

Abstract
It has been reported that hepatitis C virus (HCV) infection is closely associated with hepatic metabolic disorders. Hepatic steatosis and insulin resistance are both relatively common in patients with chronic hepatitis C. Recent investigations suggest that HCV infection changes the expression profile of lipid-metabolism-associated factors in the liver, conferring advantages to the life cycle of HCV. Moreover, insulin resistance and steatosis are independent predictors of impaired response to antiviral treatment in chronic hepatitis C. In this paper, we summarize our current knowledge of hepatic metabolic disorders and describe how HCV leads to and exploits these hepatic disorders. We also discuss the clinical significance of insulin sensitizers used to improve insulin resistance and lipid modulators used to manage lipid metabolism as potential treatment options for chronic hepatitis C.
1. Introduction
Hepatic steatosis is a well-documented histological characteristic of chronic hepatitis C virus (HCV) infection [1]. Insulin resistance or impaired glucose metabolism, is linked to hepatic steatosis in patients with chronic hepatitis C (CH-C). It is widely considered that hepatic steatosis in patients with CH-C is caused by lipid metabolic disorders, in which insulin resistance plays an important role [2]. Fat accumulation promotes oxidative stress and inflammatory reactions. A considerable number of studies have also suggested that various HCV proteins lead to alterations in lipid synthesis, catabolism and transport. In particular, HCV core protein was reported to contribute to these metabolic changes and induce reactive oxygen species generation [3, 4]. Clinically, hepatic steatosis and insulin resistance in CH-C patients are associated with hepatic fibrosis, an increased frequency of hepatocellular carcinoma, and a poor response to pegylated interferon (peg-IFN) plus ribavirin combination therapy [5].
2. HCV Infection and Insulin Resistance
It has been reported that hepatic steatosis is correlated with viral load; approximately 50% of patients with CH-C have hepatic steatosis, which enhances disease progression [6, 7]. Recent studies have shown that, as in nonalcoholic fatty liver disease (NAFLD), insulin resistance and an increased fatty acid supply to the liver are important pathogeneses of steatosis in CH-C [8]. In CH-C patients, the occurrence of insulin resistance is independent of visceral adipose tissue and hepatic steatosis and irrespective of the HCV genotype [9]. In our experience, insulin resistance is frequently observed in nonobese patients, and 36.8% patients with CH-C had a homeostasis model assessment-insulin resistance (HOMA-IR) index ≥2.5 [10]. Even though the association between the severity of insulin resistance and HCV viral load or genotype is controversial, viral eradication by antiviral therapy actually improves insulin sensitivity [11–13]. Despite the close association between chronic HCV infection and the presence of insulin resistance, the pathogenic basis of this interaction remains to be elucidated. Increasing epidemiological and experimental data suggest that the HCV core protein impairs insulin signaling, mostly by activating tumor necrosis factor α (TNFα) and members of the suppressor of cytokine signaling (SOCS) family [9, 14, 15].
SOCS proteins, which are induced by proinflammatory cytokines, induce proteasomal degradation of their target proteins, including insulin receptor substrate (IRS). Experimentally, upregulation of SOCS-1 and -3 in the liver leads to insulin resistance through several mechanisms, including degradation of IRS1 and IRS2 inhibition of insulin receptor kinase activity, and downregulation of IFN-associated innate immunity [16–18]. Activated TNFα inhibits tyrosine phosphorylation of IRS1 and IRS2, and impairs glucose transporter (GLUT)-4 translocation to the cell membrane, leading to insulin resistance and hyperinsulinemia, which can increase glycogenolysis and fatty acid synthesis [19, 20].
These changes may lead to hepatic steatosis by increasing the influx of free fatty acids via peripheral lipolysis, activation of lipogenesis-associated factors, reduced fatty acid oxidation, and decreased formation of very low-density lipoprotein (VLDL) [21]. IRS1 and IRS2 are closely linked to the regulation of glucokinase expression and lipogenic enzymes, such as sterol-regulatory element-binding protein 1c (SREBP-1c), respectively.
HCV infection, mainly through activity of the HCV core protein, decreases the expression and activity of peroxisome proliferator-activating receptor (PPAR)-α/γ in hepatocytes [22]. These effects may constitute strategies for viral survival and proliferation. PPARα and PPARγ transcriptionally regulate fatty acid β-oxidation and insulin sensitivity, respectively. Indeed, PPARγ agonists, thiazolidinediones, improve insulin sensitivity in CH-C patients. In our earlier study, we found that telmisartan, an angiotensin II receptor blocker and a potential partial PPARγ agonist, had significant therapeutic effects by attenuating insulin resistance and liver injury in patients with CH-C [10].
3. Lipid Metabolic Disorders in HCV-Infected Liver
A close association between HCV infection and lipid metabolism has been reported, and host metabolic factors and viral factors are likely to be involved in the pathogenesis of hepatic steatosis (see Figure 1). HCV core protein, which is localized to the membrane of lipid vesicles, induces hepatic fat accumulation by activating SREBP-1c [23, 24]. It also inhibits microsomal triglyceride transfer protein (MTP) activity, which is needed for VLDL assembly and excretion [25]. HCV infection decreases hepatic expression of PPARα, which negatively regulates fatty acid uptake and positively regulates β-oxidation, and promotes de novo lipid and cholesterol generation by enhancing the activities of SREBP-1 and -2 [24, 26].
Figure 1.
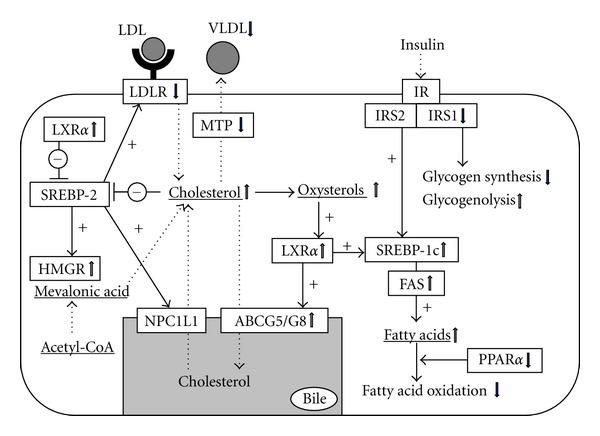
Expression profile of lipid metabolism-associated factors in chronic hepatitis C. ABCG5/G8, ATP-binding cassette G5/G8; FAS, fatty acid synthase; HMGR, HMG-CoA reductase; IR, insulin receptor; IRS, insulin receptor substrate; LDLR, LDL receptor; LXRα, liver X receptor α; MTP, microsomal triglyceride transfer protein; NPC1L1, Niemann-Pick C1-like 1; PPARα, peroxisome proliferator-activating receptor α; SREBP, sterol regulatory element-binding protein.
In our evaluation of the expression of lipid metabolism-associated genes, the regulation of lipid metabolism was impaired in HCV-infected liver [27, 28]. The expression profiles revealed that HCV infection induced intrahepatic accumulation of cholesterol as well as triglycerides, resulting in a marked reduction of low-density lipoprotein receptor (LDLR) to decrease LDL-cholesterol uptake, and upregulated ATP-binding cassette G5 to increase cholesterol output. However, de novo cholesterol and fatty acid synthesis continued to increase, perhaps because of disrupted negative feedback pathways. This uncontrolled expression pattern is almost similar in NAFLD [29, 30]. However, HCV core protein interferes with the assembly and secretion of VLDL via inactivation of MTP, leading to hypobetalipoproteinemia, whereas, in NAFLD, MTP activity is enhanced and hyperlipidemia is common [8]. This expression pattern was also apparent in a preliminary evaluation of an HCV replicon system. Cholesterol is synthesized in hepatocytes through the mevalonate pathway, which is promoted by several enzymes, including HMG-CoA reductase (HMGR). Normally, the expression of LDLR and HMGR is regulated by the transcription factor SREBP-2 according to the intracellular cholesterol load. However, despite marked cholesterol accumulation, HMGR expression was greatly enhanced in HCV-infected liver [27, 28]. During cholesterol overload, the levels of cholesterol metabolites increase, including oxysterols, which act as agonistic ligands of liver X receptor-α (LXRα). These metabolites activate the LXRα-SREBP1c axis, which ultimately leads to activation of fatty acid synthesis. Notably, LXRα expression was also enhanced in HCV-infected liver [27, 28].
In addition to the core and nonstructural HCV proteins, the activation of cholesterol and fatty acid biosynthesis play a critical role in viral assembly, release, and infectivity [31–33]. Accordingly, viral interactions with the host's lipid metabolic pathways appear to be essential for the life cycle of HCV. Attachment of the virus to the cell surface LDLR represents the first stage of HCV entry into hepatocytes, and β-lipoproteins influence HCV proliferation [34, 35]. Serum HCV antigen levels are negatively correlated with serum β-lipoprotein levels [36]. The resulting lipid droplets supply lipoproteins and lipids and provide a site for viral assembly. These changes seem to be necessary or beneficial for HCV replication. The mevalonate pathway of de novo cholesterol synthesis, in which HMGR is a rate-limiting enzyme, is also responsible for the synthesis of farnesyl pyrophosphate and geranylgeranyl pyrophosphate that are essential for viral replication [37]. These molecules are needed to activate small GTPases, such as Rho and Ras. Therefore, HCV may need lipids not only as components of virus particles but also to modulate the host's cell signaling pathways.
4. Role of the Cannabinoid System in HCV-Infected Liver
Endocannabinoids, such as anandamide and 2-arachidonoylglycerol (2-AG), are synthesized from lipid precursors in cellular membranes and specifically target cannabinoid receptors (CB) 1 and CB2 [38]. Recent studies have suggested that the hepatic cannabinoid system is involved in the pathogenesis of NAFLD by activating CB1 and that steatogenic factors, such as a high-fat diet, induce the synthesis of endocannabinoids and CB1 [39–41]. Although the signal transduction pathways have not been fully characterized, CB1 activation enhances the expression of several lipogenic factors, including SREBP-1c and fatty acid synthase (FAS) and downregulates factors involved in fatty acid oxidation, such as carnitine palmitoyltransferase I, resulting in steatosis and insulin resistance. Experimentally, steatogenic factors appear to activate CB2, but CB2 is mostly found in immune system cells. Therefore, CB2 activation may play a protective role against the inflammatory and fibrogenic responses in steatohepatitis [42, 43].
Because daily cannabis use was proposed as a risk factor for the severity of steatosis and progression of fibrosis in patients with CH-C [44], we determined the role of the hepatic cannabinoid system in HCV infection using HCV subgenomic replicon cells, which stably express viral nonstructural proteins (NS3, NS4A/4B, NS5A, and NS5B). Although the tested cannabinoid, anandamide, cannot be detected in culture medium, CB1 expression and triglyceride accumulation increased in replicon cells, as did the expression levels of several lipid synthesis-associated genes (SREBP-1c, FAS, and HMGR). IFNα treatment downregulates the expression of viral proteins and reduces triglyceride accumulation and gene expression of CB1, SREBP-1c, FAS, and HMGR. Meanwhile, treatment with a CB1 agonist increased, and a CB1 antagonist treatment decreased triglyceride accumulation in replicon cells.
These findings support the possibility that HCV infection activates the hepatic cannabinoid system and enhances steatotic changes in the liver. In healthy human liver, the hepatocytic expression of CB1 and CB2 is very low or even absent, as are endocannabinoid levels [45–48]. Marked upregulation of these receptors and endocannabinoid levels (anandamide and 2-AG) has been reported in the cirrhotic liver [45, 49–51]. Moreover, in acute liver damage, the expression of CB1 and CB2 is enhanced, and the degree and duration of inflammation may be an important factor for controlling CB1 expression or activation of the cannabinoid system. However, in a quantification assay using real-time polymerase chain reaction, CB1 gene expression was very low in liver samples from CH-C patients and healthy individuals (unpublished data). In patients with CH-C, more severe inflammation or fibrosis may be needed to activate the cannabinoid system.
As described above, there is a discrepancy between in vitro data in HCV replicon cells and findings in the liver of patients with CH-C. Therefore, it is questionable whether activation of the cannabinoid system significantly affects metabolic disorders in the HCV-infected liver. Of note, some researchers have proposed the existence of cannabinoid receptors other than CB1 and CB2 and endocannabinoids other than anandamide and 2-AG [52], although these have not yet been clearly detected. Therefore, still unknown cannabinoids and/or receptors may play a leading role in hepatic metabolic disorders in patients with CH-C.
5. Therapeutic Strategies Using Metabolic Modulators
Clinically, antioxidants, such as ursodeoxycholic acid and vitamin E, have been commonly used for NAFLD and CH-C patients as a liver supporting therapy. In many patients with insulin resistance, insulin sensitizers, such as metformin and thiazolidinediones, have shown improving effect of liver biochemistry. Nowadays, IFN-based radical antiviral treatments are generally accepted for patients with CH-C. Hepatic steatosis and insulin resistance are negative predictors for sustained virological response (SVR) in patients with CH-C treated with peg-IFNα plus ribavirin combination therapy [53, 54]. In recent meta-analyses, HOMA-IR, a marker of insulin resistance, is negatively correlated with SVR, irrespective of viral genotype [55, 56]. Therefore, lifestyle modifications, such as weight reduction by exercise and nutritional management, are recommended to enhance the effects of antiviral treatments. Moreover, it is justifiable that the use of agents targeting insulin resistance and dyslipidemia can improve the SVR rate achieved with IFN-based antiviral treatments.
Insulin sensitizers, metformin and thiazolidinediones, may increase the response to antiviral treatments [57]. In a recent clinical trial of insulin-resistant patients with CH-C genotype 1, adding metformin to standard peg-IFNα plus ribavirin therapy improved insulin sensitivity. Metformin also tended to improve SVR, particularly in females, although a statistically significant difference was not seen compared with patients receiving placebo [58]. Meanwhile, the effects of pioglitazone on SVR in patients with CH-C and insulin resistance are controversial. Pioglitazone combined with peg-IFNα plus ribavirin therapy was used as retreatment and in treatment-naïve patients, but results of two pilot trials were disappointing [59, 60]. However, some reports have described that the addition of pioglitazone to standard therapy improves SVR and insulin sensitivity [61, 62]. This discrepancy may be explained, at least in part, by genotype dependency and host characteristics.
As described above, the synthesis of cholesterol and fatty acids is still activated in the liver of patients with CH-C, despite lipid overaccumulation. Therefore, correcting cholesterol and fatty acid synthesis by lipid modulators may help to reduce steatosis and improve SVR with antiviral treatments. Considering that cholesterol synthesis is enhanced in HCV-infected liver, it is plausible that HMGR inhibitors (statins) could have antiviral effects, because statins were recently reported to suppress HCV replication [63]. In fact, it was reported that statins do impede HCV replication by inhibiting host protein geranylgeranylation, and FBL2 has been identified as a geranylgeranylated cellular protein required for HCV RNA replication [64]. Retrospective analyses of patients with CH-C treated with peg-IFN plus ribavirin combination therapy have shown that serum cholesterol and the use of statins are positive predictors of SVR [65, 66]. In clinical trials, SVR was improved by adding fluvastatin or pitavastatin to peg-IFN plus ribavirin treatment [67–69]. Although antiviral activity has been experimentally demonstrated in most statins without pravastatin [63], a statin with a more activity may achieve better SVR rates. Of note, protease inhibitors, such as telaprevir, and statins taken together may raise the blood levels of statins and increase the risk of myopathy, kidney damage, and kidney failure. It was also reported that polyunsaturated fatty acids (PUFAs) inhibit HCV replication by a still unclear mechanism, independent of their roles in regulating lipogenesis and that eicosapentaenoic acid (EPA), an n-3 PUFA, inhibits HCV replication in the replicon system and suppresses SREBP-1c activity [70–72]. Additionally, administration of EPA allows maintenance of the original ribavirin dose in patients with CH-C during peg-IFN plus ribavirin combination therapy [73]. Using HCV replicon systems, it was reported that statins and EPA have suppressive effects against HCV replication and synergistic antiviral action with IFN [37, 70, 71, 74, 75].
Based on experimental and therapeutic evidence, concomitant administration of a statin and EPA with peg-IFN plus ribavirin therapy is pathophysiologically promising for patients with CH-C. Accordingly, we are now performing a clinical trial using a new antiviral strategy for patients with CH-C genotype 1b in which pitavastatin (2 mg/day) and EPA (1,800 mg/day) are added to standard peg-IFN plus ribavirin therapy. According to recent clinical studies of patients with CH-C genotype-1b, mutation of amino acids 70 and 91 in the core region of HCV-1b, as a virus-related factor, and genomic variation of the IL28B gene (rs8099917), as a host-related factor, are strong predictors of the outcome of peg-IFN plus ribavirin combination therapy [76–79]. Within the core protein, substitution of amino acid 70 seems to be more influential on the outcome than substitution of amino acid 91 [79–81]. At present, our trial has yielded several important findings (unpublished data). First, add-on pitavastatin and EPA therapy conferred significantly higher SVR rates than did standard therapy. Second, add-on therapy significantly improved SVR rates, particularly in patients with the minor variant (TG + GG) of IL28B (rs8099917), in whom SVR is expected to be poor. Of note, among patients treated with add-on therapy, genomic variation of IL28B still predicts clinical outcomes, because SVR rates were significantly higher in patients with the major variant (TT) than in those with minor variants. Third, mutation of core amino acid 70, which is a strong negative predictor of SVR in standard peg-IFN plus ribavirin therapy, did not diminish the outcomes of add-on therapy.
6. Conclusions
Steatosis and insulin resistance induced by HCV infection are, at least in part, critical factors for the progression of CH-C and can influence the outcome of antiviral treatments. Therefore, managing these metabolic disorders by administering insulin sensitizers and lipid modulators has been examined to increase the therapeutic response of standard treatments. In particular, concomitant treatment with pitavastatin and EPA may achieve considerable improvements in the efficacy of peg-IFN plus ribavirin combination therapy, particularly in patients with CH-C resistant to standard peg-IFN plus ribavirin therapy.
Conflict of Interests
The authors have no conflict of interests to declare.
References
- 1.Scheuer PJ, Ashrafzadeh P, Sherlock S, Brown D, Dusheiko GM. The pathology of hepatitis C. Hepatology. 1992;15(4):567–571. doi: 10.1002/hep.1840150402. [DOI] [PubMed] [Google Scholar]
- 2.Hwang SJ, Lee SD. Hepatic steatosis and hepatitis C: still unhappy bedfellows? Journal of Gastroenterology and Hepatology. 2011;26(1):96–101. doi: 10.1111/j.1440-1746.2010.06542.x. [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi A, Tazuma S, Nishioka T, et al. Hepatitis C virus core protein modulates fatty acid metabolism and thereby causes lipid accumulation in the liver. Digestive Diseases and Sciences. 2005;50(7):1361–1371. doi: 10.1007/s10620-005-2788-1. [DOI] [PubMed] [Google Scholar]
- 4.Pazienza V, Clément S, Pugnale P, et al. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology. 2007;45(5):1164–1171. doi: 10.1002/hep.21634. [DOI] [PubMed] [Google Scholar]
- 5.Hui JM, Sud A, Farrell GC, et al. Insulin resistance is associated with chronic hepatitis C virus infection and fibrosis progression. Gastroenterology. 2003;125(6):1695–1704. doi: 10.1053/j.gastro.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 6.Lonardo A, Adinolfi LE, Loria P, Carulli N, Ruggiero G, Day CP. Steatosis and hepatitis C virus: mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology. 2004;126(2):586–597. doi: 10.1053/j.gastro.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 7.Powell EE, Jonsson JR, Clouston AD. Steatosis: co-factor in other liver diseases. Hepatology. 2005;42(1):5–13. doi: 10.1002/hep.20750. [DOI] [PubMed] [Google Scholar]
- 8.Miquilena-Colina ME, Lima-Cabello E, Sánchez-Campos S, et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut. 2011;60(10):1394–1402. doi: 10.1136/gut.2010.222844. [DOI] [PubMed] [Google Scholar]
- 9.Bugianesi E, Salamone F, Negro F. The interaction of metabolic factors with HCV infection: does it matter? Journal of Hepatology. 2012;56(supplement 1):S56–S65. doi: 10.1016/S0168-8278(12)60007-5. [DOI] [PubMed] [Google Scholar]
- 10.Enjoji M, Kotoh K, Kato M, et al. Therapeutic effect of ARBs on insulin resistance and liver injury in ptients with NAFLD and chronic hepatitis C: a pilot study. International Journal of Molecular Medicine. 2008;22(4):521–527. [PubMed] [Google Scholar]
- 11.Moucari R, Asselah T, Cazals-Hatem D, et al. Insulin resistance in chronic hepatitis C: association with genotypes 1 and 4, serum HCV RNA level, and liver fibrosis. Gastroenterology. 2008;134(2):416–423. doi: 10.1053/j.gastro.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 12.Simó R, Lecube A, Genescà J, Esteban JI, Hernández C. Sustained virological response correlates with reduction in the incidence of glucose abnormalities in patients with chronic hepatitis C virus infection. Diabetes Care. 2006;29(11):2462–2466. doi: 10.2337/dc06-0456. [DOI] [PubMed] [Google Scholar]
- 13.Kawaguchi T, Ide T, Taniguchi E, et al. Clearance of HCV improves insulin resistance, beta-cell function, and hepatic expression of insulin receptor substrate 1 and 2. American Journal of Gastroenterology. 2007;102(3):570–576. doi: 10.1111/j.1572-0241.2006.01038.x. [DOI] [PubMed] [Google Scholar]
- 14.Alberstein M, Zornitzki T, Zick Y, Knobler H. Hepatitis C core protein impairs insulin downstream signalling and regulatory role of IGFBP-1 expression. Journal of Viral Hepatitis. 2012;19(1):65–71. doi: 10.1111/j.1365-2893.2011.01447.x. [DOI] [PubMed] [Google Scholar]
- 15.Pascarella S, Clément S, Guilloux K, Conzelmann S, Penin F, Negro F. Effects of hepatitis C virus on suppressor of cytokine signaling mRNA levels: comparison between different genotypes and core protein sequence analysis. Journal of Medical Virology. 2011;83(6):1005–1015. doi: 10.1002/jmv.22072. [DOI] [PubMed] [Google Scholar]
- 16.Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Molecular and Cellular Biology. 2004;24(12):5434–5446. doi: 10.1128/MCB.24.12.5434-5446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. Journal of Biological Chemistry. 2002;277(44):42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 18.Kawaguchi T, Yoshida T, Harada M, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. American Journal of Pathology. 2004;165(5):1499–1508. doi: 10.1016/S0002-9440(10)63408-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knobler H, Zhornicky T, Sandler A, Haran N, Ashur Y, Schattner A. Tumor necrosis factor-α-induced insulin resistance may mediate the hepatitis C virus-diabetes association. American Journal of Gastroenterology. 2003;98(12):2751–2756. doi: 10.1111/j.1572-0241.2003.08728.x. [DOI] [PubMed] [Google Scholar]
- 20.Fernández-Veledo S, Hernandez R, Teruel T, Mas J, Ros M, Lorenzo M. Ceramide mediates TNF-α-induced insulin resistance on GLUT4 gene expression in brown adipocytes. Archives of Physiology and Biochemistry. 2006;112(1):13–22. doi: 10.1080/13813450500508137. [DOI] [PubMed] [Google Scholar]
- 21.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. Journal of Clinical Investigation. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dharancy S, Lemoine M, Mathurin P, Serfaty L, Dubuquoy L. Peroxisome proliferator-activated receptors in HCV-related infection. PPAR Research. 2009;2009:5 pages. doi: 10.1155/2009/357204. Article ID 357204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McPherson S, Jonsson JR, Barrie HD, O’Rourke P, Clouston AD, Powell EE. Investigation of the role of SREBP-1c in the pathogenesis of HCV-related steatosis. Journal of Hepatology. 2008;49(6):1046–1054. doi: 10.1016/j.jhep.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 24.Kim KH, Hong SP, Kim K, Park MJ, Kim KJ, Cheong J. HCV core protein induces hepatic lipid accumulation by activating SREBP1 and PPARγ . Biochemical and Biophysical Research Communications. 2007;355(4):883–888. doi: 10.1016/j.bbrc.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 25.Perlemuter G, Sabile A, Letteron P, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB Journal. 2002;16(2):185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 26.De Gottardi A, Pazienza V, Pugnale P, et al. Peroxisome proliferator-activated receptor-α and -γ mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Alimentary Pharmacology and Therapeutics. 2006;23(1):107–114. doi: 10.1111/j.1365-2036.2006.02729.x. [DOI] [PubMed] [Google Scholar]
- 27.Nakamuta M, Yada R, Fujino T, et al. Changes in the expression of cholesterol metabolism-associated genes in HCV-infected liver: a novel target for therapy? International Journal of Molecular Medicine. 2009;24(6):825–828. doi: 10.3892/ijmm_00000299. [DOI] [PubMed] [Google Scholar]
- 28.Fujino T, Nakamuta M, Yada R, et al. Expression profile of lipid metabolism-associated genes in hepatitis C virus-infected human liver. Hepatology Research. 2010;40(9):923–929. doi: 10.1111/j.1872-034X.2010.00700.x. [DOI] [PubMed] [Google Scholar]
- 29.Nakamuta M, Fujino T, Yada R, et al. Impact of cholesterol metabolism and the LXRα-SREBP-1c pathway on nonalcoholic fatty liver disease. International Journal of Molecular Medicine. 2009;23(5):603–608. doi: 10.3892/ijmm_00000170. [DOI] [PubMed] [Google Scholar]
- 30.Enjoji M, Yasutake K, Kohjima M, Nakamuta M. Nutrition and nonalcoholic fatty liver disease: the significance of cholesterol. International Journal of Hepatology. 2012;2012:6 pages. doi: 10.1155/2012/925807. Article ID 925807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends in Endocrinology and Metabolism. 2010;21(1):33–40. doi: 10.1016/j.tem.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLauchlan J. Lipid droplets and hepatitis C virus infection. Biochimica et Biophysica Acta. 2009;1791(6):552–559. doi: 10.1016/j.bbalip.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Tang H, Grisé H. Cellular and molecular biology of HCV infection and hepatitis. Clinical Science. 2009;117(2):49–65. doi: 10.1042/CS20080631. [DOI] [PubMed] [Google Scholar]
- 34.Nakamuta M, Fujino T, Yada R, et al. Expression profiles of genes associated with viral entry in HCV-infected human liver. Journal of Medical Virology. 2011;83(5):921–927. doi: 10.1002/jmv.22042. [DOI] [PubMed] [Google Scholar]
- 35.Enjoji M, Nakamuta M, Kinukawa N, et al. Beta-lipoproteins influence the serum level of hepatitis C virus. Medical Science Monitor. 2000;6(5):841–844. [PubMed] [Google Scholar]
- 36.Molina S, Castet V, Fournier-Wirth C, et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. Journal of Hepatology. 2007;46(3):411–419. doi: 10.1016/j.jhep.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 37.Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2561–2566. doi: 10.1073/pnas.0409834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parfieniuk A, Flisiak R. Role of cannabinoids in chronic liver diseases. World Journal of Gastroenterology. 2008;14(40):6109–6114. doi: 10.3748/wjg.14.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osei-Hyiaman D, DePetrillo M, Pacher P, et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. Journal of Clinical Investigation. 2005;115(5):1298–1305. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeong WI, Osei-Hyiaman D, Park O, et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metabolism. 2008;7(3):227–235. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Purohit V, Rapaka R, Shurtleff D. Role of cannabinoids in the development of fatty liver (steatosis) AAPS Journal. 2010;12(2):233–237. doi: 10.1208/s12248-010-9178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lotersztajn S, Teixeira-Clerc F, Julien B, et al. CB2 receptors as new therapeutic targets for liver diseases. British Journal of Pharmacology. 2008;153(2):286–289. doi: 10.1038/sj.bjp.0707511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teixeira-Clerc F, Belot MP, Manin S, et al. Beneficial paracrine effects of cannabinoid receptor 2 on liver injury and regeneration. Hepatology. 2010;52(3):1046–1059. doi: 10.1002/hep.23779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hézode C, Zafrani ES, Roudot-Thoraval F, et al. Daily cannabis use: a novel risk factor of steatosis severity in patients with chronic hepatitis C. Gastroenterology. 2008;134(2):432–439. doi: 10.1053/j.gastro.2007.11.039. [DOI] [PubMed] [Google Scholar]
- 45.Mallat A, Hezode C, Lotersztajn S. Environmental factors as disease accelerators during chronic hepatitis C. Journal of Hepatology. 2008;48(4):657–665. doi: 10.1016/j.jhep.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Julien B, Grenard P, Teixeira-Clerc F, et al. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology. 2005;128(3):742–755. doi: 10.1053/j.gastro.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 47.Teixeira-Clerc F, Julien B, Grenard P, et al. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nature Medicine. 2006;12(6):671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 48.Mallat A, Lotersztajn S. Endocannabinoids and Liver Disease. I. Endocannabinoids and their receptors in the liver. American Journal of Physiology. 2007;294(1):G9–G12. doi: 10.1152/ajpgi.00467.2007. [DOI] [PubMed] [Google Scholar]
- 49.Bátkai S, Járai Z, Wagner JA, et al. Endocannabinoids acting at vascular CB1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nature Medicine. 2001;7(7):827–832. doi: 10.1038/89953. [DOI] [PubMed] [Google Scholar]
- 50.Fernández-Rodriguez CM, Romero J, Petros TJ, et al. Circulating endogenous cannabinoid anandamide and portal, systemic and renal hemodynamics in cirrhosis. Liver International. 2004;24(5):477–483. doi: 10.1111/j.1478-3231.2004.0945.x. [DOI] [PubMed] [Google Scholar]
- 51.Siegmund SV, Qian T, De Minicis S, et al. The endocannabinoid 2-arachidonoyl glycerol induces death of hepatic stellate cells via mitochondrial reactive oxygen species. FASEB Journal. 2007;21(11):2798–2806. doi: 10.1096/fj.06-7717com. [DOI] [PubMed] [Google Scholar]
- 52.Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacological Reviews. 2006;58(3):389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poynard T, Ratziu V, McHutchison J, et al. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology. 2003;38(1):75–85. doi: 10.1053/jhep.2003.50267. [DOI] [PubMed] [Google Scholar]
- 54.Patton HM, Patel K, Behling C, et al. The impact of steatosis on disease progression and early and sustained treatment response in chronic hepatitis C patients. Journal of Hepatology. 2004;40(3):484–490. doi: 10.1016/j.jhep.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 55.Eslam M, Aparcero R, Kawaguchi T, et al. Meta-analysis: insulin resistance and sustained virological response in hepatitis C. Alimentary Pharmacology and Therapeutics. 2011;34(3):297–305. doi: 10.1111/j.1365-2036.2011.04716.x. [DOI] [PubMed] [Google Scholar]
- 56.Deltenre P, Louvet A, Lemoine M, et al. Impact of insulin resistance on sustained response in HCV patients treated with pegylated interferon and ribavirin: a meta-analysis. Journal of Hepatology. 2011;55(6):1187–1194. doi: 10.1016/j.jhep.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 57.Adinolfi LE, Restivo L, Zampino R, Lonardo A, Loria P. Metabolic alterations and chronic hepatitis C: treatment strategies. Expert Opinion on Pharmacotherapy. 2011;12(14):2215–2234. doi: 10.1517/14656566.2011.597742. [DOI] [PubMed] [Google Scholar]
- 58.Romero-Gómez M, Diago M, Andrade RJ, et al. Treatment of insulin resistance with metformin in naïve genotype 1 chronic hepatitis C patients receiving peginterferon alfa-2a plus ribavirin. Hepatology. 2009;50(6):1702–1708. doi: 10.1002/hep.23206. [DOI] [PubMed] [Google Scholar]
- 59.Overbeck K, Genné D, Golay A, Negro F. Pioglitazone in chronic hepatitis C not responding to pegylated interferon-α and ribavirin. Journal of Hepatology. 2008;49(2):295–298. doi: 10.1016/j.jhep.2008.03.033. [DOI] [PubMed] [Google Scholar]
- 60.Harrison SA, Hamzeh FM, Han J, Pandya PK, Sheikh MY, Vierling JM. Chronic hepatitis C genotype 1 patients with insulin resistance treated with pioglitazone and peginterferon alfa-2a plus ribavirin. doi: 10.1002/hep.25661. Hepatology. In press. [DOI] [PubMed] [Google Scholar]
- 61.Khattab M, Emad M, Abdelaleem A, et al. Pioglitazone improves virological response to peginterferon α-2b/ribavirin combination therapy in hepatitis C genotype 4 patients with insulin resistance. Liver International. 2010;30(3):447–454. doi: 10.1111/j.1478-3231.2009.02171.x. [DOI] [PubMed] [Google Scholar]
- 62.Serfaty L, Fartoux L, Poupon R. Pioglitazone as adjuvant therapy in chronic hepatitis C: sequential rather than concomitant administration with pegylated interferon and ribavirin? Journal of Hepatology. 2009;50(6):1269–1271. doi: 10.1016/j.jhep.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 63.Ikeda M, Abe KI, Yamada M, Dansako H, Naka K, Kato N. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology. 2006;44(1):117–125. doi: 10.1002/hep.21232. [DOI] [PubMed] [Google Scholar]
- 64.Wang C, Gale M, Keller BC, et al. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Molecular Cell. 2005;18(4):425–434. doi: 10.1016/j.molcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 65.Harrison SA, Rossaro L, Hu KQ, et al. Serum cholesterol and statin use predict virological response to peginterferon and ribavirin therapy. Hepatology. 2010;52(3):864–874. doi: 10.1002/hep.23787. [DOI] [PubMed] [Google Scholar]
- 66.Rao GA, Pandya PK. Statin therapy improves sustained virologic response among diabetic patients with chronic hepatitis C. Gastroenterology. 2011;140(1):144–152. doi: 10.1053/j.gastro.2010.08.055. [DOI] [PubMed] [Google Scholar]
- 67.Sezaki H, Suzuki F, Akuta N, et al. An open pilot study exploring the efficacy of fluvastatin, pegylated interferon and ribavirin in patients with hepatitis C virus genotype 1b in high viral loads. Intervirology. 2009;52(1):43–48. doi: 10.1159/000213504. [DOI] [PubMed] [Google Scholar]
- 68.Bader T, Fazili J, Madhoun M, et al. Fluvastatin inhibits hepatitis C replication in humans. American Journal of Gastroenterology. 2008;103(6):1383–1389. doi: 10.1111/j.1572-0241.2008.01876.x. [DOI] [PubMed] [Google Scholar]
- 69.Shimada M, Yoshida S, Masuzaki R, Schuppan D. Pitavastatin enhances antiviral efficacy of standard pegylated interferon plus ribavirin in patients with chronic hepatitis C: a prospective randomized pilot study. Journal of Hepatology. 2012;56(1):299–300. doi: 10.1016/j.jhep.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 70.Leu GZ, Lin TY, Hsu JTA. Anti-HCV activities of selective polyunsaturated fatty acids. Biochemical and Biophysical Research Communications. 2004;318(1):275–280. doi: 10.1016/j.bbrc.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 71.Huang H, Chen Y, Ye J. Inhibition of hepatitis C virus replication by peroxidation of arachidonate and restoration by vitamin E. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(47):18666–18670. doi: 10.1073/pnas.0708423104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zaima N, Sugawara T, Goto D, Hirata T. Trans geometric isomers of EPA decrease LXRα-induced cellular triacylglycerol via suppression of SREBP-1c and PGC-1β . Journal of Lipid Research. 2006;47(12):2712–2717. doi: 10.1194/jlr.M600273-JLR200. [DOI] [PubMed] [Google Scholar]
- 73.Takaki S, Kawakami Y, Imamura M, et al. Eicosapentaenoic acid could permit maintenance of the original ribavirin dose in chronic hepatitis C virus patients during the first 12 weeks of combination therapy with pegylated interferon-α and ribavirin: a prospective randomized controlled trial. Intervirology. 2008;50(6):439–446. doi: 10.1159/000114718. [DOI] [PubMed] [Google Scholar]
- 74.Ye J, Wang C, Sumpter R, Brown MS, Goldstein JL, Gale M. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(26):15865–15870. doi: 10.1073/pnas.2237238100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ikeda M, Kato N. Life style-related diseases of the digestive system: Cell culture system for the screening of anti-hepatitis C virus (HCV) reagents: suppression of HCV replication by statins and synergistic action with interferon. Journal of Pharmacological Sciences. 2007;105(2):145–150. doi: 10.1254/jphs.fm0070050. [DOI] [PubMed] [Google Scholar]
- 76.Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nature Genetics. 2009;41(10):1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 77.Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-α and ribavirin therapy for chronic hepatitis C. Nature Genetics. 2009;41(10):1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 78.Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461(7262):399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 79.Hayes CN, Kobayashi M, Akuta N, et al. HCV substitutions and IL28B polymorphisms on outcome of peg-interferon plus ribavirin combination therapy. Gut. 2011;60(2):261–267. doi: 10.1136/gut.2010.223495. [DOI] [PubMed] [Google Scholar]
- 80.Akuta N, Suzuki F, Kawamura Y, et al. Prediction of response to pegylated interferon and ribavirin in hepatitis C by polymorphisms in the viral core protein and very early dynamics of viremia. Intervirology. 2007;50(5):361–368. doi: 10.1159/000107707. [DOI] [PubMed] [Google Scholar]
- 81.El-Shamy A, Kim SR, Ide YH, et al. Polymorphisms of hepatitis C virus non-structural protein 5A and core protein and clinical outcome of pegylated-interferon/ribavirin combination therapy. Intervirology. 2012;55(1):1–11. doi: 10.1159/000322219. [DOI] [PubMed] [Google Scholar]