

Abstract
Drugs targeting the renin-angiotensin-aldosterone system (RAAS) are the mainstay of therapy to retard the progression of proteinuric chronic kidney disease (CKD) such as diabetic nephropathy. However, diabetic nephropathy is still the first cause of end-stage renal disease. New drugs targeted to the pathogenesis and mechanisms of progression of these diseases beyond RAAS inhibition are needed. There is solid experimental evidence of a key role of oxidative stress and its interrelation with inflammation on renal damage. However, randomized and well-powered trials on these agents in CKD are scarce. We now review the biological bases of oxidative stress and its role in kidney diseases, with focus on diabetic nephropathy, as well as the role of the Keap1-Nrf2 pathway and recent clinical trials targeting this pathway with bardoxolone methyl.
1. Background
Chronic kidney disease (CKD) is a serious public health problem, which carries a high morbidity and mortality [1]. CKD is characterized by a progressive loss of renal function, chronic inflammation, oxidative stress, vascular remodeling, and glomerular and tubulointerstitial scarring. CKD treatment still represents a clinical challenge. Diabetic nephropathy (DN) is the leading cause of CKD and end-stage renal disease (ESRD) [2]. The renin-angiotensin-aldosterone system (RAAS) is a major pathway involved in the pathogenesis and progression of DN [3, 4], and RAAS blockade is an effective therapeutic strategy to reduce proteinuria and slow progression of diabetic and nondiabetic CKD. However targeting the system sets off compensatory mechanisms that may increase angiotensin II, aldosterone, or renin, and partial RAAS blockade does not prevent progression in all CKD patients. Angiotensin II (AT II) is the key mediator of the RAAS [5–7]. Animal models of experimental diabetes, clinical trials, and metanalysis have clearly demonstrated the effectiveness of angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) therapy to improve glomerular/tubulointerstitial damage, reduce proteinuria, and decrease CKD progression, independently of blood pressure (BP) control [8–13]. Dual RAAS blockade with ACEI plus ARB inhibits compensatory AT II activity resulting from ACE-independent pathways and limits compensatory AT production induced by AT1 receptor blockade. This combination reduced proteinuria by 25–45% in DN [14–16]. Results are worse for DN with diminished kidney function or nonproteinuric CKD with ischemic renal injury, probably due to advanced structural renal changes [13, 17, 18] and adverse effects; such acute deterioration of renal function or hyperkalemia is more frequent. The aldosterone antagonists spironolactone and eplerenone reduce albuminuria by 30–60% and slow CKD progression in experimental models [19–21] and clinical studies [22–25] in DN. These agents abrogated the “aldosterone breakthrough” phenomenon and its proinflammatory and profibrotic effects. ACEI/ARB therapy increases renin. Aliskiren, a direct renin inhibitor, was beneficial in animal models of diabetic/hypertensive nephropathy [26, 27] and reduced albuminuria in clinical DN [28]. In a multicenter and double-blind, randomized clinical trial in hypertensive type 2 DM patients with nephropathy, aliskiren plus losartan at maximal dose was 20% more effective than losartan/placebo to reduce albuminuria without adverse effects, independent of BP control [29].
A number of other strategies have been tried. Adequate BP and glucose control are part of standard care of DN patients. Intensive glucose control has more impact on GFR if early instituted in patients with type 1 DM but this may not necessarily apply to patients with type 2 DM or with advanced CKD [30]. A trial of the vitamin D activator paricalcitol missed the primary endpoint of albuminuria reduction in DN and caused a transient decrease in eGFR [31]. The nephroprotective effect of statins on CKD found in experimental models has not been conclusively proven in clinical studies [32]. A 1-year dose-ranging study of pirfenidone suggested better preservation of eGFR by pirfenidone in a small number of diabetic nephropathy patients [33]. The selective endothelin antagonist atrasentan reduced albuminuria in a short-term (8 weeks) study in a small number of diabetic patients while receiving RAS inhibitors but did not assess long-term renal function [34]. Heart failure patients or with peripheral edema were excluded.
In spite of all this experimental and clinical evidence, there are 35–40% of patients with DN that progress to advanced renal disease or ESRD. The risk of progression to ESRD is still clinically relevant in other proteinuric nephropathies [35, 36]. Novel therapeutic targets are needed in CKD that are based on a clear understanding of the pathogenesis of CKD progression beyond the RAAS.
2. Oxidative Stress and Kidney Disease
Oxidative stress and inflammation promote kidney and vascular injury [37–42]. Several factors induce ROS in renal cells, such as inflammatory cytokines, Toll-like receptors, Angiotensin II, bradykinin, arachidonic acid, thrombin, growth factors, and mechanical pressure. NADPH oxidases, now renamed Nox enzymes, are key ROS generators in response to these stimuli [43, 44].
In acute kidney injury (AKI) induced by ischemic-reperfusion injury, sepsis or acute rejections ROS contribute of to endothelial and tubular injury [45, 46]. In murine models of AKI, bardoxolone methyl decreased functional and structural renal injury and increased the expression of protective genes (Nrf2, PPARγ, HO-1) on glomerular endothelium, cortical peritubular capillaries, tubules, and interstitial leukocytes [47].
ROS contribute to hypertension-induced kidney and vascular injury [41, 43, 48, 49]. The chronic complications of diabetes are characterized by a defect in Nrf2 signaling and its adaptive response to oxidative stress. This is a potential mechanism for cellular stress hypersensitivity and tissue damage [50]. Diabetic nephropathy is characterized by initial hyperfiltration, albuminuria and subsequent loss of renal function, thickening of basement membranes, expansion of mesangial matrix and interstitial fibrosis, and podocytes and renal cell damage [51]. ROS production in response to hyperglycemia, protein kinase C (PKC), advanced glycosylation end products (AGEs), free fatty acids, inflammatory cytokines, and TGF-beta1 contributes to these changes [43, 44, 52–54]. These stimuli activate the NADPH/NADPH oxidase system in renal cells. Oxidative stress induced by hyperglycemia or glucose degradation products may cause leukocyte or renal cell apoptosis and release of extracellular matrix [52, 55–59]. PKC activates NF-kappaB, extending the inflammatory response [60]. TGF-beta signaling is key to the excessive matrix formation [61, 62]. The activation of Nrf2 is increased in diabetic nephropathy and can ameliorate mesangial damage via partial inhibition of TGF-beta1 and reduction of extracellular matrix deposition [63]. ACE inhibitors lower TGF-beta in urine from DN patients. In rat DN glomerular HO-1 is increased, evidencing oxidative stress [52, 64]. ROS can activate several transcription factors such as NF-kappaB, AP-1, Sp1, which in turn affect the expression of mediators of inflammation, fibrosis, and cell death [52, 65] (Figure 1(a)).
Figure 1.
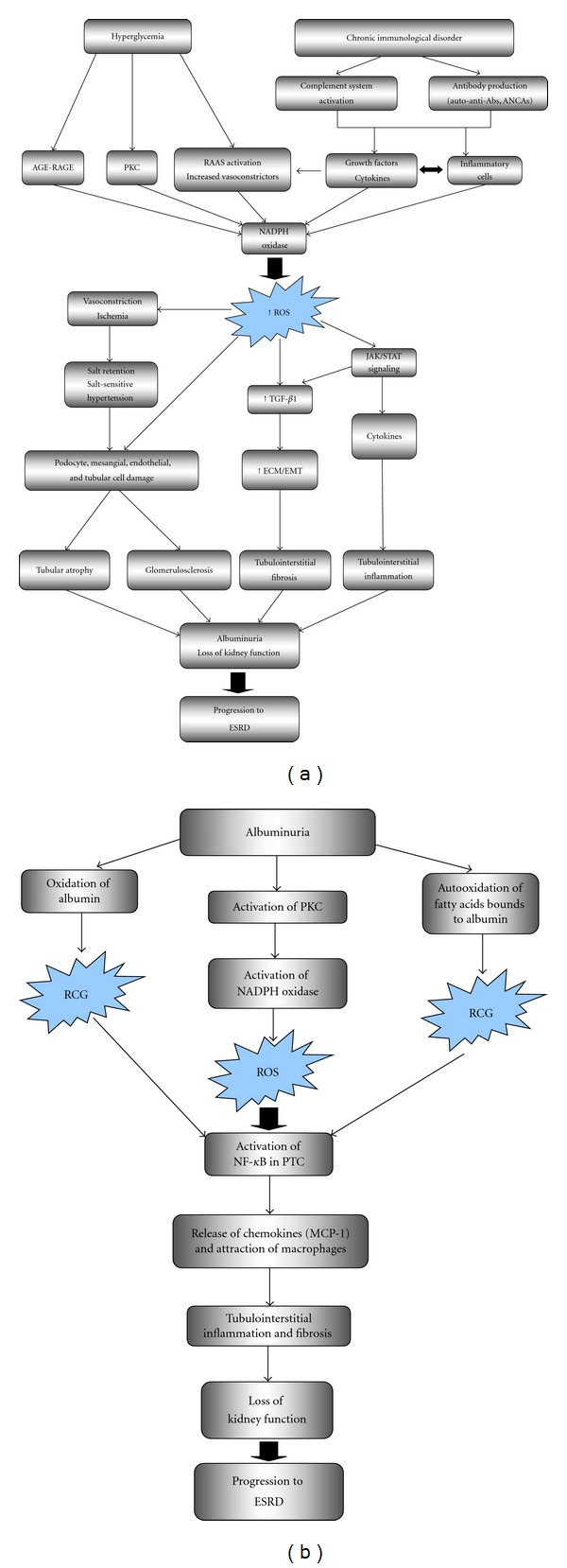
Overview of interrelation of ROS with other key pathogenic factors in kidney disease. (a) Role of ROS in diabetic nephropathy and immune-mediated glomerulonephritis. ROS are induced in renal cells in response to high glucose, AGE, and cytokines. PKC, NADPH oxidase, and mitochondrial metabolism are key to ROS generation. ROS activate signal transduction cascade and transcription factors, leading to upregulation of genes and proteins involved in renal cell injury, glomerular and interstitial extracellular matrix deposition, and recruitment of inflammatory cells, promoting albuminuria and progression of chronic kidney disease. (b) Role of albuminuria and ROS in tubular damage and progression of CKD. Albuminuria injures PTC and activates them to release chemokines that attract macrophages and promote tubulointerstitial fibrosis. Membrane NADPH oxidase is the main source of the ROS. It is possible the generation of other reactive species, as carbonyl groups derived from abnormal oxidation of albumin and fatty acids bound to albumin. Abs: antibodies, AGE: advanced glycation end products, ANCA: antineutrophil cytoplasmic antibodies, ECM: extracellular matrix, EMT: epithelial-mesenchymal transition, ESRD: end-stage renal disease, MCP-1: monocyte chemoattractant protein-1, NADPH: nicotinamide adenine dinucleotide phosphate, NF-kappa B: nuclear factor kappa B, PKC: protein kinase C, PTC: proximal tubular cell, RAAS: renin-angiotensin-aldosterone system, ROS: reactive oxygen species, RCG: reactive carbonyl groups, TGF-β1: transforming growth factor beta 1.
ROS also contribute to renal injury in experimental glomerulonephritis. In experimental anti-Thy 1 glomerulonephritis, ROS enhance cell proliferation and matrix accumulation and fibrosis and this is improved by the antioxidant alpha-lipoic acid [66, 67]. ROS also regulate the immune response [68]. In nephrotoxic nephritis, neutrophils promote glomerular TNF-alpha expression via H2O2 production [69]. TNF-alpha is a key mediator of glomerular injury [70]. Interstitial inflammatory leukocytes in proliferative glomerulonephritis locally generate ROS and contribute to sodium retention [71]. Angiotensin II promotes ROS-mediated F-actin cytoskeleton rearrangement, resulting in podocyte injury [72]. In cultured podocytes AT1R signaling activates Rac-1 and NADPH oxidase to produce additional ROS and downregulates the antioxidant protein peroxiredoxin (Prdx2) [73]. In experimental passive Heymann nephritis, a model of membranous nephropathy, C5b-9 activation promotes ROS-mediated injury in glomerular cells [74, 75]. In this regard, evidence for oxidative stress, the glomerular neoexpression of aldose reductase (AR) and SOD2, and the appearance of anti-AR and anti-SOD2 autoantibodies was recently reported in human membranous nephropathy suggesting that oxidative stress may generate new autoimmune targets [76].
In lupus nephritis, multiple abnormalities in T and B cells lead to autoimmune renal inflammation and ROS production [77]. Nrf2-knockout mice showed impaired antioxidant activity, increased oxidative stress, and a lupus-like autoimmune nephritis with glomerular injury, impaired kidney function, and a shortened lifespan. Thus, Nrf2 deficiency could lead to systemic autoimmune inflammation with enhanced lymphoproliferation [78]. In antineutrophil cytoplasmic antibodies (ANCAs-) associated vasculitis, ANCA-activated neutrophils and monocytes release MPO and generate ROS, producing endothelial and tissue damage [79, 80]. ANCAs also promote ROS-dependent dysregulation of neutrophil apoptosis [81] (Figure 1(a)).
In proteinuric nephropathies and independently of etiology, the presence of albumin in urine activates proximal tubules to release chemokines that promote interstitial inflammation [82]. Albumin uptake by tubular cells activates PKC and ROS generation via NADPH oxidase which activates NF-kappaB and production of inflammatory mediators [83, 84] (Figure 1(b)).
Accelerated atherogenesis is common in early and advanced stages of CKD [40, 85]. Oxidative stress contributes to the Immune inflammation-Renal injury-Atherosclerosis complex (IRA paradigm), a condition present in AKI and in CKD, and accelerated atherogenesis [86, 87].
A simplified overview of several components of oxidative stress and its interrelations with other key elements of pathogenesis and progression in kidney disease is shown in Figure 1(a).
3. Oxidative Stress and the Keap1-Nrf2 Pathway
Reactive oxygen species (ROS) include superoxide anion (SOA), hydrogen peroxide (H2O2), and hydroxyl radical. ROS are formed continuously as by-product of aerobic metabolism. Sources of ROS include the mitochondrial electron transport chain, metabolism of arachidonate by cyclooxygenases or lipoxygenases, cytochrome P450 enzymes, NADPH oxidases, or nitric oxide synthetases [88]. ROS contribute to killing bacteria, and genetic defects of NADPH oxidase cause chronic granulomatosis [89]. However, ROS may cause chemical damage to DNA, proteins, and unsaturated lipids and lead to cell death. ROS contribute to multiple pathologic processes [48, 90]. In this regard, homeostasis is maintained through a complex set of antioxidants mechanisms that prevent oxidative stress-induced injury. The main mechanisms are enzymes that catalyze antioxidant reactions: glutathione peroxidase, superoxide dismutase, catalase, Hem-oxygenase (HO-1), NADPH-quinone oxidoreductase and glutamate-cysteine ligase. These enzymes are encoded by stress-response genes or phase 2 genes that contain antioxidant response elements (AREs) in their regulatory regions [88, 91]. Nrf2 is the principal transcription factor that binds to the ARE promoting transcription. Actin-tethered Keap1 is a cytosolic repressor that binds to and retains Nrf2 in the cytoplasm, promoting its proteasomal degradation. Inducers of phase 2 genes modify specific cysteine residues of Keap1 resulting in conformational changes that render Keap1 unable to repress Nrf2. Consequently, Nrf2 activates the transcription of phase 2 genes. Oleanolic acid activates the ARE-Keap1-Nrf2 pathway, resulting in reduced proinflammatory activity of the IKK-beta/NF-kappaB pathway, increases production of antioxidant/reductive molecules, and decreases oxidative stress, thereby restoring redox homeostasis in areas of inflammation. In various cell lines, this results in inhibition of proliferation, promotion of differentiation and apoptosis induction [91–93]. Synthetic analogues of oleanolic acid, named triterpenoids, are potent anti-inflammatory agents that activate the ARE-Keap1-Nrf2 pathway [91]. Bardoxolone methyl, also known as CDDOMe, is a triterpenoid whose nephroprotective action has been recently explored in humans.
4. Antioxidant Agents in Kidney Disease
Epidemiological studies have demonstrated association between inflammatory and oxidative stress markers with cardiovascular and renal outcomes in CKD and ESRD [40, 94–96]. Experimental data in animal models of renal disease suggest beneficial effects of antioxidants agents, but results in human studies are limited and controversial.
In early experimental diabetes mellitus in hypertensive rats, the administration of tempol, an antioxidant SOD mimetic, corrected the oxidative imbalance and improved oxidative stress-induced renal injury, decreasing albuminuria and fibrosis [97]. Similar protection was afforded by the antioxidants N-acetyl-L-cysteine (thiol) and kallistatin in Dahl salt-sensitive rats [98, 99]. In spontaneously hypertensive rats, a lifelong antioxidant-rich diet diminished the severity of hypertension, improved oxidative stress and ameliorated abnormalities of antioxidant enzyme expressions and activities in contrast to regular diet [100]. In summary, in models of hypertensive rats, synthetic and natural antioxidants induced renal and endothelial protection with reduction of oxidative stress. In a model of ischaemia reperfusion and cyclosporin toxicity after unilateral nephrectomy, the blockage of the mitochondrial enzymes monoamine oxidases with pargyline 28 days following surgery prevented H2O2 production and improved renal function and renal inflammation (lower IL-1β and TNF-α gene expression) [101]. Pargyline administrated before ischemia reperfusion significantly reduced apoptosis, necrosis, and fibrosis. This effect was associated to decreased expression of TGF-β1, collagen types I, III, and IV and to the normalization of SOD1, catalase, and inflammatory gene expression. In models of renal chronic failure (5/6 nephrectomy rats) [102], AST-120, an oral carbonic adsorbent, improved the oxidative stress in endothelial cells, measured as oxidized/unoxidized albumin ratio. This effect was reached reducing the blood levels of indoxyl sulfate, an uremic toxin that induces ROS. In another model of remnant kidney, the administration of omega-3 fatty acids, an effective compound in mitigating atherosclerosis, significantly lowered several components of oxidative stress and markers of inflammatory and fibrotic response. Furthermore, it attenuated tubulointerstitial fibrosis and inflammation in the remnant kidney [103]. In anti-Thy1 glomerulonephritis, the treatment with parthenolide, an anti-inflammatory agent related to the triterpenoid family, diminished renal inflammation via NF-kappaB inhibition, decreased MCP-1 and iNOS, and improved proteinuria, tubular, and glomerular damage [104]. The beneficial effect of exogenous antioxidants shown in animal models with hypertension or chronic renal failure has not been demonstrated in people with clinical hypertension [96, 105] or CKD.
5. Nephroprotection by Bardoxolone Methyl
Bardoxolone was initially described as an agent that protected cells from radiation-induced damage (radiation mitigator) through Nrf2-dependent and -independent pathways [106]. In humans, its potential antineoplasic activity was evaluated. In phase 1 trials in oncologic patients, bardoxolone unexpectedly improved kidney function, assessed as serum creatinine and creatinine clearance, especially in patients with previous CKD. These findings lead to evaluate potential nephroprotective actions in patients with CKD and type 2 DM, first in an exploratory phase II open-label trial and then in a larger randomized clinical trial. In the first trial [107], 20 patients older than 18 years, with moderate-severe diabetic CKD, were evaluated after 8 weeks of bardoxolone at increasing oral doses of 25 to 75 mg/day. Notably, there was a significant increase in estimated GFR at 4 weeks (+2.8 mL/min/1.73 m2) with 25 mg/day and at 8 weeks (+7.2 mL/min/1.73 m2) with 75 mg/day. Serum creatinine and BUN decreased and creatinine clearance increased, without changes in total excretion or tubular secretion of creatinine. Unfortunately, GFR was not measured. Blood pressure did not change, and albuminuria had a small albeit not statistically significant increase. Markers of vascular injury and inflammation were improved by treatment with bardoxolone, suggesting a potential beneficial effect on endothelial injury. There were not changes in urine NGAL or NAG adjusted for creatinine concentration, suggesting lack of significant renal toxicity associated to bardoxolone. There were few adverse effects, mainly muscle spasms and a self-limited increase of hepatic enzymes without a true hepatic toxicity. A short followup and an open-label design without control group are major limitations of this study, and they do not allow drawing solid conclusions about the efficacy and long-term safety of this drug on relevant renal outcomes.
The beneficial effect on eGFR was confirmed in a larger, multicenter, double-blind, randomized trial [108]. This trial randomized 227 patients with moderate-severe CKD and type 2 DM, with stable treatment with ACEI/ARB, to bardoxolone 25, 75 or 150 mg/day or placebo for 52 weeks. Patients were categorized by GFR, urinary albumin-creatinine ratio (UACR), and HbA1c. Patients with hepatic dysfunction or recent cardiovascular events were excluded. A significant improvement in the primary endpoint (change of GFR at 24 weeks) was observed in all bardoxolone groups (+8.2, +11.4 and +10.4 mL/min/1.73 m2 resp.) versus 0 mL/min/1.73m2 in the placebo group. The secondary endpoint (change of GFR at 52 weeks) also was significantly improved in bardoxolone groups (+5.8, +10.5 and +9.3 mL/min/1.73 m2 resp. versus 0). More patients in the placebo group had a GFR decrease ≥25% with respect to baseline value at 24 and 52 weeks. Additionally, serum BUN, phosphorus, and uric acid were significantly lower at 24 and 52 weeks in all bardoxolone groups when compared to placebo.
Potential unwanted effects included a mild but significant increase of UACR and decreased serum magnesium. UACR increased in patients receiving 75 or 150 mg/day bardoxolone versus placebo. This was observed at 24 and 52 weeks of treatment, but UACR decreased when patients stopped the therapy, suggesting that this effect is reversible. There was an inverse correlation between changes in serum BUN, phosphorus, uric acid, magnesium, and changes in eGFR, as well as a direct correlation between changes in eGFR and changes in UACR, suggesting that changes in eGFR may be the basis for the other observed changes. Interestingly, there was a trend toward higher systolic BP values in the 75 mg bardoxolone group, which was observed despite weight loss and that will merit close attention in further trials. The main adverse effects were muscle spasms (63% of patients in the 75 mg group) and nausea (25%).
Another significant effect was loss of body weight. This appears to be related to decreased appetite and/or nausea and may be a welcome addition to the therapeutic armamentarium for patients with increased body mass index (BMI). Indeed, weight loss was more evident in patients with higher (>35 kg/m2) BMI (mean change −10 kg). However, and perhaps worryingly, it was also observed in patients with normal BMI (−3 kg over 52 weeks).
The increased eGFR and effects on systolic BP and albuminuria are interesting results on surrogates renal variables which requires more long-term studies. These parameters and, more importantly, cardiac and renal hard end-points (cardiovascular death and progression to ESRD) will be studied at 2 years of followup in an ongoing randomized clinical trial in 1600 patients older than 18 years with advanced CKD (stage 4) and type 2 DM [109]. This study will compare bardoxolone versus placebo in patients receiving standard of care.
6. Conclusions and Recommendations
RAAS blockade is the mainstay of current therapy to slow progression of diabetic and nondiabetic CKD, but this strategy is frequently not enough. Consequently, an important number of patients progress to ESRD. There is solid experimental evidence for a key role of ROS and oxidative stress and their interplay with RAAS and inflammation, in the pathogenesis of CKD. Bardoxolone methyl, a novel synthetic triterpenoid with antioxidant and anti-inflammatory properties, has shown to improve kidney function in patients with advanced DN already receiving RAAS blockers, with few adverse events. This may be a welcomed addition to the therapeutic armamentarium if data are confirmed in larger, longer trials (Figure 2). However, the relative importance and eventual management of the observed influence of bardoxolone on UACR, magnesium, and body weight must be further studied.
Figure 2.
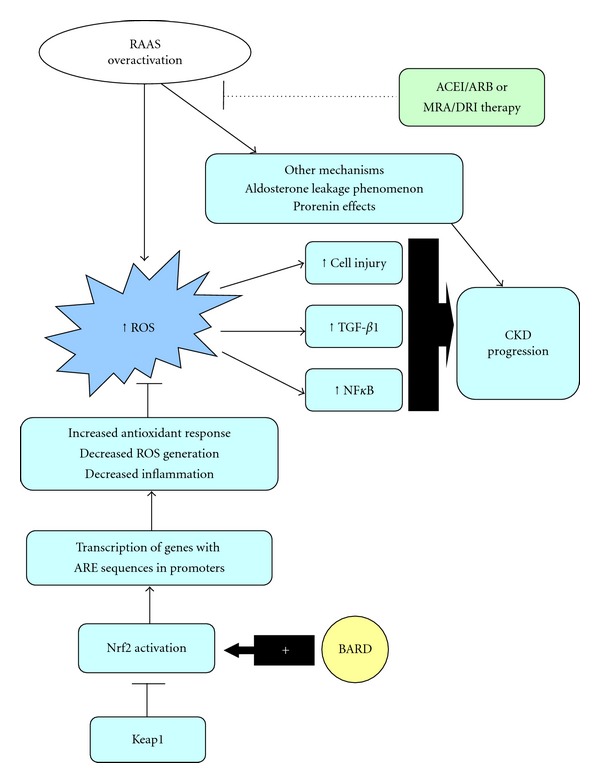
Overview of RAAS blockers and bardoxolone in pathogenic pathways in kidney diseases. RAAS blockers target several pathogenic pathways in kidney injury, including those generating ROS. However, there are several escape mechanisms (aldosterone breakthrough, increased prorenin effects) as well as less sensitive lesions (significant loss of kidney function, ischemic disease, persistent immune activity). BARD promotes activation of the Nrf2 transcription factor, that is released of the inhibitory Keap1 protein and migrates to the nucleus where it regulates transcription of genes containing ARE sequences in their promoters. These phase 2 response genes are collectively involved in the reduction of ROS and inhibition of NF-kappaB. Thus, BARD could promote renal protection through antioxidants and anti-inflammatory effects be promoting the activity of the Nrf2 transcription factor and inhibiting the activity of the NF-kappaB transcription factor. ACE/ACEIs: angiotensin converting enzyme/angiotensin converting enzyme inhibitors, ARBs: angiotensin receptor blockers, AREs: antioxidant response elements, BARD: bardoxolone methyl, CKD: chronic kidney disease, DRI: direct renin inhibitor, mineralocorticoid receptor antagonists, Keap1: Kelch-like ECH-associated protein 1, MRA: mineralocorticoid receptor antagonists, NF-kappaB: nuclear factor kappa B, Nrf2: nuclear factor (erythroid derive 2)-like 2, RAAS: renin-angiotensin-aldosterone system, ROS: reactive oxygen species, TGFβ-1: transforming growth factor beta 1.
Acknowledgments
This paper is supported by the following Grants: ISCIII and FEDER funds CP04/00060, FIS PS09/00447, Sociedad Española de Nefrologia, ISCIII-RETIC REDinREN/RD06/0016, Comunidad de Madrid/FRACM/S-BIO0283/2006, S2010/BMD-2378, Programa Intensificación Actividad Investigadora (ISCIII/Agencia Laín-Entralgo/CM) to AO, and ISCIII-Redes RECAVA (RD06/0014/0035), ISCIII funds PI10/00072, EUS2008/03565 to JE and Fundacion Lilly, cvREMOD.
References
- 1.Center for Disease Control and Prevention. Prevalence of chronic kidney disease and associated risk factors: United States, 1999–2004. Morbidity and Mortality Weekly Report . 2007;56:161–165. . [PubMed] [Google Scholar]
- 2.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care . 2004;27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. . [DOI] [PubMed] [Google Scholar]
- 3.Ruggenenti P, Cravedi P, Remuzzi G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nature Reviews Nephrology . 2010;6(6):319–330. doi: 10.1038/nrneph.2010.58. . [DOI] [PubMed] [Google Scholar]
- 4.Morgan T. Renin, angiotensin, sodium and organ damage. Hypertension Research . 2003;26(5):349–354. doi: 10.1291/hypres.26.349. . [DOI] [PubMed] [Google Scholar]
- 5.Ferrario CM. Role of angiotensin II in cardiovascular disease—therapeutic implications of more than a century of research. Journal of the Renin-Angiotensin-Aldosterone System . 2006;7(1):3–14. doi: 10.3317/jraas.2006.003. . [DOI] [PubMed] [Google Scholar]
- 6.Cooper ME. The role of the renin-angiotensin-aldosterone system in diabetes and its vascular complications. American Journal of Hypertension . 2004;17(11):16S–20S. doi: 10.1016/j.amjhyper.2004.08.004. . [DOI] [PubMed] [Google Scholar]
- 7.Brewster UC, Perazella MA. The renin-angiotensin-aldosterone system and the kidney: effects on kidney disease. American Journal of Medicine . 2004;116(4):263–272. doi: 10.1016/j.amjmed.2003.09.034. . [DOI] [PubMed] [Google Scholar]
- 8.Siragy HM, Carey RM. Protective role of the angiotensin AT2 receptor in a renal wrap hypertension model. Hypertension . 1999;33(5):1237–1242. doi: 10.1161/01.hyp.33.5.1237. . [DOI] [PubMed] [Google Scholar]
- 9.Ondetti MA, Rubin B, Cushman DW. Design of specific inhibitors of angiotensin converting enzyme: new class of orally active antihypertensive agents. Science . 1977;196(4288):441–444. doi: 10.1126/science.191908. . [DOI] [PubMed] [Google Scholar]
- 10.Remuzzi G. Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. The Lancet . 1996;349(9069):1857–1863. . [PubMed] [Google Scholar]
- 11.Taguma Y, Kitamoto Y, Futaki G. Effect of captopril on heavy proteinuria in azotemic diabetics. The New England Journal of Medicine . 1985;313(26):1617–1620. doi: 10.1056/NEJM198512263132601. . [DOI] [PubMed] [Google Scholar]
- 12.Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. The New England Journal of Medicine . 2001;345(12):861–869. doi: 10.1056/NEJMoa011161. . [DOI] [PubMed] [Google Scholar]
- 13.Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. The New England Journal of Medicine . 2001;345(12):851–860. doi: 10.1056/NEJMoa011303. . [DOI] [PubMed] [Google Scholar]
- 14.Jacobsen P, Andersen S, Rossing K, Hansen BV, Parving HH. Dual blockade of the renin-angiotensin system in type 1 patients with diabetic nephropathy. Nephrology Dialysis Transplantation . 2002;17(6):1019–1024. doi: 10.1093/ndt/17.6.1019. . [DOI] [PubMed] [Google Scholar]
- 15.Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. British Medical Journal . 2000;321(7274):1440–1444. doi: 10.1136/bmj.321.7274.1440. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobsen P, Andersen S, Jensen BR, Parving HH. Additive effect of ACE inhibition and angiotensin II receptor blockade in type I diabetic patients with diabetic nephropathy. Journal of the American Society of Nephrology . 2003;14(4):992–999. doi: 10.1097/01.asn.0000054495.96193.bf. . [DOI] [PubMed] [Google Scholar]
- 17.Ruggenenti P, Mosconi L, Sangalli F, et al. Glomerular size-selective dysfunction in NIDDM is not ameliorated by ACE inhibition or by calcium channel blockade. Kidney International . 1999;55(3):984–994. doi: 10.1046/j.1523-1755.1999.055003984.x. . [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z, Shahinfar S, Keane WF, et al. Importance of baseline distribution of proteinuria in renal outcomes trials: lessons from the reduction of endpoints in NIDDM with the angiotensin II antagonist losartan (RENAAL) study. Journal of the American Society of Nephrology . 2005;16(6):1775–1780. doi: 10.1681/ASN.2004080632. . [DOI] [PubMed] [Google Scholar]
- 19.Huang W, Xu C, Kahng KW, Noble NA, Border WA, Huang Y. Aldosterone and TGF-β1 synergistically increase PAI-1 and decrease matrix degradation in rat renal mesangial and fibroblast cells. American Journal of Physiology . 2008;294(6):F1287–F1295. doi: 10.1152/ajprenal.00017.2008. . [DOI] [PubMed] [Google Scholar]
- 20.Nagase M, Shibata S, Yoshida S, Nagase T, Gotoda T, Fujita T. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension . 2006;47(6):1084–1093. doi: 10.1161/01.HYP.0000222003.28517.99. . [DOI] [PubMed] [Google Scholar]
- 21.Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system. Nature Reviews Nephrology . 2010;6(5):261–273. doi: 10.1038/nrneph.2010.30. . [DOI] [PubMed] [Google Scholar]
- 22.Struthers A, Krum H, Williams GH. A comparison of the aldosterone-blocking agents eplerenone and spironolactone. Clinical Cardiology . 2008;31(4):153–158. doi: 10.1002/clc.20324. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney International . 2006;70(12):2116–2123. doi: 10.1038/sj.ki.5001854. . [DOI] [PubMed] [Google Scholar]
- 24.Mehdi UF, Adams-Huet B, Raskin P, Vega GL, Toto RD. Addition of angiotensin receptor blockade or mineralocorticoid antagonism to maximal angiotensin-converting enzyme inhibition in diabetic nephropathy. Journal of the American Society of Nephrology . 2009;20(12):2641–2650. doi: 10.1681/ASN.2009070737. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Navaneethan SD, Nigwekar SU, Sehgal AR, Strippoli GFM. Aldosterone antagonists for preventing the progression of chronic kidney disease: a systematic review and meta-analysis. Clinical Journal of the American Society of Nephrology . 2009;4(3):542–551. doi: 10.2215/CJN.04750908. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y, Wongamorntham S, Kasting J, et al. Renin increases mesangial cell transforming growth factor-β1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney International . 2006;69(1):105–113. doi: 10.1038/sj.ki.5000011. . [DOI] [PubMed] [Google Scholar]
- 27.Ichihara A, Suzuki F, Nakagawa T, et al. Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor-deficient mice. Journal of the American Society of Nephrology . 2006;17(7):1950–1961. doi: 10.1681/ASN.2006010029. . [DOI] [PubMed] [Google Scholar]
- 28.Persson F, Rossing P, Reinhard H, et al. Renal effects of aliskiren compared with and in combination with irbesartan in patients with type 2 diabetes, hypertension, and albuminuria. Diabetes Care . 2009;32(10):1873–1879. doi: 10.2337/dc09-0168. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. The New England Journal of Medicine . 2008;358(23):2433–2446. doi: 10.1056/NEJMoa0708379. . [DOI] [PubMed] [Google Scholar]
- 30.De Boer IH, Sun W, Cleary PA, et al. Intensive diabetes therapy and glomerular filtration rate in type 1 diabetes. The New England Journal of Medicine . 2011;265:2366–2376. doi: 10.1056/NEJMoa1111732. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Zeeuw D, Agarwal R, Amdahl M, et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. The Lancet . 2010;376(9752):1543–1551. doi: 10.1016/S0140-6736(10)61032-X. . [DOI] [PubMed] [Google Scholar]
- 32.Rutter MK, Prais HR, Charlton-Menys V, et al. Protection against nephropathy in diabetes with atorvastatin (PANDA): a randomized double-blind placebo-controlled trial of high- vs. low-dose atorvastatin (1) Diabetic Medicine . 2011;28(1):100–108. doi: 10.1111/j.1464-5491.2010.03139.x. . [DOI] [PubMed] [Google Scholar]
- 33.Sharma K, Ix JH, Mathew AV, et al. Pirfenidone for diabetic nephropathy. Journal of the American Society of Nephrology . 2011;22(6):1144–1151. doi: 10.1681/ASN.2010101049. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohan DE, Pritchett Y, Molitch M, et al. Addition of atrasentan to renin-angiotensin system blockade reduces albuminuria in diabetic nephropathy. Journal of the American Society of Nephrology . 2011;22(4):763–772. doi: 10.1681/ASN.2010080869. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trachtman H, Fervenza FC, Gipson DS, et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-β antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney International . 2011;79(11):1236–1243. doi: 10.1038/ki.2011.33. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waldman M, Austin HA., III Controversies in the treatment of idiopathic membranous nephropathy. Nature Reviews Nephrology . 2009;5(8):469–479. doi: 10.1038/nrneph.2009.101. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruiz-Ortega M, Ortiz A. Angiotensin II and reactive oxygen species. Antioxidants and Redox Signaling . 2005;7(9-10):1258–1260. doi: 10.1089/ars.2005.7.1258. . [DOI] [PubMed] [Google Scholar]
- 38.Lazaro A, Gallego-Delgado J, Justo P, et al. Long-term blood pressure control prevents oxidative renal injury. Antioxidants and Redox Signaling . 2005;7(9-10):1285–1293. doi: 10.1089/ars.2005.7.1285. . [DOI] [PubMed] [Google Scholar]
- 39.Neria F, Castilla MA, Sanchez RF, et al. Inhibition of JAK2 protects renal endothelial and epithelial cells from oxidative stress and cyclosporin A toxicity. Kidney International . 2009;75(2):227–234. doi: 10.1038/ki.2008.487. . [DOI] [PubMed] [Google Scholar]
- 40.Cachofeiro V, Goicochea M, de Vinuesa SG, Oubía P, Lahera V, Lũo J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney International . 2008;74(supplement 111):S4–S9. doi: 10.1038/ki.2008.516. . [DOI] [PubMed] [Google Scholar]
- 41.Vaziri ND, Rodriguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nature Clinical Practice Nephrology . 2006;2(10):582–593. doi: 10.1038/ncpneph0283. . [DOI] [PubMed] [Google Scholar]
- 42.Kim HJ, Sato T, Vaziri ND, Rodriguez-Iturbe B. Role of intrarenal angiotensin system activation, oxidative stress, inflammation, and impaired nuclear factor-erythroid-2-related factor 2 activity in the progression of focal glomerulosclerosis. Journal of Pharmacology and Experimental Therapeutics . 2011;337(3):583–590. doi: 10.1124/jpet.110.175828. . [DOI] [PubMed] [Google Scholar]
- 43.Wardle EN. Cellular oxidative processes in relation to renal disease. American Journal of Nephrology . 2005;25(1):13–22. doi: 10.1159/000083477. . [DOI] [PubMed] [Google Scholar]
- 44.Pleskova M, Beck KF, Behrens MH, et al. Nitric oxide down-regulates the expression of the catalytic NADPH oxidase subunit Nox1 in rat renal mesangial cells. The FASEB Journal . 2006;20(1):139–141. doi: 10.1096/fj.05-3791fje. . [DOI] [PubMed] [Google Scholar]
- 45.Molitoris BA, Marrs J. The role of cell adhesion molecules in ischemic acute renal failure. American Journal of Medicine . 1999;106(5):583–592. doi: 10.1016/s0002-9343(99)00061-3. . [DOI] [PubMed] [Google Scholar]
- 46.Daemen MARC, Van’t Veer C, Denecker G, et al. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. Journal of Clinical Investigation . 1999;104(5):541–549. doi: 10.1172/JCI6974. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alonso de Vega JM, Diaz J, Serrano E, Carbonell LF. Oxidative stress in critically ill patients with systemic inflammatory response syndrome. Critical Care Medicine . 2002;30(8):1782–1786. doi: 10.1097/00003246-200208000-00018. . [DOI] [PubMed] [Google Scholar]
- 48.Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension . 2003;42(6):1075–1081. doi: 10.1161/01.HYP.0000100443.09293.4F. . [DOI] [PubMed] [Google Scholar]
- 49.Ruiz-Ortega M, Egido J, Lorenzo O, Rupérez M, König S, Wittig B. Angiotensin II activates nuclear transcription factor κB through AT1 and AT2 in vascular smooth muscle cells molecular mechanisms. Circulation Research . 2000;86(12):1266–1272. doi: 10.1161/01.res.86.12.1266. . [DOI] [PubMed] [Google Scholar]
- 50.Bittar MS, Al-Mulla F. A defect in Nrf2 signaling constitutes a mechanism for cellular stress hypersensitivity in type 2 diabetes. American Journal of Physiology . 2011;301(6):E1119–E1129. doi: 10.1152/ajpendo.00047.2011. . [DOI] [PubMed] [Google Scholar]
- 51.Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. Journal of the American Society of Nephrology . 2003;14(5):1358–1373. doi: 10.1097/01.asn.0000065640.77499.d7. . [DOI] [PubMed] [Google Scholar]
- 52.Lee HB, Yu MR, Yang Y, Jiang Z, Ha H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. Journal of the American Society of Nephrology . 2003;14(8, supplement 3):S241–S245. doi: 10.1097/01.asn.0000077410.66390.0f. . [DOI] [PubMed] [Google Scholar]
- 53.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature . 2001;414(6865):813–820. doi: 10.1038/414813a. . [DOI] [PubMed] [Google Scholar]
- 54.Lal MA, Brismar H, Eklof AC, Aperia A. Role of oxidative stress in advanced glycation end product-induced mesangial cell activation. Kidney International . 2002;61(6):2006–2014. doi: 10.1046/j.1523-1755.2002.00367.x. . [DOI] [PubMed] [Google Scholar]
- 55.Justo P, Sanz AB, Egido J, Ortiz A. 3,4-Dideoxyglucosone-3-ene induces apoptosis in renal tubular epithelial cells. Diabetes . 2005;54(8):2424–2429. doi: 10.2337/diabetes.54.8.2424. . [DOI] [PubMed] [Google Scholar]
- 56.Kang BPS, Frencher S, Reddy V, Kessler A, Malhorta A, Meggs LG. High glucose promotes mesangial cell apoptosis by oxidant-dependent mechanism. American Journal of Physiology . 2003;284(3):F455–F466. doi: 10.1152/ajprenal.00137.2002. . [DOI] [PubMed] [Google Scholar]
- 57.Iglesias-de La Cruz MC, Ruiz-Torres P, AlcamI J, et al. Hydrogen peroxide increases extracellular matrix mRNA through TGF-β in human mesangial cells. Kidney International . 2001;59(1):87–95. doi: 10.1046/j.1523-1755.2001.00469.x. . [DOI] [PubMed] [Google Scholar]
- 58.Catalan MP, Santamaría B, Reyero A, Ortiz A, Egido J, Ortiz A. 3,4-Di-deoxyglucosone-3-ene promotes leukocyte apoptosis. Kidney International . 2005;68(3):1303–1311. doi: 10.1111/j.1523-1755.2005.00528.x. . [DOI] [PubMed] [Google Scholar]
- 59.Ortiz A, Lorz C, Justo P, Catalan MP, Egido J. Contribution of apoptotic cell death to renal injury. Journal of Cellular and Molecular Medicine . 2001;5(1):18–32. doi: 10.1111/j.1582-4934.2001.tb00135.x. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inoguchi T, Sonta T, Tsubouchi H, et al. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. Journal of the American Society of Nephrology . 2003;14(8, supplement 3):S227–S232. doi: 10.1097/01.asn.0000077407.90309.65. . [DOI] [PubMed] [Google Scholar]
- 61.Zyyadeh FN. Mediators of diabetic renal disease: the case for TGF-β as a major mediator. Journal of the American Society of Nephrology . 2004;15(supplement 1):S55–S57. doi: 10.1097/01.asn.0000093460.24823.5b. . [DOI] [PubMed] [Google Scholar]
- 62.Weigert C, Sauer U, Brodbeck K, Pfeiffer A, Häring HU, Schleicher ED. AP-1 proteins mediate hyperglycemia-induced activation of the human TGF-β1 promoter in mesangial cells. Journal of the American Society of Nephrology . 2000;11(11):2007–2016. doi: 10.1681/ASN.V11112007. . [DOI] [PubMed] [Google Scholar]
- 63.Jiang T, Huang Z, Lin Y, Zhang Z, Fang D, Zhang DD. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes . 2010;59(4):850–860. doi: 10.2337/db09-1342. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li JM, Shah AM. ROS generation by nonphagocytic NADPH oxidase: potential relevance in diabetic nephropathy. Journal of the American Society of Nephrology . 2003;14(3):S221–S226. doi: 10.1097/01.asn.0000077406.67663.e7. . [DOI] [PubMed] [Google Scholar]
- 65.Sanz AB, Sanchez-Niño MD, Ramos AM, et al. NF-κB in renal inflammation. Journal of the American Society of Nephrology . 2010;21(8):1254–1262. doi: 10.1681/ASN.2010020218. . [DOI] [PubMed] [Google Scholar]
- 66.Shah SV. Oxidants and iron in chronic kidney disease. Kidney International, Supplement . 2004;66(91):S50–S55. doi: 10.1111/j.1523-1755.2004.09108.x. . [DOI] [PubMed] [Google Scholar]
- 67.Budisavljevic MN, Hodge L, Barber K, et al. Oxidative stress in the pathogenesis of experimental mesangial proliferative glomerulonephritis. American Journal of Physiology . 2003;285(6):F1138–F1148. doi: 10.1152/ajprenal.00397.2002. . [DOI] [PubMed] [Google Scholar]
- 68.Kantengwa S, Jornot L, Devenoges C, Nicod LP. Superoxide anions induce the maturation of human dendritic cells. American Journal of Respiratory and Critical Care Medicine . 2003;167(3):431–437. doi: 10.1164/rccm.200205-425OC. . [DOI] [PubMed] [Google Scholar]
- 69.Suzuki Y, Gómez-Guerrero C, Shirato I, et al. Pre-existing glomerular immune complexes induce polymorphonuclear cell recruitment through an Fc receptor-dependent respiratory burst: potential role in the perpetuation of immune nephritis. Journal of Immunology . 2003;170(6):3243–3253. doi: 10.4049/jimmunol.170.6.3243. . [DOI] [PubMed] [Google Scholar]
- 70.Ortiz A, Gonzalez-Cuadrado S, Bustos C, et al. Tumor necrosis factor as a mediator of glomerular damage. Journal of Nephrology . 1995;8(1):27–34. . [Google Scholar]
- 71.Rodriguez-Iturbe B, Herrera-Acosta J, Johnson RJ. Interstitial inflammation, sodium retention, and the pathogenesis of nephrotic edema: a unifying hypothesis. Kidney International . 2002;62(4):1379–1384. doi: 10.1111/j.1523-1755.2002.kid561.x. . [DOI] [PubMed] [Google Scholar]
- 72.Rodriguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: all for one and one for all. American Journal of Physiology . 2004;286(4):F606–F616. doi: 10.1152/ajprenal.00269.2003. . [DOI] [PubMed] [Google Scholar]
- 73.Hsu HH, Hoffmann S, Di Marco GS, et al. Downregulation of the antioxidant protein peroxiredoxin 2 contributes to angiotensin II-mediated podocyte apoptosis. Kidney International . 2011;80:959–969. doi: 10.1038/ki.2011.250. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neale TJ, Ojha PP, Exner M, et al. Proteinuria in passive Heymann nephritis is associated with lipid peroxidation and formation of adducts on type IV collagen. Journal of Clinical Investigation . 1994;94(4):1577–1584. doi: 10.1172/JCI117499. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Couser WG, Nangaku M. Cellular and molecular biology of membranous nephropathy. Journal of Nephrology . 2006;19(6):699–705. . [PubMed] [Google Scholar]
- 76.Prunotto M, Carnevali ML, Candiano G, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. Journal of the American Society of Nephrology . 2010;21(3):507–519. doi: 10.1681/ASN.2008121259. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Foster MH. T cells and B cells in lupus nephritis. Seminars in Nephrology . 2007;27(1):47–58. doi: 10.1016/j.semnephrol.2006.09.007. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoh K, Itoh K, Enomoto A, et al. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney International . 2001;60(4):1343–1353. doi: 10.1046/j.1523-1755.2001.00939.x. . [DOI] [PubMed] [Google Scholar]
- 79.Falk RJ, Jennette JC. ANCA are pathogenicoh—oh yes they are! Journal of the American Society of Nephrology . 2002;13(7):1977–1979. doi: 10.1681/ASN.V1371977. . [DOI] [PubMed] [Google Scholar]
- 80.Gaut JP, Byun J, Tran HD, et al. Myeloperoxidase produces nitrating oxidants in vivo. Journal of Clinical Investigation . 2002;109(10):1311–1319. doi: 10.1172/JCI15021. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harper L, Ren Y, Savill J, Adu D, Savage COS. Antineutrophil cytoplasmic antibodies induce reactive oxygen-dependent dysregulation of primed neutrophil apoptosis and clearance by macrophages. American Journal of Pathology . 2000;157(1):211–220. doi: 10.1016/S0002-9440(10)64532-4. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gómez-Chiarri M, Ortiz A, González-Cuadrado S, et al. Interferon-inducible protein-10 is highly expressed in rats with experimental nephrosis. American Journal of Pathology . 1996;148(1):301–311. . [PMC free article] [PubMed] [Google Scholar]
- 83.Tang S, Leung JCK, Abe K, et al. Albumin stimulates interleukin-8 expression in proximal tubular epithelial cells in vitro and in vivo. Journal of Clinical Investigation . 2003;111(4):515–527. doi: 10.1172/JCI16079. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morigi M, Macconi D, Zoja C, et al. Protein overload-induced NF-κB activation in proximal tubular cells requires H2O2 through a PKC-dependent pathway. Journal of the American Society of Nephrology . 2002;13(5):1179–1189. . [PubMed] [Google Scholar]
- 85.Stocker R, Keaney JF. Role of oxidative modifications in atherosclerosis. Physiological Reviews . 2004;84(4):1381–1478. doi: 10.1152/physrev.00047.2003. . [DOI] [PubMed] [Google Scholar]
- 86.Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim RM. Perspectives in renal medicine: the elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney International . 2002;62(5):1524–1538. doi: 10.1046/j.1523-1755.2002.00600.x. . [DOI] [PubMed] [Google Scholar]
- 87.Swaminathan S, Shah SV. Novel inflammatory mechanisms of accelerated atherosclerosis in kidney disease. Kidney International . 2011;80:453–463. doi: 10.1038/ki.2011.178. . [DOI] [PubMed] [Google Scholar]
- 88.Champe P, Harvey RA. Pentose Phosphate Pathway and NADPH in Biochemistry . 4th edition. Philadelphia, Pa, USA: Lippincott Williams & Wilkins; 2008. . [Google Scholar]
- 89.Sen CK. Antioxidant and redox regulation of cellular signaling: Introduction. Medicine and Science in Sports and Exercise . 2001;33(3):368–370. doi: 10.1097/00005768-200103000-00005. . [DOI] [PubMed] [Google Scholar]
- 90.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circulation Research . 2000;86(5):494–501. doi: 10.1161/01.res.86.5.494. . [DOI] [PubMed] [Google Scholar]
- 91.Dinkova-Kostova AT, Liby KT, Stephenson KK, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proceedings of the National Academy of Sciences of the United States of America . 2005;102(12):4584–4589. doi: 10.1073/pnas.0500815102. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li W, Khor TO, Xu C, et al. Activation of Nrf2-antioxidant signaling attenuates NFκB-inflammatory response and elicits apoptosis. Biochemical Pharmacology . 2008;76(11):1485–1489. doi: 10.1016/j.bcp.2008.07.017. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thimmulappa RK, Lee H, Rangasamy T, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. Journal of Clinical Investigation . 2006;116(4):984–995. doi: 10.1172/JCI25790. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lacy F, Kailasam MT, O’Connor DT, Schmid-Schönbein GW, Parmer RJ. Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension . 2000;36(5):878–884. doi: 10.1161/01.hyp.36.5.878. . [DOI] [PubMed] [Google Scholar]
- 95.Dixon LJ, Hughes SM, Rooney K, et al. Increased superoxide production in hypertensive patients with diabetes mellitus: role of nitric oxide synthase. American Journal of Hypertension . 2005;18(6):839–843. doi: 10.1016/j.amjhyper.2005.01.004. . [DOI] [PubMed] [Google Scholar]
- 96.Roggensack AM, Zhang Y, Davidge ST. Evidence for peroxynitrite formation in the vasculature of women with preeclampsia. Hypertension . 1999;33(1 I):83–89. doi: 10.1161/01.hyp.33.1.83. . [DOI] [PubMed] [Google Scholar]
- 97.Quiroz Y, Ferrebuz A, Vaziri ND, Rodriguez-Iturbe B. Effect of chronic antioxidant therapy with superoxide dismutase-mimetic drug, tempol, on progression of renal disease in rats with renal mass reduction. Nephron . 2009;112(1):e31–e42. doi: 10.1159/000210577. . [DOI] [PubMed] [Google Scholar]
- 98.Zhang L, Fujii S, Igarashi J, Kosaka H. Effects of thiol antioxidant on reduced nicotinamide adenine dinucleotide phosphate oxidase in hypertensive Dahl salt-sensitive rats. Free Radical Biology and Medicine . 2004;37(11):1813–1820. doi: 10.1016/j.freeradbiomed.2004.08.019. . [DOI] [PubMed] [Google Scholar]
- 99.Shen B, Hagiwara M, Yao YY, Chao L, Chao J. Salutary effect of kallistatin in salt-induced renal injury, inflammation, and fibrosis via antioxidative stress. Hypertension . 2008;51(5):1358–1365. doi: 10.1161/HYPERTENSIONAHA.107.108514. . [DOI] [PubMed] [Google Scholar]
- 100.Zhan CD, Sindhu RK, Pang J, Ehdaie A, Vaziri ND. Superoxide dismutase, catalase and glutathione peroxidase in the spontaneously hypertensive rat kidney: effect of antioxidant-rich diet. Journal of Hypertension . 2004;22(10):2025–2033. doi: 10.1097/00004872-200410000-00027. . [DOI] [PubMed] [Google Scholar]
- 101.Chaaya R, Alfarano C, Guilbeau-Frugier C, et al. Pargyline reduces renal damage associated with ischaemia-reperfusion and cyclosporin. Nephrology Dialysis Transplantation . 2011;26(2):489–498. doi: 10.1093/ndt/gfq445. . [DOI] [PubMed] [Google Scholar]
- 102.Shimoishi K, Anraku M, Kitamura K, et al. An oral adsorbent, AST-120 protects against the progression of oxidative stress by reducing the accumulation of indoxyl sulfate in the systemic circulation in renal failure. Pharmaceutical Research . 2007;24(7):1283–1289. doi: 10.1007/s11095-007-9248-x. . [DOI] [PubMed] [Google Scholar]
- 103.An WS, Kim HJ, Cho KH, Vaziri ND. Omega-3 fatty acid supplementation attenuates oxidative stress, inflammation, and tubulointerstitial fibrosis in the remnant kidney. American Journal of Physiology . 2009;297(4):F895–F903. doi: 10.1152/ajprenal.00217.2009. . [DOI] [PubMed] [Google Scholar]
- 104.López-Franco O, Suzuki Y, Sanjuán G, et al. Nuclear factor-κB inhibitors as potential novel anti-inflammatory agents for the treatment of immune glomerulonephritis. American Journal of Pathology . 2002;161(4):1497–1505. doi: 10.1016/s0002-9440(10)64425-2. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roberts CK, Won D, Pruthi S, et al. Effect of a short-term diet and exercise intervention on oxidative stress, inflammation, MMP-9, and monocyte chemotactic activity in men with metabolic syndrome factors. Journal of Applied Physiology . 2006;100(5):1657–1665. doi: 10.1152/japplphysiol.01292.2005. . [DOI] [PubMed] [Google Scholar]
- 106.Kim SB, Eskiocak U, Ly P, et al. Bardoxolone-methyl (CDDO-Me): an antioxidant, antiinflammatory modulator is a novel radiation countermeasure and mitigator. In: Proceedings of the 22nd Annual NASA Space Radiation Investigators’ Workshop; 2011; . [Google Scholar]
- 107.Pergola PE, Krauth M, Huff JW, et al. Effect of bardoxolone methyl on kidney function in patients with T2D and stage 3b-4 CKD. American Journal of Nephrology . 2011;33(5):469–476. doi: 10.1159/000327599. . [DOI] [PubMed] [Google Scholar]
- 108.Pergola PE, Raskin P, Toto RD, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. The New England Journal of Medicine . 2011;365(4):327–336. doi: 10.1056/NEJMoa1105351. . [DOI] [PubMed] [Google Scholar]
- 109.Bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes: the ocurrence of renal events (BEACON) Identifier: NCT01351675, http://www.clinicaltrials.gov/ [DOI] [PubMed]