

ABSTRACT
Susceptibility to mouse adenovirus type 1 (MAV-1) is mouse strain dependent; susceptible mice die from hemorrhagic encephalomyelitis. The MAV-1 susceptibility quantitative trait locus Msq1 accounts for ~40% of the phenotypic (brain viral load) variance that occurs between resistant BALB/c and susceptible SJL mice after MAV-1 infection. Using an interval-specific congenic mouse strain (C.SJL-Msq1SJL), in which the SJL-derived allele Msq1SJL is present in a BALB/c background, we demonstrate that Msq1SJL controls the development of high brain viral titers in response to MAV-1 infection, yet does not account for the total extent of brain pathology or mortality in SJL mice. C.SJL-Msq1SJL mice had disruption of the blood-brain barrier and increased brain water content after MAV-1 infection, but these effects occurred later and were not as severe, respectively, as those noted in infected SJL mice. As expected, BALB/c mice showed minimal pathology in these assays. Infection of SJL- and C.SJL-Msq1SJL-derived primary mouse brain endothelial cells resulted in loss of barrier properties, whereas BALB/c-derived cells retained their barrier properties despite being equally capable of supporting MAV-1 infection. Finally, we provide evidence that organ pathology and inflammatory cell recruitment to the brain following MAV-1 infection were both influenced by Msq1. These results validate Msq1 as an important host factor in MAV-1 infection and refine the major role of the locus in development of MAV-1 encephalitis. They further suggest that additional host factors or gene interactions are involved in the mechanism of pathogenesis in MAV-1-infected SJL mice.
IMPORTANCE
A successful viral infection requires both host and viral factors; identification of host components involved in viral replication and pathogenesis is important for development of therapeutic interventions. A genetic locus (Msq1) controlling mouse adenovirus type 1 (MAV-1) brain infection was previously identified. Genes in Msq1 belong to the same family of genes associated with susceptibility to other encephalitic viruses, HIV-1 and West Nile virus. We constructed an interval-specific congenic mouse strain to examine the contribution of Msq1 to MAV-1 susceptibility and brain morbidity. We compared infected resistant, susceptible, and congenic mice regarding known MAV-1 disease manifestations in the brain (survival, viral loads, blood-brain barrier disruption, edema, mouse brain endothelial cell barrier properties, pathology, and inflammatory cell recruitment) to determine the extent to which Msq1 influences MAV-1 infection outcome. Our results showed that Msq1 is a critical host genetic factor that controls many aspects of MAV-1 infection.
Introduction
Viruses from at least 11 different virus families can cause encephalitis (1). These include DNA viruses, RNA viruses, and retroviruses. The mechanisms of pathogenesis by encephalitic viruses are not well understood and likely multifactorial (2-7). However, many encephalitic viral infections share certain common features, including recruitment of inflammatory cells, altered production of cytokines and chemokines, and modulation of expression of tight junction protein and cell adhesion molecules, leading to blood-brain barrier (BBB) disruption (2-7).
The BBB is composed of a specialized layer of microvascular endothelial cells joined by complex tight cell-cell junctions, a basement membrane, and the foot processes of perivascular astrocytes (8-10). It is a highly regulated physical, transport, and biochemical interface that functions to maintain and protect normal brain activity by controlling the passage of ions, macromolecules, and other solutes from the peripheral circulation to the central nervous system (CNS). The BBB also strictly restricts infiltration of immune cells into the CNS; consequently, accumulation of leukocytes in the CNS is usually a sign of pathological inflammatory processes.
Viral infection and inflammation of the CNS can lead to perturbations in the function of the BBB, compromising its ability to exclude harmful substances and immune cells from the brain parenchyma. Changes in BBB permeability can also have significant effects on CNS tissue homeostasis, including changes in intracellular and extracellular water content that may lead to electrolyte imbalance (11). In some instances of CNS viral infection, these disruptions have devastating outcomes, including acute neuroinflammation and neurodegeneration (4, 12-14).
Human adenoviruses can infect the CNS of immunocompromised individuals who suffer from disseminated infections (15-17). However, the study of human adenovirus brain pathogenesis has been limited by the species specificity of adenoviruses and inherent difficulties in collecting samples from ongoing human CNS infections. In contrast, mouse adenovirus type 1 (MAV-1) is a well-characterized non-human-infecting adenovirus that enables in vivo study of a natural encephalitic viral infection in a convenient small-animal model. MAV-1 infection causes fatal hemorrhagic encephalomyelitis with BBB disruption in susceptible mouse strains (5, 18, 19). The MAV-1/mouse model enables comparison of mouse strains to identify host factors that play a role in MAV-1 infection and encephalitis. Knowledge of the role that host factors play in viral encephalitis would inform future design of therapeutic strategies. Antiviral drugs that interfere with host factors essential for viral replication are under development for several viruses. For example, inhibitors that interfere with cyclophilins are being developed to counter hepatitis C virus (HCV) infection. A cyclophilin A inhibitor, currently in phase III studies, has potent antiviral activity with a low incidence of adverse effects (20). Another example of host-targeting drugs in development are CCR5 antagonists, which have been shown to be effective at decreasing HIV’s entry into host cells (21). Thus, identifying host cell pathways that are important for viral replication is important for treatment of viral diseases.
Susceptibility to MAV-1 is mouse strain dependent and is inherited as a dominant trait (18, 19, 22-24); SJL mice are highly susceptible to MAV-1 infection, while BALB/c mice are resistant (23). Using a positional cloning approach to identify host genes that contribute to MAV-1 infection and encephalitis, we identified a 0.75-Mb locus on mouse chromosome 15 that is strongly linked to brain viral loads, mouse adenovirus type 1 susceptibility quantitative trait locus 1 (Msq1) (23-25). This locus contributes ~40% of the variation in brain viral loads of (BALB/c × SJL) F1 × BALB/c backcross mice and is the single most significant determinant of susceptibility distinguishing resistant BALB/c and susceptible SJL mice.
In this study, we tested the genetic contribution of Msq1 to the outcome of MAV-1 infection using an interval-specific congenic strain. We show that Msq1 controls MAV-1 brain infection and contributes significantly to strain susceptibility. These findings corroborate our positional cloning results identifying Msq1 as a major genetic host factor for MAV-1 infection. We also compared BALB/c, SJL, and C.SJL-Msq1SJL mice and primary mouse brain endothelial cells (pmBECs) derived from these strains for a number of parameters following MAV-1 infection, including BBB exclusion of small- and large-molecule dyes from the CNS, edema, transendothelial electrical resistance (TEER), brain pathology, and leukocyte recruitment. Our data demonstrate that Msq1 is an important factor for a subset of physiological components of MAV-1-induced encephalitis. Additionally, differences between infected C.SJL-Msq1SJL and SJL mice suggest that other host factors are involved in MAV-1-induced brain pathology.
RESULTS
Construction of the congenic mouse strain C.SJL-Msq1SJL.
To determine the in vivo contribution of Msq1 to the outcome of MAV-1 infection, we bred a congenic mouse strain (C.SJL-Msq1SJL) that contains the SJL-derived Msq1 susceptibility locus on the BALB/c background. After 11 generations of backcrossing, >99.9% of the C.SJL-Msq1SJL congenic mouse genome consisted of the recipient (BALB/c) genome, while the remaining <0.1% includes Msq1SJL and other scattered loci from the donor (SJL) strain that may remain in the congenic background. Progeny homozygous for Msq1 were used to initiate a homozygous congenic strain. Heterozygotes were also mated to produce littermates of the following genotypes: heterozygous for Msq1 (C.SJL-Msq1BALB/SJL), homozygous for BALB-derived Msq1 (C.SJL-Msq1BALB), and homozygous for SJL-derived Msq1 (C.SJL-Msq1SJL). Because Msq1 is an important contributor to MAV-1 susceptibility and because susceptibility to MAV-1 is dominant, we expected both C.SJL-Msq1SJL and C.SJL-Msq1BALB/SJL mice to be susceptible.
C.SJL-Msq1SJL mice have brain viral loads comparable to those of SJL mice but increased survival rates.
We performed infection experiments with the congenic mice to determine their susceptibility to MAV-1. Previous studies showed that susceptible mouse strains develop high brain viral loads at 8 days postinfection (dpi) at 102 PFU of MAV-1; brain viral loads at this time point were used in our positional mapping studies to identify the Msq1 locus (23, 24). We used the 102-PFU MAV-1 dose and assayed congenic mice and controls for brain virus loads 8 dpi.
MAV-1-infected C.SJL-Msq1SJL mice had significantly higher brain viral titers than their C.SJL-Msq1BALB littermates (Fig. 1A) (P <0.0001). These data indicate that the high viral loads seen in C.SJL-Msq1SJL mice were due to the contribution of Msq1 and not to other minor scattered loci. C.SJL-Msq1BALB mice had viral loads similar to those of resistant parental BALB/c mice (data not shown). The brain viral loads of C.SJL-Msq1SJL mice were comparable to those of SJL mice infected in parallel. SJL brain viral loads were consistent with levels assayed in previous experiments (24) and were also significantly higher (P < 0.0001) than those seen in C.SJL-Msq1BALB mice. Congenic littermate mice heterozygous for Msq1, C.SJL-Msq1BALB/SJL, had brain viral loads comparable to C.SJL-Msq1SJL brain viral loads. This demonstrates that the Msq1 locus acts as a dominant trait and that a single copy of Msq1 is sufficient to confer the brain viral load phenotype on an otherwise resistant mouse background. On the other hand, C.SJL-Msq1BALB/SJL brain viral loads were significantly different from SJL brain viral loads (P = 0.0017). Infrequently, MAV-1-infected SJL, C.SJL-Msq1SJL, and C.SJL-Msq1BALB/SJL mice had low brain viral titers. This is consistent with previous experimental results for MAV-1 infection of SJL and other MAV-1-susceptible mouse strains. Anomalous infections among these genetically uniform animals are likely due to variability in the infection process or environmental factors. Mice that survived MAV-1 infection showed no clinical signs of disease, and virus was not detected in their brains at 21 dpi by capture enzyme-linked immunosorbent assays (ELISAs) (data not shown).
FIG 1 .

Congenic mice are susceptible to MAV-1 infection. (A) Mice of the indicated strains were infected with 102 PFU of MAV-1, and brains were harvested 8 dpi. Viral loads in brain homogenates were measured using capture ELISA. Each symbol represents the mean of three measurements per homogenate for an individual mouse; the number of mice is indicated below the axis. The means and standard deviations (SD) are indicated. Statistical significance was calculated by a two-tailed Mann-Whitney test. OD450, optical density at 450 nm. (B) SJL and C.SJL-Msq1SJL (designated C.SJL-Msq1 here and in subsequent figures) mice were infected with either 102 PFU or 104 PFU of virus. The numbers of mice in each group are indicated. For SJL mice infected at 102 PFU versus C.SJL-Msq1 mice infected at 102 PFU, P = 0.0001; for SJL mice infected at 104 PFU versus C.SJL-Msq1 infected at 104 PFU, P = 0.0022. Statistical significance for survival curves was calculated by the log-rank (Mantel-Cox) test.
We challenged C.SJL-Msq1SJL and SJL mice with either 102 or 104 PFU to compare survival rates over time (Fig. 1B). Survival rates of C.SJL-Msq1SJL and SJL mice were significantly different at doses of both 102 and 104 PFU. At 104 PFU, all SJL mice died by 8 dpi, while more than half of C.SJL-Msq1SJL mice survived. At 102 PFU, C.SJL-Msq1SJL mice were resistant to MAV-1 infection up to 21 dpi, while all SJL mice died between 8 and 10 dpi.
C.SJL-Msq1 mice were then infected with MAV-1 doses ranging from 102 to 107 PFU to determine the 50% lethal dose (LD50). The LD50 of C.SJL-Msq1SJL mice was determined to be 103.5 PFU; C.SJL-Msq1BALB/SJL mice, which have only a single copy of Msq1SJL, had an LD50 of 105.6 PFU. The time of death was dose dependent for both strains of mice. C.SJL-Msq1SJL and C.SJL-Msq1BALB/SJL mice given 107 PFU died by 3 to 4 dpi, while those that succumbed to lower doses died later, by 9 dpi. All of the C.SJL-Msq1SJL mice and 7 of 8 C.SJL-Msq1BALB/SJL mice survived MAV-1 infection at the lowest dose tested (102 PFU). Mice that survived MAV-1 infection showed no clinical signs of disease, and virus was not detected in their brains at 21 dpi by capture ELISA (data not shown).
BALB/c mice were previously assayed to show an LD50 value of >104.4 PFU; we hypothesized that the actual LD50 value is likely higher, but 104.4 PFU was the maximum dose achievable at the time of the study (23). In order to accurately determine the LD50 for BALB/c mice, we infected them with MAV-1 doses that exceed 104.4 PFU (i.e., up to 106.9 PFU). The LD50 for BALB/c mice was calculated to be 106.4 PFU. With this new calculation of the BALB/c LD50, we determined that the LD50 for C.SJL-Msq1SJL mice was ~3 log units lower than that for BALB/c mice. These results confirm that Msq1 is a major genetic determinant of brain viral loads and also of host susceptibility to MAV-1.
Increased BBB permeability in infected SJL and C.SJL-Msq1SJL mice.
MAV-1 causes encephalitis in susceptible mouse strains (18, 19, 22, 23). MAV-1 infection results in dose-dependent encephalomyelitis and breakdown of the BBB in susceptible C57BL/6 mice (5, 18). Similarly, SJL mice infected with 102 PFU of virus at 8 dpi also show changes to the endothelial cell vasculature, including mild perivascular edema and inflammatory cell infiltration (23). In addition, positive endothelial cell staining in both SJL and C3H/HeJ mice is seen using in situ hybridization with a MAV-1 early region 3 riboprobe (23), indicating that MAV-1 replicates in the vascular endothelium of both susceptible and resistant mice. However, high brain viral loads and brain pathology following MAV-1 infection are seen only in susceptible mouse strains. We investigated the role of Msq1 in MAV-1-induced BBB pathology, since the locus contributes significantly to the brain viral load phenotype.
We infected BALB/c, C.SJL-Msq1BALB, SJL, and C.SJL-Msq1SJL mice to examine whether there was evidence of increased BBB permeability. BALB/c mice were included in this study to act as a negative control; these mice are resistant to MAV-1-induced encephalitis and BBB disruption (18). Sodium fluorescein was used as a small tracer molecule to probe the integrity of the BBB in SJL and C.SJL-Msq1SJL mice after MAV-1 infection (5). A functional BBB would restrict entry of small molecules into the brain parenchyma, while a compromised BBB would result in sodium fluorescein dye leakage into the CNS.
Increased BBB permeability was seen in MAV-1-infected SJL and C.SJL-Msq1SJL mice but not infected BALB/c or C.SJL-Msq1BALB mice at 6 dpi (Fig. 2). The brains of infected SJL mice had a 3.9 (± 2.5)-fold increase over mock-infected mice in the amount of brain sodium fluorescein, while infected C.SJL-Msq1SJL mouse brains had a 2.7 (± 1.6)-fold increase over mock-infected mice. At that time point, the difference between the results determined for SJL and C.SJL-Msq1SJL mice was not statistically significant (P = 0.17). The increase in BBB permeability was significantly higher in infected SJL and C.SJL-Msq1SJL mice than in infected BALB/c mice and C.SJL-Msq1BALB mice. Neither BALB/c nor C.SJL-Msq1BALB infected littermate mice had increased BBB permeability that could be detected by this assay. These data corroborate prior studies that showed degenerative vascular changes and increased permeability in susceptible mice (5, 18, 23).
FIG 2 .

Increased BBB (blood-brain barrier) permeability seen in SJL and C.SJL-Msq1SJL mice. BALB/c, C.SJL-Msq1BALB, SJL, and C.SJL-Msq1SJL mice were either mock infected or infected with 104 PFU of MAV-1, and brains were harvested 6 dpi. Sodium fluorescein levels in the brains and serum were measured in duplicate; the average amount of brain sodium fluorescein was normalized to average levels in serum for each mouse. After normalization, the amount of sodium fluorescein in brains of infected mice of the appropriate strain was determined and is represented as a ratio to the amount of dye in mock-infected brains. Each data point represents an individual mouse; numbers of mice in each group are indicated below the respective sample data. For SJL versus C.SJL-Msq1BALB mice, P = 0.008; for C.SJL-Msq1SJL versus C.SJL-Msq1BALB mice, P = 0.034. Means and SDs are indicated. Statistical significance was calculated by one-way analysis of variance (ANOVA) (Kruskal-Wallis) with Dunn’s multiple comparison test.
BBB disruption is delayed in C.SJL-Msq1SJL mice compared to SJL mice.
To investigate when BBB disruption begins following MAV-1 infection and whether strain differences in timing and/or severity exist, we measured BBB permeability at 1-day intervals after infection. Brain viral loads of these mice were also measured in parallel. In the time course studies, Evans blue, which is a larger molecule than sodium fluorescein (Mr = 961 and 376, respectively), was used to evaluate the permeability of BBB to macromolecules (26). In addition to its higher molecular weight, Evans blue has a high affinity for albumin in the serum and forms large dye-albumin complexes, which under normal circumstances would be excluded from the CNS (27). Accordingly, the presence of Evans blue dye in the brain indicates substantial BBB disruption and is a better indicator of damage that could have biological relevance.
We did not detect increased viral loads or BBB permeability with Evans blue following MAV-1 infection in BALB/c mice from 3 to 6 dpi (Fig 3; 6 dpi data not shown). These results are consistent with our previous data determined using sodium fluorescein to measure BBB permeability (Fig. 2). Virus was detectable in SJL mouse brains at 3 dpi (Fig. 3A), and SJL mice had progressive BBB disruption beginning at 4 dpi (Fig. 3B); BBB permeability results were significantly different between BALB/c and SJL mice by 4 dpi. In contrast, BBB disruption was seen 1 day later (at 5 dpi) in infected C.SJL-Msq1SJL mice. We do not show data determined for 6 dpi using this assay, because when we attempted to measure BBB permeability in SJL mice at that time, 9 of 10 SJL mice died or had to be euthanized following Evans blue injection. The deaths of these mice were not surprising given our survival data and previous findings (Fig. 1B) (23).
FIG 3 .

Onset of BBB disruption is delayed in C.SJL-Msq1SJL mice. Mice were either mock infected or MAV-1 infected at 104 PFU and assayed for BBB disruption using Evans blue dye at 3, 4, and 5 dpi. (A) Viral loads of individual infected mouse brains were measured by capture ELISA. Each symbol represents the mean of three measurements per homogenate. (B) The amount of Evans blue dye in infected mouse brains is represented as a ratio to the amount of dye found in mock-infected brains for each individual strain. Each symbol represents the average of duplicate measurements for an individual mouse; numbers of mice in each group are indicated. Means and SDs are indicated. Statistical significance was calculated by a two-tailed Mann-Whitney test.
Despite the 1-day difference between the SJL and C.SJL-Msq1SJL mouse BBB results, the levels of BBB disruption reached were similar. The levels of increased BBB permeability seen in SJL mice at 4 dpi (1.2-fold increase) and C.SJL-Msq1SJL mice at 5 dpi (1.7-fold increase) were not statistically significantly different, and neither were the differences between the levels of increased BBB permeability seen in SJL (1.7-fold increase) and C.SJL-Msq1SJL mice at 5 dpi. These results indicate that this pathological change (BBB permeability to large macromolecules) is in part controlled by Msq1 following MAV-1 infection.
Edema seen in MAV-1-infected SJL and C.SJL-Msq1SJL mice.
Significant alterations in vascular permeability can lead to cerebral edema. Edema occurs when there is an increase in brain water content due to accumulation of intracellular or extracellular fluid leading to an increase in brain volume (11, 28). The healthy BBB strictly limits exchange of fluid between brain tissues and blood, providing an optimal homeostatic environment for the brain. However, this regulation can be disrupted due to BBB damage during pathological conditions such as viral CNS infections. Since increased BBB permeability was seen in susceptible mice but not resistant mice, it is possible that the development of cerebral edema can contribute to differences in strain susceptibility. We tested whether the delay in the onset of increased BBB permeability in C.SJL-Msq1SJL mice compared to SJL mice resulted in reduced edema, thus helping to explain the observed susceptibility difference.
The brains of both SJL and C.SJL-Msq1SJL mice infected with MAV-1 had significantly increased percentages of water content compared to mock-infected brains at 8 dpi, whereas infected BALB/c brains had no measurable change in water content (Fig. 4A). However, the increase in water content was significantly greater in SJL mice than C.SJL-Msq1SJL mice. We also measured Na+ and K+ ion contents of these brains. Alteration of the levels and ratios of these major brain cations can be an important indicator of edema. In particular, leakage of high-sodium, low-potassium plasma contents into the brain following damage to the vasculature is commonly associated with vascular edema (11).
FIG 4 .

Edema in MAV-1-infected mouse strains. Mice of the indicated strains were either mock infected or MAV-1 infected (Inf) with 104 PFU of MAV-1 and assayed 6 dpi. (A) Brain weights were determined prior to and after dehydration. Percent water content was calculated as described in Materials and Methods. (B and C) Ion contents of the brains represented in panel A were measured by flame photometry. Ion contents were then normalized to individual mouse brain weights. Each symbol represents a single mouse. Numbers of mice and means and SDs are indicated. Statistical significance was calculated by a two-tailed Mann-Whitney test.
We observed brain sodium levels that were significantly higher in infected than mock-infected SJL mice (Fig. 4B) (P = 0.0004). Conversely, no significant difference in the sodium level was measured for either C.SJL-Msq1SJL or BALB/c mice upon infection. Potassium levels were essentially unchanged for all tested strains (Fig. 4C). These data are consistent with development of vascular edema following MAV-1 infection for SJL mice but not C.SJL-Msq1SJL or BALB/c mice.
MAV-1 infection of C.SJL-Msq1SJL- and SJL-derived pmBECs results in loss of barrier properties.
Endothelial cells and the tight junctions they form are critical for BBB function (8). MAV-1 infects endothelial cells (18, 29), and MAV-1 infection decreases barrier properties of C57BL/6 primary mouse brain endothelial cells (pmBECs) by altering tight junction protein expression (5). To determine whether brain endothelial cells from resistant and susceptible mouse strains respond differently to MAV-1 infection, pmBECs were prepared from BALB/c, SJL, and C.SJL-Msq1SJL mice and cultured on transwells, allowed to form tight junctions, and infected with MAV-1. We used TEER values as a measure of tight junction integrity in the pmBECs; a monolayer of pmBECs with intact tight junctions has high TEER, while a compromised barrier has decreased TEER (30).
pmBECs from all tested strains were able to establish equally high TEER levels (Fig. 5). By 2 dpi, there was a drop in TEER in both infected SJL- or C.SJL-Msq1-derived pmBECs (Fig. 5B and 5C). Infected BALB/c-derived pmBECs did not have a drop in TEER, indicating that tight junctions in the BALB/c wells remained intact up to 3 dpi (Fig. 5A). Mock-infected pmBECs derived from all the different strains maintained their TEER levels up to 3 dpi.
FIG 5 .

SJL- and C.SJL-Msq1-derived pmBECs (primary mouse brain endothelial cells) lose BBB properties after MAV-1 infection. pmBECs from (A) BALB, (B) SJL, or (C) C.SJL-Msq1 mice were isolated, grown to confluence, and infected with MAV-1 at an MOI of 5. TEER (transendothelial electrical resistance) measured on the day of infection was normalized to 100%. After infection, measurements were taken at 24-h intervals in each sample well. Means and SDs at each time point are shown. Data for each graph were combined from the results of 3 independent experiments.
Increased MAV-1-associated pathology is not due to a difference in the ability of virus to replicate in cells from different mouse strains.
To determine whether the difference in abilities of pmBECs to maintain TEER was due to a difference in ability of virus to grow in endothelial cells from different mouse strains, we isolated pmBECs and tested their capabilities to support MAV-1 growth. Differences in viral growth would suggest that there are strain differences at the cellular level in abilities to support MAV-1 infection, such as expression levels of virus receptors or host factors involved in viral replication. MAV-1 replicated to similar levels in C.SJL-Msq1SJL-, SJL-, and BALB/c-derived pmBECs (Fig. 6). This suggests that cell-autonomous differences between strains in the ability to support replication do not account for the differences in susceptibility. These results are consistent with previous data suggesting that strain differences in susceptibility to MAV-1 are systemic (e.g., at the level of the tissue) rather than cellular (23).
FIG 6 .
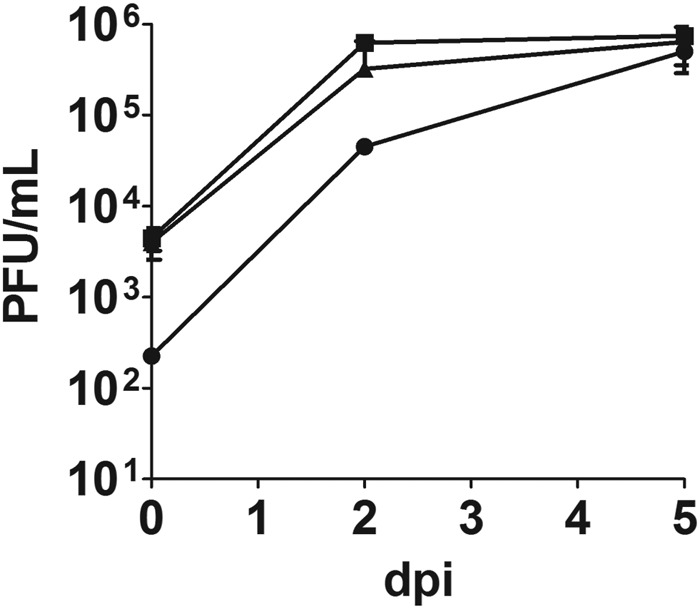
Growth curves of MAV-1 in SJL-, C.SJL-Msq1-, and BALB/c-derived pmBECs. pmBECs from SJL, C.SJL-Msq1, or BALB/c mice were isolated and infected at an MOI of 5. Cells were harvested at 0, 2, and 5 dpi and assayed for viral titers. Data are combined from the results of at least 2 experiments. Virus yields were determined by plaque assays performed in triplicate. Means and SDs are shown.
Brain lesions are more severe in SJL than in C.SJL-Msq1SJL mice.
We examined the pathology of MAV-1 infection in BALB/c, SJL, and C.SJL-Msq1SJL mice to investigate whether strain differences could be seen in the distribution or severity of lesions in different organs. We observed no significant lesions in BALB/c mice for any of the organs analyzed. The majority of lesions found in infected SJL and C.SJL-Msq1SJL mice were in the brain. Infected SJL and C.SJL-Msq1SJL mice had similar brain lesions, consisting of lymphoid meningitis, vasculitis, scattered microhemorrhages, and perivascular edema, which mostly affected the cerebellum. Molecular and granular layer capillaries were surrounded by scattered red blood cells and infrequent small numbers of neutrophils and mononuclear cells.
Although the types of lesions were similar, the extents and distributions of pathology seen in the brains of SJL mice were more severe than those seen in C.SJL-Msq1SJL mice (Fig. 7A). Only 2 of 5 infected SJL mice survived to 6 dpi. Both surviving mice had perivascular hemorrhages and edema. In one mouse, the vascular lesions were centered primarily in the cerebellum and brain stem. In the other, the vascular lesions were present throughout the brain. In contrast, 5 of 5 infected C.SJL-Msq1SJL mice survived, and 3 of 5 had only mild lymphoid meningitis. The fourth mouse had multifocal vasculitis and microhemorrhages in all brain regions, and another had vasculitis with microhemorrhages in the cerebellum and brain stem. When brains were scored, SJL brains showed more severe signs of disease than C.SJL-Msq1SJL brains. Because the numbers of mice per group used in these studies were low, we did not determine statistical significance.
FIG 7 .

Increased pathology in brains of C.SJL-Msq1SJL and SJL mice after MAV-1 infection. (A) Cerebellum sections from BALB/c, SJL, and C.SJL-Msq1 mice infected with 104 PFU of virus at 6 dpi were stained with hematoxylin and eosin. Top panel, cerebellum from mock-infected BALB, SJL, and C.SJL-Msq1 mice. Bars, 100 µm. Middle panel, cerebellum from virus-infected BALB, SJL, and C.SJL-Msq1 mice. Bars, 50 µm. Bottom panel, high magnification of MAV-1-infected BALB, SJL, and C.SJL-Msq1 mice, showing vasculitis in the latter two images. Bars, 20 µm. (B) Histological changes were assigned scores and then totaled. Presence of vasculitis, microhemorrhages, perivascular edema, focal lesions, and meningeal lymphoid infiltrates each received a score of 1. Multifocal lesions received a score of 2. Each symbol represents the score of a single mouse. There were 5 mice per infected group, but 3 SJL mice died prior to analysis. Due to the small sample size, we did not determine whether differences between groups were statistically significant.
Increased inflammatory cell recruitment to brains of infected SJL and C.SJL-Msq1SJL mice.
We used flow cytometry to identify and quantify immune cells whose recruitment to the brain in response to MAV-1 infection is directly or indirectly affected by the presence of Msq1. After MAV-1 infection, there was a large increase in the number of infiltrating leukocytes [CD45(hi)] in SJL and C.SJL-Msq1SJL mice and minimal recruitment in BALB/c mice (Fig. 8). We further characterized the CD45(hi) cell population for cell-type-specific markers. There was an obvious increase in the total numbers of T cells (CD3+) and of both CD4+ (CD3+, CD4+) and CD8+ (CD3+, CD8+) T cells recruited to the brain of SJL mice following MAV-1 infection. There were also increases in the CD3+, CD4+, and CD8+ T cell populations in infected C.SJL-Msq1SJL mice and BALB/c mice but to lesser extents. The same was true for neutrophils (CD11b+, LY6G+) and inflammatory monocytes [CD11b+, LY6C(hi)]. However, for macrophages (F4/80+), the increased levels of recruitment of cells following MAV-1 infection were similar for SJL and C.SJL-Msq1SJL mice. No increase was seen in macrophage recruitment in brains of infected versus mock-infected BALB/c mice. No change in the numbers of brain B cells (major histocompatibility complex [MHC] class II+, CD19+) was seen for any of the mouse strains after MAV-1 infection.
FIG 8 .

Increased inflammatory cell recruitment to brains of C.SJL-Msq1SJL and SJL mice after MAV-1 infection. Mice from the indicated strains were infected with 102 PFU of MAV-1, and mice were euthanized 8 dpi. Cells isolated from brains from either mock-infected or MAV-1-infected mice were isolated and pooled and stained with relevant antibodies. The numbers of experiments for each group are indicated on the graphs. For the CD45(hi) population, the numbers of cells were determined independently in at least 2 separate tubes per experiment.
DISCUSSION
We demonstrate here that Msq1 is a major genetic determinant of susceptibility to MAV-1, confirming the localization of the quantitative trait locus. Not only do C.SJL-Msq1SJL mice develop high brain viral loads after MAV-1 infection, but also the presence of Msq1 alone on a resistant strain background is sufficient to confer a ~3-log-unit decrease in LD50 compared to the parental BALB/c strain. However, Msq1 does not account for the entire difference in susceptibility between the two strains, since the LD50s for SJL and C.SJL-Msq1SJL mice are different. This is consistent with previous results from genetic mapping studies, from which it was calculated that Msq1 contributes to ~40% of the phenotypic variation between BALB/c and SJL mice (24). The data indicate that susceptibility to MAV-1 is a polygenic trait; there are additional quantitative trait loci for susceptibility that were identified in the mapping study. It is likely that a combination of genetic and environmental factors is needed to deliver the full susceptibility phenotype of SJL mice.
We observed that SJL and C.SJL-Msq1SJL mice differ significantly in susceptibility (survival and LD50), despite detection of similar levels of brain viral loads in the two strains. In previous studies, death and low LD50 values correlated well with high viral loads in the brain for a number of mouse strains, making viral loads a good predictor of susceptibility (23-25). Strains with low LD50 values have high brain viral loads after 8 dpi with 102 PFU of MAV-1; strains with high LD50 values have low brain viral loads. Mice that displayed signs of disease (including ruffled fur, lethargy, squinty eyes, hunched posture, and seizures) and those that were discovered dead also had high brain titers of MAV-1. Therefore, in our identification of Msq1, we used brain viral loads as a quantifiable measure of susceptibility (24, 25), because it is not possible to use LD50 assays to determine individual mouse susceptibility. Our data demonstrate that high brain viral titers are not sufficient to cause death. Thus, it is likely that other host factors contribute to mortality. It is also possible that the capture enzyme-linked immunosorbent assay (ELISA) method used is not sensitive enough to distinguish modest differences in viral loads between infected SJL and C.SJL-Msq1SJL mouse brains that are crucial in determining survival. These possibilities are not mutually exclusive.
MAV-1 infection of susceptible C57BL/6 mice results in BBB disruption (5), and we found here that BBB permeability also occurred in infection of two other susceptible mouse strains, SJL and 129SvEv/S6 (data not shown), but not in BALB/c mice. Msq1 contributed critically to the breakdown of the BBB in the susceptible strains. BBB disruption in C.SJL-Msq1SJL mice as assayed by sodium fluorescein measurements was similar to that seen in SJL mice at the peak of infection, either 6 dpi at 104 PFU or 8 dpi at 102 PFU (data not shown). Strain differences regarding the timing of the onset of BBB permeability to macromolecules were detected through time course experiments using Evans blue. We attempted to measure BBB disruption using Evans blue at peak viral infection; however, at 6 dpi, 9 of 10 SJL mice died or had to be euthanized after being injected with Evans blue. For both SJL and C.SJL-Msq1SJL mice, there was a wide range of dye permeability results and viral loads in response to MAV-1 infection. Phenotypic variability is also seen in other mouse models of virus-induced BBB damage (31, 32) and may result from variability in biological response due to environmental causes. Comparable high levels of brain viral loads and BBB breakdown were seen in SJL and C.SJL-Msq1SJL mice, yet there were differences in MAV-1-induced mortality and edema between the strains. The correlation we see between brain viral loads and BBB disruption is consistent with previous published observations (5). However, there is not a clear connection of BBB disruption or viral loads with mortality.
It is possible that mortality following MAV-1 infection is associated with development of cerebral edema as a consequence of BBB disruption, since cerebral edema was observed in histopathological assessments of MAV-1-infected brains (18, 23). For another virus, lymphocytic choriomeningitis virus, data suggest that mortality in mice is caused by edema and ventricular failure rather than BBB damage (31). We found evidence of vasogenic edema in the brains of MAV-1-infected SJL mice but only a modest increase in brain water content of C.SJL-Msq1SJL mice. Our observations also suggest that mice that are sicker have more severe edema; edema appeared to develop close to the time of death for SJL mice (data not shown). These results are significant, because SJL and C.SJL-Msq1SJL had similar brain viral loads and levels of BBB permeability at the time of assay. These data suggest that viral loads and the extent of BBB permeability at 6 dpi in C.SJL-Msq1SJL mice are insufficient to trigger the more severe increase in brain water content seen in SJL mice and that additional host factors outside the Msq1 locus contribute to the development of vasogenic edema in SJL mice during MAV-1 infection. The data support the hypothesis that vasogenic edema correlates with mortality following MAV-1 infection.
MAV-1 infects endothelial cells, which are important structural components of the BBB (22, 23). We observed a loss of endothelial cell barrier integrity upon MAV-1 infection, as demonstrated by a drop in TEER in infected SJL- and C.SJL-Msq1-derived pmBECs. Loss of TEER during MAV-1 infection correlates with reduced tight junction protein mRNA and protein expression in pmBECs (5). Msq1 includes 14 members of the Ly6 gene family (25) which may play a role in MAV-1 BBB pathogenesis at the level of barrier structure. In Drosophila, members of the Ly6/CD49 family, Coiled and Boudin, mediate cell-cell adhesion by controlling structural organization of septate junction proteins in the BBB and the trachea, respectively (33). It is possible that MAV-1 causes a reorganization of tight junction proteins in C57BL/6-, SJL-, and C.SJL-Msq1SJL-derived pmBECs, leading to increased barrier permeability (5). The observed decrease in TEER in SJL- and C.SJL-Msq1-derived pmBECs upon infection is consistent with increases we observed in BBB permeability in vivo but does not correlate with the marked alterations in vasogenic edema that we noted only in SJL mice. Identification of genetic loci that control vasogenic edema in MAV-1 infections and the molecular processes involved requires additional experiments and backcrosses.
Ly6 gene products are cell surface proteins, which makes them potential candidate viral receptors. We observed differences in the expression levels of two of the three LY6 proteins that we examined on pmBECs from BALB/c and SJL mice (data not shown). Such expression variation might lead to differences in the ability to support viral infection. However, results from infecting endothelial cells ex vivo suggest that differences in susceptibility to MAV-1 seen in vivo are not attributable to LY6 protein expression in endothelial cells. Viral yields were similar between pmBECs derived from SJL, C.SJL-Msq1SJL, and BALB/c mice. This is consistent with infection data of other cell types; MAV-1 infection of mouse embryonic fibroblasts and primary bone marrow macrophages (another target of MAV-1 infection in vivo) derived from resistant (C3H/HeJ) and susceptible (SJL) mouse strains also yield equivalent amounts of virus (23). We cannot, however, rule out strain-specific differences in additional cell types we have not tested.
The difference in strain susceptibility to MAV-1 in vivo was not reflected in the ability of pmBECs to support virus growth ex vivo, leading us to investigate strain differences at the systemic level. We did not find differences between SJL and C.SJL-Msq1SJL mice in the distribution of lesions in organs we collected, and in both strains of mice, the majority of lesions were found in the brain. In comparison, in infected BALB/c mice, no significant lesions were found in any of the organs collected. These data suggest that Msq1 is responsible for the development and organ localization of MAV-1 pathology. However, we observed that the pathology found in the infected SJL brains was more severe than that found in C.SJL-Msq1SJL brains, suggesting that other host factors likely contribute to the severity of MAV-1 brain pathology.
Our studies revealed that SJL and C.SJL-Msq1SJL mice had large increases in the numbers of infiltrating cells, whereas BALB/c mice had a comparatively smaller infiltrate of cells. LY6 proteins have known and putative immune functions; therefore, it is likely that Msq1 is involved in immune cell recruitment to the brain. LY6A and LY6C proteins affect lymphocyte development, activation, adhesion, and homing (34-37). In addition, upregulation of LY6 proteins, in particular, LY6C, is linked to increased chemokine secretion (38). Finally, a human LY6 antigen, E48 (highly homologous to mouse LY6D), regulates the release of chemoattractant factors from human umbilical vein endothelial cells that induce monocytes to migrate across an endothelial cell layer (39).
MAV-1 infects cells of the monocytic lineage (29, 40), which could lead to the hypothesis that these cells are involved in viral spread. We observed an increase in macrophage and inflammatory monocyte recruitment for both C.SJL-Msq1SJL and SJL mice, but not BALB/c mice, after MAV-1 infection. However, clodronate-loaded liposome depletion of liver, spleen, lymph node, and peritoneal macrophages does not affect brain viral titers in either SJL or BALB/c mice (40). It is therefore unlikely that infected macrophages are responsible for dissemination of the virus to the brain. The depletion treatment also results in increased spleen viral loads in BALB/c mice, suggesting that macrophages have a role in controlling MAV-1 infection. However, despite the large numbers of recruited macrophages in SJL and C.SJL-Msq1SJL mouse brains, viral loads were high. It is possible that the recruitment of macrophages was delayed in SJL and C.SJL-Msq1SJL mice and thus unable to control the infection. Alternatively, we hypothesize that the large increase in numbers of inflammatory monocytes at the BBB results in increased secretion of chemokines or proteases, such as matrix metalloproteinases (41), and in opening of the BBB, leading to further cell recruitment. A systematic examination of the kinetics of cell recruitment after MAV-1 infection would be informative with respect to strain differences in susceptibility.
Numbers of all analyzed cell types, with the exception of B cells, were increased in the brain following MAV-1 infection of all strains. B cells are crucial for survival of acute MAV-1 infection (42). Infected mice lacking Bruton’s tyrosine kinase, and B cell-deficient mice on both C57BL/6 and BALB/c backgrounds, suffer from disseminated infection, with high brain and spleen viral loads and death. Our data suggest that B cells do not play a role in MAV-1-induced brain pathology in SJL mice and that B cell recruitment is not influenced by Msq1. However, given the crucial role of B cells in controlling MAV-1 infection, it is likely that they influence the progression of MAV-1 infection of SJL mice in other organs or in the brain at different time points.
T cells are responsible for much of the acute pathology attributed to MAV-1 infection, and they are important for survival and control of MAV-1 replication and clearance (43). Mice on a C57BL/6 background are intermediate in susceptibility between BALB and SJL mice (18, 23), and the T-cell immune-deficient mice we have previously examined are on this background. Mice that lack T cells do not develop encephalomyelitis but have detectable brain viral loads following MAV-1 infection (43). Furthermore, MAV-1-infected T cell-deficient mice have little or no immunopathology, suggesting that the acute histopathology is due to T cells. More T cells were recruited to the brains of SJL than C.SJL-Msq1SJL MAV-1-infected mice, suggesting that the large number of recruited T cells in SJL mice causes irreparable damage, leading to higher mortality. The fact that we see higher numbers of recruited T cells after MAV-1 infection in SJL than C.SJL-Msq1SJL mice, but similar high brain viral loads in the two strains, is consistent with previous data indicating that T cells do not control brain viral loads (43).
Although the total numbers of CD45(hi) cells were similar between SJL and C.SJL-Msq1SJL mice, there were significant differences between strains in the numbers of cells in each of the immune cell subpopulations we examined. The numbers of cells for all subpopulations except macrophages and B cells were lower in C.SJL-Msq1SJL mice than in SJL mice. As a result, the combined number of immune cells from the subpopulations that we looked at corresponded well with the total number of infiltrating cells [CD45(hi)] in SJL but not C.SJL-Msq1SJL mice. There appears to be a population(s) of missing cells that do not express any of the markers that we examined, such as NK cells, dendritic cells, and mast cells, which we did not analyze.
In addition to our results here, Ly6 gene family members have also been identified as important host factors influencing viral replication in several other viruses, some of which cause viral encephalitis (44-47). Chicken LY6E is a candidate for susceptibility to Marek’s disease, an avian herpesvirus, and interacts with viral protein US10 in two-hybrid assays (48, 49). Four mouse Ly6 genes are more highly upregulated during infection by highly neuroinvasive West Nile virus strains than by strains that are less neuroinvasive (50). More recently, human LY6E was shown to affect West Nile virus infection in a small interfering RNA screen (51). In addition, human LY6E increases viral replication of another flavivirus, yellow fever virus, in vitro (52). Finally, the human Ly6 locus, Ly6, and Ly6-related genes were also implicated in two separate screens for HIV susceptibility factors (53, 54). These results indicate that Ly6 genes are susceptibility factors for diverse RNA and DNA viruses. Future studies of the function and interaction partners of LY6 proteins may result in the identification and characterization of common pathways leading to vascular damage and/or mortality in encephalitis and may yield new therapeutic targets.
In summary, Msq1 is an important genetic contributor to the pathogenesis of MAV-1 encephalitis but does not account for the full susceptibility phenotype seen in SJL mice. Our data confirmed that Msq1 regulated brain viral loads and that it contributed significantly to strain susceptibility. We demonstrated that the presence of Msq1SJL correlated with increased permeability of the BBB to both small and large tracer molecules and with differences in abilities of infected pmBECs to maintain high TEER. Msq1 also controlled the development of brain lesions caused by MAV-1 infection. The data reveal that other factors (environmental or genetic) likely contributed to the onset of increased BBB permeability and the development of vasogenic edema. We also show that Msq1 mediated recruitment of certain subsets of inflammatory cells to the brain but not others. This difference in inflammatory cell infiltrates may help explain the differences we see in levels of MAV-1 susceptibility and pathogenesis in SJL and C.SJL-Msq1SJL mice. Experiments are in progress to identify the major susceptibility gene(s) within Msq1.
MATERIALS AND METHODS
Generation of congenic strain.
C.SJL-Msq1SJL congenic mice were developed by backcrossing the SJL-derived Msq1 locus onto a BALB/c background for 11 generations (N11). Progeny mice heterozygous for Msq1 were then intercrossed to initiate a homozygous congenic strain. Genotyping was performed by PCR with tail DNA. Simple sequence length polymorphism markers were used to differentiate between SJL- and BALB/c-derived Msq1 genomic intervals. The flanking region loci genotyped were D15Spn101 and D15Spn54 (23). PCR products were electrophoresed in 7% acrylamide gels and visualized by ethidium bromide staining.
Virus and mice.
Wild-type MAV-1 virus was obtained from S. Larsen (55). BALB/c and SJL male mice (3 to 4 weeks old and 4 to 5 weeks old) were obtained from Jackson Laboratory and used for primary cell preparations and infection experiments, respectively. Congenic mice (3 to 4 weeks old and 3 to 8 weeks old) were used for primary cell preparations and infection experiments, respectively. Mice were infected via intraperitoneal (i.p.) injection in volumes of 100 µl, with doses that ranged from 102 to 107 PFU of virus. Virus was diluted in 10-fold serial dilutions in endotoxin-free Dulbecco’s phosphate-buffered saline (DPBS; Lonza). Mock-infected mice were injected with conditioned media similarly diluted in DPBS. Mice were monitored at least twice daily for signs of disease and were euthanized by CO2 asphyxiation if moribund. The animal care and use complied with both federal and university guidelines. Food and water were provided to the mice ad libitum.
Determination of MAV-1 loads by capture ELISA.
Whole brains were aseptically collected from euthanized mice. Brain homogenates were prepared as previously described (24) and assayed for MAV-1 viral load by capture ELISA (56). In each assay, an undiluted MAV-1 virus stock, PBS, and conditioned media were included as controls. Quantification of virus particles by ELISA correlates with infectious virus levels measured by plaque assay (24).
Determination of LD50.
Experiments were carried out with 4- to 6-week-old C.SJL-Msq1SJL mice. Serial dilutions of virus were prepared, and groups of 5 to 21 mice were infected with each dose; three and six doses were used in LD50 determinations for BALB/c and C.SJL-Msq1 mice, respectively. LD50s were determined using the Reed and Muench method (57). Mice were euthanized if moribund or at the conclusion of the experiment.
Measurement of brain water and ion content.
After euthanasia, brains were rapidly removed and weighed to obtain wet weight. Dry weight was determined after drying the brains for 24 h at 100°C. Percent water content was calculated as [(wet weight – dry weight)/wet weight] × 100. Na+ and K+ contents were measured using a flame photometer (Model IL 943; Instrumentation Laboratory, Inc.) after reconstituting the dried brains in 0.1 M nitric acid for 72 to 144 h at room temperature.
BBB permeability assay.
Sodium fluorescein and Evans blue are small-molecule tracers used to assess permeability of the BBB. Mice were injected with either 200 µl of 2% Evans blue dye (Sigma-Aldrich) in DPBS 4 h prior to euthanasia or 100 µl of 10% sodium fluorescein (Sigma) in DPBS 10 min prior to euthanasia. Mice were euthanized by CO2 asphyxiation. Cardiac blood was collected in heparinized syringes into tubes containing 50 µl of heparin (1 mg/ml) and placed on ice. Mice were immediately perfused transcardially with 30 ml of ice-cold PBS. Brains were collected and snap-frozen until use.
Evans blue staining in brains was quantified as previously described (31). Briefly, brains were thawed and photographed and then homogenized in N,N-dimethylformamide (Sigma) at a concentration of 300 mg/ml and incubated at 50°C for 48 h. Samples were centrifuged (3,000 × g, 10 min), and absorbance of the supernatant was measured on a multidetection microplate reader (Biotek Instruments) at 620 nm. The amount of Evans blue in the brain of each infected mouse is represented as a fold change from the average measurement in brains of mock-infected mice of the respective strain.
Sodium fluorescein levels in brain and plasma were determined as previously described (5, 58). Fluorescence levels were measured on a multidetection microplate reader (Bio-Tek) with 485-nm excitation and 530-nm emission. Standards were used to calculate the sodium fluorescein content of brain and plasma samples. Brain values were normalized to their respective plasma dye values to allow comparisons among mice. The amount of sodium fluorescein in each infected mouse brain is represented as a fold change from the average uptake in brains of mock-infected mice of the respective strain.
pmBEC preparation.
pmBECs were isolated similarly to methods previously described (5). For each primary cell preparation, we used 30 mouse brains from BALB/c, SJL, or C.SJL-Msq1SJL mice (3 to 4 weeks old). The cortexes were isolated, and major blood vessels were removed based on visual inspection. Brains were minced and mechanically homogenized in Hanks’ balanced salt solution (HBSS; Gibco Life Technologies) using a Dounce homogenizer. Cells were pelleted (205 × g, 5 min) and resuspended in 18% dextran solution (USB Products). The suspension was centrifuged (11,726 × g, 10 min, 4°C) using a Sorvall SA-600 rotor. Myelin and liquid were aspirated, and erythrocytes and microvessels were resuspended in HBSS. The suspension was layered onto precentrifuged (SW41 rotor, Sorvall ultracentrifuge) (26,793 × g, 1 h) 46.5% Percoll (GE Healthcare) gradients and then centrifuged (1,800 × g, 10 min) in a Jouan benchtop centrifuge. Microvessels formed a distinct red band in the density gradient and were carefully transferred to a 50-ml tube and washed with HBSS. Microvessels were centrifuged (205 × g, 5 min) to again form a pellet. The pellet was resuspended in collagenase/dispase (Roche Diagnostics) (1 mg/ml) and incubated at 37°C for 20 to 40 min. The enzyme solution was then inactivated with the addition of 2 to 3 volumes of HBSS. Microvessels were centrifuged (205 × g, 5 min) to again form a pellet. Supernatant was aspirated, and the remaining cells were resuspended in growth media, which consisted of DMEM (Gibco) (pH 7.2) containing 10% fetal bovine serum, 10% newborn calf serum, endothelial cell growth supplement (BD Biosciences) (0.1 mg/ml), heparin (Sigma) (0.1 mg/ml), 2 mM glutamine, penicillin and streptomycin (Pen/Strep) (Gibco), antimycotic/antibiotic (Gibco), nonessential amino acids (Sigma), and 20 mM HEPES (Sigma). The cells were plated onto collagen IV-coated plates (BD). Puromycin (4 µg/ml) was added 24 h after isolation for 48 h total to inhibit growth of fibrocytes. Before use, cells were subjected to one passage at a dilution of 1:2 or 1:3 with Accutase (EMD Millipore Corporation) to release them from the plate.
Growth curve.
pmBECs were isolated and plated in collagen IV-coated wells of a 12-well transwell plate (Corning Inc.) and infected at a multiplicity of infection (MOI) of 5. After 1 h of adsorption at 37°C, the cells were washed 2 times with PBS before media was replenished. At 0, 2, and 4 or 5 dpi, supernatant, scraped cells, and membrane were collected. Samples were freeze-thawed 3 times to release intracellular virus; infectious virus was quantified by plaque assay.
Measurement of TEER.
After initial isolation, pmBECs were plated onto collagen IV-coated 12-well 0.4-µm-pore-size transwells (Corning). Media was supplemented with hydrocortisone (500 ng/ml) and 50% to 80% astrocyte-conditioned media to aid in the formation of tight junctions. Astrocyte-conditioned media were obtained from primary astrocytes prepared as previously described (59). The percentage of astrocyte-conditioned media was gradually reduced to 20% once tight junctions had formed. TEER values (Ω·cm2) were determined by subtraction of a blank well containing media alone and multiplying by the surface area of the transwell membrane. Cells were allowed to reach confluence over 7 to 10 days from the time of plating, until TEER levels reached 20 to 50 Ω·cm2. TEER was measured using an Endohm-12 electrical resistance apparatus (World Precision Instruments Inc.). Cells were either mock infected or infected with MAV-1 at an MOI of 5. TEER was measured on the day of infection and at 24-h intervals thereafter. Data are presented as percentages of the initial (day 0) TEER reading.
Histology.
Mice were either mock infected or infected with MAV-1. Mice were perfused with PBS following euthanasia, and organs were collected for histopathology. Organs (thymus, lung, heart, brain, liver, kidney, and spleen) were subjected to immersion fixation in 10% neutral buffered formalin for 24 h, embedded in paraffin, sectioned at 5 µm, and stained with hematoxylin and eosin. Blinded and randomized samples were scored by a board-certified pathologist.
Isolation and staining of cells for flow cytometry.
Mice were mock infected or infected with MAV-1 in groups of 5. On the day of harvest, mice were euthanized and immediately perfused with 30 ml of PBS prior to organ collection. Brains were pooled in DMEM (Gibco) containing 10% fetal bovine calf serum, and cells were mechanically isolated using a cell strainer. Cells were pelleted and resuspended in 70% isotonic Percoll (GE) in a final volume of 15 ml; 37% isotonic Percoll in DMEM and 30% isotonic Percoll in HBSS were sequentially overlaid on top of the cells (15 ml each) in a 50-ml conical tube. Samples were centrifuged for 20 min at 500 × g, and cells at the 37:70 interface were collected. Cells were stained with fluorescently labeled antibodies to CD45.1/CD45.2 (PerCP-Cy5.5; clones A20 and 104 for CD45.1 and CD45.2, respectively), CD3 (fluorescein isothiocyanate [FITC]; clone 145-2C11), CD8 (phycoerythrin [PE]; clone 53-6.7), CD4 (biotin; clone GK1.5), CD44 (PE-Cy7; clone IM7), CD62L (allophycocyanin [APC]; clone MEL-14), CD19 (APC; clone 1D3), MHC class II (PE; clones M5/114.15.2 and OX-6 for BALB/c and SJL, respectively), CD11b (FITC; clone M1/70), CD11c (PE-Cy7; clone N418), PDCA (biotin; clone eBio927), LY6G (PE; clone 1A8), LY6C (biotin; clone AL-21), F4/80 (APC; clone BM8), and biotinylated antibodies were detected with APC-eFluor 780 strepavidin. All antibodies were obtained from eBioSciences, except CD3, CD8, CD19, MHC class II clone M5/114.15.2, LY6C, and LYG, which were obtained from BD; MHC class II clone OX-6 was from Abcam and F4/80 was from Caltag Laboratories Invitrogen. Stained samples were analyzed using a FACSCanto flow cytometer (BD) and FlowJo software (Tree Star, Inc.).
Statistical analysis.
Statistical analysis was carried out using Microsoft Office Excel and Graph Pad Prism 5 software. Specific statistical tests are denoted in each figure legend.
ACKNOWLEDGMENTS
This work was supported by NIH R01 AI068645 to K.R.S. T.-H.H has been supported by NIH National Research Service award T32 GM07544, a University of Michigan (U.-M.) Rackham Graduate School Merit Fellowship and two Rackham graduate student research grants, a U.-M. Frances Wang Chin fellowship, and the U.-M. Endowment for the Development of Graduate Education award. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank Amanda Welton, Shanna Ashley, and Jenny Imperiale for husbandry and genotyping of the early congenic mouse crosses. We thank Richard Keep for technical assistance, loan of equipment, and advice regarding edema experiments. We are also grateful to Anuska Andjelkovic, Jason Weinberg, and Yasmina Laouar for technical assistance. In addition, we thank David Burke, Michael Imperiale, David Miller, and Bethany Moore for helpful discussion and comments on the manuscript.
Footnotes
Citation Hsu T, Althaus IW, Foreman O, Spindler KR. 2012. Contribution of a single host genetic locus to mouse adenovirus type 1 infection and encephalitis. mBio 3(3):e00131-12. doi:10.1128/mBio.00131-12.
REFERENCES
- 1. Fields BN, Knipe DM, Howley PM. 2007. Fields virology, 5th ed. Wolters Kluwer Health/Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 2. Chaturvedi UC, Dhawan R, Khanna M, Mathur A. 1991. Breakdown of the blood-brain barrier during dengue virus infection of mice. J. Gen. Virol. 72(Pt 4):859–866 [DOI] [PubMed] [Google Scholar]
- 3. Dallasta LM, et al. 1999. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am. J. Pathol. 155:1915–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Getts DR, et al. 2008. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J. Exp. Med. 205:2319–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gralinski LE, Ashley SL, Dixon SD, Spindler KR. 2009. Mouse adenovirus type 1-induced breakdown of the blood-brain barrier. J. Virol. 83:9398–9410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ivey NS, et al. 2009. Association of FAK activation with lentivirus-induced disruption of blood-brain barrier tight junction-associated ZO-1 protein organization. J. Neurovirol. 15:312–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Verma S, Kumar M, Gurjav U, Lum S, Nerurkar VR. 2010. Reversal of West Nile virus-induced blood-brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology 397:130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. 2010. Structure and function of the blood-brain barrier. Neurobiol. Dis. 37:13–25 [DOI] [PubMed] [Google Scholar]
- 9. Hawkins BT, Davis TP. 2005. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 57:173–185 [DOI] [PubMed] [Google Scholar]
- 10. Coisne C, Engelhardt B. 2011. Tight junctions in brain barriers during central nervous system inflammation. Antioxid. Redox Signal. 15:1285–1303 [DOI] [PubMed] [Google Scholar]
- 11. Klatzo I. 1987. Pathophysiological aspects of brain edema. Acta Neuropathol. 72:236–239 [DOI] [PubMed] [Google Scholar]
- 12. Kanmogne GD, Primeaux C, Grammas P. 2005. HIV-1 gp120 proteins alter tight junction protein expression and brain endothelial cell permeability: implications for the pathogenesis of HIV-associated dementia. J. Neuropathol. Exp. Neurol. 64:498–505 [DOI] [PubMed] [Google Scholar]
- 13. Lee YR, et al. 2006. MCP-1, a highly expressed chemokine in dengue haemorrhagic fever/dengue shock syndrome patients, may cause permeability change, possibly through reduced tight junctions of vascular endothelium cells. J. Gen. Virol. 87:3623–3630 [DOI] [PubMed] [Google Scholar]
- 14. Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. 1992. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin. Microbiol. Rev. 5:49–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Echavarría M. 2008. Adenoviruses in immunocompromised hosts. Clin. Microbiol. Rev. 21:704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tebruegge M, Curtis N. 2010. Adenovirus infection in the immunocompromised host. Adv. Exp. Med. Biol. 659:153–174 [DOI] [PubMed] [Google Scholar]
- 17. Carrigan DR. 1997. Adenovirus infections in immunocompromised patients. Am. J. Med. 102:71–74 [DOI] [PubMed] [Google Scholar]
- 18. Guida JD, Fejer G, Pirofski LA, Brosnan CF, Horwitz MS. 1995. Mouse adenovirus type 1 causes a fatal hemorrhagic encephalomyelitis in adult C57BL/6 but not BALB/c mice. J. Virol. 69:7674–7681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kring SC, King CS, Spindler KR. 1995. Susceptibility and signs associated with mouse adenovirus type 1 infection of adult outbred Swiss mice. J. Virol. 69:8084–8088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Flisiak R, et al. 2008. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 47:817–826 [DOI] [PubMed] [Google Scholar]
- 21. Gilliam BL, Riedel DJ, Redfield RR. 2011. Clinical use of CCR5 inhibitors in HIV and beyond. J. Transl. Med. 9(Suppl 1):S9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Charles PC, Guida JD, Brosnan CF, Horwitz MS. 1998. Mouse adenovirus type-1 replication is restricted to vascular endothelium in the CNS of susceptible strains of mice. Virology 245:216–228 [DOI] [PubMed] [Google Scholar]
- 23. Spindler KR, et al. 2001. SJL/J mice are highly susceptible to infection by mouse adenovirus type 1. J. Virol. 75:12039–12046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Welton AR, et al. 2005. Identification of quantitative trait loci for susceptibility to mouse adenovirus type 1. J. Virol. 79:11517–11522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spindler KR, et al. 2010. The major locus for mouse adenovirus susceptibility maps to genes of the hematopoietic cell surface-expressed LY6 family. J. Immunol. 184:3055–3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patterson CE, Rhoades RA, Garcia JG. 1992. Evans blue dye as a marker of albumin clearance in cultured endothelial monolayer and isolated lung. J. Appl. Physiol. 72:865–873 [DOI] [PubMed] [Google Scholar]
- 27. Rawson RA. 1943. The binding of T-1824 and structurally related diazo dyes by the plasma proteins. Am. J. Physiol. 138:708–717 [Google Scholar]
- 28. Kempski O. 2001. Cerebral edema. Semin. Nephrol. 21:303–307 [DOI] [PubMed] [Google Scholar]
- 29. Kajon AE, Brown CC, Spindler KR. 1998. Distribution of mouse adenovirus type 1 in intraperitoneally and intranasally infected adult outbred mice. J. Virol. 72:1219–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gaillard PJ, et al. 2001. Establishment and functional characterization of an in vitro model of the blood-brain barrier, comprising a co-culture of brain capillary endothelial cells and astrocytes. Eur. J. Pharm. Sci. 12:215–222 [DOI] [PubMed] [Google Scholar]
- 31. Matullo CM, O’Regan KJ, Hensley H, Curtis M, Rall GF. 2010. Lymphocytic choriomeningitis virus-induced mortality in mice is triggered by edema and brain herniation. J. Virol. 84:312–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morrey JD, et al. 2008. Increased blood-brain barrier permeability is not a primary determinant for lethality of West Nile virus infection in rodents. J. Gen. Virol. 89:467–473 [DOI] [PubMed] [Google Scholar]
- 33. Hijazi A, et al. 2009. Boudin is required for septate junction organisation in Drosophila and codes for a diffusible protein of the Ly6 superfamily. Development 136:2199–2209 [DOI] [PubMed] [Google Scholar]
- 34. Cray C, Keane RW, Malek TR, Levy RB. 1990. Regulation and selective expression of Ly-6A/E, a lymphocyte activation molecule, in the central nervous system. Brain Res. Mol. Brain Res. 8:9–15 [DOI] [PubMed] [Google Scholar]
- 35. Hänninen A, Jaakkola I, Salmi M, Simell O, Jalkanen S. 1997. Ly-6C regulates endothelial adhesion and homing of CD8(+) T cells by activating integrin-dependent adhesion pathways. Proc. Natl. Acad. Sci. U. S. A. 94:6898–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Flood PM, Dougherty JP, Ron Y. 1990. Inhibition of Ly-6A antigen expression prevents T cell activation. J. Exp. Med. 172:115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bamezai A, et al. 1995. Regulated expression of Ly-6A.2 is important for T cell development. J. Immunol. 154:4233–4239 [PubMed] [Google Scholar]
- 38. Flanagan K, et al. 2008. Intestinal epithelial cell up-regulation of LY6 molecules during colitis results in enhanced chemokine secretion. J. Immunol. 180:3874–3881 [DOI] [PubMed] [Google Scholar]
- 39. Eshel R, et al. 2002. Human Ly-6 antigen E48 (Ly-6D) regulates important interaction parameters between endothelial cells and head-and-neck squamous carcinoma cells. Int. J. Cancer 98:803–810 [DOI] [PubMed] [Google Scholar]
- 40. Ashley SL, Welton AR, Harwood KM, Van Rooijen N, Spindler KR. 2009. Mouse adenovirus type 1 infection of macrophages. Virology 390:307–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goetzl EJ, Banda MJ, Leppert D. 1996. Matrix metalloproteinases in immunity. J. Immunol. 156:1–4 [PubMed] [Google Scholar]
- 42. Moore ML, McKissic EL, Brown CC, Wilkinson JE, Spindler KR. 2004. Fatal disseminated mouse adenovirus type 1 infection in mice lacking B cells or Bruton's tyrosine kinase. J. Virol. 78:5584–5590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moore ML, Brown CC, Spindler KR. 2003. T cells cause acute immunopathology and are required for long-term survival in mouse adenovirus type 1-induced encephalomyelitis. J. Virol. 77:10060–10070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kenzy SG, Cho BR, Kim Y. 1973. Oncogenic Marek’s disease herpesvirus in avian encephalitis (temporary paralysis). J. Natl. Cancer Inst. 51:977–982 [DOI] [PubMed] [Google Scholar]
- 45. Turtle L, Griffiths MJ, Solomon T. 2012. Encephalitis caused by flaviviruses. QJM 105:219–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wiley CA, et al. 1991. Pathogenesis of HIV encephalitis. Acta Pathol. Jpn. 41:192–196 [DOI] [PubMed] [Google Scholar]
- 47. Mims CA. 1957. The invasion of the brain by yellow fever virus present in the blood of mice. Br. J. Exp. Pathol. 38:329–338 [PMC free article] [PubMed] [Google Scholar]
- 48. Liu HC, Niikura M, Fulton JE, Cheng HH. 2003. Identification of chicken lymphocyte antigen 6 complex, locus E (LY6E, alias SCA2) as a putative Marek’s disease resistance gene via a virus-host protein interaction screen. Cytogenet. Genome Res. 102:304–308 [DOI] [PubMed] [Google Scholar]
- 49. Morgan RW, et al. 2001. Induction of host gene expression following infection of chicken embryo fibroblasts with oncogenic Marek’s disease virus. J. Virol. 75:533–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Venter M, et al. 2005. Gene expression in mice infected with West Nile virus strains of different neurovirulence. Virology 342:119–140 [DOI] [PubMed] [Google Scholar]
- 51. Krishnan MN, et al. 2008. RNA interference screen for human genes associated with West Nile virus infection. Nature 455:242–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schoggins JW, et al. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Loeuillet C, et al. 2008. In vitro whole-genome analysis identifies a susceptibility locus for HIV-1. PLoS Biol. 6:e32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brass AL, et al. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921–926 [DOI] [PubMed] [Google Scholar]
- 55. Ball AO, Beard CW, Villegas P, Spindler KR. 1991. Early region 4 sequence and biological comparison of two isolates of mouse adenovirus type 1. Virology 180:257–265 [DOI] [PubMed] [Google Scholar]
- 56. Welton AR, Spindler KR. 2007. Capture ELISA quantitation of mouse adenovirus type 1 in infected organs. Methods Mol. Med. 130:215–221 [DOI] [PubMed] [Google Scholar]
- 57. Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 27:493–497 [Google Scholar]
- 58. Phares TW, Kean RB, Mikheeva T, Hooper DC. 2006. Regional differences in blood-brain barrier permeability changes and inflammation in the apathogenic clearance of virus from the central nervous system. J. Immunol. 176:7666–7675 [DOI] [PubMed] [Google Scholar]
- 59. Stamatovic SM, et al. 2005. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 25:593–606 [DOI] [PubMed] [Google Scholar]