

Abstract
FBW7 (F-box and WD repeat domain-containing 7) has been characterized as an onco-suppressor protein in human cancers. Recent studies have also shown that FBW7 exerts its anti-tumor function primarily by promoting the degradation of various oncoproteins, through which FBW7 regulates cellular proliferation, differentiation and causes genetic instability. In this review, we will discuss the role of FBW7 downstream substrates and how dysregulation of Fbw7-mediated proteolysis of these substrates contributes to tumorigenesis. Additionally, we will also summarize the currently available various Fbw7-knockout mouse models that support Fbw7 as a tumor suppressor gene in the development and progression of human malignancies.
Keywords: cancer, tumor suppressor, FBW7, SCF, ubiquitination, oncoproteins
1. Introduction
The abundance of many cellular proteins, which are involved in diverse cellular processes including cell cycle progression, cell proliferation, and cell apoptosis, is governed by the ubiquitin proteasome system (UPS) through ubiquitination-mediated degradation by the 26S proteasome [1]. Due to mutated components of the UPS, elevated degradation of certain tumor suppressor proteins or impaired destruction of oncoproteins appears to lead to tumor development [1]. It is well documented that the UPS is rather complex and consists of the ubiquitin-activating enzyme (E1), the ubiquitin-conjugating enzyme (E2), and ubiquitin-protein ligases (E3). The E1 utilizes ATP to activate ubiquitin for conjugation and transfers it to E2. The E2 enzyme interacts with a specific E3 partner and transfers the ubiquitin to the target protein, guiding the substrate for degradation [1]. It is worth mentioning that the specificity of target protein selection is mainly dependent on the individual E3 enzyme. In this regard, there are hundreds of different E3 enzyme identified in human, which theoretically provides the necessary specificity for the extending list of targets to be degraded by the UPS [1].
Among the E3 enzymes, the SCF (SKP1-CUL1-F-box protein) E3 ligase complex, which consists of Skp1 (S-phase kinase-associated protein 1), Cul1, Rbx1/Roc1, and a variable subunit denoted as the F-box protein, has been well characterized [2]. Importantly, the F-box protein determines target specificity through recognition and binding of target proteins for ubiquitination and degradation. So far, more than 70 putative F-box proteins have been identified in human genome, although the function and their substrates of most F-box proteins remain elusive [3]. One well-studied F-box-containing protein is FBW7 (F-box and WD repeat domain-containing 7) also known as FBXW7. The first member of the FBW7 gene family was originally identified in budding yeast and called Cdc4 [4]. It is known that human FBW7 is located on chromosome 4 and encodes three transcripts (isoforms α, β and γ) derived from the same gene locus by alternative splicing. All three isoforms vary at the N-terminal region but contain conserved interaction domains in the C-terminus (F-box and WD40 repeats). Three FBW7 α, β and γ isoforms localize to the nucleoplasm, cytoplasm and nucleolus, respectively [3]. The F-box motif is a 40 amino acid region within each F-box protein that recruits the SCF complex by directly interacting with Skp1 to form a functional E3 ligase complex [3]. Also present at the C-terminal region of FBW7 is a stretch of eight WD40 repeats that bind phosphorylated substrates (Figure 1). Recently, FBW7 is believed to serve as a tumor suppressor through negative regulation of many oncogenic proteins [3]. Therefore, in the following section, we will discuss what substrate has been identified thus far as the major FBW7 substrates as well as discuss the roles of its upstream regulatory factors, highlighting the novel functions of FBW7 in carcinogenesis.
Figure 1. Schematic illustration of a SCF-type of E3 ubiquitin ligase complex.

The SCF (Skp1-Cullin 1-F-box) complex consists of four components: the invariable component Skp1, Rbx1 and Cullin1, and the interchangeable F-box protein that functions as a receptor for target proteins. FBW7 recognizes the targeted substrates, which are presented closely to the E2 enzyme to ensure consequent conjugation of ubiquitin. The addition of polyubiquitin targets proteins to the 26S-proteasome for degradation.
2. The downstream substrates of FBW7
Recent studies have identified multiple specific substrates of FBW7 including Cyclin E [5], c-Myc [6, 7], c-Jun [8, 9], Notch [10, 11], Mcl-1 (Myeloid cell leukemia-1) [12, 13], SREBP (Sterol regulatory element-binding proteins) [14, 15], mTOR (mammalian target of rapamycin) [16], KLF5 (Kruppel-like factor 5) [17, 18], c-Myb [19–21], Aurora A [22], NF1 (Neurofibromatosis type 1) [23], NRF1 (Nuclear factor E2-related factor 1) [24], and HIF-1α (Hypoxia inducible factor-1α) [25, 26]. More recently, Busino and colleague identified that p100 is a new FBW7 substrate [27]. FBW7α targets nuclear p100 for proteasomal degradation on phosphorylation of p100 by GSK3 (glycogen synthase kinase 3) [27]. Here, we will briefly discuss several key substrates that would help us to understand the critical role of FBW7 in cell cycle progression, apoptosis, tumor metastasis, and drug resistance.
2.1 Myeloid cell leukemia-1
Mcl-1, a pro-survival member of the Bcl-2 family, is frequently elevated in various human tumors, suggesting that Mcl-1 is an attractive and potential therapeutic target for human malignancies [28]. Targeted inactivation of Mcl-1 has been shown to therapeutically important [28]. It has been identified that two E3 ubiquitin ligases, namely MULE (Mcl-1 ubiquitin ligase E3) and β-TRCP (beta-transducin repeat containing protein), polyubiquitinate and target Mcl-1 for degradation [29, 30]. Conversely, deubiquitinase USP9X (Ubiquitin specific peptidase 9 X-linked) stabilizes Mcl-1 through reversing its polyubiquitination [31]. Recently, we and others have identified that FBW7 regulates cellular apoptosis by controlling the ubiquitination and destruction of the Mcl-1 in a GSK3 phosphorylation-dependent manner [12, 13].
In the following paragraph, we will discuss the regulation of Mcl-1 by FBW7 in more detail than other substrates, which serves as an example for initiating cutting-edge research for better understanding of how FBW7, an E3 ligase, could execute its anti-tumor functions mediated via promoting the degradation of its substrates. First, FBW7 substrates typically contain the conserved CPD (Cdc4 phospho-degron) sequence (L)-X-pT/pS-P-(P)-X-pS/pT/E/D (X=any amino acid) [12]. Second, proper phosphorylation of the substrate is required for FBW7 to recognize and target its substrate for ubiquitylation. Indeed, we identified the putative FBW7 phosphodegron sequence in Mcl-1, which is conserved across different species [12]. Moreover, we identified S159 and T163 within the phosphodegron as the major GSK3-mediated phosphorylation sites as well as S121 as a minor GSK3-mediated phosphorylation site. Not surprisingly, inactivation of these GSK3-mediated phosphorylation sites impairs the interaction between Mcl-1 and FBW7 [12]. To further support this result, pharmacological inhibition of GSK3 activity blocked the interaction between FBW7 and Mcl-1, suggesting that GSK3-dependent phosphorylation of Mcl-1 is required for the interaction of Mcl-1 with FBW7 [12]. Another group led by Dr. Dixit and Dr. Wertz also reported similar results [13]. They identified similar Mcl-1 degron motifs and protein kinases that direct recruitment to FBW7 during mitotic arrest [13]. Furthermore, they discovered S64, S121, S159 and T163 sites in Mcl-1 as GSK3-mediated phosphorylation sites [13]. Taken together, results from both groups consistently showed that FBW7-mediated degradation of Mcl-1 was dependent on GSK3-mediated Mcl-1 phosphorylation at multiple Ser/Thr sites within the phosphodegron sequence. More importantly, these two independent groups have shown that FBW7 plays important roles in the regulation of drug sensitivity via governing Mcl-1 degradation.
2.2 Hypoxia inducible factor-1α
HIF-1α is a major transcription factor involved in cellular response to hypoxia (low oxygen) [32]. Hypoxia is a common feature of human malignancies that contributes to progression of cancer such as tumor metastasis [33]. HIF-1α has been found to stimulate the expression of genes associated with angiogenesis including VEGF (vascular endothelial growth factor) and Glut1 [33]. Overexpression of HIF-1α has been identified in various types of human cancers including prostate, breast, and ovarian cancer [34]. Moreover, a number of studies from various laboratories have demonstrated that increased levels of HIF-1α correlate with poor prognosis such as reduced patient survival in many types of human cancers [32, 34]. Therefore, it is critical to understand the molecular mechanisms governing the functional role of HIF1α overexpression and the regulation of its degradation.
HIF-1α protein is tightly regulated by post-translational modification such as phosphorylation, nitrosylation, and sumoylation, leading to its proper expression levels [33, 34]. Dysregulation of HIF-1α could contribute to angiogenesis, which is a process known to promote tumor growth and metastasis [32]. Although earlier studies have clearly demonstrated that the VHL (von Hippel-Lindau) tumor suppressor, which is frequently mutated/deleted in the kidney tumors, is the major E3 ligase governing HIF1α stability [35, 36], it remains largely unknown how HIF1α destruction is deregulated in other types of human tumors. Recently, two separated groups have identified HIF-1α as new FBW7 substrate [25, 26]. Under normoxia, HIF-1α is ubiquitinated and degraded by VHL [37] whereas under hypoxia, HIF-1α is targeted for ubiquitin-dependent degradation through FBW7 interactions and subsequent phosphorylation by GSK3β [25]. Moreover, GSK3β and FBW7-dependent HIF-1α degradation can be antagonized by the USP28 (ubiquitin specific peptidase-28), suggesting that FBW7 and USP28 could reciprocally regulate cell migration and angiogenesis in a HIF-1α–dependent manner [26].
2.3 Kruppel-like factor 5
KLF5 transcription factor has been shown to play important roles in tumorigenesis [38], but its exact function remains debateable. Overexpression of KLF5 has been found in breast cancer, and it serves as a prognostic factor for disease-free survival and overall survival in patients diagnosed with breast cancer [39]. Moreover, KLF5 was reported to promote cell proliferation and tumorigenesis in a variety of human cancers such as breast cancer and bladder cancer [40–42]. Furthermore, inhibition of KLF5 significantly suppressed tumor growth in vivo, suggesting that KLF5 may function as an oncogenic transcription factor, thereby it is a potential therapeutic target for the treatment of human cancers [40, 41]. However, several studies have demonstrated frequent loss of function of KLF5 by deletion and/or because of its loss of expression in breast cancer, AML (acute myeloid leukemia), and prostate cancer, arguing that in certain cellular contexts, KLF5 might be a possible tumor suppressor [43–45]. Therefore further investigation is needed to delineate the exact role of KLF5 in human tumorigenesis.
Recently, Zhao et al. found that FBW7 targets KLF5 for ubiquitin-mediated proteasomal degradation [18]. Initially, this group observed that KLF5 contains two putative evolution-conserved CPD motifs that could recruit FBW7 [18]. Next, the authors showed that the GSK3β kinase is involved in the phosphorylation of KLF5 at the S303 site, which is required for FBW7 to target KLF5 for ubiquitination and degradation [18]. Additionally, overexpression of FBW7 decreases the KLF5 protein level and its half-life, while inactivation of FBW7 increases the KLF5 protein and half-life [18]. More importantly, FBW7 inhibits breast cancer cell proliferation, at least, partly due to promotion of KLF5 proteolysis. Consistent with this finding, Liu et al. also found that FBW7 specifically promoted the degradation and ubiquitination of KLF5 in a GSK3β-phosphorylation dependent manner [17]. Taken together, studies from these two groups support the notion that FBW7 is a key negative regulator controlling KLF5-mediated cell proliferation.
2.4 Nuclear Factor E2-related Factor 1
NRF1 belongs to the CNC (cap-n-collar) subfamily of basic-leucine zipper transcription factors [46]. It has been reported that NRF1 can direct ARE (antioxidant response element)-mediated expression of target genes, which respond to oxidative stress [47]. Specifically, NRF1 regulates the genes encoding enzymes involved in glutathione biosynthesis and other oxidative defense enzymes [47]. NRF1 can induce the expression of MT-1 and MT-2 (metallothionein-1 and -2), protecting cells from heavy metal-induced damage [48]. Aside from response to oxidative stress, NRF1 has also been found to play a pivotal role in controlling various other cellular processes [47]. For example, it has been demonstrated that NRF1 interacts with C/EBP-β (CCAAT enhancer-binding protein beta) to negatively regulate the odontoblast differentiation [49]. Unfortunately, although multiple groups have tried to investigate the biological roles of NRF1, physiological functions of NRF1 remain largely elusive. It has been reported that the loss of NRF1 function in mice leads to anemia and embryonic lethality, suggesting that NRF1 as an essential gene during development [50]. Moreover, liver-specific inactivation of the NRF1 gene in adult mouse leads to steatohepatitis and hepatic neoplasia, indicating that NRF1 possibly functions as a tumor suppressor [51]. However, the molecular mechanism underlying NRF1’s anti-tumor activity require further investigations.
Recently, it was found that FBW7 regulates the turnover of NRF1 transcription factor through proteasome-mediated proteolysis [24]. The group led by Dr. Chan provides further evidence to support this notion. First, NRF1 is an unstable protein since it has shorter half-life. Second, using MG132 treatment and in vivo ubiquitination assay, they showed that NRF1 is degraded by ubiquitin-dependent pathway. Third, NRF1 and FBW7α interact with each other. Fourth, knockdown of FBW7 by shRNA stabilizes NRF1. Fifth, FBW7 regulates NRF1 via a CPD motif at residues 350–354. Sixth, activation of NRF1-dependent transcription is governed by FBW7 expression [24]. Taken together, NRF1 is an ubiquitin substrate of FBW7. Since both FBW7 and NRF1 are tumor suppressor proteins, therefore it is difficult to appreciate the significance of FBW7-mediated NRF1 destruction in tumorigenesis. In this regard, the author argued that increased NFR1 due to FBW7 deficiency might promote cells to undergo cellular transformation, thus providing a growth advantage mediated by up-regulation of stress response genes [24]. It is also possible that up-regulation of NRF1, together with c-Myc, c-Jun, and Cyclin E, which are known FBW7 substrates, promote tumorigenesis [24]; however, further studies are warranted before reaching a solid conclusion.
2.5 Neurofibromatosis type 1
NF1 is a tumor suppressor gene that is frequently mutated in a variety of human cancers, including lung cancer, glioblastoma multiforme, neurofibrosarcomas, and pheochromocytomas [52–54]. In mouse models, the homozygous NF1−/− embryos die between E 11.0 and E 13.5 days with many defects such as retarded eye development and malformation of the heart outflow vessels [55]. NF1+/− mice are also predisposed to the formation of phaeochomocytoma and myeloid leukemia [56]. Moreover, loss of NF1 function has been found in human malignancies. Since NF1 accelerates the conversion of activated GTP-bound Ras to inactive GDP-Ras, thus the loss of NF1 exhibits elevated GTP-bound Ras (the active form of Ras) thereby promoting tumorigenesis [53]. Therefore, NF1 is a well-characterized tumor suppressor gene.
Recently, multiple studies have demonstrated that NF1 stability is governed by the ubiquitin-proteasome pathway [57–59]. Cichowski et al. found that NF1 is degraded through ubiquitin-mediated proteolysis by 26S proteasome in response to serum and growth factors such as EGF (epithelial growth factor), PDGF (platelet derived growth factor) and LPA (lysophosphatidic acid) [57]. Moreover, sequences adjacent to the NF1 GRD (GAP-related domain) are required for NF1 degradation [57], suggesting that the dynamic proteasomal regulation of NF1 controls Ras-mediated signaling pathway. This group recently demonstrated that NF1 is inactivated in sporadic gliomas in part through excessive proteasomal degradation of NF1 [58]. Specifically, NF1 is destabilized by PKC (protein kinase C) and the proteasome, leading to the activation of Ras and tumorigenesis [58]. In addition, Phan et al. showed that ETEA, which contains both UBA and UBX domains, ubiquitinates NF1 [59]. More importantly, a recent study led by Dr. Sun discovered that NF1 is a physiological substrate of the SAG (sensitive to apoptosis gene)-Cul1-FBW7 E3 ubiquitin ligase [23]. NF1 contains an FBW7-binding motif at the C terminus, which is necessary for its ubiquitination. Furthermore, SAG-Cul1-FBW7 binds to NF1 as a complex and promotes NF1 ubiquitination. As a result of FBW7-mediated ubiquitination of NF1, FBW7 and SAG significantly shorten NF1 protein half-life. Moreover, FBW7 and SAG are required for degradation of NF1 upon mitogen stimulation [23]. Therefore, this study provides an FBW7-dependent regulatory mechanism for the NF1-Ras pathway.
2.6 Cyclin E
Cyclin E is a critical regulator of the cell cycle progression that is frequently upregulated in cancer [60]. The function of Cyclin E is to bind and activate the CDK2 (cyclin-dependent kinase 2), leading to phosphorylation of various downstream substrates involved in cell cycle progression [60]. It has been discovered that the amount of Cyclin E protein is tightly controlled and destructed by FBW7 [5]. FBW7 can associate specifically with phosphorylated Cyclin E and catalyze its ubiquitination [5]. Cyclin E has been considered as a key mediator for the ability of FBW7 to inhibit tumorigenisis. Specifically, Lengauer et al. reported that depletion of Cyclin E abrogated genomic instability that caused by loss of FBW7 in HCT116 colon cancer cells [61]. In support this notion, Minella et al. demonstrated that mutant Cyclin E, which cannot be degraded by FBW7, whereas deletion of FBW7 can be achieved by inducing genomic instability [62]. Furthermore, transgenic mice expressing the non-degradable version of Cyclin E mutant display increased susceptibility to Ras-induced lung cancer due to elevated genetic instability [63]. However, in-depth investigation is needed to elucidate the exact molecular mechanism underlying the anti-tumor activity of FBW7 that is mediated through degradation of Cyclin E.
2.7 Notch
Most studies have shown oncogenic functions of Notch in many human carcinomas with up-regulation of Notch expression [64]. It is known that Notch signaling pathway plays pivotal roles in many key cellular processes such as cell proliferation, differentiation and apoptosis, thus deregulation of Notch may contribute to carcinogenesis [65]. Thus far, four Notch receptors (Notch 1–4) and five ligands (Dll-1, 3, 4, Jagged-1, 2) have been identified in mammals. Notch signaling is activated after ligand binding to an adjacent Notch receptor, subsequently leading to a series of proteolytic cleavages by the metalloprotease, tumor necrosis factor-α-converting enzyme and the γ-secretase complex (presenilin, nicastrin, Pen-2, and Aph-1) [64]. Oberg et al. firstly identified that Notch is ubiquitinated and negatively regulated by FBW7 through a genetic screen [66]. In support of this finding, mice lacking the FBW7 exhibited elevated Notch expression and subsequently impaired cardiovascular development [10]. Moreover, FBW7 mutations in leukemic cells were found to result in Notch pathway activation through inhibition of Notch degradation [67]. Recently, it has been demonstrated that SGK1 (serum- and glucocorticoid-inducible protein kinase 1) significantly reduced the Notch1 stability through FBW7 [68]. Moreover, the intracellular domain of Jagged-1 was also found to interact with Notch1 intracellular domain and promote its degradation through FBW7-dependent proteasomal pathway [69]. Interestingly, presenilin, one of the components of the γ-secretase complex was found to regulate the ubiquitin ligase FBW7 to modulate EGFR signaling and cell transformation [70]. These and other findings suggest the importance of FBW7 in the regulation of Notch signaling, and as such calls for further in-depth research.
2.8 Other substrates
In addition to these substrates as we have discussed above, some oncoproteins have also been identified as FBW7 substrates including c-Jun [8, 9], c-Myc [6, 7], mTOR [16], c-Myb [19, 20], and Aurora A [22]. It has been shown previously that c-Jun, induced by many mitogens, has positive effects on cell proliferation. We have discovered that FBW7 regulates the degradation of c-Jun in a GSK3 phosphorylation-dependent manner [9]. Moreover, the point mutation in the v-Jun protein allows v-Jun to escape GSK3-dependent recognition and destruction by the FBW7 [9]. Similar to c-Jun, c-Myc is an oncoprotein that was found routinely activated in approximately 70% of all human cancers. Several studies have coherently shown that FBW7 regulates GSK3 phosphorylation-dependent c-Myc protein degradation [6, 7]. Interestingly, later studies showed that FBW7-mediated degradation of c-Myc can partially be counteracted by the USP28 [71].
Furthermore, the mTOR protein has emerged as an oncoprotein for controlling many cellular processes, such as cell growth and cell division. Interestingly, the mTOR protein was also found to be degraded through the GSK3/FBW7-dependent mechanism [16, 72]. Moreover, tumor cell lines harboring inactivation of FBW7 are particularly sensitive to mTOR inhibitor rapamycin treatment, indicating that loss of FBW7 could be a biomarker of human cancer susceptibility to mTOR inhibitor treatment [72]. Additionally, c-Myb, a transcription factor with elevated expression in many human cancers, was found to be ubiquitinated and degraded by FBW7 in a NIK/GSK3-dependent manner [19, 21]. Thus, it is clear that most of these oncoproteins are identified as FBW7 substrates, suggesting that FBW7 mediated degradation of oncoproteins may serve as anti-tumor mechanism in human cancers.
3. FBW7 knockout mouse models
To better understand the underlying mechanisms of development and tumor formation due to deregulated FBW7, several FBW7 knockout mouse models have been developed using gene knockout strategies in ES (embryonic stem) cells. In the following paragraphs, we will focus our discussion on these mouse models to understand the anti-cancer function of FBW7.
3.1 FBW7−/− mice
Several groups including Dr. Nakayama’s laboratory have generated mice deficient in FBW7 [10, 11]. Accumulated evidence has shown that FBW7−/− embryos die in utero at embryonic day 10.5 [11]. Specifically, it has growth retardation, especially in the head region in all homozygous mutant embryos [11]. Moreover, impaired vascular development was found in the FBW7−/− mice. Formation of the major veins such as the anterior cardinal vein was severely impaired in FBW7−/− embryos [11]. Furthermore, accumulation of Notch-4 and enhanced expression of Hey-1 were found in FBW7−/− mice, suggesting that Notch-4 and its downstream gene Hey-1 could play an important role in the repression of venous cell fate determination as well as determination of vascular network formation [11]. Consistent with this notion, the group led by Dr. Elledge demonstrated similar results where they found that FBW7−/− mice have defective cardiovascular development and elevated Notch proteins [10]. Further studies revealed that FBW7−/− mice die due to a combination of deficiencies in vascular and hematopoietic, and atrial as well as ventricular chamber formation development [10]. Abnormalities in cardiovascular development in FBW7−/− mice are due to the results of dysregulation of Notch-1 and Notch-4 [10]. Interestingly, these two independent groups showed different expression levels of Cyclin E in FBW7−/− mice, suggesting that FBW7-mediated destruction of substrates may vary in different cellular contexts. Obviously, further investigation is required to determine the exact functions of Cyclin E in FBW7 knockout mice.
3.2 Conditional FBW7−/− mice
To better understand the physiological functions of FBW7 in tumorigenesis, specifically because of the limitation of embryonic mortality associated with the FBW7−/− mice, the conditional ablation of FBW7 in various adult tissues with tissue specific expression of Cre could be a practical strategy. So far, conditional inactivation of FBW7 in the T cell lineage, bone marrow, intestine, liver, breast and brain has been reported [73–80]. In the following sections, we will briefly discuss these conditional FBW7−/− mouse models, which coherently support a pivotal role for FBW7 in suppressing tumorigenesis in vivo.
3.2.1 FBW7−/− in T cell lineage
It has been shown that mice with conditional inactivation of FBW7 in the T cell lineage are predisposed to thymic lymphoma [78]. Specifically, mice with ablation of FBW7 in the T cell lineage have massive thymic enlargement. Moreover, thymus in these mice contains an uniform population of immature lymphoid cells with necrosis [78]. Both CD4 and CD8 are expressed on thymic lymphoma cells, suggesting an accumulation of immature T cells in the lymphomas [78]. Furthermore, loss of FBW7 in T cells develops thymic lymphoma in part due to excessive accumulation of c-Myc [78]. More importantly, the double mutant mice that have knockout FBW7 in T cells combination with ablation of p53 develop thymic lymphomas at a markedly increased frequency and with a reduced latency [78], indicating a potential synergistic interaction between loss of FBW7 and p53 tumor suppressors in facilitating tumorigenesis.
3.2.2 FBW7−/− in bone marrow
To investigate the role of FBW7 in hematopoietic cells, the bone marrow (BM)-specific FBW7 knockout mice were generated [75]. These mice have markedly lower levels of hemoglobin and platelets at early age; however, these levels stopped decreasing around 12 weeks [75]. About 30% of FBW7-BM-deficient mice exhibited extremely severe pancytopenia in 12 weeks post-depletion of FBW7. Specifically, the numbers of BM mononuclear cells including all lineages of hematopoietic cells are significantly decreased [75]. Moreover, p53 expression levels were increased in BM cells derived from FBW7-deficient mice at 4 weeks, while p53 protein levels were down-regulated in BM mononuclear cells of FBW7-deficient mice at 12 weeks, suggesting that the fate of FBW7-deficient hematopoietic cells is determined by p53 expression [75]. Furthermore, more than half of FBW7-BM-deficient mice developed T-ALL within 16 weeks. Lymphoid blasts invaded BM and many organs including liver, spleen, thymus, and kidney in the leukemic mice [75]. More importantly, leukemic cells of FBW7-deficient mice showed marked accumulation of Notch-1 and c-Myc proteins, indicating that high expression of Notch-1 and c-Myc in FBW7-deficient BM cells may lead to the development of T-ALL [75]. Additionally, deletion of p53 in FBW7-BM-deficient mice caused T-cell malignancies with a much shorter latency and died within 12 weeks, demonstrating that p53 could suppress leukemogenesis in FBW7-BM deficient mice [75]. In support of the critical role of FBW7 in tumorigenesis, similar results in BM-specific FBW7 knockout mice were obtained by the Alfantis group [80] as well, echoing the frequently identification of FBW7 mutation and deletion in human T-ALL patients.
3.2.3 FBW7−/− in the intestine
Recently, Sancho et al. generated conditional gut-specific knockout mice lacking FBW7 specifically in the intestinal tissue and investigated the function of FBW7 in intestine [79]. Mice with FBW7 deletion in the gut were viable and fertile and did not exhibit any gross phenotypic alteration. Moreover, these mice have a drastic decrease of goblet and paneth cells, as well as an increased enteroendocrine numbers [79], suggesting that FBW7 is a regulator of intestinal cell fate decisions. Furthermore, accumulation of progenitor cells was found in the absence of FBW7 in the gut. More importantly, it showed a significant increase in Notch-1 and c-Jun in the conditional FBW7 deletion mice in the intestines [79]. Although FBW7-intestine-deficient mice showed the accumulation of actively proliferating progenitor cells, these mice did not develop intestinal tumors. However, genetic loss of FBW7 in intestine affects the APC (Adenomatous polyposis coli-mediated tumorigenesis)-mediated intestinal tumorigenesis. Specifically, the complete absence of FBW7 caused an increase in both tumor number and tumor size, thus leading to decreased survival rate in APCmin/+ mice [79]. Moreover, both c-Jun and Notch-1 are significantly upregulated in APCmin/+; FBW7−/− mice. Deletion of c-Jun in APCmin/+; FBW7−/− mice reduced tumor size but not tumor number, indicating that c-Jun might be an important factor to control tumor size, instead of tumor initiation [79]. Taken together, these results suggest that FBW7 functions as a haploinsufficient tumor suppressor of intestinal tumors induced by APC mutation that is frequently observed in human intestinal tumors.
3.2.4 FBW7−/− in the liver
Recently, Onoyama et al. generated mice with liver-specific null mutations of FBW7 [77]. They found that the livers of these mice were enlarged and lighter in color with massive lipid deposition and nonalcoholic steatohepatitis-like lesions in the FBW7-deficient liver [77]. Consistent with this observation, they showed that the accumulation of SREBP, which is one of many target of FBW7, led to triglyceride deposition in the liver, which in turn affects the expression of other adipogenic and lipogenic genes such as ChREBP (carbohydrate response element-binding protein), PPARγ (peroxisome proliferator-activated receptor gamma) as well as their downstream targets including Fas (fatty acid synthase), SCD1 (stearoyl CoA desaturase-1), LDLR (LDL receptor) and HMGCS1 (HMG-CoA synthase) through a negative feedback loop [77]. Moreover, cell proliferation was increased in FBW7-deficient liver. As a result, FBW7 deficient liver eventually led to marked proliferation of the biliary system and development of hamartomas [77]. More importantly, skewed hepatic differentiation in the FBW7-deficient liver is possibly due to Notch-1 accumulation and upregulation of its target genes such as Hes-1 and Hey-1 [77]. Taken together, FBW7 plays a pivotal role in governing lipogenesis, cell proliferation and differentiation in the liver.
3.2.5 FBW7−/− in the brain and breast
To examine the consequences of FBW7 deficiency in the brain, the group led by Dr. Nakayama generated mice with conditional ablation of FBW7 in the brain [74]. Mice with brain-specific deletion of FBW7 died within 24 hours after birth with smaller body and absence of milk in the stomach, indicating that their death could be due to a defect in suckling behavior [74]. Moreover, the mutant mice have substantial changes in their brain structure. For example, the third ventricle was dilated and distorted with horizontal sulcus in their brains [74]. The cellularity of the cortex was also reduced in the brain of FBW7−/− mice. Furthermore, accumulation of Notch-1 and Notch-3 was found in the FBW7-deficient brain [74]. They further demonstrated that FBW7-dependent degradation of Notch is required for normal brain development [74]. Another group also independently showed similar results that mice with conditional inactivation of FBW7 in the nervous system did not survive to weaning age [73]. More importantly, Hoeck et al. used this mouse model to show that FBW7 controls neural stem cell differentiation and progenitor cell apoptosis by antagonizing the Notch and c-Jun signaling [73]. Lastly, in line with a role of FBW7 in tumorigenesis, mammary epithelium-specific FBW7 knockout mice quickly developed breast cancer [76].
3.2.6 FBW7−/− in mouse embryonic fibroblasts
To study the physiological role of FBW7 in a tissue culture model, Dr. Nakayama’s laboratory conditionally ablated FBW7 in MEFs (mouse embryonic fibroblasts) [81]. They reported that FBW7−/− MEFs were flattened and detached spontaneously from the culture dish under conventional culture conditions [81]. Additionally, the proliferation of FBW7−/− MEFs was significantly decreased. Moreover, ablation of FBW7 reduced cell growth via induction of cell cycle arrest at G1 phase and increased frequency of apoptosis [81]. Surprisingly, inhibition of cell growth by loss of FBW7 in MEFs was accompanied by increased abundance of Notch-1. Furthermore, both induction of cell cycle arrest and increased apoptosis in FBW7−/− MEFs require Notch-RBP-J (recombination signal binding protein for immunoglobulin kappa J region) signaling pathway [81]. Interestingly, the cell cycle arrest by ablation of FBW7 in MEFs was dependent on the p53 pathway, whereas increased apoptosis in these MEFs was mediated via a p53-independent pathway [81]. Recently, Dr. Nakayama’s group further demonstrated that ablation of FBW7 led to aberrant activation of Notch-1, which in turn inhibited the expression of p27 and p57 but promoted the expression of p21 and p53 [82]. The p19 expression was dependent on c-Myc, whereas accumulation of p16 was found to be independent on Notch and c-Myc in FBW7−/− MEFs [82]. Therefore, these unexpected results argued that FBW7 may not only play a tumor suppressor role in MEFs, it also suggests that FBW7 could have different effects in different tissues in a context-dependent manner.
3.3 FBW7+/− mice
Although homozygous FBW7−/− embryos die, heterozygous mice appeared normal and were fertile [11]. FBW7+/− mice have no obvious phenotypes and do not develop spontaneous tumors up to 12 months of age [11]. It is possible that FBW7+/− mice will develop the tumors later in life. Therefore, loss of FBW7 could contribute to cancer in mice under the proper conditions, such as combination with overexpression of oncogenes or deletion of tumor suppressor genes. For example, Dr. Mao et al. found that FBW7+/− mice have susceptibility to radiation-induced tumorigenesis, but most tumors have the wild-type allele, suggesting that FBW7 has the properties of a haploinsufficient tumor suppressor gene [83]. In line with this concept, both FBW7−/− and FBW7+/− cells from embryonic day 9.5 embryos showed almost the same degree of expression of increased Aurora-A and Notch-4 [83]. Additionally, lung adenocarcinoma, hepatocarcinoma and haemangiosarcoma were found in the irradiated FBW7+/− mice [83]. To further investigate the function of FBW7 in tumorigenesis, this group detected the radiation-induced tumorigenesis in p53-deficient, FBW7-deficient mice, and double deficient mice. As expected, morn than 30% of irradiated mice developed tumors in FBW7+/− mice, whereas no spontaneous tumors were found in unirradiated FBW7+/− mice [83]. Moreover, it showed accelerated tumorigenesis in irradiated FBW7+/−/p53+/− mice compared with p53+/− mice. Furthermore, the FBW7+/−/p53+/− mice developed a wide range of tumors in epithelial tissues including lung, liver and ovary [83]. Thus, these results suggest that FBW7 is a haploinsufficient tumor suppressor gene.
Recently, Sancho et al. generated conditional knockout FBW7+/− mice lacking one allele of FBW7 in the intestine and investigated the effect of FBW7 absence in APC-mediated tumorigenesis [79]. These FBW7+/− mice have no other phenotypic alteration and no accumulation of progenitor cells. However, FBW7+/− heterozygous mutation increased the number of tumors in APCmin/+ mice [79]. Interestingly, unlike FBW7 homozygous mutation, heterozygosity for FBW7 did not increase tumor size [79]. More importantly, the survival rate was decreased in APCmin/+ mice with an FBW7 heterozygous situation. In addition, Notch-1 was remarkably increased in FBW7+/−/APCmin/+ tumors [79], suggesting that Notch-1 could contribute to colon cancer in these mice.
4. FBW7 in human tumor tissues
In line with the genetic information obtained from the various Fbw7 knockout mouse models, it has been well documented that FBW7 serves as a tumor suppressor due to its negative regulation of oncoproteins that are frequently overexpressed in human cancers [3]. Therefore, loss of FBW7 by means of mutation and/or deletion is often observed in a variety of tumors including breast cancer, colon cancer, T-ALL (T-cell acute lymphoblastic leukemia), prostate cancer, pancreatic cancer, and gastric cancer [3]. To evaluate the suppressive role of FBW7 in human tumorigenesis, Akhoondi et al. preformed a comprehensive genetic screen of primary tumors [84]. This study led to the discovery that FBW7 is inactivated by mutation in various types of human malignances with an overall mutation frequency of approximately 6% [84]. Specifically, the mutation frequencies identified in tumors of cholangiocarcinomas, T-ALL, endometrium, colon, stomach are 35%, 31%, 9%, 9%, and 6%, respectively [84].
In separate studies from multiple other groups have confirmed the frequent FBW7 mutations in human cancers [61, 85, 86]. For example, Spruck et al. found that FBW7 is mutated in at least 16% of human endometrial tumors [85]. Moreover, FBW7 mutations were also identified in both human colorectal cancers and their precursor lesions [61]. Consistent with this notion, Yakobori et al. identified that lower expression of FBW7 contributed to lymph node metastasis, tumor size, and poor prognosis in gastric cancer [86]. Furthermore, this group found that patients diagnosed with gastric cancer with low FBW7 expression and p53 mutation had a distinct poor prognosis, suggesting that synergistic disruption of both p53 and FBW7 contributes to poor prognosis for cancer patients [86]. Additionally, genetic alterations in the FBW7 locus was found in 6% of human prostate cancer while differential expression of FBW7 isoforms was inversely correlated with advanced stage and recurrence in prostate cancer [87]. Study from another group also demonstrated that FBW7 mutations, which lead to high expression of Cyclin E, play a critical role in the development of human pancreatic cancer [88]. Recently, it has also been shown that loss of FBW7 in the gut altered homeostasis of the intestinal epithelium, leading to elevated Notch and c-Jun expression that subsequently induced the development of adenomas [89]. In further support of the tumor suppressor role of FBW7 in carcinogenesis, FBW7 mutations were observed in adult T-cell and B-cell acute lymphocytic leukemias [90]. Taken together, these results suggest that FBW7 functions as a tumor suppressor in the genesis of various types of human cancers. Interestingly, mutations of FBW7 were infrequent in ovarian cancer and breast cancer, suggesting the existence of other ways for inactivation of FBW7 in facilitating human malignances [91, 92]. Indeed, Akhoondi et al. identified that FBW7 promoter hypermethylation also contributes to inactivation of FBW7 in human breast cancer, which is associated with poorly differentiated tumors [91]. However, further in-depth investigation is required to define the exact molecular mechanism underlying the anti-tumor activity of FBW7 as well as the mechanism by which the expression of FBW7 is regulated, and how the loss of FBW7 contributes to the development of human cancers.
5. Conclusions
In conclusion, FBW7, a well-characterized tumor suppressor, is frequently mutated or depleted in a variety of human malignances. FBW7 exerts its tumor suppressor function through regulation of many key substrates including Mcl-1, Notch, c-Jun, c-Myc, Cyclin E, HIF1α, mTOR, and KLF5, most of which possess strong oncogenic roles (Figure 2). Furthermore, the observations from the multiple FBW7 knockout mouse models as shown in Figure 3 suggested that accumulation of FBW7 substrates in response to ablation of FBW7 occurs in a tissue-specific manner, leading to the differences in the phenotypic consequence of FBW7 deletion [76]. More importantly, FBW7 specifically recognizes phosphorylated substrates, which is dependent on the balance of various kinase activities in a cell type- or developmental stage-specific manner [81]. In addition, the findings from FBW7+/−/p53+/− mice and FBW7+/−/APCmin/+ mice indicated that the FBW7 heterozygote may have some effect on tumorigenesis, but there is selective pressure for a second hit [93, 94].
Figure 2. Illustrated signaling pathways for FBW7-mediated degradation of its major downstream targets as well as the identified FBW7 upstream regulators.
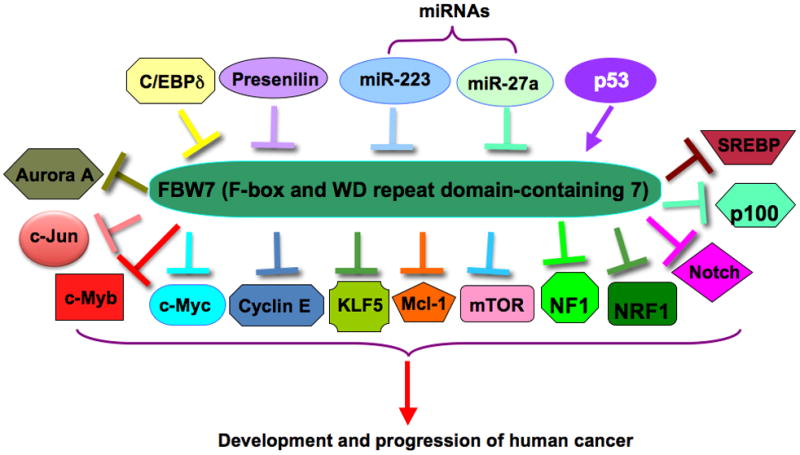
The specific substrates of FBW7 include Mcl-1, c-Jun, c-Myc, Cyclin E, HIF1α, Notch, c-Myb, KLF5, mTOR, and SREBP. Several proteins such as p53 and C/EBP-δ, and microRNAs including miR-27a and miR-223 are found to regulate the expression of FBW7. C/EBP-δ: CCAAT/enhancer-binding protein-δ; HIF1α: Hypoxia inducible factor 1α; KLF5: Krüppel-like factor 5; Mcl-1: Myeloid cell leukemia 1; mTOR: Mammalian target of rapamycin; NF1: Neurofibromatosis type 1; NRF1: Nuclear factor E2-related factor 1; SREBP: Sterol Regulatory Element-Binding Proteins.
Figure 3.
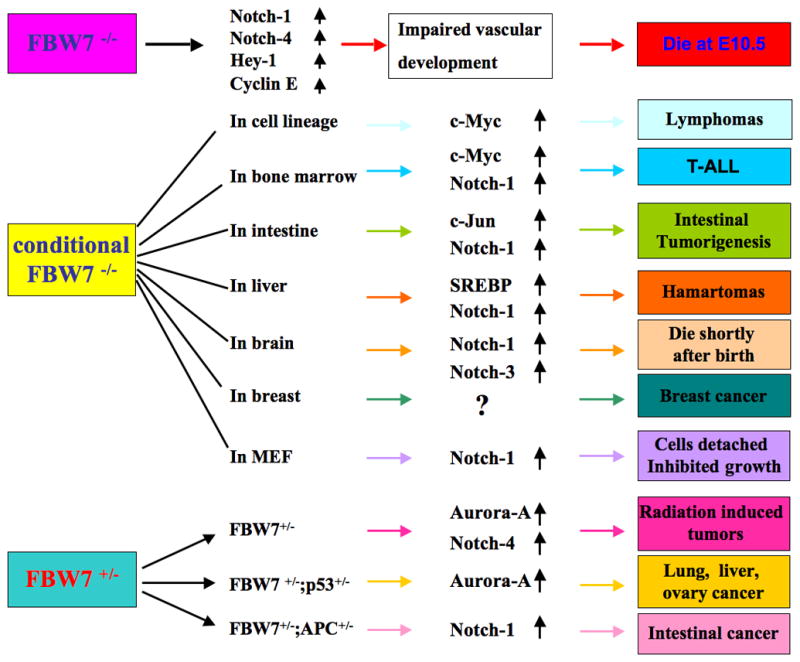
Illustrated presentation of the various FBW7 knockout mouse models including FBW7−/−, conditional FBW7−/− and FBW7+/− mouse models, which coherently support the tumor suppressor role of Fbw7 in vivo.
Since most studies focus on identifying the targets of FBW7 ubiquitin ligase pathway, relatively little is known currently about the regulation of FBW7 expression itself that lead to inactivation of FBW7 in human malignances. Recently, accumulated evidence has shown that several molecules such as p53 and C/EBP-δ (CCAAT/enhancer-binding protein-δ), as well as miRNAs (microRNAs) including miR-27a and miR-223 can regulate FBW7’s tumor suppressor functions [83, 95–99] (Figure 2). In this regard, a better understanding of the upstream regulator for FBW7 could provide important insights for scientists and physicians to design better strategies to restore the FBW7 tumor suppressor function as an efficient means to treat cancer patients. Furthermore, FBW7 was recently found to be involved in drug resistance [12, 13], suggesting that targeting FBW7 may open a newer therapeutic window for drug administration. However, a critical concern is how to design and find specific compounds to restore only FBW7 activity, but not affecting other signaling pathways without causing unwanted side effects. Recently, natural compound oridonin has been reported to activate FBW7 E3 ubiquitin ligase, leading to inhibition of c-Myc pathway [100]. Obviously, the more knowledge we gain in terms of the upstream regulator and downstream targets of FBW7, the better we can achieve more targeted activation of the FBW7 signaling pathway to suppress tumorigenesis. In addition, it will be useful to profile the FBW7 status of individual tumor, such as mRNA and protein levels, in guiding the optimization of personalized medicines to achieve better clinical treatment. We anticipate that directly or indirectly targeting FBW7 could offer novel strategies to benefit better treatment of cancer patients in the near future.
Acknowledgments
We would like to sincerely apologize to all those colleagues whose important work has not been cited here due to space limitations. The authors’ work cited in this review was funded by grants from the National Institute of General Medicines, NIH (GM089763) to W.W., and Massachusetts Life Science Center New Investigator award (W.W.), and Department of Defense Prostate New Investigator award to W.W. W.W is an American Cancer Society Scholar. Z. W is supported by NIH NRSA fellowship.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10:29–46. doi: 10.1038/nrd3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weissman AM, Shabek N, Ciechanover A. The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat Rev Mol Cell Biol. 2011;12:605–620. doi: 10.1038/nrm3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 4.Simchen G, Hirschberg J. Effects of the mitotic cell-cycle mutation cdc4 on yeast meiosis. Genetics. 1977;86:57–72. doi: 10.1093/genetics/86.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- 6.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. Embo J. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303:1374–1378. doi: 10.1126/science.1092880. [DOI] [PubMed] [Google Scholar]
- 9.Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Tetzlaff MT, Yu W, Li M, Zhang P, Finegold M, Mahon K, Harper JW, Schwartz RJ, Elledge SJ. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proc Natl Acad Sci U S A. 2004;101:3338–3345. doi: 10.1073/pnas.0307875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsunematsu R, Nakayama K, Oike Y, Nishiyama M, Ishida N, Hatakeyama S, Bessho Y, Kageyama R, Suda T, Nakayama KI. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J Biol Chem. 2004;279:9417–9423. doi: 10.1074/jbc.M312337200. [DOI] [PubMed] [Google Scholar]
- 12.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 14.Punga T, Bengoechea-Alonso MT, Ericsson J. Phosphorylation and ubiquitination of the transcription factor sterol regulatory element-binding protein-1 in response to DNA binding. J Biol Chem. 2006;281:25278–25286. doi: 10.1074/jbc.M604983200. [DOI] [PubMed] [Google Scholar]
- 15.Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7) Cell Metab. 2005;1:379–391. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Fu L, Kim YA, Wang X, Wu X, Yue P, Lonial S, Khuri FR, Sun SY. Perifosine inhibits mammalian target of rapamycin signaling through facilitating degradation of major components in the mTOR axis and induces autophagy. Cancer Res. 2009;69:8967–8976. doi: 10.1158/0008-5472.CAN-09-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu N, Li H, Li S, Shen M, Xiao N, Chen Y, Wang Y, Wang W, Wang R, Wang Q, et al. The Fbw7/human CDC4 tumor suppressor targets proproliferative factor KLF5 for ubiquitination and degradation through multiple phosphodegron motifs. J Biol Chem. 2010;285:18858–18867. doi: 10.1074/jbc.M109.099440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao D, Zheng HQ, Zhou Z, Chen C. The Fbw7 tumor suppressor targets KLF5 for ubiquitin-mediated degradation and suppresses breast cell proliferation. Cancer Res. 2010;70:4728–4738. doi: 10.1158/0008-5472.CAN-10-0040. [DOI] [PubMed] [Google Scholar]
- 19.Kitagawa K, Hiramatsu Y, Uchida C, Isobe T, Hattori T, Oda T, Shibata K, Nakamura S, Kikuchi A, Kitagawa M. Fbw7 promotes ubiquitin-dependent degradation of c-Myb: involvement of GSK3-mediated phosphorylation of Thr-572 in mouse c-Myb. Oncogene. 2009;28:2393–2405. doi: 10.1038/onc.2009.111. [DOI] [PubMed] [Google Scholar]
- 20.Kitagawa K, Kotake Y, Hiramatsu Y, Liu N, Suzuki S, Nakamura S, Kikuchi A, Kitagawa M. GSK3 regulates the expressions of human and mouse c-Myb via different mechanisms. Cell Div. 2010;5:27. doi: 10.1186/1747-1028-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanei-Ishii C, Nomura T, Takagi T, Watanabe N, Nakayama KI, Ishii S. Fbxw7 acts as an E3 ubiquitin ligase that targets c-Myb for nemo-like kinase (NLK)-induced degradation. J Biol Chem. 2008;283:30540–30548. doi: 10.1074/jbc.M804340200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finkin S, Aylon Y, Anzi S, Oren M, Shaulian E. Fbw7 regulates the activity of endoreduplication mediators and the p53 pathway to prevent drug-induced polyploidy. Oncogene. 2008;27:4411–4421. doi: 10.1038/onc.2008.77. [DOI] [PubMed] [Google Scholar]
- 23.Tan M, Zhao Y, Kim SJ, Liu M, Jia L, Saunders TL, Zhu Y, Sun Y. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev Cell. 2011;21:1062–1076. doi: 10.1016/j.devcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biswas M, Phan D, Watanabe M, Chan JY. The Fbw7 tumor suppressor regulates nuclear factor E2-related factor 1 transcription factor turnover through proteasome-mediated proteolysis. J Biol Chem. 2011;286:39282–39289. doi: 10.1074/jbc.M111.253807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cassavaugh JM, Hale SA, Wellman TL, Howe AK, Wong C, Lounsbury KM. Negative regulation of HIF-1alpha by an FBW7-mediated degradation pathway during hypoxia. J Cell Biochem. 2011;112:3882–3890. doi: 10.1002/jcb.23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flugel D, Gorlach A, Kietzmann T. Glycogen synthase kinase-3beta regulates cell growth, migration and angiogenesis via Fbw7 and USP-28-dependent degradation of hypoxia-inducible factor-1alpha. Blood. 2011 doi: 10.1182/blood-2011-08-375014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Busino L, Millman SE, Scotto L, Kyratsous CA, Basrur V, O’Connor O, Hoffmann A, Elenitoba-Johnson KS, Pagano M. Fbxw7a- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol. 2012 doi: 10.1038/ncb2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584:2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 29.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 30.Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O’Rourke K, Bazan F, Eastham-Anderson J, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–107. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 32.Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145–173. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- 36.Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 37.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 38.Dong JT, Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol Life Sci. 2009;66:2691–2706. doi: 10.1007/s00018-009-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tong D, Czerwenka K, Heinze G, Ryffel M, Schuster E, Witt A, Leodolter S, Zeillinger R. Expression of KLF5 is a prognostic factor for disease-free survival and overall survival in patients with breast cancer. Clin Cancer Res. 2006;12:2442–2448. doi: 10.1158/1078-0432.CCR-05-0964. [DOI] [PubMed] [Google Scholar]
- 40.Zheng HQ, Zhou Z, Huang J, Chaudhury L, Dong JT, Chen C. Kruppel-like factor 5 promotes breast cell proliferation partially through upregulating the transcription of fibroblast growth factor binding protein 1. Oncogene. 2009;28:3702–3713. doi: 10.1038/onc.2009.235. [DOI] [PubMed] [Google Scholar]
- 41.Chen C, Benjamin MS, Sun X, Otto KB, Guo P, Dong XY, Bao Y, Zhou Z, Cheng X, Simons JW, et al. KLF5 promotes cell proliferation and tumorigenesis through gene regulation and the TSU-Pr1 human bladder cancer cell line. Int J Cancer. 2006;118:1346–1355. doi: 10.1002/ijc.21533. [DOI] [PubMed] [Google Scholar]
- 42.Nandan MO, Chanchevalap S, Dalton WB, Yang VW. Kruppel-like factor 5 promotes mitosis by activating the cyclin B1/Cdc2 complex during oncogenic Ras-mediated transformation. FEBS Lett. 2005;579:4757–4762. doi: 10.1016/j.febslet.2005.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen C, Bhalala HV, Qiao H, Dong JT. A possible tumor suppressor role of the KLF5 transcription factor in human breast cancer. Oncogene. 2002;21:6567–6572. doi: 10.1038/sj.onc.1205817. [DOI] [PubMed] [Google Scholar]
- 44.Chen C, Bhalala HV, Vessella RL, Dong JT. KLF5 is frequently deleted and down-regulated but rarely mutated in prostate cancer. Prostate. 2003;55:81–88. doi: 10.1002/pros.10205. [DOI] [PubMed] [Google Scholar]
- 45.Diakiw SM, Kok CH, To LB, Lewis ID, Brown AL, D’Andrea RJ. The granulocyte-associated transcription factor Kruppel-like factor 5 is silenced by hypermethylation in acute myeloid leukemia. Leuk Res. 2012;36:110–116. doi: 10.1016/j.leukres.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 46.Chan JY, Han XL, Kan YW. Cloning of Nrf1, an NF-E2-related transcription factor, by genetic selection in yeast. Proc Natl Acad Sci U S A. 1993;90:11371–11375. doi: 10.1073/pnas.90.23.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biswas M, Chan JY. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol Appl Pharmacol. 2010;244:16–20. doi: 10.1016/j.taap.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, Hayes JD, Yamamoto M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem. 2008;283:33554–33562. doi: 10.1074/jbc.M804597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Narayanan K, Ramachandran A, Peterson MC, Hao J, Kolsto AB, Friedman AD, George A. The CCAAT enhancer-binding protein (C/EBP)beta and Nrf1 interact to regulate dentin sialophosphoprotein (DSPP) gene expression during odontoblast differentiation. J Biol Chem. 2004;279:45423–45432. doi: 10.1074/jbc.M405031200. [DOI] [PubMed] [Google Scholar]
- 50.Chan JY, Kwong M, Lu R, Chang J, Wang B, Yen TS, Kan YW. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 1998;17:1779–1787. doi: 10.1093/emboj/17.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Z, Chen L, Leung L, Yen TS, Lee C, Chan JY. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc Natl Acad Sci U S A. 2005;102:4120–4125. doi: 10.1073/pnas.0500660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parada LF. Neurofibromatosis type 1. Biochim Biophys Acta. 2000;1471:M13–19. doi: 10.1016/s0304-419x(00)00014-7. [DOI] [PubMed] [Google Scholar]
- 54.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 56.Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 57.Cichowski K, Santiago S, Jardim M, Johnson BW, Jacks T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev. 2003;17:449–454. doi: 10.1101/gad.1054703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, Beroukhim R, De Raedt T, Johnson BW, Williams SM, Nghiemphu P, Liau LM, et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell. 2009;16:44–54. doi: 10.1016/j.ccr.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phan VT, Ding VW, Li F, Chalkley RJ, Burlingame A, McCormick F. The RasGAP proteins Ira2 and neurofibromin are negatively regulated by Gpb1 in yeast and ETEA in humans. Mol Cell Biol. 2010;30:2264–2279. doi: 10.1128/MCB.01450-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stamatakos M, Palla V, Karaiskos I, Xiromeritis K, Alexiou I, Pateras I, Kontzoglou K. Cell cyclins: triggering elements of cancer or not? World J Surg Oncol. 2010;8:111. doi: 10.1186/1477-7819-8-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- 62.Minella AC, Swanger J, Bryant E, Welcker M, Hwang H, Clurman BE. p53 and p21 form an inducible barrier that protects cells against cyclin E-cdk2 deregulation. Curr Biol. 2002;12:1817–1827. doi: 10.1016/s0960-9822(02)01225-3. [DOI] [PubMed] [Google Scholar]
- 63.Loeb KR, Kostner H, Firpo E, Norwood T, KDT, Clurman BE, Roberts JM. A mouse model for cyclin E-dependent genetic instability and tumorigenesis. Cancer Cell. 2005;8:35–47. doi: 10.1016/j.ccr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 64.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 65.Wang Z, Li Y, Sarkar FH. Notch signaling proteins: legitimate targets for cancer therapy. Curr Protein Pept Sci. 2010;11:398–408. doi: 10.2174/138920310791824039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J Biol Chem. 2001;276:35847–35853. doi: 10.1074/jbc.M103992200. [DOI] [PubMed] [Google Scholar]
- 67.O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mo JS, Ann EJ, Yoon JH, Jung J, Choi YH, Kim HY, Ahn JS, Kim SM, Kim MY, Hong JA, et al. Serum- and glucocorticoid-inducible kinase 1 (SGK1) controls Notch1 signaling by downregulation of protein stability through Fbw7 ubiquitin ligase. J Cell Sci. 2011;124:100–112. doi: 10.1242/jcs.073924. [DOI] [PubMed] [Google Scholar]
- 69.Kim MY, Jung J, Mo JS, Ann EJ, Ahn JS, Yoon JH, Park HS. The intracellular domain of Jagged-1 interacts with Notch1 intracellular domain and promotes its degradation through Fbw7 E3 ligase. Exp Cell Res. 2011;317:2438–2446. doi: 10.1016/j.yexcr.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 70.Rocher-Ros V, Marco S, Mao JH, Gines S, Metzger D, Chambon P, Balmain A, Saura CA. Presenilin modulates EGFR signaling and cell transformation by regulating the ubiquitin ligase Fbw7. Oncogene. 2010;29:2950–2961. doi: 10.1038/onc.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Popov N, Herold S, Llamazares M, Schulein C, Eilers M. Fbw7 and Usp28 regulate myc protein stability in response to DNA damage. Cell Cycle. 2007;6:2327–2331. doi: 10.4161/cc.6.19.4804. [DOI] [PubMed] [Google Scholar]
- 72.Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321:1499–1502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoeck JD, Jandke A, Blake SM, Nye E, Spencer-Dene B, Brandner S, Behrens A. Fbw7 controls neural stem cell differentiation and progenitor apoptosis via Notch and c-Jun. Nat Neurosci. 2010;13:1365–1372. doi: 10.1038/nn.2644. [DOI] [PubMed] [Google Scholar]
- 74.Matsumoto A, Onoyama I, Sunabori T, Kageyama R, Okano H, Nakayama KI. Fbxw7-dependent degradation of Notch is required for control of “stemness” and neuronal-glial differentiation in neural stem cells. J Biol Chem. 2011;286:13754–13764. doi: 10.1074/jbc.M110.194936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, Mashimo Y, Oguro H, Nitta E, Ito K, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 2008;22:986–991. doi: 10.1101/gad.1621808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Onoyama I, Nakayama KI. Fbxw7 in cell cycle exit and stem cell maintenance: insight from gene-targeted mice. Cell Cycle. 2008;7:3307–3313. doi: 10.4161/cc.7.21.6931. [DOI] [PubMed] [Google Scholar]
- 77.Onoyama I, Suzuki A, Matsumoto A, Tomita K, Katagiri H, Oike Y, Nakayama K, Nakayama KI. Fbxw7 regulates lipid metabolism and cell fate decisions in the mouse liver. J Clin Invest. 2011;121:342–354. doi: 10.1172/JCI40725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Onoyama I, Tsunematsu R, Matsumoto A, Kimura T, de Alboran IM, Nakayama K, Nakayama KI. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J Exp Med. 2007;204:2875–2888. doi: 10.1084/jem.20062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sancho R, Jandke A, Davis H, Diefenbacher ME, Tomlinson I, Behrens A. F-box and WD repeat domain-containing 7 regulates intestinal cell lineage commitment and is a haploinsufficient tumor suppressor. Gastroenterology. 2010;139:929–941. doi: 10.1053/j.gastro.2010.05.078. [DOI] [PubMed] [Google Scholar]
- 80.Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, Zavadil J, Nimer SD, Aifantis I. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med. 2008;205:1395–1408. doi: 10.1084/jem.20080277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ishikawa Y, Onoyama I, Nakayama KI, Nakayama K. Notch-dependent cell cycle arrest and apoptosis in mouse embryonic fibroblasts lacking Fbxw7. Oncogene. 2008;27:6164–6174. doi: 10.1038/onc.2008.216. [DOI] [PubMed] [Google Scholar]
- 82.Masuda K, Ishikawa Y, Onoyama I, Unno M, de Alboran IM, Nakayama KI, Nakayama K. Complex regulation of cell-cycle inhibitors by Fbxw7 in mouse embryonic fibroblasts. Oncogene. 2010;29:1798–1809. doi: 10.1038/onc.2009.469. [DOI] [PubMed] [Google Scholar]
- 83.Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–779. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- 84.Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 85.Spruck CH, Strohmaier H, Sangfelt O, Muller HM, Hubalek M, Muller-Holzner E, Marth C, Widschwendter M, Reed SI. hCDC4 gene mutations in endometrial cancer. Cancer Res. 2002;62:4535–4539. [PubMed] [Google Scholar]
- 86.Yokobori T, Mimori K, Iwatsuki M, Ishii H, Onoyama I, Fukagawa T, Kuwano H, Nakayama KI, Mori M. p53-Altered FBXW7 expression determines poor prognosis in gastric cancer cases. Cancer Res. 2009;69:3788–3794. doi: 10.1158/0008-5472.CAN-08-2846. [DOI] [PubMed] [Google Scholar]
- 87.Koh MS, Ittmann M, Kadmon D, Thompson TC, Leach FS. CDC4 gene expression as potential biomarker for targeted therapy in prostate cancer. Cancer Biol Ther. 2006;5:78–83. doi: 10.4161/cbt.5.1.2290. [DOI] [PubMed] [Google Scholar]
- 88.Calhoun ES, Jones JB, Ashfaq R, Adsay V, Baker SJ, Valentine V, Hempen PM, Hilgers W, Yeo CJ, Hruban RH, et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol. 2003;163:1255–1260. doi: 10.1016/S0002-9440(10)63485-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Babaei-Jadidi R, Li N, Saadeddin A, Spencer-Dene B, Jandke A, Muhammad B, Ibrahim EE, Muraleedharan R, Abuzinadah M, Davis H, et al. FBXW7 influences murine intestinal homeostasis and cancer, targeting Notch, Jun, and DEK for degradation. J Exp Med. 2011;208:295–312. doi: 10.1084/jem.20100830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song JH, Schnittke N, Zaat A, Walsh CS, Miller CW. FBXW7 mutation in adult T-cell and B-cell acute lymphocytic leukemias. Leuk Res. 2008;32:1751–1755. doi: 10.1016/j.leukres.2008.03.040. [DOI] [PubMed] [Google Scholar]
- 91.Akhoondi S, Lindstrom L, Widschwendter M, Corcoran M, Bergh J, Spruck C, Grander D, Sangfelt O. Inactivation of FBXW7/hCDC4-beta expression by promoter hypermethylation is associated with favorable prognosis in primary breast cancer. Breast Cancer Res. 2010;12:R105. doi: 10.1186/bcr2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kwak EL, Moberg KH, Wahrer DC, Quinn JE, Gilmore PM, Graham CA, Hariharan IK, Harkin DP, Haber DA, Bell DW. Infrequent mutations of Archipelago (hAGO, hCDC4, Fbw7) in primary ovarian cancer. Gynecol Oncol. 2005;98:124–128. doi: 10.1016/j.ygyno.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 93.Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011;476:163–169. doi: 10.1038/nature10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Davis H, Tomlinson I. CDC4/FBXW7 and the “just enough” model of tumorigenesis. J Pathol. 2012 doi: 10.1002/path.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kimura T, Gotoh M, Nakamura Y, Arakawa H. hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer Sci. 2003;94:431–436. doi: 10.1111/j.1349-7006.2003.tb01460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Balamurugan K, Wang JM, Tsai HH, Sharan S, Anver M, Leighty R, Sterneck E. The tumour suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis. EMBO J. 2010;29:4106–4117. doi: 10.1038/emboj.2010.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Feng DD, Zhang H, Zhang P, Zheng YS, Zhang XJ, Han BW, Luo XQ, Xu L, Zhou H, Qu LH, et al. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukemia. J Cell Mol Med. 2010 doi: 10.1111/j.1582-4934.2010.01213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lerner M, Lundgren J, Akhoondi S, Jahn A, Ng HF, Moqadam FA, Oude Vrielink JA, Agami R, Den Boer ML, Grander D, et al. miRNA-27a controls FBW7/hCDC4-dependent cyclin E degradation and cell cycle progression. Cell Cycle. 2011:10. doi: 10.4161/cc.10.13.16248. [DOI] [PubMed] [Google Scholar]
- 99.Xu Y, Sengupta T, Kukreja L, Minella AC. MicroRNA-223 regulates cyclin E activity by modulating expression of F-box and WD-40 domain protein 7. J Biol Chem. 2010;285:34439–34446. doi: 10.1074/jbc.M110.152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Qu LH, Huang HL, Weng HY, Wang LQ, Yu CH, Huang QJ, Zhao PP, Wen JZ, Zhou H. Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth inhibition and apoptosis. Mol Cancer Ther. 2012 doi: 10.1158/1535-7163.MCT-12-0066. [DOI] [PubMed] [Google Scholar]