

Abstract
The general thermodynamic principles behind pH driven conformational transitions of biological macromolecules are well understood. What is less obvious is how they can be used to engineer pH switches in proteins. The acid unfolding of staphylococcal nuclease (SNase) was used to illustrate different factors that can affect pH-driven conformational transitions. Acid unfolding is a structural transition driven by preferential H+ binding to the acid unfolded state (U) over the native (N) state of a protein. It is the result of carboxylic groups that titrate with more normal pKa values in the U state than in the N state. Acid unfolding profiles of proteins reflect a balance between electrostatic and non-electrostatic contributions to stability. Several strategies were used in attempts to turn SNase into an acid insensitive protein: (1) enhancing global stability of the protein with mutagenesis or with osmolytes, (2) use of high salt concentrations to screen Coulomb interactions, (3) stabilizing the N state through specific anion effects, (4) removing Asp or Glu residues that titrate with depressed pKa values in the N state, and (5) removing basic residues that might have strong repulsive interactions in the N state at low pH. The only effective way to engineer acid resistance in SNase is not through modulation of pKa values of Asp/Glu but by enhancing the global stability of the protein. Modulation of pH-driven conformational transitions by selective manipulation of the electrostatic component of the switch is an extremely difficult undertaking.
Keywords: acid unfolding, pKa, electrostatics, pH effects, staphylococcal nuclease, protein engineering
This issue of Biophysical Chemistry celebrates the 25th Anniversary of the Gibbs Conference in Biothermodynamics. One of the original goals of the Gibbs Conference was to promote the rigorous application of equilibrium thermodynamics and kinetics to examine molecular mechanism in proteins, nucleic acids, membranes, and their assemblies. In our lab, efforts in this area are focused on improving understanding of fundamental structural and physical origins of electrostatic effects in proteins. This effort is warranted by the wealth of essential biochemical processes (e.g. catalysis, H+ coupled e− transfer reactions, ion homeostasis, H+ transport, e− transfer, etc) governed by electrostatics, where the structural basis of biological function cannot be established without deep understanding of contributions from electrostatic factors.
Biochemical processes governed by electrostatics are unique in one important respect: electrostatic contributions to the changes in Gibbs free energy (ΔG) during a process are related to the product of the electrostatic potential (ψ, an intensive variable) and charge (q, an extensive variable): ΔG = ψ q. To the extent that ψ and q can be calculated using molecular structures, ΔG can be calculated from structure. This is one of the few situations where structure-based calculations of ΔG starting from first principles are possible, and where it is in principle possible to connect structure with biological function by examination of how fundamental physical forces (i.e. ψ, the electrostatic potential) determine the Gibbs free energy function.
Most efforts in studies of protein electrostatics are focused on the related problems of understanding molecular determinants of pKa values and understanding the structural and physical bases of pH driven conformational transitions. Until recently, progress in understanding determinants of pKa values has been stymied by a lack of experimental data that could be used to identify the different electrostatic factors (hydration, Coulomb interactions, hydrogen bonding, conformational reorganization, etc) that can affect the pKa of an ionizable group in a protein. Much progress has been made recently in this area through the study of ionizable groups with highly anomalous pKa values; it is starting to become possible to identify and quantitate contributions from different electrostatic factors 1; 2; 3. In contrast, the important problem of pH-driven conformational transitions of proteins has received relatively little attention.
We have used the acid unfolding of a globular protein to illustrate fundamental thermodynamic concepts of ligand-linked conformational transitions in proteins in general, to demonstrate how subtle and complex electrostatic contributions to stability can be, and to illustrate how difficult it is to manipulate them rationally. The ultimate goal of this study was to engineer a variant of staphylococcal nuclease (SNase) that did not unfold under acidic conditions. The design of proteins that resist unfolding by acid or base is of biotechnological importance, but our goal here was simply to test how well-understood principles of equilibrium thermodynamics and classical electrostatics could be exploited to alter the solution properties of a protein. This exercise illustrates the fine balance between electrostatic and non-electrostatic contributions to stability and the problems inherent to the design of pH-sensitive conformational switches in proteins.
RESULTS and DISCUSSION
Staphylococcal nuclease (SNase) is ideally suited for studies of acid unfolding for several reasons: (1) the thermodynamic stability of variants with substitution of every single position with Ala and Gly has been measured 4; 5; 6; therefore, it is possible to enhance global thermodynamic stability with a rational strategy that would be impractical with other proteins. Variants have been engineered previously with thermodynamic stability more than double that of the wild-type protein 7; 8; 9; 10. (2) the pKa of every Asp, Glu and His (all the residues that titrate at pH below 8) has been measured with NMR spectroscopy 11; 12. Thus, it is possible to know, at least in principle, exactly how much each ionizable group contributes to the H+ binding reactions that drive acid unfolding. (3) The acid unfolding reaction of the wild-type protein has been studied extensively, and it is known to be a highly cooperative transition between two states involving the preferential binding of many H+ to the U state 13; 14. All these factors were exploited in attempts to engineer an acid insensitive variant of SNase.
Thermodynamic basis of pH-driven conformational transitions
pH-driven conformational transitions are a special case of ligand-driven conformational transitions in which H+ acts as the ligand and the most immediate consequence of ligand binding or release is a change in the charge of the macromolecule. It is because pH-driven processes involve a change in the charge state of the macromolecule that these processes can be understood in terms of electrostatic forces.
A change in pH can drive a conformational transition in a protein only if the pKa of at least one ionizable group is different in the different conformational states. The effects of pH on the equilibrium between two conformational states can be expressed in terms of a pH-independent term and the difference in pKa of the ionizable group. Expressions to describe the effects of pH on the equilibrium between two states are straightforward. The example that will be used to this end is that of a transition between the native (N) and unfolded (U) states of a protein under the influence of a single ionizable group that titrates with different pKa values in the N and in the U states. This is illustrated by Scheme I: The pKa that describes the H+ titration of an ionizable group in the N state describes the equilibrium between N− and No:
| (1) |
and that of the ionizable group in the U state is related to:
| (2) |
The equilibrium between the folded (N) and unfolded (U) states of a protein at a pH where the ionizable group is deprotonated is given by:
| (3) |
and under conditions of pH where the ionizable group is protonated is given by:
| (4) |
The observed equilibrium between the N and U states at any pH can include 4 different macromolecular species:
| (5) |
and because
| (6) |
then
| (7) |
In this expression and account for the pH independent component of ΔGo at a reference pH where the ionizable group in both N and U conformations is completely deprotonated or protonated, respectively. The term inside the parentheses in Equation 7 accounts for the pH dependent component governed by the difference in the pKa values of one ionizable group between the N and U states. Equation 7 can be generalized for the case of multiple H+ binding sites and multiple conformational states. The probability that conformational state i is populated is:
| (8) |
where N refers to the total number of conformational states j. If the different conformational states can bind H+ with different affinity, then pH will affect the probability distribution of states according to:
| (9) |
The rightmost term in this expression accounts for the effects of pH and the term accounts for the part of the equilibrium process that is insensitive to pH. In the modulating term (Ai)
| (10) |
Kij refers to the binding constant of site j in conformation i, Kok refers to the binding constant for site k in the reference conformational state o, Ni refers to the number of H+ binding sites in state i and No to the number of H+ binding sites in reference state o. States for which the modulating term Ai > 1 are stabilized with respect to the reference state, and states where Ai < 1 are destabilized with respect to the reference state. The partial derivative of this modulating term with respect to pH is equal to the difference in the extent of H+ binding (ν) in all binding sites in the protein in conformational states i and o:
| (11) |
Scheme I.
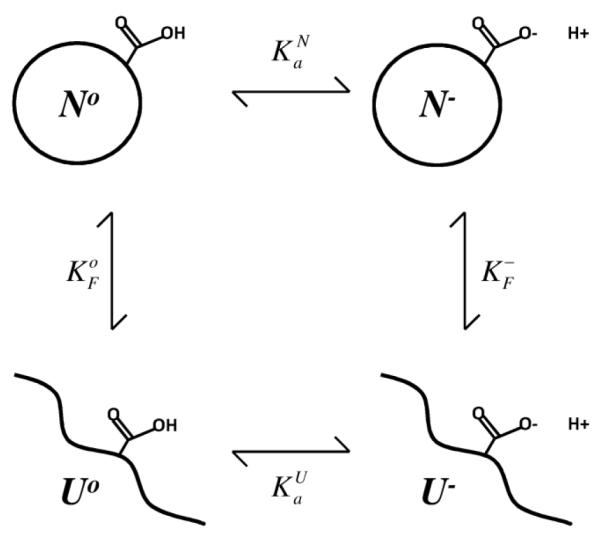
An increase in H+ concentration will stabilize state i if the amount of H+ bound to state i is greater than the amount bound to the reference state o. This can happen if the H+ binding affinity is higher in state i than in state o, or if there are more H+ binding sites in states i than in state o. Under conditions of pH where all binding sites are saturated, the state with the largest number of H+ binding sites will be the one that is stabilized preferentially.
The physical meaning of areas under H+ titration curves
The Gibbs free energy of H+ binding at a single site can be obtained by integration of the H+ titration curve described by the extent of H+ binding (νH+) vs pH:
| (12) |
When pH1 = 0 and pH2 is 2 pH units or more above the pKa of the ionizable group, the area described by the integral in Equation 12 corresponds to the same ΔGo that would have been calculated with the well-known relationship ΔGo = −RT lnK, which for the case of pKa values and for ΔGo in units of kcal/mol is ΔGo = 1.36 pKa. Notice that by fixing one of the limits of integration in equation 12 to pH = 0, the ΔGo is being defined relative to a reference concentration of 1 M [H+] (i.e. a 1 M standard state). Equation 12 can also be used to calculate the pH dependence of the Gibbs free energy difference stored in a difference in pKa values for an ionizable group in two different conformational states (A and B):
| (13) |
Figure 1A shows H+ titration curves for a carboxylic group with a normal pKa of 4.5 (thin), and titration curves for cases of pKa values of 4.0, 3.5, 3.0, 2.5, 2.0, 1.5 and 1.0 (thickest). Figure 1B shows ΔΔGo obtained by integration of the area between the reference curve corresponding to the titration of a group with pKa = 4.5 and the other titration curves. For a ΔpKa = 1 the largest possible ΔΔGo is 1.36 kcal/mol (corresponding to 2.303 RT at 298 K, obtained from ΔΔGo = 1.36 ΔpKa where ΔpKa = 1). The curves in Figure 1B illustrate how the N state of a protein that has a Glu residue with pKa depressed below the normal value of 4.5 begins to become destabilized as the pH is lowered below pH 5.5 owing to the depression in the pKa of the Glu residue. The magnitude of the destabilization depends both on the pH and on the magnitude of the depression to the pKa in the N state relative to the value in the alternative conformational state. Destabilization of the N state relative to the alternative state increases with decreasing pH. For a protein with a carboxylic group that has a highly depressed pKa = 1 in the N state and a normal pKa = 4.5 in the U state, the protein would be destabilized by nearly 5 kcal/mol between pH 6 and pH 0 (thickest curve in Fig. 1B).
FIGURE 1.

Gibbs free energy related to shifts in pKa values of a single H+ binding site (A) Simulated H+ binding curves for a carboxylic group with pKa = 4.5 (thinnest), 4.0, 3.5, 3.0, 2.5, 2.0, 1.5 and 1.0 (thickest). (B) ΔΔG° reflected in shifts in pKa values, calculated by integration (with Equation 13) of the area between the reference curve with pKa of 4.5 and the other curves in 1A for groups with depressed pKa values.
Why can proteins be unfolded by acid?
Acid unfolding of proteins is primarily a consequence of the difference in pKa values of Asp/Glu residues in the N and in the U states (His residues can also contribute). Asp/Glu in water normally begin to bind H+ at pH values below 5.5. If the pKa values of Asp/Glu are depressed in the N state and normal in the U state, then the U state becomes preferred at pH values below 5.5 because H+ binds preferentially to the U state under those conditions. Although the pKa values of ionizable groups in the U state are usually quite similar to the normal pKa values in water, in general the pKa values of most surface Asp/Glu residues in proteins in the N state tend to be depressed slightly, either because of net favorable Coulomb interactions with basic residues or because of hydrogen bonding15.
The destabilization of the N state relative to the U state increases with decreasing pH (Fig. 1B). The destabilizing contributions to the N state by Asp/Glu residues with depressed pKa values can be very significant when many residues have depressed pKa values in the N state and normal ones in the U state (Figure 2). Proteins become acid unfolded when the destabilization of the N state originating from differences in the pKa values of ionizable groups in N and U states becomes comparable to the net contributions by all other non-covalent interactions that affect thermodynamic stability. It should be emphasized that, from the thermodynamic perspective, the mechanism of acid unfolding need not invoke anything more than the shift in conformational state driven by the energy stored in differences in pKa values. Specifically, the mechanism of acid unfolding need not invoke the neutralization of strongly stabilizing ion pairs, or the accruement of repulsive interactions between positive charged groups in the N state when Asp/Glu become neutralized at low pH. These factors, and any others that affect the pKa values of the carboxylic groups, affect the acid-unfolding equilibrium only to the extent that they affect pKa values of ionizable groups. Histidines can also contribute to acid-unfolding reactions, but while carboxylic groups in net favorable electrostatic environments are the ones that actively contribute to the acid unfolding process, the histidines that contribute to the acid unfolding reaction are those that are in net destabilizing electrostatic environments in the N state.
FIGURE 2.

pH dependence of Gibbs free energy related to shifts in pKa values. (A) Simulated H+ binding curves for the native (N) state (solid line) calculated with pKa values for Asp, Glu and His residues measured with NMR spectroscopy. Simulated H+ binding curves for the unfolded (U) state (dashed line) calculated with pKa values of 3.7, 4.2 and 6.3 for Asp, Glu and His, respectively. These values were obtained by analysis of the H+ binding curve of unfolded SNase in water measured with direct potentiometric methods 30. Contributions from Lys, Arg and N and C termini were not included in these calculations. (B) Preferential H+ binding (Δmoles H+ bound, dashed line with reference to left axis)) calculated as moles H+ bound in U – moles H+ bound in N). ΔΔG° (solid line with reference to right axis) calculated as the area under the Δmoles H+ bound vs pH curve.
Acid unfolding of staphylococcal nuclease
The unfolding free energy (ΔG°H2O) of wild-type SNase measured with chemical denaturation is 5.4 kcal/mol at pH 74. It decreases with decreasing pH14. The midpoint (pHmid) of the transition between the N and the U state is 3.8. Acid unfolding of SNase is a highly cooperative process, involving the uptake of a large number of H+ that bind preferentially to the U state 14; 16; 17. The number of H+ bound upon unfolding has been measured previously with direct potentiometric methods14. It can also be estimated (Fig. 2A) using the pKa values for His, Asp and Glu measured by NMR spectroscopy and assuming that in the U state these ionizable groups titrate with the normal pKa values. The pHmid of unfolding of wild-type SNase is relatively close to the normal pKa value of Asp and Glu residues in water; therefore, the acid unfolding of this protein must be driven by small contributions from a large number of Asp/Glu residues with pKa values below the normal values of 4.0 and 4.5. Only small depressions in pKa values are necessary to explain the high cooperativity in the acid unfolding transition.
The pKa values of Asp, Glu and His residues measured with NMR spectroscopy in the N state and in model compounds for the U state can be used to simulate the overall H+ titration curves for the N and U states of SNase as the sum of the H+ titration curves of the individual H+ binding groups (Fig 2A). These titration curves can be used to calculate the total contributions made by preferential H+ binding to the energetics of acid unfolding. By integrating the area between the H+ titration curves of N and U states relative to a 1 M H+ concentration (i.e. pH = 0) it is possible to show that at pH 7 the total difference in pKa values of ionizable groups in N and U states is equivalent to 14 kcal/mol (Fig. 2B). Consider that the total ΔG°H2O of wild-type SNase measured with chemical denaturation at pH 7 is 5.4 kcal/mol. As the pH begins to decrease below 7, the ΔG° stored in the difference in pKa begins to become a factor that destabilizes the N state relative to the U state at the rate described by the solid line in Fig. 2B. In other words, if the ΔΔG° curve in Figure 2B were shifted along the Y-axis until the point were reached where ΔΔG° = 5.4 kcal/mol at pH 7, the midpoint of the acid unfolding transition would correspond to the pH where ΔG° = 0 (at the pHmid, half the molecules are in the N state and half in the U state). The pHmid for acid unfolding of wild-type SNase obtained from H+ titrations monitored with Trp fluorescence is 3.8, and by analysis of the ΔG° curve (Fig. 2B) obtained from pKa values measured by NMR spectroscopy it is 3.6.
Strategies for the engineering of acid resistant proteins
The simulated H+ titration curves and the Gibbs free energies based on experimental pKa values (Fig. 2) suggest strategies that can be used to engineer an acid insensitive variant of SNase. Specifically, the simulations in Figure 2B suggest that by increasing the global thermodynamic stability of the N state the pHmid of the acid unfolding transition (i.e. the pH where ΔG° = 0 in the solid curve in Fig. 2B) would shift towards lower pH values. In fact, the maximum ΔG° stored in the sum of ΔpKa values is approximately 14 kcal/mol (Fig 2B). If the stability of SNase were increased by any means (mutagenesis, use of osmolytes, use of specific ion binding) to slightly above 14 kcal/mol at pH 7, SNase would be almost completely resistant to acid.
The pHmid of unfolding of SNase could also be shifted to lower pH values by diminishing the differences in pKa values of Asp/Glu residues in N and U states. This could be achieved by removal of Asp/Glu residues that titrate with depressed pKa values in the N state, by removal of basic residues with favorable Coulomb interactions that depress pKa values of Asp/Glu residues in the N state, by removal of basic residues with strong repulsive interactions in the N state, or by the addition of salt to screen Coulomb interactions with the intent of normalizing the pKa values of Asp or Glu residues that are under the influence of Lys, His or Arg residues.
Dependence of acid unfolding on global thermodynamic stability
Several strategies have been used previously to increase the thermodynamic stability of wild-type SNase. The PHS variant engineered with P117G, H124L and S128A substitutions increases the ΔG°H2O of the wild-type from 5.4 kcal/mol to 8.5 kcal/mol in 100 mM ionic strength, 298 K 9; 10. The Δ+PHS variant, which includes additional G50F and V51N substitutions and a 44-49 deletion that eliminates the unstructured Ω loop, further increases the stability to 11.8 kcal/mol 18. The Δ+VIAGLA variant, which consists of Δ+PHS with additional T33V, T41I, S59A substitutions increases the stability even further to 12.5 kcal/mol 8. The variant structure of the Δ+PHS variant and the location of all Asp and Glu residues is shown in Figure 3.
FIGURE 3.

Stereo image of Δ+PHS variant of SNase 11 showing ionizable moieties of Lys, Arg and His residues (dark grey) and of Asp and Glu residues (light grey). Thickness of the Asp and Glu residues is proportional to the magnitude in the difference in pKa values in the N state and in model compounds in water.
Acid titration of these stable variants of SNase, monitored by Trp fluorescence, showed two things (Fig. 4A). First, the more stable variants (Δ+PHS and Δ+VIAGLA) exhibit a predenaturational transition centered near pH 4.5. The origins of this transition are not known but are likely to involve an electrostatic field effect on the Trp-140 moiety used to monitor the acid titrations; this pre-denaturational transition is not as pronounced in titrations monitored with CD spectroscopy at 222 nm (data not shown). The more significant trend illustrated by the H+ titration curves in Figure 4A is the dramatic decrease in the midpoint of acid unfolding with increasing thermodynamic stability. Whereas the pHmid of wild-type SNase was 3.8, for the highly stable Δ+VIAGLA variant it was 2.0. The pHmid values measured by H+ titrations monitored with fluorescence are comparable to what the ΔG° vs pH curves in Fig. 2B show. An increase in thermodynamic stability beyond that achieved with Δ+VIAGLA would likely translate into a completely acid insensitive form of SNase but it has been difficult to increase the stability of the protein beyond 12.5 kcal/mol.
FIGURE 4.

H+ titration curves of SNase and SNase variants monitored with Trp fluorescence. All data measured at 298 K in 100 mM KCl unless otherwise noted. Solid lines correspond to fits with 2 or 3 state unfolding models, as described previously7. (A) Dependence of acid unfolding on global thermodynamic stability. Data for the wild-type (Black), PHS (Blue), Δ+PHS (Green) and Δ+VIAGLA (Red) variants are shown. All data measured at 298 K in 100 mM KCl. (B) Effects of stabilizing osmolytes on acid unfolding of Δ+PHS SNase in 100 mM KCl (Black), 300 mM TMAO (Blue), 1 M glycerol (Green) and 1.5 M sucrose (Red) are shown. (C) Effects of salt on acid unfolding of wild-type and Δ+PHS SNase. Curves for the wild-type (solid symbols) and Δ+PHS SNase (open symbols) in 10 mM KCl (Black), 100 mM (Blue) and 1 M (Red) are shown. (D) Specific salt effects on the acid unfolding of wild-type and Δ+PHS SNase. Curves for the wild-type (solid symbols) and Δ+PHS SNase (open symbols) in 100 mM KCl (Black), with 100 mM KClO4 (Blue), 50 mM (NH4) 2SO4 (Red) and 300 mM (NH4)2SO4 (Green) are shown. (E) Acid unfolding in variants of wild-type SNase where Asp or Glu residues have been neutralized through substitution to Ala. Curves for the wild-type (Black), E10A (Purple), D19A (Blue), D77A (Green), D83A (Orange) and D95A (Red) proteins are shown. (F) Acid unfolding of variants of Δ+VIAGLA SNase where Asp or Glu residues have been neutralized through substitution to Asn or Gln. Curves for Δ+VIAGLA (Black), E10Q (Purple), D19N (Dark Blue), D83N (Light Blue), D95N (Green), D77N/D83N (Orange), E10Q/D19N/D77N/D83N (Red) and E10Q/D19N/D77N/D83N/D95N (Red) variants are shown. (G) Acid unfolding of variants of Δ+VIAGLA SNase where Lys or His residues have been neutralized through substitution to Gln. Curves for Δ+VIAGLA (Black), H8Q (Dark Blue), K16Q (Light Blue), K63Q(Green), K71Q (Orange), and K134Q (Red) variants are shown. (H) H+ titration curves monitored with Trp fluorescence in variants of Δ+PHS SNase where various residues have been neutralized through substitution to Asn or Gln. Curves for Δ+PHS (Black), D19N (Purple), D21N (Blue), E57Q (Light Blue), E135Q (Green), R35Q (Orange), R87Q (Red), and R35Q/R87Q (Brown) variants are shown.
Increased resistance to acid unfolding in the presence of stabilizing osmolytes
The stability of the N state of proteins can usually be effectively increased by the addition of stabilizing osmolytes such as sucrose, glycerol and TMAO 19; 20. The effects of different types and concentrations of stabilizing osmolytes on pHmid values for the Δ+PHS variant of SNase were tested (Fig. 4B). The concentrations of osmolytes that were studied were dictated by what was practical from the experimental perspective – a detailed survey of the dependence of acid unfolding on osmolyte concentration was beyond the scope of this study. TMAO in concentrations as high as 300 mM had no detectable impact on the acid unfolding transition. Higher concentrations above 2 M are known to be necessary to stabilize other proteins but these high concentrations were not tested. Glycerol at 1 M concentration appears to have had an effect on the structural properties of the U state. The acid unfolding transition in the presence of glycerol had the same pHmid as in its absence, but the spectroscopic properties of the U state in glycerol were quite different from those in water and consistent with the presence of more structure than in water. In sharp contrast with the inability of TMAO and glycerol to protect SNase against unfolding with acid, Δ+PHS nuclease in 1.5 M sucrose was almost totally insensitive to acid. Although the ΔG°H2O for Δ+PHS nuclease in 1.5 M sucrose has not been measured, the data suggest it must stabilize the protein by at least 2 kcal/mol. It is likely that TMAO and glycerol at higher concentrations would be as efficient as sucrose in stabilizing SNase.
High salt concentration increases acid resistance
The pKa values of all Asp and Glu residues were measured previously by NMR spectroscopy at both high (1.0 M) and low (0.1 M) concentrations of KCl 11 With increasing salt concentration the pKa values of the Asp/Glu residues with the most depressed pKa values moved closer to the normal values of 4.0 and 4.5, respectively, suggesting that the depression is the result of medium and long-range stabilizing interactions with basic residues. Increasing salt concentration was expected to affect the pHmid of acid unfolding significantly through two mechanisms: (1) by bringing the depressed pKa values of carboxylic groups in the N state closer to the normal values in the U state, and (2) by screening repulsive interactions between basic groups in the N state. At low pH, when most H+ binding sites are protonated and the repulsive interactions between basic groups are the dominant interactions in the protein, this effect could be significant.
The pHmid of the wild-type protein was rather insensitive to salt concentration in the range 10 mM to 1 M (Fig. 4C). In contrast, the pHmid of the more stable Δ+PHS protein was very sensitive to salt concentration; at salt concentrations of 1 M full unfolding of this protein was not achieved and a significant decrease in the apparent pHmid was observed. Under these conditions of high salt the Δ+PHS protein was very acid resistant. Several factors contribute to the difference in the effects of salt on the pHmid of the wild-type and the Δ+PHS forms of SNase. First, the effects of high salt on the pKa values of Asp/Glu residues are relatively mild in cases where the pKa values in the N state are depressed only slightly (less than 0.5 pKa units) from the normal values in water. Because the pKa values of most of the 20 Asp/Glu residues are depressed, even at high salt concentration the preferential binding of H+ to the U state distributed over many groups is sufficient to drive acid unfolding of the wild-type protein. High salt concentration has a significant effect on the most depressed pKa values of Asp/Glu. These are the groups that govern the unfolding of the more stable variants of SNase that unfold under more acidic conditions. Under conditions of high salt concentration, fewer Asp/Glu residues in these highly stable proteins end up being active in preferential H+ binding to the N state; therefore, at high salt concentrations the pHmid of the more stable forms of SNase shifts towards lower values.
The difference in the salt sensitivity of the acid unfolding transitions of wild-type and of the Δ+PHS variant suggests that the mechanism of acid unfolding of these two proteins is different. The wild-type acid unfolds owing to small differences in pKa values of groups distributed over the entire protein – small effects add up to produce a very large and highly cooperative acid unfolding transition. In the case of the Δ+PHS protein, at pH values below 2 most of the carboxylic groups are already protonated. The few key ionizable groups that remain largely unprotonated in the N state under these highly acidic conditions contribute to the acid unfolding reaction. At the same time, under acidic conditions, the N state must experience considerable strain from net repulsive interactions between the basic Lys, His and Arg residues. It appears that screening of these repulsive interactions with salt contributes significantly to preventing unfolding at low pH. The effects of salt acting on pKa values and on repulsive interactions cannot be teased apart with these data alone.
Increased acid resistance through specific salt effects
To test the extent to which specific, preferential interactions of ions with the N state could affect the energetics of acid unfolding, the H+ titration properties of the wild-type and of the Δ+PHS protein were measured in the presence of perchlorate (ClO4−), and sulfate (SO42-), which are known to stabilize SNase at neutral pH and to stabilize proteins in general against acid unfolding 21; 22; 23; 24. The acid unfolding of the wild-type protein was insensitive to the addition of ClO4− (Fig. 4D). A minor drop in pHmid was detected in the presence of SO42-. In contrast, in the Δ+PHS protein both ClO4− and SO42- had a detectable effect on the acid unfolding behavior. SO42- at 50 mM was enough to prevent total unfolding of the protein at low pH values, and the effect was even more pronounced with SO42- concentrations of 300 mM. The reason behind the difference in the stabilizing effect of SO42- in the wild-type and in the Δ+PHS is not obvious, but it must be related to the appearance of competent SO42- binding sites in the N state at low pH, where the net positive charge of the protein is much higher than at the pH where the acid unfolding of the wild-type protein takes place. Much higher concentrations of SO42- might have a more noticeable effect on the properties of the wild-type protein, but this was not tested. Previous studies in our laboratory have employed mutagenesis for selective removal of basic residues that might be responsible for the stabilization of SNase by specific interactions. No critical groups have ever been identified that could explain the ability of SO42- and other anions to stabilize SNase through site-specific anion binding.
Removal of acidic residues is not sufficient to prevent acid unfolding
Because the acid unfolding of SNase is driven by the preferential binding of H+ to Asp/Glu residues with higher pKa values in the U than in the N state, it was of interest to attempt to modify the acid unfolding profile by selective removal of Asp/Glu residues that have depressed pKa values in the N state relative to the normal pKa values of 4.0 and 4.5 in water, respectively. According to NMR spectroscopy studies published previously 11, the groups with the most anomalous pKa values in 100 mM KCl, where ΔpKa is defined as pKa (in N state of protein) – pKa (model compound) was larger than 1.0 are: Asp-19 (ΔpKa = −1.69), Asp-77 and Asp-83 (ΔpKa > −1.7), Asp-95 (ΔpKa = −1.74), Glu-10 (ΔpKa = −1.53) and Glu-75 (ΔpKa = −1.09). Asp-21 also has a highly shifted pKa value but it is higher (pKa = 6.54) than the normal pKa of 4.0, so H+ binding to this group actually promotes the N state over the U state.
In the wild-type protein the acid unfolding of variants with E10A, D19A, D77A, D83A or D95A substitutions occurred at pH values higher than the pHmid of 3.8 of the wild-type protein (Fig. 4E). In all of these cases the removal of an ionizable group with a depressed pKa actually accomplished the opposite of what was intended: the variants in which an Asp or Glu with a depressed pKa in the N state was removed actually became more acid labile than the reference protein. H+ titrations were also measured in variants of the more stable Δ+VIAGLA form of SNase with substitutions at E10Q, D19N, D83N or D95N. The D19N substitution decreased the pHmid slightly from 2.02 to 1.8 (Fig. 4F) and there was evidence for the presence of structure in the acid unfolded form of this protein. Quadruple (E10Q, D19N, D77N, and D83N) and quintuple (E10Q, D19N, D77N, D83N, and D95N) variants of the Δ+VIAGLA increased the acid sensitivity of SNase significantly (Fig. 4F).
Removal of Asp and Glu residues with depressed pKa values was the obvious thing to do to attempt to shift the acid unfolding transition of SNase to even lower pH values. With the exception of the D19N variant, not only did the substitutions fail to lower the pHmid, the variants in which the Asp or Glu residues with the most depressed pKa values were removed were more acid labile than the reference protein used to engineer them. The basis of this result is illustrated by the simulations in Figures 1 and 2 and by the ΔG° in Table 1. The removal of an Asp or a Glu either by substitution to Ala or to Asn or Gln is destabilizing at all pH values, for two reasons. First, because the Asp or Glu residues with depressed pKa values have net favorable electrostatic interactions that stabilize the N state and removal of these charges eliminates these interactions. Second, surface side chains have other types of favorable non-covalent interactions that usually stabilize the N state5. In the case of the variants studied, the substitution of Asp/Glu with Ala/Asn/Gln was always destabilizing (Table 1). The net Gibbs free energy stored in differences in pKa values in a single group is quite modest: for a ΔpKa = 2 the maximum ΔΔGo = 2.72 kcal/mol, which is comparable to the ΔΔGo experienced by the variants upon substitution of Asp/Glu residues. In other words, the loss of stability related to factors not related to ΔpKa are of magnitude comparable to the gains related to ΔpKa.
Table I.
Thermodynamic parameters of acid denaturation and chemical denaturation of SNase and SNase variants.
| Reference | Variant | [KCl] mM |
Osmolyte/Salt | pHmid | an | ΔG°H2O kcal/mol pH 7 |
|---|---|---|---|---|---|---|
| WT | 10 | 3.82 ± 0.02 | 3.60 ± 0.27 | |||
| Δ+PHS | 10 | 2.35 ± 0.02 | 3.77 ± 0.06 | |||
| WT | 100 | 3.79 ± 0.01 | 3.40 ± 0.12 | 5.4 | ||
| PHS | 100 | 2.91 ± 0.07 | 4.22 ± 0.18 | 8.5 | ||
| Δ+PHS | 100 | 2.06 ± 0.01 | 2.80 ± 0.12 | 11.8 ± 0.1 | ||
| Δ+VIAGLA | 100 | 2.02 ± 0.02 | 2.72 ± 0.12 | 12.5± 0.2 | ||
| WT SNase | 1000 | 3.68 ± 0.28 | 2.59 ± 0.13 | |||
| Δ+PHS | 1000 | n/d | ||||
| Δ+PHS | 100 | 50mM SO4 | n/d | |||
| Δ+PHS | 100 | 300mM SO4 | n/d | |||
| Δ+PHS | 100 | 100mM NaClO4 | 2.02 ± 0.03 | 1.99 ± 0.04 | ||
| Δ+PHS | 100 | 1.5M Sucrose | n/d | |||
| Δ+PHS | 100 | 300mM TMAO | 1.90 ± 0.03 | 1.98 ± 0.07 | 13.1 ± 0.3 | |
| Δ+PHS | 100 | 1M Glycerol | 1.92 ± 0.03 | 2.41 ± 0.10 | 11.8 ± 0.3 | |
| Δ+PHS | D19N | 100 | 1.95 ± 0.04 | 2.33 ± 0.25 | ||
| Δ+PHS | D21N | 100 | 2.19 ± 0.02 | 2.88 ± 0.11 | ||
| Δ+PHS | R35Q | 100 | 2.26 ± 0.02 | 2.37 ± 0.11 | ||
| Δ+PHS | E57Q | 100 | 3.46 ± 0.72 | 1.82 ± 0.98 | ||
| Δ+PHS | R87Q | 100 | 2.96 ± 0.05 | 2.86 ± 0.29 | ||
| Δ+PHS | R35Q | R87Q | 100 | 3.05 ± 0.17 | 3.49 ± 0.21 | |
| Δ+PHS | E135Q | 100 | 2.03 ± 0.02 | 2.72 ± 0.12 | ||
| Δ+VIAGLA | 100 | 2.02 ± 0.02 | 2.72 ± 0.12 | 12.5 ± 0.2 | ||
| Δ+VIAGLA | E10Q | 100 | 2.03 ± 0.01 | 2.55 ± 0.10 | 11.7 ± 0.2 | |
| Δ+VIAGLA | D19N | 100 | 1.8 ± 0.03 | 2.20 ± 0.11 | 13.2 ± 0.2 | |
| Δ+VIAGLA | D83N | 100 | 2.32 ± 0.03 | 2.26 ± 0.06 | 10.8 ± 0.2 | |
| Δ+VIAGLA | D95N | 100 | 2.2 ± 0.02 | 2.54 ± 0.14 | 10.8 ± 0.1 | |
| Δ+VIAGLA | H8Q | 100 | 1.95 ± 0.01 | 2.60 ± 0.11 | 12.3 ± 0.2 | |
| Δ+VIAGLA | K16Q | 100 | 2.03 ± 0.02 | 2.86 ± 0.23 | 11.9 ± 0.3 | |
| Δ+VIAGLA | K63Q | 100 | 2.11 ± 0.02 | 2.67 ± 0.16 | 11.7 ± 0.2 | |
| Δ+VIAGLA | K71Q | 100 | 1.98 ± 0.02 | 2.61 ± 0.13 | 12.2 ± 0.2 | |
| Δ+VIAGLA | K134Q | 100 | 2.02 ± 0.01 | 2.78 ± 0.11 | 12.1 ± 0.2 | |
| Δ+VIAGLA | D77N | |||||
| D83N | 100 | 2.73 ± 0.04 | 2.07 ± 0.06 | 8.8 ± 0.1 | ||
| Δ+VIAGLA | E10Q | |||||
| D19N | ||||||
| D77N | ||||||
| D83N | 100 | 2.83 ± 0.05 | 1.72 ± 0.09 | 8.7 ± 0.1 | ||
| Δ+VIAGLA | E10Q | |||||
| D19N | ||||||
| D77N | ||||||
| D83N | ||||||
| D95N | 100 | 3.25 ± 0.20 | 1.35 ± 0.25 | 6.5 ± 0.1 | ||
| WT | 100 | 100mM NaClO4 | 3.79 ± 0.02 | 2.73 ± 0.12 | ||
| WT | 100 | 50mM SO4 | 3.6 ± 0.03 | 2.77 ± 0.13 | ||
| WT | E10A | 100 | 4.02 ± 0.01 | 2.89 ± 0.11 | 3.7 ± 0.3 | |
| WT | D19A | 100 | 4.07 ± 0.01 | 3.27 ± 0.18 | 4.6 ± 0.1 | |
| WT | D77A | 100 | 4.08 ± 0.02 | 2.31 ± 0.08 | 2.1 ± 0.2 | |
| WT | D83A | 100 | 4.42 ± 0.11 | 2.35 ± 0.48 | 1.3 ± 0.5 | |
| WT | D95A | 100 | 4.61± 0.35 | 3.25± 0.45 | 1.3 ± 0.3 |
Slope of the pH titration curves, which is proportional to the net number of H+ bound preferentially to the U state over the N state upon acid unfolding.
Removal of basic residues has no impact on acid unfolding
Attempts were made to increase the resistance of SNase against acid by eliminating basic residues that might influence the pKa values of Asp/Glu residues through favorable Coulomb interactions or decrease the stability of SNase through repulsive interactions in the N state at low pH. The H8Q, K16Q, K63Q, K71Q and K134Q variants were engineered, one at a time, in the Δ+VIAGLA background (Table 1). The acid unfolding profiles of these variants were comparable to that of the background protein (Fig. 4G). The H8Q and the K71Q variants had slightly lower pHmid than the reference protein but the effects were minor. Overall, the results with these variants suggest that the effect of substitution of basic residues on global ΔG°H2O was negligible. If the removal of these positive charges decreased the repulsive force experienced by the N state at low pH, this trend, which should have decreased the pHmid value, was almost perfectly balanced by destabilizing contribution to ΔG°H2O, which would increase the pHmid value of these variants. The possibility remains that the simultaneous removal of many basic side chains could effectively decrease the acid sensitivity of the protein, but this was not tested.
CONCLUSIONS
The unfolding of proteins by acid is not the most subtle pH-driven conformational transition in the protein world. In some mammals and probably other species, acid unfolding of proteins is physiologically significant as it helps ensure efficient digestion of proteins in the stomach by stabilizing the acid unfolded state to enhance access of the backbone by acid-insensitive proteases. However, the acid unfolding of proteins is also an unavoidable property of most globular proteins that have ionizable groups on their surface. These groups enhance solubility, prevent non-specific associations with other macromolecules, and guide and stabilize specific interactions. The charged moieties of surface ionizable groups tend to be in environments where they experience net stabilizing Coulomb interactions 15; 25. This is something that is either tuned evolutionarily or a consequence of the influence of charges during the folding reaction proper; ionizable groups are driven towards environments where they can avoid dehydration and interact with bulk water, and they have considerable latitude to guide the placement of charged moieties in environments where they maximize contacts with charges of the opposite polarity. As a consequence of this maximization of favorable Coulomb interactions among surface residues, most surface carboxylic groups in proteins have pKa values that are slightly lower than the normal pKa values in water and this renders folded globular proteins sensitive to acid.
pH is tightly regulated in living systems. Not surprisingly, many proteins have evolved to interpret changes in pH as regulatory signals of physiological relevance. Examples include the well-known Bohr effect in human hemoglobin, a mechanism whereby exposure of oxygenated hemoglobin to the relatively acidic environment of tissues performing respiration under anaerobic conditions triggers a conformational change that stabilizes preferentially the deoxygenated form of the molecule, thereby ensuring delivery of O2 to exercised tissues. The hemagglutinin protein in the influenza virus is another example of a protein that undergoes a substantial conformational rearrangement in response to transient exposure to the relatively acidic environment of the endosome 26. The conformational transition between the inactive and the fusogenic form of hemagglutinin ensures uncoating of the virus necessary for the release of nucleoprotein complexes into the cytoplasm and for successful infection. The molecular mechanisms and thermodynamic principles that govern how a change in pH triggers a conformational transition in hemoglobin or in hemagglutinin are the same ones that apply to all pH-driven conformational transitions, including the acid unfolding reaction.
Like the acid-unfolding reaction, the physiologically essential pH-driven conformational transitions of proteins are rarely encoded in a single ionizable group with different pKa values in the initial and final states of the transition. Instead, the mechanism involves small contributions from many different ionizable groups, a recurring motif in many processes governed by electrostatics. The necessity to avoid encoding an important regulatory ligand-driven conformational transition in a single trigger group ensures robustness against evolutionary fluctuations that could eliminate the capacity to respond to changes in pH entirely by chance elimination of the key trigger group.
It is possible to engineer proteins where changes in the ionization state of a single group is sufficient to regulate a conformational transition. This is possible when, for example, an ionizable group plays a key role stabilizing an element of secondary structure through a capping interaction that is made or broken depending on the ionization state of the group 27. The E57Q variant of Δ+PHS SNase illustrates how a relatively innocuous substitution, in this case one that removes a Glu with a helix capping function, can have substantial conformational consequences (Fig. 4H). A pH-driven conformational transition can also be encoded in a single ionizable group when the ionizable group has a highly anomalous pKa, as in the case of ionizable groups buried in hydrophobic environments in a protein 28. Large shifts in pKa values can store a large ΔG°, enough to drive a substantial conformational transition between two states. Another situation where a single ionizable group in a protein is sufficient to drive a large change in conformation is in highly symmetric assemblies such as helical or icosahedral viral capsids. Small shifts in the pKa value of a single group present in a large number of identical, symmetry-related copies of the protein can serve to sense and amplify an environmental signal such as a small change in pH, and to transduce that information into a large, cooperative structural transition.
This study of the acid unfolding properties of SNase illustrates the complexity inherent to pH-driven conformational transitions and the difficulties in rational engineering of conformational switches driven by changes in pH. The acid unfolding of SNase is highly cooperative in both the wild-type protein that acid unfolds between pH 3.5 and 4, and in the highly stable variants that acid unfold near pH 2. The cooperativity of the unfolding transition arises in both regimes of pH because a large number of groups are involved in the H+ binding reactions that drive the acid-unfolding transition. The experimental data show that the most important determinant of the acid unfolding profile of SNase is not in the details of the electrostatic properties of individual Asp/Glu residues, but in the high number of Asp/Glu with preferential H+ binding to the U state, and in the global balance between the electrostatic (i.e. H+ coupled) and non-electrostatic components of thermodynamic stability. This is likely to be the case for most proteins that are naturally sensitive to pH, where changes in pH drive a conformational transition that activates or inhibits a biochemical state or process.
Tuning the acid sensitivity of SNase by modest manipulation of the constellation of naturally occurring surface charges is not straightforward. Any substitution that eliminated a carboxylic group that had a depressed pKa in the N state had a secondary destabilizing consequence that was greater than the contribution related to the elimination of the charge. The only effective way of enhancing the resistance of SNase against acid required the engineering of enhanced thermodynamic stability. It was possible to do this with SNase with mutagenesis only because extensive previous characterization of the effects of substitutions on thermodynamic stability has allowed the design of variants that are more than twice as stable as the wild-type. Rational engineering of increased thermodynamic stability in proteins remains a challenging task that cannot be completed successfully with most proteins.
The experimental data show that acid unfolding is not driven by the elimination of key ion pairs essential for the structural integrity of the native state. The data illustrate that nothing more sophisticated than preferential H+ binding between the N and the U states need be invoked to understand the acid unfolding process, or any other pH driven conformational switch in proteins. The data show that the mechanism of acid unfolding of SNase is different for the wild-type protein that acid unfolds between pH 4 and 3.5 and for the stable Δ+PHS and Δ+VIAGLA variants that acid unfold near pH 2.0. The wild-type protein was insensitive to high salt concentration whereas the stable variants were highly sensitive to increasing salt concentrations. Detailed mechanistic interpretation of the acid unfolding process was possible only because pKa values of Asp and Glu residues in the Δ+PHS variant of SNase measured previously showed that, in 0.1 M KCl, 14 out of 20 Asp and Glu residues titrate with pKa values depressed by 0.4 pKa units or more. The wild-type protein is unfolded by acid near pH 4, close to the where Asp and Glu in water normally titrate, because the thermodynamic stability of the protein at pH values above 4 is already quite low, and preferential binding of H+ distributed over these 14 groups represents a ΔGo greater than the global stability of the protein at pH 4. The acid-unfolding transition is extremely steep and cooperative because it is driven by H+ binding processes distributed across many ionizable groups throughout the protein. The acid unfolding of the wild-type was acid insensitive because even in 1 M salt more than 10 Asp and Glu residues have pKa values that are depressed relative to the normal values in water, and this is enough to drive the protein from the N to the U state. In contrast, the acid unfolding transition in the stable variants that unfold near pH 2 was highly salt sensitive and could be almost totally eliminated with 1 M salt. At pH 2 in 0.1 M KCl, only five carboxylic groups remain fully or partially unprotonated (Asp-19, Asp-77, Asp-83, Asp-94 and Glu-10). Substitution of these groups with non-ionizable amino acids lowered the global stability of the protein thereby rendering the protein more acid labile. On the other hand, normalizing the pKa values of these 5 carboxylic groups through the use of high salt concentration to screen favorable Coulomb interactions with basic groups effectively eliminated the preferential H+ binding required to drive the acid unfolding reaction. The fact that the pHmid of the acid unfolding transition at high salt moved towards more acidic pH values shows that the global stability of the protein was not diminished in high salt. This suggests that the elimination of favorable Coulomb interactions between Asp, Glu and the basic residues in the protein is more than compensated by the elimination of the repulsive interactions between basic residues under these highly acidic conditions. The observation that elimination of even a single acidic residue can enhance the propensity of the more stable forms of SNase to be unfolded by acid suggests that for proteins with naturally occurring low pHmid of acid unfolding, the strategy for engineering acid insensitivity might have to focus on the removal of repulsive interactions through the selective elimination of certain basic residues.
MATERIALS AND METHODS
Protein Engineering
All variants of SNase were purified following procedures described previously7; 29. Variants were engineered in either the wild-type or the stable Δ+PHS or Δ+VIAGLA backgrounds using QuickChange kits, as described previously 7.
H+ Titrations Monitored with Trp Fluorescence
H+ titrations were monitored with Trp fluorescence using an Aviv ATF-105 automated titration fluorometer using protocols described previously 7; 14. To extract midpoints and slopes of pH titrations they were analyzed with either 2 or 3 state models, as described previously 7. All measurements were performed at 298 K and in 100 mM KCl unless explicitly mentioned otherwise.
Thermodynamic stability
Thermodynamic stability was measured with GdnHCl titrations monitored with Trp fluorescence using an Aviv ATF-105 automated titration fluorometer using protocols described previously 7; 14. Chemical denaturation curves were analyzed with the linear extrapolation model assuming a two state equilibrium process, as described previously 7. All measurements were performed at 298 K and in 100 mM KCl unless explicitly mentioned otherwise.
Acknowledgments
This work was supported by NIH Grants ROI GM-073838 to B. G-M. E.
Abbreviations
- SNase
staphylococcal nuclease
- PHS
stable variant of SNase with P117G, H124L, and S128A substitutions
- Δ+PHS
stable variant of SNase consisting of PHS with a 44-49 deletion and additional G50F and V51N substitutions
- Δ+VIAGLA
stable variant of SNase consisting of Δ+PHS with additional T33V, T41I, S59A substitutions
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Peregrine Bell-Upp, Department of Biophysics, Johns Hopkins University, 3400 N. Charles St., Baltimore, MD 21218.
Aaron C. Robinson, Department of Biophysics, Johns Hopkins University, 3400 N. Charles St., Baltimore, MD 21218
Erika L. Wheeler, Department of Biophysics, Johns Hopkins University, 3400 N. Charles St., Baltimore, MD 21218
Janine Lin, Department of Biophysics, Johns Hopkins University, 3400 N. Charles St., Baltimore, MD 21218.
Bertrand García-Moreno E, Department of Biophysics, Johns Hopkins University, 3400 N. Charles St., Baltimore, MD 21218.
REFERENCES
- 1.Isom DG, Cannon BR, Castañeda CA, Robinson A, Garcia-Moreno E,B. High tolerance for ionizable residues in the hydrophobic interior of proteins. Proc. Natl. Acad. Sci. USA. 2008;105:17784–17788. doi: 10.1073/pnas.0805113105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isom DG, Castaneda CA, Cannon BR, Garcia-Moreno E,B. Large shifts in pKa values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. USA. 2011;108:5260–5265. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isom DG, Castañeda CA, Cannon BR, Velu PD, García-Moreno B. Charges in the hydrophobic interior of a protein. Proc. Natl. Acad. Sci. USA. 2010;107:16096–16100. doi: 10.1073/pnas.1004213107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green SM, Meeker AK, Shortle D. Contributions of the polar, uncharged amino acids to the stability of staphylococcal nuclease: evidence for mutational effects on the free energy of the denatured state. Biochemistry. 31(1992):5717–5728. doi: 10.1021/bi00140a005. [DOI] [PubMed] [Google Scholar]
- 5.Meeker AK, García-Moreno E,B, Shortle D. Contributions of the ionizable amino acids to the stability of staphylococcal nuclease. Biochemistry. 1996;35:6443–6449. doi: 10.1021/bi960171+. [DOI] [PubMed] [Google Scholar]
- 6.Shortle D, Stites WE, Meeker AK. Contributions of the large hydrophobic amino acids to the stability of staphylococcal nuclease. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 7.Karp DA, Gittis AG, Stahley MR, Fitch CA, Stites WE, García-Moreno E,B. High Apparent Dielectric Constant Inside a Protein Reflects Structural Reorganization Coupled to the Ionization of an Internal Asp. Biophysical Journal. 2007;92:2041–2053. doi: 10.1529/biophysj.106.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Lu Z, Sakon J, Stites WE. Increasing the Thermostability of Staphylococcal Nuclease: Implications for the Origin of Protein Thermostability. Journal of Molecular Biology. 2000;303:125–130. doi: 10.1006/jmbi.2000.4140. [DOI] [PubMed] [Google Scholar]
- 9.Dwyer J, Gittis A, Karp D, Lattman E, Spencer D, Stites W, García-Moreno E,B. High apparent dielectric constants in the interior of a protein reflect water penetration. Biophysical Journal. 2000;79:1610–20. doi: 10.1016/S0006-3495(00)76411-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.García-Moreno E,B, Dwyer J, Gittis A, Lattman E, Spencer D, Stites W. Experimental measurement of the effective dielectric in the hydrophobic core of a protein. Biophysical Chemistry. 1997;64:211–24. doi: 10.1016/s0301-4622(96)02238-7. [DOI] [PubMed] [Google Scholar]
- 11.Castañeda CA, Fitch CA, Majumdar A, Khangulov V, Schlessman JL, Garcia-Moreno E,B. Molecular determinants of the pKa values of Asp and Glu residues in staphylococcal nuclease. Proteins: Struct. Funct. Bioinf. 2009;77:570–588. doi: 10.1002/prot.22470. [DOI] [PubMed] [Google Scholar]
- 12.Lee KK, Fitch CA, Lecomte JTJ, García-Moreno E,B. Electrostatic effects in highly charged proteins: salt sensitivity of pKa values of histidines in staphylococcal nuclease. Biochemistry. 2002;41:5656–5667. doi: 10.1021/bi0119417. [DOI] [PubMed] [Google Scholar]
- 13.Fitch CA, Whitten ST, Hilser VJ, Garcia-Moreno E,B. Molecular mechanisms of pH-driven conformational transitions of proteins. Insights from continuum electrostatics calculations of acid unfolding. Proteins: Structure, Function, and Bioinformatics. 2006;63:113–126. doi: 10.1002/prot.20797. [DOI] [PubMed] [Google Scholar]
- 14.Whitten ST, García-Moreno E,B. pH dependence of stability of staphylococcal nuclease: Evidence of substantial electrostatic interactions in the denatured state. Biochemistry. 2000;39:14292–14304. doi: 10.1021/bi001015c. [DOI] [PubMed] [Google Scholar]
- 15.Forsyth WR, Antosiewicz JJ, Robertson AD. Empirical relationships between protein structure and carboxyl pKa values in proteins. Proteins: Structure, Function, and Genetics. 2002;48:388–403. doi: 10.1002/prot.10174. [DOI] [PubMed] [Google Scholar]
- 16.Goto Y, Calciano LJ, Fink AL. Acid-induced unfolding of proteins. Proceedings of the National Academy USA. 1990;87:573–577. doi: 10.1073/pnas.87.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goto Y, Takahashi N, Fink AL. Mechanism of Acid-Induced Folding of Proteins. Biochemistry. 1990;29:3480–3488. doi: 10.1021/bi00466a009. [DOI] [PubMed] [Google Scholar]
- 18.Fitch CA, Karp DA, Lee KK, Stites WE, Lattman EE, García-Moreno E,B. Experimental pKa values of buried residues: analysis with continuum methods and role of water penetration. Biophysical Journal. 2002;82:3289–3304. doi: 10.1016/s0006-3495(02)75670-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Auton M, Bolen DW. Predicting the energetics of osmolyte-induced protein folding/unfolding. Proceedings of the National Academy of Sciences USA. 2005;102:15065–15068. doi: 10.1073/pnas.0507053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolen DW, Baskakov IV. Forcing thermodynamically unfolded proteins to fold. The Journal of Biological Chemistry. 1998;273:4831–4834. doi: 10.1074/jbc.273.9.4831. [DOI] [PubMed] [Google Scholar]
- 21.Nishimura C, Uversky VN, Fink AL. Effect of salts on the stability and folding of staphylococcal nuclease. Biochemistry. 2001;40:2113–2128. doi: 10.1021/bi000861k. [DOI] [PubMed] [Google Scholar]
- 22.Fink AL, Calciano LJ, Goto Y, Kurotsu T, Palleros DR. Classification of acid denaturation of proteins: Intermediates and unfolded states. Biochemistry. 1994;33:12504–12511. doi: 10.1021/bi00207a018. [DOI] [PubMed] [Google Scholar]
- 23.Fink AL, Calciano LJ, Goto Y, Nishimura M, Swedberg SA. Characterization of the stable, acid-induced, molten globule-like state of staphylococcal nuclease. Protein Science. 1993;2:1155–1160. doi: 10.1002/pro.5560020710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goto Y, Nishikiori S. Role of electrostatic repulsion in the acidic molen globule of cytochrome c. Journal of Molecular Biology. 1991;222:679–686. doi: 10.1016/0022-2836(91)90504-y. [DOI] [PubMed] [Google Scholar]
- 25.Edgecomb SP, Murphy KP. Variability in the pKa of Histidine Side-Chains Correlates With Burial Within Proteins. Proteins: Structure, Function, and Genetics. 2002;49:1–6. doi: 10.1002/prot.10177. [DOI] [PubMed] [Google Scholar]
- 26.Baker D, Agard DA. Influenza Hemagglutinin: kinetic control of protein function. Structure. 1994;2:907–910. doi: 10.1016/s0969-2126(94)00091-3. [DOI] [PubMed] [Google Scholar]
- 27.Lecomte JTJ, Moore CD. Helix formation in apocytochrome b5: The role of a neutral histidine at the N-Cap Poition. J. Am. Chem. Soc. 1991;113:9663–9665. [Google Scholar]
- 28.Chimenti MS, Castaneda CA, Majumdar A, Garcia-Moreno E,B. Structural origins of high apparent dielectric constants experienced by ionizable groups in the hydrophobic core of a protein. J. Mol. Biol. 2011;405:361–377. doi: 10.1016/j.jmb.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shortle D, Meeker A. Mutant forms of staphylococcal nuclease with altered patterns of guanidine hydrochloride and urea denaturation. Proteins: Structure, Function, and Genetics. 1986;1:81–89. doi: 10.1002/prot.340010113. [DOI] [PubMed] [Google Scholar]
- 30.Fitzkee NC, Garcia-Moreno E,B. Electrostatic effects in unfolded staphylococcal nuclease. Protein Science. 2008;17:216–227. doi: 10.1110/ps.073081708. [DOI] [PMC free article] [PubMed] [Google Scholar]