

Abstract
To explore the mechanism underlying antileukaemia effect of sodium valproate, the growth and survival of the K562 cell line were investigated. Global profiles of gene expression in K562 cells exposed to sodium valproate were assessed and validated. The differentially expressed genes identified were further used to query the connectivity map database to retrieve a ranked list of compounds that act on the same intracellular targets as sodium valproate. A significant increase in cell apoptosis and a change in gene expression profile were observed in valproate-exposed K562 cells. The significant enrichment analysis of gene ontology terms for the differentially expressed genes showed that these genes were involved in many important biological processes. Eight differentially expressed genes involved in apoptosis were verified by quantitative real-time PCR. The connectivity map analysis showed gene expression profile in K562 cells exposed to sodium valproate was most similar to that of HDACi and PI3K inhibitors, suggesting that sodium valproate might exert antileukaemic action by inhibiting HDAC as well as inhibiting PI3K pathway. In conclusion, our data might provide clues to elucidate the molecular and therapeutic potential of VPA in leukaemia treatment, and the connectivity map is a useful tool for exploring the molecular mechanism of drug action.
1. Introduction
Sodium valproate (VPA) is a well-known antiepileptic drug, also used to treat bipolar disorder, migraine, and neuropathic pain. Recently, VPA has been described as an HDAC inhibitor, resulting in an increased interest for its use in cancer therapy. Studies show that VPA, alone or in combination with other antileukaemic compounds, possesses significant antileukaemic actions on acute myeloid leukemia (AML) cells [1], chronic myeloid leukaemia (CML) cells [2–4], acute lymphoblastic leukemia (ALL) cells [5], and chronic lymphocytic leukemia (CLL) cells [6, 7]. Clinical trials show that VPA therapy is of clinical benefit in patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) [8–11]. However, the exact molecular mechanisms of VPA action on leukemia treatment remain poorly understood.
Microarray is a high-throughput tool which allows for the analysis of global gene expression profile in a single experiment and has been widely used for exploring molecular mechanisms of pathogenesis and drug treatment. Based on this high-throughput technology, the molecular mechanisms underlying the observed antileukaemic activity of VPA in CLL cells [12] and AML cells [13, 14] have been described. So far, however, there has been no study exploring genomewide gene expression changes in CML.
The Connectivity Map (CMAP) is a collection of genomewide transcriptional expression data from cultured human cells treated with bioactive small molecules. It connects drugs, genes, and diseases together through the transitory feature of common gene-expression changes. By comparing gene-expression signatures, this tool can be used to find connections among small molecules drugs affecting common molecular pathways and putative mechanisms of action of unknown drugs. CMAP has previously been used to discover the mechanisms of drug action [15–18] and disease pathogenesis [19]. In the current study, we first investigated effects of VPA on apoptosis and gene expression profiles of K562 cells, a model for CML, and then mined the CMAP database to explore the molecular mechanism underlying the observed anti-CML effect of VPA.
2. Methods and Materials
2.1. Culture of K562 Cell Line
K562 cells, human chronic myelogenic leukemic cell line, were procured from Sun Yat-sen University Cancer Center. The cells were grown in RPMI (Life Technology Corporation, Camarillo, CA, USA), supplemented with 15% fetal bovine serum, 100 U/mL penicillin, and 1 mg/mL streptomycin. Cultures were incubated at 37°C in 5% CO2.
2.2. Assessment of Apoptosis by Annexin V/PI Dual Staining Method
In order to determine the effect of VPA treatment on apoptosis rate of K562 cells, FACS analysis was carried out as previously described [20].
2.3. Analysis of Gene Expression Profile
To analyze gene expression profile, K562 cells were first cultured for 12 hours with 2 mM VPA or without VPA as a control. Then, the K562 cells were harvested and total RNA was extracted using the RNeasy Mini Kit (QIAGen, USA) according to the manufacturer's instructions. Total RNA quality was evaluated using formaldehyde agarose gel electrophoresis and was quantified via spectrophotometry (Nanodrop, Wilmington, DE). RNA was amplified and labelled according to a previous protocol [21]. Briefly, 100 ng of total RNA was used to synthesize the double-strand cDNA. RNA was amplified by in vitro transcription using Ambion's MessageAmp II aRNA Amplification Kits (Life Technologies, Austin, TX, USA). Then, aRNA was reverse-transcribed into cDNA and further labelled with Klenow enzyme. cDNA from VPA-pretreated K562 cells was labelled with Cy3-deoxycytidine triphosphate and cDNA from control K562 cells was labelled with Cy5-deoxycytidine triphosphate. Fluorescent dye-labelled cDNA was hybridised to an Agilent SurePrint G3 Human GE 8 × 60 K Microarray. Hybridisation, scanning, and washing were carried out on Agilent's Microarray Platform according to Agilent's standard protocols. The array data were extracted with Agilent Feature Extraction software. After global mean normalization, probes with an intensity <400 were filtered out for further analysis. Differentially expressed genes were further analysed based on a significant enrichment of GO terms using hypergeometric distribution in the R language package software.
2.4. Validation of Differentially Expressed Genes
To explore the mechanism of the observed VPA-inducing apoptosis, eight differentially expressed genes involved in apoptosis were selected for validation of the microarray results by quantitative real-time PCR (qRT-PCR). Total RNAs were first separated from K562 cell line treated with or without VPA. The isolated total RNAs were treated with DNase and exclusively used for validation of the microarray results. Following DNase treatment, 1 μg of DNase-treated RNA was reverse-transcribed cDNA with oligo (dT)15 using M-MLV reverse transcriptase (Life Technologies) in a total volume of 20 μL reaction volume. After reverse transcription reaction, 1 μL of the reaction mixture was used for a qPCR program of 45 cycles consisting of melting (30 s at 94°C), annealing (30 s at 58°C), and extension (30 s at 72°C). The 20 μL reaction mixture contained 1 × PCR Buffer (Mg2+ Plus), 0.5 μM of forward primer, 0.5 μM of reverse primer (Table 1), 200 μM of each dNTP, and Eva Green Master Mix in a LightCycler 480 Real-Time PCR System (Roche Applied Science). Data were analysed by the 2−ΔΔCt method [22]. Glyceraldehyde 3-phosphate dehydrogenase (GADPH) was used as a reference gene. All other results were shown as fold change relative to GADPH control.
Table 1.
Primer pairs for qRT-PCR validation of differentially expressed genes involved in apoptosis.
| Gene symbol | GenBank Accession | Primer (5′→3′) | |
|---|---|---|---|
| GSN | NM_198252 | F | CGAAGAGGTTCCTGGTGAGC |
| R | GCTGGGTCCGTCTCGATGTA | ||
| TP53INP1 | NM_033285 | F | CTTCTCATTGAACATCCCAGCA |
| R | TGGGTTGTTCCAGAAAAGTTGTA | ||
| TNFSF9 | NM_003811 | F | GAGCTTTCGCCCGACGAT |
| R | GCTCCTTCGTGTCCTCTTTGTAG | ||
| DDIT4 | NM_019058 | F | CCGAACTCCCACCCCAGA |
| R | GCAACGACACCCCATCCA | ||
| POLR2G | NM_002696 | F | AAGATTGTGGGGACCCGTGT |
| R | GGATGAGTTGCCTTCCTTGC | ||
| LGALS1 | NM_033101 | F | CAACCCTCGCTTCAACGC |
| R | GGCGGTTGGGGAACTTGA | ||
| BBC3 | NM_014417 | F | CGGACGACCTCAACGCAC |
| R | GCACCTAATTGGGCTCCATCT | ||
| FOXO3 | NM_001455 | F | GGGGAACTTCACTGGTGCTAA |
| R | AACGGTATCACTGTCCACTTGC | ||
| PUF60 | NM_014281 | F | CTGTTTCCCGAGTCAGAGCG |
| R | CTCCTCTGTCACCTCCCCTTC | ||
| RRAGA | NM_006570 | F | TGGTCATGTCAGATCCGTCG |
| R | AAGGCAATTCAGCATTTTCAGA |
2.5. Statistics
The microarray data have been deposited in NCBI's Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO series accession number GSE32189. The significance between groups was analyzed by the Student's t-test (Microsoft Excel, Microsoft Corporation, Seattle, WA). A P value of less than 0.05 (P < 0.05) was considered statistically significant.
2.6. Comparison of Gene Expression Profiles with the CMAP Database
To further explore the mechanisms underlying the observed effects of VPA, the gene expression profile of VPA treatments was used to query the CMAP database (build 02), which contains more than 7,000 expression profiles representing 1,309 compounds [23]. The similarity between the gene expression profiles of the query signature and a CMAP instance is measured by the connectivity score, ranging from −1 to 1. A high positive connectivity score indicates inducement of the expression of the query signature by the corresponding drug, a high negative, inhibition. Prior to the query in the CMAP database via the Internet (http://www.broad.mit.edu/cmap/), the probe ID of Agilent SurePrint G3 Human GE 8 × 60 K Microarray was transformed into the probe ID defined by the Affymetrix GeneChip Human Genome U133A array according to the probe corresponding gene.
3. Results
3.1. Analysis of Apoptosis Using the Annexin V/PI Dual Staining Method
Using the Annexin V/PI dual staining method just as we previously reported, we measured apoptosis rate of K562 cells. In this study the similar increase in apoptosis was also observed in K562 cells exposed to 2 mM VPA for 48 hours (11.47 ± 0.25; P < 0.05, n = 3), compared to the K562 control (4.77 ± 0.40) [20].
3.2. Differentially Expressed Genes in K562 Cells Pretreated with VPA
Considering that the detection of differentially expressed genes is more easily in cells during the primary response phase of a drug exposure at certain doses that result in a 10–20% decrease in cell viability, we pretreated K562 cells with 2 mM VPA for 12 hours. This treatment resulted in an approximately 12% decrease in cell viability. The transcription profiles of K562 cells pretreated with or without VPA were analyzed using Agilent gene expression microarray. A total of 706 transcripts were identified as being significantly differentially expressed. The significant enrichment analysis of GO terms for the differentially expressed genes using the R language software package showed that apoptosis was the mostly enrichment function genes, and 34 genes involved in apoptosis were listed in Table 2.
Table 2.
Differentially expressed genes involved in apoptosis detected by significant enrichment analysis of GO terms using the R language software package.
| Genes | GenBank accession | Fold change | Gene description |
|---|---|---|---|
| PDCD7 | NM_005707 | −2.20 | Programmed cell death 7 |
| RRAGA | NM_006570 | 2.03 | Ras-related GTP binding A |
| DAPK3 | NM_001348 | 2.04 | Death-associated protein kinase 3 |
| PUF60 | NM_014281 | −2.00 | Poly-U binding splicing factor 60kda isoform c |
| FOXO3 | NM_001455 | 2.16 | Forkhead box O3A |
| ARHGEF18 | NM_015318 | 2.20 | Rho/Rac guanine nucleotide exchange factor 18 |
| BBC3 | NM_014417 | −2.18 | BCL2 binding component 3 isoform 1 |
| GSN | NM_198252 | 5.02 | Gelsolin isoform c |
| LGALS1 | NM_033101 | 2.58 | Galectin-1 |
| NFKB1 | NM_003998 | −3.33 | Nuclear factor kappa-B, subunit 1 isoform 1 |
| SH3GLB1 | NM_016009 | 2.82 | SH3-containing protein SH3GLB1 |
| POLR2G | NM_002696 | 2.02 | DNA directed RNA polymerase II polypeptide G |
| DDIT4 | NM_019058 | −2.23 | Homo sapiens DNA-damage-inducible transcript 4 |
| STEAP3 | NM_182915 | −2.52 | Dudulin 2 isoform a |
| DRAM1 | NM_018370 | 2.29 | DNA-damage regulated autophagy modulator 1 |
| RNF130 | NM_018434 | 2.07 | Ring finger protein 130 precursor |
| AVEN | NM_020371 | −2.62 | Cell death regulator Aven |
| TRIB3 | NM_021158 | −2.19 | Tribbles 3 |
| BAX | NM_138764 | 2.06 | BCL2-associated X protein isoform beta |
| BID | NM_197966 | −2.76 | BH3 interacting domain death agonist isoform 1 |
| PERP | NM_022121 | 9.42 | PERP, TP53 apoptosis effector |
| SPTAN1 | NM_003127 | 2.63 | Spectrin, alpha, nonerythrocytic 1 |
| BUB1B | NM_001211 | −2.20 | Budding uninhibited by benzimidazoles 1 beta |
| VDAC1 | NM_003374 | −2.37 | Voltage-dependent anion channel 1 |
| BCL2L12 | NM_138639 | 2.19 | BCL2-like 12 isoform 1 |
| CUL1 | NM_003592 | −2.08 | Cullin 1 |
| YARS | NM_001040436 | −2.30 | Tyrosyl-TRNA synthetase |
| TNFSF9 | NM_003811 | 2.38 | Tumor necrosis factor (ligand) superfamily |
| IER3 | NM_003897 | 2.17 | Immediate early response 3 |
| ARHGEF2 | NM_004723 | −2.10 | Rho/Rac guanine nucleotide exchange factor 2 |
| TP53INP1 | NM_033285 | 3.03 | Tumor protein p53 inducible nuclear protein 1 |
| ITM2B | NM_021999 | 2.32 | Integral membrane protein 2B |
| LITAF | NM_004862 | 2.25 | Lipopolysaccharide-induced TNF-alpha factor |
| ESPL1 | NM_012291 | −2.11 | Separase |
−: indicate downregulation.
3.3. Validation of the Differentially Expressed Genes Using qRT-PCR
Based on the microarray results, eight differentially expressed genes involved in apoptosis were selected for qRT-PCR confirmation. Consistent with the results obtained from microarray analysis, PERP, GSN, TP53INP1, NXA4, FI6, ERPINB9, and CDKN2D were upregulated and NFKB1 was downregulated (Figure 1).
Figure 1.
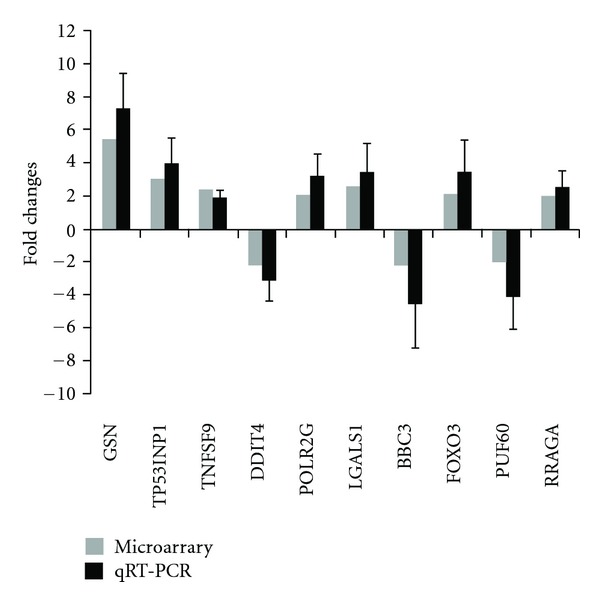
Alterations in the expression levels identified by microarray were confirmed using qRT-PCR. Fold changes between K562 cell line treated with and without valproate detected by microarray were compared with those measured by qRT-PCR. In qRT-PCR assay, RNA was isolated from K562 cell line treated with or without valproate exclusively for validation of differentially expressed genes. mRNA levels were normalized with GADPH and fold changes were calculated by dividing mRNA level of each K563 cell line treated with valproate by mean mRNA level from control samples in triplicate. Data are mean values ± standard deviations of the means from three independent experiments performed in triplicate.
3.4. Comparison of Gene Expression Profiles with the CMAP Database
To gain a better understanding of the anticancer and proapoptotic mechanisms of VPA exposure in a CML model, the gene expression profile of differentially expressed genes related to VPA treatment was used to compare with those of 1,309 compounds in the CMAP database [23, 24]. The similarity of the gene expression profile between the query signature and a CMAP instance were measured by the connectivity score, which ranges from −1 to 1. Only drugs with positive connectivity scores, which induce the expression of the query signature, are listed (Table 3). The results showed that the top ten drug hits belong to HDACi PI3K inhibitor, selective estrogen receptor modulator, antipsychotic or antimycobacterial agent, consistent with VPA's pharmaceutical actions including recently reported action of inhibiting HDAC. Gene expression profile in K562 cells exposed to VPA observed in this study was most similar to that of HDACi and PI3K inhibitor in CMAP database.
Table 3.
Top CMAP hits correlated with gene expression signature induced by VPA.
| Rank | CMAP drug | P | Mean score | Enrichment | Specificity | Percent nonnull | Function |
|---|---|---|---|---|---|---|---|
| 1 | Vorinostat | 0.00000 | 0.918 | 0.974 | 0.0201 | 100 | HDACi |
| 2 | Trichostatin A | 0.00000 | 0.817 | 0.957 | 0.0000 | 96 | HDACi |
| 3 | Wortmannin | 0.00000 | 0.335 | 0.695 | 0.0065 | 77 | PI3K inhibitor |
| 4 | Ly-294002 | 0.00000 | 0.325 | 0.564 | 0.0201 | 65 | PI3K inhibitor |
| 5 | VPA | 0.00000 | 0.370 | 0.516 | 0.0000 | 61 | Anti-epileptic; HDACi; Mood stabilizer |
| 6 | Raloxifene | 0.00002 | 0.302 | 0.789 | 0.0000 | 85 | SERM |
| 7 | Trifluoperazine | 0.00002 | 0.303 | 0.594 | 0.1154 | 68 | Antipsychotic |
| 8 | Scriptaid | 0.00004 | 0.728 | 0.968 | 0.0185 | 100 | HDACi |
| 9 | Rifabutin | 0.00004 | 0.802 | 0.967 | 0.0052 | 100 | Antimycobacterial agent |
| 10 | Thioridazine | 0.00008 | 0.260 | 0.486 | 0.2831 | 65 | Antipsychotic |
The connectivity score, similarity measurement of the gene expression profile between the query signature and that of a CMAP instance, ranges from −1 to 1. Only drugs with positive connectivity scores, which induce the expression of the query signature, are listed. HDACi: histone deacetylase inhibitor. SERM: selective estrogen receptor modulator. VPA: valproic acid.
4. Discussion
VPA is a short-chain, branched fatty acid that has recently been described as a potent HDACi at therapeutic concentrations. Unlike other HDAC inhibitors, which are associated with various toxic side effects, VPA is clinically available. It can be taken orally, can cross the blood-brain barrier, and can be used for extended periods. HDAC inhibition is responsible for the acetylation of histones and nonhistone proteins such as hsp90 [25] and p53 [26]. This event has been associated with the induction of apoptosis in many models of leukaemia [27, 28]. Thus, the use of VPA prior to or concurrently with anticancer drugs may prove to be a beneficial treatment of leukaemia [8, 29]. The K562 cell line, originating from a patient diagnosed with CML in terminal blast crisis, is highly undifferentiated and of the granulocytic series. In this current study, we investigated the apoptosis rate of K562 cells exposed to VPA. The observed apoptosis result was in agreement with our previous reports [20].
Although the molecular mechanisms underlying antileukaemic activity of VPA in CLL [12] and AML [13, 14] models have been reported, genomewide gene expression changes in CML, however, have not been described. CML is a type of myeloproliferative disease associated with a characteristic chromosomal translocation called the Philadelphia chromosome. Conversely, CLL affects B-cell lymphocytes, and AML is characterized by the rapid growth of abnormal white blood cells that accumulate in the bone marrow and interfere with the production of normal blood cells. The observed clinical features and pathology of CML patients, as well as the therapy options, are quite different from those of CLL and AML patients. Consequently, the effect of VPA treatment on CML patients and the underlying molecular mechanisms may be different from effects observed in CLL and AML patients. Based on this information, we investigated the global gene expression profile of K562 cells exposed to VPA using gene expression microarray. Our microarray data demonstrated that VPA exposure resulted in widespread gene expression changes in this cell line. The significant enrichment analysis of GO terms revealed that genes involved in apoptosis were significantly altered by VPA treatment. This alteration was in accordance with the increase in cell apoptosis observed in VPA-pretreated K562 cells using flow cytometry.
To gain a better understanding of the antileukaemic mechanisms of VPA exposure in a CML model, the gene expression profile of differentially expressed genes related to VPA treatment was utilised to compare with those of 1,309 compounds in the CMAP database. CMAP analysis showed that the top ten drug hits included HDACi, PI3K inhibitor and VPA itself. These results were consistent with VPA's pharmaceutical actions including recently reported action of inhibiting HDAC, indicating the reliability of our result of gene expression profile. In this study, VPA ranked in the top five drug hits. This result not only reflects the reliability of our result of gene expression profile, but also indicates that the connectivity map is a reliable tool for exploring molecular mechanism of drug action. The top four drug hits were vorinostat (suberoylanilide hydroxamic acid, ZolinzaTM), trichostatin A, Wortmannin, and LY-294002. Vorinostat and trichostatin A belong to HDACi. HDACi has a multitude of effects on cancer cells. It can induce cell-cycle arrest, promote differentiation, and stimulate tumor cell death [30]. It activates the apoptosis of cancer cells by the death-receptor (extrinsic) pathway and/or the mitochondrial (intrinsic) pathway [31]. The latter is induced by stress stimuli that disrupt the mitochondrial membrane. Recent studies have showed that HDACi treatment generates reactive oxygen species (ROS) and causes DNA damage in AML cell line, AML patient-derived blasts cultured ex vivo [32], and CML cell line [33], suggesting that ROS inducing intrinsic apoptosis might be one of important mechanisms underlying pro-apoptotic effect of HDACi. The intrinsic apoptosis may be dependent or independent on p53 pathway. We found that expressions of many genes involved in p53-dependent apoptotic pathway were up-regulated by VPA, for example, TP53INP1, PERP, and BAX (Table 2). TP53INP1 promotes p53/TP53 phosphorylation in response to double-strand DNA breaks. PERP plays a role as an effector in the TP53-dependent apoptotic pathway. BAX is regulated by p53 gene. It forms the mitochondrial pore leading to mitochondrial depolarization by oligomerization with BAK [34] and confers an essential gateway for the activation of caspases in the intrinsic apoptosis. Moreover, GO analysis showed that the genes involved in release of cytochrome c from mitochondria (GO: 0001836) were significantly enriched (P = 0.00066) (data not shown). It has been reported that VPA produces DNA oxidative damage in liver cells [35, 36], increases ROS formation, and induces apoptosis in postimplantation embryos [37]. Taken together, VPA might induce ROS and activate p53-dependent apoptosis of k562.
The expression of the BCR/ABL oncogene is the major pathogenic event in CML and the abnormal BCR/ABL tyrosine kinase promotes leukaemogenesis by inducing the phosphorylation of multiple downstream protein targets that mediate growth promoting and antiapoptotic signals, including MAPK/ERK and PI3K pathways [38–40]. PI3K is required only for the proliferation of BCR/ABL-dependent leukemic cells but not of normal hematopoietic cells [41, 42]. Wortmannin and LY294002 are the earliest and still widely employed inhibitors of PI3K. Studies have shown they are able to block proliferation of Ph1-positive cells [38, 43, 44] and enhance the antileukemia effect of Bcr-Abl tyrosine-kinase inhibitor [45–47], the initial choice of treatment for most CML patients. Based on CMAP analysis, we speculate that VPA might inhibit PI3K pathway too, and such inhibition might be one important mechanism by which VPA exerts antileukaemic action. We found that one member of the forkhead transcription factor O (FOXO) family, FOXO3, was upregulated by VPA (Table 2). FOXO family plays an important role in stress defence and it is negatively regulated by AKT-dependent phosphorylation [48].
In conclusion, we observed that VPA exposure altered mRNA expression in the K562 cell line. The CMAP analysis suggests that these alterations might be associated with inhibiting effect of VPA on HDAC as well as PI3K pathway. The data obtained in this study might provide clues to elucidate the molecular and therapeutic potential of VPA in leukaemia treatment, and the CMAP, by which we confirmed GO analysis and analyze the possible mechanisms of VPA antileukaemia, is a useful tool for exploring molecular mechanisms of drug actions.
Conflict of Interests
The authors declare no potential conflict of interests.
Authors' Contribution
These authors contributed equally to this work X. Z. Zhang and A. H. Yin.
Acknowledgment
This work was supported by Guangdong Province Science and Technology Fund no. 2009B030803033.
Abbreviations
- CML:
Chronic myelogenous leukaemia
- VPA:
Sodium valproate
- GO:
Gene ontology
- CMAP:
Connectivity Map.
References
- 1.Wang AH, Wei L, Chen L, et al. Synergistic effect of bortezomib and valproic acid treatment on the proliferation and apoptosis of acute myeloid leukemia and myelodysplastic syndrome cells. Annals of Hematology. 2011;90(8):917–931. doi: 10.1007/s00277-011-1175-6. [DOI] [PubMed] [Google Scholar]
- 2.Kircher B, Schumacher P, Petzer A, et al. Anti-leukemic activity of valproic acid and imatinib mesylate on human Ph+ ALL and CML cells in vitro. European Journal of Haematology. 2009;83(1):48–56. doi: 10.1111/j.1600-0609.2009.01242.x. [DOI] [PubMed] [Google Scholar]
- 3.Buchi F, Pastorelli R, Ferrari G, et al. Acetylome and phosphoproteome modifications in imatinib resistant chronic myeloid leukaemia cells treated with valproic acid. Leukemia Research. 2011;35(7):921–931. doi: 10.1016/j.leukres.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 4.Iacomino G, Medici MC, Russo GL. Valproic acid sensitizes K562 erythroleukemia cells to TRAIL/Apo2L-induced apoptosis. Anticancer Research A. 2008;28(2):855–864. [PubMed] [Google Scholar]
- 5.Sung ES, Kim A, Park JS, Chung J, Kwon MH, Kim YS. Histone deacetylase inhibitors synergistically potentiate death receptor 4-mediated apoptotic cell death of human T-cell acute lymphoblastic leukemia cells. Apoptosis. 2010;15(10):1256–1269. doi: 10.1007/s10495-010-0521-9. [DOI] [PubMed] [Google Scholar]
- 6.Lagneaux L, Gillet N, Stamatopoulos B, et al. Valproic acid induces apoptosis in chronic lymphocytic leukemia cells through activation of the death receptor pathway and potentiates TRAIL response. Experimental Hematology. 2007;35(10):1527–1537. doi: 10.1016/j.exphem.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 7.Stieglmaier J, Bremer E, Kellner C, et al. Selective induction of apoptosis in leukemic B-lymphoid cells by a CD19-specific TRAIL fusion protein. Cancer Immunology, Immunotherapy. 2008;57(2):233–246. doi: 10.1007/s00262-007-0370-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raffoux E, Cras A, Recher C, et al. Phase 2 clinical trial of 5-azacitidine, valproic acid, and all-trans retinoic acid in patients with high-risk acute myeloid leukemia or myelodysplastic syndrome. Oncotarget. 2010;1:34–42. doi: 10.18632/oncotarget.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bug G, Ritter M, Wassmann B, et al. Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer. 2005;104(12):2717–2725. doi: 10.1002/cncr.21589. [DOI] [PubMed] [Google Scholar]
- 10.Kuendgen A, Schmid M, Schlenk R, et al. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer. 2006;106(1):112–119. doi: 10.1002/cncr.21552. [DOI] [PubMed] [Google Scholar]
- 11.Pilatrino C, Cilloni D, Messa E, et al. Increase in platelet count in older, poor-risk patients with acute myeloid leukemia or myelodysplastic syndrome treated with valproic acid and all-trans retinoic acid. Cancer. 2005;104(1):101–109. doi: 10.1002/cncr.21132. [DOI] [PubMed] [Google Scholar]
- 12.Stamatopoulos B, Meuleman N, De Bruyn C, et al. Antileukemic activity of valproic acid in chronic lymphocytic leukemia B cells defined by microarray analysis. Leukemia. 2009;23(12):2281–2289. doi: 10.1038/leu.2009.176. [DOI] [PubMed] [Google Scholar]
- 13.Khanim FL, Bradbury CA, Arrazi J, et al. Elevated FOSB-expression; a potential marker of valproate sensitivity in AML. British Journal of Haematology. 2009;144(3):332–341. doi: 10.1111/j.1365-2141.2008.07449.x. [DOI] [PubMed] [Google Scholar]
- 14.Cheng YC, Lin H, Huang MJ, Chow JM, Lin S, Liu HE. Downregulation of c-Myc is critical for valproic acid-induced growth arrest and myeloid differentiation of acute myeloid leukemia. Leukemia Research. 2007;31(10):1403–1411. doi: 10.1016/j.leukres.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 15.Fayad W, Fryknäs M, Brnjic S, Olofsson MH, Larsson R, Linder S. Identification of a novel topoisomerase inhibitor effective in cells overexpressing drug efflux transporters. Plos ONE. 2009;4(10) doi: 10.1371/journal.pone.0007238. Article ID e7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gheeya J, Johansson P, Chen QR, et al. Expression profiling identifies epoxy anthraquinone derivative as a DNA topoisomerase inhibitor. Cancer Letters. 2010;293(1):124–131. doi: 10.1016/j.canlet.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hieronymus H, Lamb J, Ross KN, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10(4):321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Wen Z, Wang Z, Wang S, et al. Discovery of molecular mechanisms of traditional Chinese medicinal formula Si-Wu-Tang using gene expression Microarray and Connectivity Map. Plos ONE. 2011;6(3) doi: 10.1371/journal.pone.0018278. Article ID e18278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slonim DK, Koide K, Johnson KL, et al. Functional genomic analysis of amniotic fluid cell-free mRNA suggests that oxidative stress is significant in Down syndrome fetuses. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(23):9425–9429. doi: 10.1073/pnas.0903909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang XZ, Yin AH, Zhu XY, Ding Q, Wang CH, Chen YX. Using an exon microarray to identify a global profile of gene expression and alternative splicing in K562 cells exposed to sodium valproate. Oncology Reports. 2012;27:1258–1265. doi: 10.3892/or.2011.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patterson TA, Lobenhofer EK, Fulmer-Smentek SB, et al. Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nature Biotechnology. 2006;24(9):1140–1150. doi: 10.1038/nbt1242. [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Lamb J, Crawford ED, Peck D, et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 24.Lamb J. The Connectivity Map: a new tool for biomedical research. Nature Reviews Cancer. 2007;7(1):54–60. doi: 10.1038/nrc2044. [DOI] [PubMed] [Google Scholar]
- 25.Kovacs JJ, Murphy PJM, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Molecular Cell. 2005;18(5):601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 26.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90(4):595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 27.Romanski A, Bacic B, Bug G, et al. Use of a novel histone deacetylase inhibitor to induce apoptosis in cell lines of acute lymphoblastic leukemia. Haematologica. 2004;89(4):419–426. [PubMed] [Google Scholar]
- 28.Morotti A, Cilioni D, Messa F, et al. Valproate enhances imatinib-induced growth arrest and apoptosis in chronic myeloid leukemia cells. Cancer. 2006;106(5):1188–1196. doi: 10.1002/cncr.21725. [DOI] [PubMed] [Google Scholar]
- 29.Fredly H, Stapnes Bjørnsen C, Gjertsen BT, Bruserud Ø. Combination of the histone deacetylase inhibitor valproic acid with oral hydroxyurea or 6-mercaptopurin can be safe and effective in patients with advanced acute myeloid leukaemia—a report of five cases. Hematology. 2010;15(5):338–343. doi: 10.1179/102453310X12647083620967. [DOI] [PubMed] [Google Scholar]
- 30.Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Letters. 2008;269(1):7–17. doi: 10.1016/j.canlet.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 31.Rosato RR, Almenara JA, Dai Y, Grant S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Molecular Cancer Therapeutics. 2003;2(12):1273–1284. [PubMed] [Google Scholar]
- 32.Petruccelli LA, Dupéré-Richer D, Pettersson F, Retrouvey H, Skoulikas S, Miller WH., Jr. Vorinostat induces reactive oxygen species and dna damage in acute myeloid leukemia cells. Plos ONE. 2011;6(6) doi: 10.1371/journal.pone.0020987. Article ID e20987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brodska B, Holoubek A. Generation of reactive oxygen species during apoptosis induced by DNA-damaging agents and/or histone deacetylase inhibitors. Oxidative Medicine and Cellular Longevity. 2011;2011:7 pages. doi: 10.1155/2011/253529. Article ID 253529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26(9):1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tong V, Teng XW, Chang TKH, Abbott FS. Valproic acid II: effects on oxidative stress, mitochondrial membrane potential, and cytotoxicity in glutathione-depleted rat hepatocytes. Toxicological Sciences. 2005;86(2):436–443. doi: 10.1093/toxsci/kfi185. [DOI] [PubMed] [Google Scholar]
- 36.Schulpis KH, Lazaropoulou C, Regoutas S, et al. Valproic acid monotherapy induces DNA oxidative damage. Toxicology. 2006;217(2-3):228–232. doi: 10.1016/j.tox.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 37.Tung EW, Winn LM. Valproic acid increases formation of reactive oxygen species and induces apoptosis in postimplantation embryos: a role for oxidative stress in valproic acid-induced neural tube defects. Molecular Pharmacology. 2011;80:979–987. doi: 10.1124/mol.111.072314. [DOI] [PubMed] [Google Scholar]
- 38.Skorski T, Kanakaraj P, Nieborowska-Skorska M, et al. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood. 1995;86(2):726–736. [PubMed] [Google Scholar]
- 39.Goldman JM, Melo JV. Mechanisms of disease: chronic myeloid leukemia—advances in biology and new approaches to treatment. The New England Journal of Medicine. 2003;349(15):1451–1464. doi: 10.1056/NEJMra020777. [DOI] [PubMed] [Google Scholar]
- 40.Zhu X, Wang L, Zhang B, Li J, Dou X, Zhao RC. TGF-β1-induced PI3K/Akt/NF-κB/MMP9 signalling pathway is activated in Philadelphia chromosome-positive chronic myeloid leukaemia hemangioblasts. Journal of Biochemistry. 2011;149(4):405–414. doi: 10.1093/jb/mvr016. [DOI] [PubMed] [Google Scholar]
- 41.Martelli AM, Nyåkern M, Tabellini G, et al. Phosphoinositide 3-kinase/ Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia. 2006;20(6):911–928. doi: 10.1038/sj.leu.2404245. [DOI] [PubMed] [Google Scholar]
- 42.Giles FJ, Albitar M. Mammalian target of rapamycin as a therapeutic target in leukemia. Current Molecular Medicine. 2005;5(7):653–661. doi: 10.2174/156652405774641034. [DOI] [PubMed] [Google Scholar]
- 43.Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102(3):972–980. doi: 10.1182/blood-2002-11-3429. [DOI] [PubMed] [Google Scholar]
- 44.Calabretta B, Skorski T. BCR/ABL regulation of PI-3 kinase activity. Leukemia and Lymphoma. 1996;23(5-6):473–476. doi: 10.3109/10428199609054856. [DOI] [PubMed] [Google Scholar]
- 45.Klejman A, Rushen L, Morrione A, Slupianek A, Skorski T. Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene. 2002;21(38):5868–5876. doi: 10.1038/sj.onc.1205724. [DOI] [PubMed] [Google Scholar]
- 46.Marley SB, Lewis JL, Schneider H, Rudd CE, Gordon MY. Phosphatidylinositol-3 kinase inhibitors reproduce the selective antiproliferative effects of imatinib on chronic myeloid leukaemia progenitor cells. British Journal of Haematology. 2004;125(4):500–511. doi: 10.1111/j.1365-2141.2004.04933.x. [DOI] [PubMed] [Google Scholar]
- 47.Weisberg E, Banerji L, Wright RD, et al. Potentiation of antileukemic therapies by the dual PI3K PDK-1 inhibitor, BAG956: effects on BCR-ABL and mutant FLT3-expressing cells. Blood. 2008;111(7):3723–3734. doi: 10.1182/blood-2007-09-114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lei H, Quelle FW. FOXO transcription factors enforce cell cycle checkpoints and promote survival of hematopoietic cells after DNA damage. Molecular Cancer Research. 2009;7(8):1294–1303. doi: 10.1158/1541-7786.MCR-08-0531. [DOI] [PubMed] [Google Scholar]