T, Arfa O, Laufs U, Fritz M, et al. Angiotensin II impairs endothelial progenitor cell number and function in vitro and in vivo: implications for vascular regeneration. Hypertension. 2011;58:394–403. doi: 10.1161/HYPERTENSIONAHA.110.169193. [DOI] [PubMed] [Google Scholar]
- Ferrario CM. Angiotensin I, angiotensin II and their biologically active peptides. J Hypertens. 2002;20:805–807. doi: 10.1097/00004872-200205000-00004. [DOI] [PubMed] [Google Scholar]
- Fukuda D, Sata M. Role of bone marrow renin-angiotensin system in the pathogenesis of atherosclerosis. Pharmacol Ther. 2008;118:268–276. doi: 10.1016/j.pharmthera.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Grothusen C, Bley S, Selle T, Luchtefeld M, Grote K, Tietge UJ, et al. Combined effects of HMG-CoA-reductase inhibition and renin-angiotensin system blockade on experimental atherosclerosis. Atherosclerosis. 2005;182:57–69. doi: 10.1016/j.atherosclerosis.2005.01.045. [DOI] [PubMed] [Google Scholar]
- Hu C, Dandapat A, Chen J, Liu Y, Hermonat PL, Carey RM, et al. Over-expression of angiotensin II type 2 receptor (agtr2) reduces atherogenesis and modulates LOX-1, endothelial nitric oxide synthase and heme-oxygenase-1 expression. Atherosclerosis. 2008;199:288–294. doi: 10.1016/j.atherosclerosis.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Imanishi T, Tsujioka H, Ikejima H, Kuroi A, Takarada S, Kitabata H, et al. Renin inhibitor aliskiren improves impaired nitric oxide bioavailability and protects against atherosclerotic changes. Hypertension. 2008;52:563–572. doi: 10.1161/HYPERTENSIONAHA.108.111120. [DOI] [PubMed] [Google Scholar]
- Iwai M, Chen R, Li Z, Shiuchi T, Suzuki J, Ide A, et al. Deletion of angiotensin II type 2 receptor exaggerated atherosclerosis in apolipoprotein E-null mice. Circulation. 2005;112:1636–1643. doi: 10.1161/CIRCULATIONAHA.104.525550. [DOI] [PubMed] [Google Scholar]
- Johansson ME, Wickman A, Fitzgerald SM, Gan LM, Bergstrom G. Angiotensin II, type 2 receptor is not involved in the angiotensin II-mediated pro-atherogenic process in ApoE-/- mice. J Hypertens. 2005;23:1541–1549. doi: 10.1097/01.hjh.0000174078.95745.77. [DOI] [PubMed] [Google Scholar]
- Johnstone MT, Perez AS, Nasser I, Stewart R, Vaidya A, Al Ammary F, et al. Angiotensin receptor blockade with candesartan attenuates atherosclerosis, plaque disruption, and macrophage accumulation within the plaque in a rabbit model. Circulation. 2004;110:2060–2065. doi: 10.1161/01.CIR.0000143627.55926.4C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Ishida J, Nagano K, Honjo K, Sugaya T, Takeda N, et al. Deterioration of atherosclerosis in mice lacking angiotensin II type 1A receptor in bone marrow-derived cells. Lab Invest. 2008;88:731–739. doi: 10.1038/labinvest.2008.42. [DOI] [PubMed] [Google Scholar]
- Koga J, Egashira K, Matoba T, Kubo M, Ihara Y, Iwai M, et al. Essential role of angiotensin II type 1a receptors in the host vascular wall, but not the bone marrow, in the pathogenesis of angiotensin II-induced atherosclerosis. Hypertens Res. 2008;31:1791–1800. doi: 10.1291/hypres.31.1791. [DOI] [PubMed] [Google Scholar]
- Koitka A, Cao Z, Koh P, Watson AM, Sourris KC, Loufrani L, et al. Angiotensin II subtype 2 receptor blockade and deficiency attenuate the development of atherosclerosis in an apolipoprotein E-deficient mouse model of diabetes. Diabetologia. 2010;53:584–592. doi: 10.1007/s00125-009-1619-x. [DOI] [PubMed] [Google Scholar]
- Lu H, Cassis LA, Daugherty A. Atherosclerosis and arterial blood pressure in mice. Curr Drug Targets. 2007a;8:1181–1189. doi: 10.2174/138945007782403829. [DOI] [PubMed] [Google Scholar]
- Lu H, Rateri DL, Daugherty A. Immunostaining of mouse atherosclerosis lesions. Methods Mol Med. 2007b;139:77–94. doi: 10.1007/978-1-59745-571-8_4. [DOI] [PubMed] [Google Scholar]
- Lu H, Rateri DL, Feldman DL, Charnigo RJ, Jr, Fukamizu A, Ishida J, et al. Renin inhibition reduces hypercholesterolemia-induced atherosclerosis in mice. J Clin Invest. 2008;118:984–993. doi: 10.1172/JCI32970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard J, Guyene TT, Peyrard S, Azizi M. Conformational changes in prorenin during renin inhibition in vitro and in vivo. J Hypertens. 2006;24:529–534. doi: 10.1097/01.hjh.0000209989.59230.2e. [DOI] [PubMed] [Google Scholar]
- Merino VF, Todiras M, Mori MA, Sales VM, Fonseca RG, Saul V, et al. Predisposition to atherosclerosis and aortic aneurysms in mice deficient in kinin B1 receptor and apolipoprotein E. J Mol Med. 2009;87:953–963. doi: 10.1007/s00109-009-0501-0. [DOI] [PubMed] [Google Scholar]
- Muller P, Flesch G, de Gasparo M, Gasparini M, Howald H. Pharmacokinetics and pharmacodynamic effects of the angiotensin II antagonist valsartan at steady state in healthy, normotensive subjects. Eur J Clin Pharmacol. 1997;52:441–449. doi: 10.1007/s002280050317. [DOI] [PubMed] [Google Scholar]
- Nishimoto M, Takai S, Kim S, Jin DN, Yuda A, Sakaguchi M, et al. Significance of chymase-dependent angiotensin II-forming pathway in the development of vascular proliferation. Circulation. 2001;104:1274–1279. doi: 10.1161/hc3601.094304. [DOI] [PubMed] [Google Scholar]
- Nussberger J, Wuerzner G, Jensen C, Brunner HR. Angiotensin II suppression in humans by the orally active renin inhibitor aliskiren (SPP100): comparison with enalapril. Hypertension. 2002;39:E1–E8. doi: 10.1161/hy0102.102293. [DOI] [PubMed] [Google Scholar]
- Nussberger J, Gradman AH, Schmieder RE, Lins RL, Chiang Y, Prescott MF. Plasma renin and the antihypertensive effect of the orally active renin inhibitor aliskiren in clinical hypertension. Int J Clin Pract. 2007;61:1461–1468. doi: 10.1111/j.1742-1241.2007.01473.x. [DOI] [PubMed] [Google Scholar]
- Nussberger J, Aubert JF, Bouzourene K, Pellegrin M, Hayoz D, Mazzolai L. Renin inhibition by aliskiren prevents atherosclerosis progression: comparison with irbesartan, atenolol, and amlodipine. Hypertension. 2008;51:1306–1311. doi: 10.1161/HYPERTENSIONAHA.108.110932. [DOI] [PubMed] [Google Scholar]
- Oates HF, Stokes GS. Renin stimulation caused by blood collection techniques in the rat. Clin Exp Pharmacol Physiol. 1974;1:495–501. doi: 10.1111/j.1440-1681.1974.tb00570.x. [DOI] [PubMed] [Google Scholar]
- Ortlepp JR, Breuer J, Eitner F, Kluge K, Kluge R, Floege J, et al. Inhibition of the renin-angiotensin system ameliorates genetically determined hyperinsulinemia. Eur J Pharmacol. 2002;436:145–150. doi: 10.1016/s0014-2999(01)01587-4. [DOI] [PubMed] [Google Scholar]
- Poss J, Werner C, Lorenz D, Gensch C, Bohm M, Laufs U. The renin inhibitor aliskiren upregulates pro-angiogenic cells and reduces atherogenesis in mice. Basic Res Cardiol. 2010;105:725–735. doi: 10.1007/s00395-010-0120-5. [DOI] [PubMed] [Google Scholar]
- Rader DJ, Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451:904–913. doi: 10.1038/nature06796. [DOI] [PubMed] [Google Scholar]
- Sales VL, Sukhova GK, Lopez-Ilasaca MA, Libby P, Dzau VJ, Pratt RE. Angiotensin type 2 receptor is expressed in murine atherosclerotic lesions and modulates lesion evolution. Circulation. 2005;112:3328–3336. doi: 10.1161/CIRCULATIONAHA.105.541714. [DOI] [PubMed] [Google Scholar]
- Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh JR, Blehm DJ, Frierdich GE, Mcmahon EG, Blaine EH. Differential effects of renin-angiotensin system blockade on atherogenesis in cholesterol-fed rabbits. J Clin Invest. 1993;91:1453–1458. doi: 10.1172/JCI116350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strawn WB, Chappell MC, Dean RH, Kivlighn S, Ferrario CM. Inhibition of early atherogenesis by losartan in monkeys with diet-induced hypercholesterolemia. Circulation. 2000;101:1586–1593. doi: 10.1161/01.cir.101.13.1586. [DOI] [PubMed] [Google Scholar]
- Tsubakimoto Y, Yamada H, Yokoi H, Kishida S, Takata H, Kawahito H, et al. Bone marrow angiotensin AT1 receptor regulates differentiation of monocyte lineage progenitors from hematopoietic stem cells. Arterioscler Thromb Vasc Biol. 2009;29:1529–1536. doi: 10.1161/ATVBAHA.109.187732. [DOI] [PubMed] [Google Scholar]
- Uresin Y, Taylor AA, Kilo C, Tschope D, Santonastaso M, Ibram G, et al. Efficacy and safety of the direct renin inhibitor aliskiren and ramipril alone or in combination in patients with diabetes and hypertension. J Renin Angiotensin Aldosterone Syst. 2007;8:190–198. doi: 10.3317/jraas.2007.028. [DOI] [PubMed] [Google Scholar]
- Wassmann S, Czech T, Van Eickels M, Fleming I, Bohm M, Nickenig G. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice. Circulation. 2004;110:3062–3067. doi: 10.1161/01.CIR.0000137970.47771.AF. [DOI] [PubMed] [Google Scholar]
- Weiss D, Taylor WR. Deoxycorticosterone acetate salt hypertension in apolipoprotein E-/- mice results in accelerated atherosclerosis: the role of angiotensin II. Hypertension. 2008;51:218–224. doi: 10.1161/HYPERTENSIONAHA.107.095885. [DOI] [PubMed] [Google Scholar]
- Weiss D, Kools JJ, Taylor WR. Angiotensin II-induced hypertension accelerates the development of atherosclerosis in apoE-deficient mice. Circulation. 2001;103:448–454. doi: 10.1161/01.cir.103.3.448. [DOI] [PubMed] [Google Scholar]
- Wiggins KJ, Kelly DJ. Aliskiren: a novel renoprotective agent or simply an alternative to ACE inhibitors? Kidney Int. 2009;76:23–31. doi: 10.1038/ki.2009.105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.